
Photostability of amine-free CsPbBr3 perovskite nanocrystals under continuous UV illumination†
Luiz Gustavo
Bonato‡
 a,
Guilherme
Dal Poggetto‡
b,
Raphael Fernando
Moral
b,
Brener Rodrigo
de Carvalho Vale
a,
José Carlos
Germino
c,
Diogo Burigo
Almeida
d,
Patrícia
Santiago
b,
Pablo Sebastian
Fernandez
b,
Claudio Francisco
Tormena
b,
Lázaro A.
Padilha
*a and
Ana Flávia
Nogueira
*b
a,
Guilherme
Dal Poggetto‡
b,
Raphael Fernando
Moral
b,
Brener Rodrigo
de Carvalho Vale
a,
José Carlos
Germino
c,
Diogo Burigo
Almeida
d,
Patrícia
Santiago
b,
Pablo Sebastian
Fernandez
b,
Claudio Francisco
Tormena
b,
Lázaro A.
Padilha
*a and
Ana Flávia
Nogueira
*b
aInstituto de Física “Gleb Wataghin”, Universidade Estadual de Campinas, Campinas, Brazil. E-mail: padilha@ifi.unicamp.br
bInstituto de Química, Universidade Estadual de Campinas, Campinas, Brazil. E-mail: anafla@unicamp.br
cDepartment of Physics, University of Aveiro, Aveiro, Portugal
dDepartamento de Física, Universidade Federal do ABC, Santo André, Brazil
First published on 6th March 2023
Abstract
Despite all the recent progress on understanding the optoelectronic and structural properties of the CsPbX3 perovskite nanocrystals (PNCs), the impact of the ligands on their photostability, under continuous UV light soaking, is still ambiguous. To address this subject, in this work, we have investigated the influence of the organic ligand on the photophysics of PNCs by comparing the emission behaviour of CsPbBr3 PNCs capped with oleate (OA) and oleylphosphate (OPA). Based on our experimental observations, we propose two different mechanisms to explain their photostability under continuous UV illumination. We hypothesize that the UV illumination promotes a reconstruction of the nanocrystals’ surface, and this process depends on the equilibrium between desorption and adsorption of the surface ligands. In the case of oleate-capped PNCs, this reconstruction is delayed, and even hampered, due to the steric hindered Pb–oleate that desorbs from the surface. In contrast, for the oleylphosphate-caped PNCs, the detached Pb–oleylphosphate can re-adsorb to the surface soon after it leaves because the steric effects are less critical. The results reported here suggest that alkylphosphate ligands can be used to improve the photostability of colloidal PNCs dispersions, an important step towards applications that require continuous excitation, such as back-illuminated LEDs and solar cells.
10th anniversary statementThe Journal of Materials Chemistry C is a leading journal for researchers in the field of metal halide perovskite materials for solar cells, light emitting diodes and other optoelectronic applications. Personally it is an honor to be part of the Journal of Materials Chemistry community, for many years as an author where we had the chance to showcase the science in emerging photovoltaics developed in South America and more recently as a member of the editorial board. |
Introduction
Lead halide perovskite nanocrystals (PNCs) have attracted much attention due to their outstanding optoelectronic properties,1 becoming one of the most studied semiconductor nanostructures of the past decade. These materials are appealing for applications in lighting devices,2–7 X-ray scintillators,8 down-shifting fluorescent panels,9 as well as can be used for fundamental studies in physics and chemistry of luminescent nanomaterials.10–14 Nonetheless, their poor thermal- and photostability are bottlenecks to their practical application in optoelectronic devices.15 Thus, much effort has been made to understand the origin of the poor photostability aiming to produce PNCs with improved stability for future utilization of such materials in technological applications.Different from metal chalcogenide quantum dots,16,17 CsPbX3 perovskite nanocrystals exhibit lower photostability, which also is strongly dependent on their chemical composition,18,19 morphology,20 temperature,21 and excitation energy.22,23 Chen et al.24 have shown that CsPbBr3 PNCs dispersed in toluene undergo a decrease in their photoluminescence (PL) intensity after long-term storage, depending on the storage conditions. Using time-resolved photoluminescence (TRPL) measurements and transmission electron microscopy (TEM) images, the authors assigned the changes in PL dynamics and the formation of larger nanocrystals to the light-induced loss of surface ligands. Thus, long-term light exposure induces surface decomposition, and, consequently, the formation of trap states. Light-induced ligand loss has also been observed by An et al.25 for CsPbI3 PNCs dispersed in hexane under continuous white-light exposure. Using ex-situ Fourier-transform infrared (FTIR) spectroscopy and TEM images, they showed that continuous light illumination induces ligand detachment and changes in the PNC morphology, promoting the formation of surface defect states (undercoordinated Pb atoms) and the deposition of metallic Pb clusters on their surfaces. Under-coordinated Pb atoms and metallic Pb clusters act like charge trap states, changing charge-carrier recombination dynamics and quenching PL emission intensity of the PNCs. Such detaching of ligands induced by light can be related to the dynamic equilibrium of the surface ligands in PNCs passivated by oleylammonium and oleate, as observed by De Roo et al.26 Thus, the so-called amine-free PNCs are prone to exhibit better photostability, since they are credited to present lowered surface ligand dynamics, as first proposed by Yassitepe et al.2 However, the surface ligand dynamics in amine-free PNCs is still not reported, which hamper the utilization of the mechanisms proposed by Chen et al.24 and An et al.25 to explain the observations about their photostability behavior during long light-exposure experiments.
Considering these still-open questions, in this work, we proposed two different mechanisms, based on light-induced ligand loss, to explain the photostability behavior of amine-free CsPbBr3 PNCs passivated by oleate (OA) and oleylphosphate (OPA) ligands. To support the proposed mechanisms, we performed experiments focusing on the dynamics of the interaction between the organic moieties at the surface of the PNCs and the inorganic core and how these interactions are perturbed under photoexcitation. Using Nuclear Magnetic Resonance Spectroscopy (one- and two-dimensional 1H NMR techniques), we demonstrate that the ligands are tightly bounded to the NC surface at dark conditions, as proposed by Yassitepe et al.2 On the other hand, using in situ Fourier-Transform Infrared Spectroscopy (FTIR), Photoluminescence (PL) Spectroscopy, and Time-Correlated Single Photon Counting (TCSPC) measurements, all as a function of UV illumination time, we demonstrate that the surface ligands become dynamic under continuous UV illumination.
The mechanisms proposed in this work, which are based on the geometry of the molecular species that detach from the NC surface, allow better descriptions of the distinct photostability behavior exhibited by OA- and OPA-capped CsPbBr3 PNCs. PL-decay and integrated-PL measurements, both as a function of illumination time, suggest, for oleate-capped PNCs, the formation of hole-trap and electron-trap states, which are dependent on the concentration of the components detached from the NC surface. In contrast, the same experiments performed for oleylphosphate-capped PNCs suggest the redistribution of the hole-trap states across the ensemble surface due to a self-healing phenomenon since the early stages of UV illumination. The results reported here suggest that alkyphosphate ligands can be used to improve the photostability of colloidal PNCs dispersions and can be used to enhance the photostability of PNCs in applications that require continuous excitation, such as back-illuminated LEDs and solar cells.
Experimental
Chemicals
1-Octadecene (ODE, tech. grade 90%, Sigma Aldrich), oleic acid (OA, tech. grade 90%, Sigma Aldrich), oleylphosphoric acid (OPA, mono- and di-ester mixture 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 wt%, TCI), Cs2CO3 (99%, Sigma Aldrich), Pb(acetate)2·3H2O (≥99%, Sigma Aldrich), tetraoctylammonium bromide (TOABr 98%, Sigma Aldrich), methylacetate (MeOAc, anhydrous 99%, Sigma Aldrich), deuterated benzene (C6D6, 99.99%, Sigma Aldrich). All chemicals were purchased and used without further purification procedures.
:40 wt%, TCI), Cs2CO3 (99%, Sigma Aldrich), Pb(acetate)2·3H2O (≥99%, Sigma Aldrich), tetraoctylammonium bromide (TOABr 98%, Sigma Aldrich), methylacetate (MeOAc, anhydrous 99%, Sigma Aldrich), deuterated benzene (C6D6, 99.99%, Sigma Aldrich). All chemicals were purchased and used without further purification procedures.
Preparation of Br-precursor solution
1 mmol of TOABr (550 mg) was loaded into a 25 mL vial, along with 1 mL of ODE and 1 mL of OA. After sealing with a rubber septum, the mixture was kept under reduced pressure for 30 minutes at 100 °C. This step is necessary to remove the water from the stock solution and avoid degradation of the perovskite nanocrystals. The stock solutions were kept under N2 atmosphere and mild temperature (about 50 °C) before use. For the synthesis of OPA-capped NCs, OA was exchanged by OPA, following the same that was described above.Synthesis of OA-capped CsPbBr3 perovskite nanocrystals
80 mg of Cs2CO3 (0.5 mmol of Cs+) and 380 mg of Pb(acetate)2·3H2O (1 mmol of Pb2+) were loaded into a 50 mL 3-neck flask, along with 2 mL of OA and 5 mL of ODE. The mixture was kept under reduced pressure for 1 h at 100 °C. After the complete removal of water and acetic acid from the mixture, the system was kept under nitrogen flowing and cooled down until the chosen reaction temperature (T = 75 °C). Then, the TOABr precursor in OA/ODE stock solution was swiftly injected. The mixture was immediately placed into a water bath until room temperature. The PNCs were purified from the crude solution by adding 7 mL of anhydrous MeOAc, followed by centrifugation at 13500 rpm for 5 minutes, and the solid was re-suspended with anhydrous hexane before further experiments.
Synthesis of OPA-capped CsPbBr3 perovskite nanocrystals
80 mg of Cs2CO3 (0.5 mmol of Cs+) and 380 mg of Pb(acetate)2·3H2O (1 mmol of Pb2+) were loaded into a 50 mL 3-neck flask, along with 2 mL of OPA and 5 mL of ODE. The mixture was kept under reduced pressure for 1 h at 100 °C. After the complete removal of water and acetic acid from the mixture, the system was kept under nitrogen flowing and cooled down until the reaction temperature of 75 °C. Then, the TOABr precursor in OPA/ODE stock solution was swiftly injected. The mixture was immediately placed into a water bath until room temperature. The PNCs were purified from the crude solution by adding 7 mL of anhydrous MeOAc, followed by centrifugation at 13500 rpm for 5 minutes, and the solid was re-suspended with anhydrous hexane. After this, the suspension was centrifuged once again at 13500 rpm for 5 minutes, then the was solid discarded, and the supernatant was stocked for further experiments.
Optical characterizations
Absorption spectroscopy was performed in a Carry 60 UV-Vis Agilent Technologies spectrophotometer, using 10 mm optical path length quartz cuvettes.Photoluminescence (PL) spectroscopy measurements were performed on an Ocean Optics QEPro spectrofluorimeter along with and 365 nm LED as an excitation light source, using 10 mm optical path length quartz cuvettes. For long-time illumination measurements, the samples were placed in a sealed cuvette along with continuous stirring over the entire experiment.
Relative PLQY values were measured using Rhodamine B in methanol as a reference chromophore (PLQYref ∼ 0.70). All PNC samples were measured using hexane as a solvent.
PL dynamics were carried out using time-correlated single photon-counting (TCSPC) collected with a Horiba Nanolog Spectrometer equipped with a photon-counting detector (Horiba PPD850) and TCSPC electronics; The excitation was performed through a femtosecond laser (Spectra-physics, Spirit), which pumps a noncolinear optical parametric amplifier (NOPA) delivering pulses of 120 fs centered at (400 ± 20) nm. The time resolution of the TCSPC system is about 70 ps. For long-time illumination measurements, an LED centered at 365 nm, 2.6 mW cm−2 was collimated and directed to the sample. The fluorescence emerging from the sample was collected by a lens and focalized into a fiber, which is coupled to a spectrometer (Ocean Optics, 2000+). Each spectrum was recorded after every two minutes. The samples were placed in a sealed cuvette along with continuous stirring over the entire experiment. Each PL-decay curve was performed by turning the LED off and collecting the signal after every 10 minutes unblocking the LED. By using this instrumentation, we were able to record both steady-state and PL decay curves without changing the sample position. Fig. S1 in the ESI,† illustrates the experimental setup.
Morphological and structural characterizations
X-ray Diffraction (XRD) data were obtained through a Shimadzu XRD 7000 Diffractometer using Cu-Kα radiation (15414 Å); Both samples were deposited by drop-cast on top of common-glass slides before data acquirement.
High-resolution transmission electron microscopy (HRTEM) images were obtained using a Jeol 2100F; both samples were placed on sample holders composed of copper grids coated with amorphous carbon.
Surface ligands characterizations
In situ Fourier-transform infrared spectroscopy (in situ FTIR) experiments were performed on a Shimadzu Prestige 21 spectrometer with a HgCdTe (MCT) detector. Attenuated total reflectance (ATR) was performed in a Teflon cell adapted to a ZnSe prism in the bottom and with a quartz window as cell cover (for further details, see Fig. S2 in the ESI†). The cover was used to avoid solvent evaporation and allowed to irradiate the solution with a 365 nm LED. The spectra of the perovskite dispersion were collected with a resolution of 4 cm−1 and an accumulation of 256 interferograms. The perovskite dispersion spectra are presented as (R/R0) where R0 is the perovskite dispersion spectra in the dark. The spectra of the capping agent, the capping agent with lead, and perovskite dispersion, all dissolved/dispersed in hexane (“blank spectra”), were performed with a 4 cm−1 resolution and accumulation of 100 interferograms. The spectra are presented as (R/R0) where R0 is the hexane.1H Nuclear Magnetic Resonance (NMR) and Nuclear Overhauser Effect Spectroscopy (NOESY) spectra were recorded in a Bruker Avance III HD 250 MHz spectrometer, using benzene-d6 (C6D6) as a solvent, except for the NOESY for OPA, which was acquired in a 500 MHz; chemical shifts were compared with residual proton signal assigned to benzene (δ = 7.16 ppm). 1H Diffusion NMR experiments (Diffusion Ordered Spectroscopy, DOSY) were recorded using a Bruker Avance III 500 MHz. DOSY data were processed using the General NMR Analysis Toolbox (GNAT).
Results and discussion
We prepared one sample using oleic acid (OA) and one sample using oleylphosphoric acid (OPA), both synthesized at the injection temperature of 75 °C. More details about the experimental procedures can be found in the Experimental section. Following previous works,1,2 bright-green colloidal dispersions that glow under UV excitation were obtained for both PNCs samples (OA- and OPA-capped CsPbBr3 NCs). Spectral and structural data for the OA- and OPA-capped CsPbBr3 PNCs synthesized at 75 °C are presented in Fig. 1.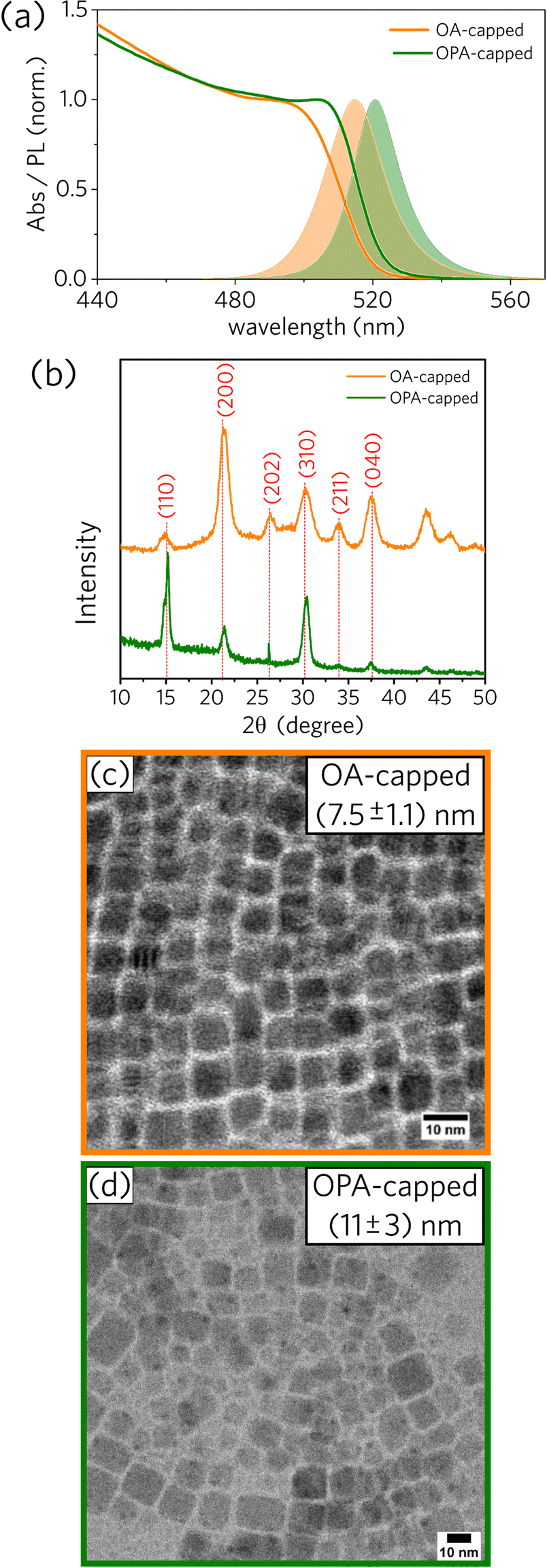 | ||
| Fig. 1 (a) Absorption and PL emission spectra for OA-capped and OPA-capped CsPbBr3 PNCs (OA: oleic acid, OPA: oleylphosphoric acid). (b) Diffractograms for OA-capped and OPA-capped CsPbBr3 PNCs with diffraction patterns corresponding to an orthorhombic. TEM images for (c) OA- and (d) OPA-capped CsPbBr3 PNCs. | ||
As depicted in Fig. 1a, absorbance and PL spectra for OPA-capped PNCs are red-shifted compared to those for OA-capped PNCs. Additionally, XRD diffractograms for both samples, shown in Fig. 1b, are consistent with an orthorhombic lattice,27 with some changes in the relative intensity and width of the main diffraction peaks between them. Both spectral red-shift and XRD divergence can be justified by the difference in size distributions as observed from TEM images (Fig. 1c and d). The mean edge size for OPA-capped PNCs is 11 ± 3 nm, while the mean value for the OA-capped sample is 7.5 ± 1.1 nm. Note that the relative size distribution for the OPA-capped PNCs is about twice as broad as for OA-capped nanoparticles, indicating that further systematic studies on the synthesis of OPA-capped PNCs is necessary to have better control over their size and size distribution. More detailed morphological analysis based on TEM data for OA- and OPA-capped PNCs can be found in the ESI† (Fig. S3 and S4).
The photostability behavior under long-term UV radiation is strikingly different depending on the type of the capping ligand, as depicted in Fig. 2. Both sample solutions were kept in a 1 mm cuvette at constant stirring under UV excitation at 365 nm. The irradiance was set to be 2.6 mW cm−2, and a new PL spectrum was recorded every 2 minutes during 240 minutes of continuous UV illumination (for further details, see Optical characterizations in the Experimental section and Fig. S1 in the ESI†). For OA-capped PNCs, a simple inspection at Fig. 2a and b reveals that the PL intensity decreases, with a subtle narrowing of the full width at half-maximum (FWHM) and no shift of the PL-peak across the illumination time. Besides, one can see important changes in the absorbance spectra, as depicted in Fig. 2c. The red-shift in the onset absorbance, together with the scattering observed at longer wavelengths, could be assigned to the formation of a solid film composed of agglomerated NCs. In contrast, for OPA-capped PNCs, PL intensity increases across the illumination time, as shown in Fig. 2d. Furthermore, the narrowing of the FWHM, along with the red-shift of the PL-peak (Fig. 2e) indicates the passivation of the surface defects.28 Finally, contrary to what was observed for OA-capped PNCs, the absorbance spectra for OPA-capped PNCs, shown in Fig. 2f is nearly unchanged (the absorption peak sifts by less than 1 nm, see ESI†), suggesting the absence of degradation or aggregation during the experiment.
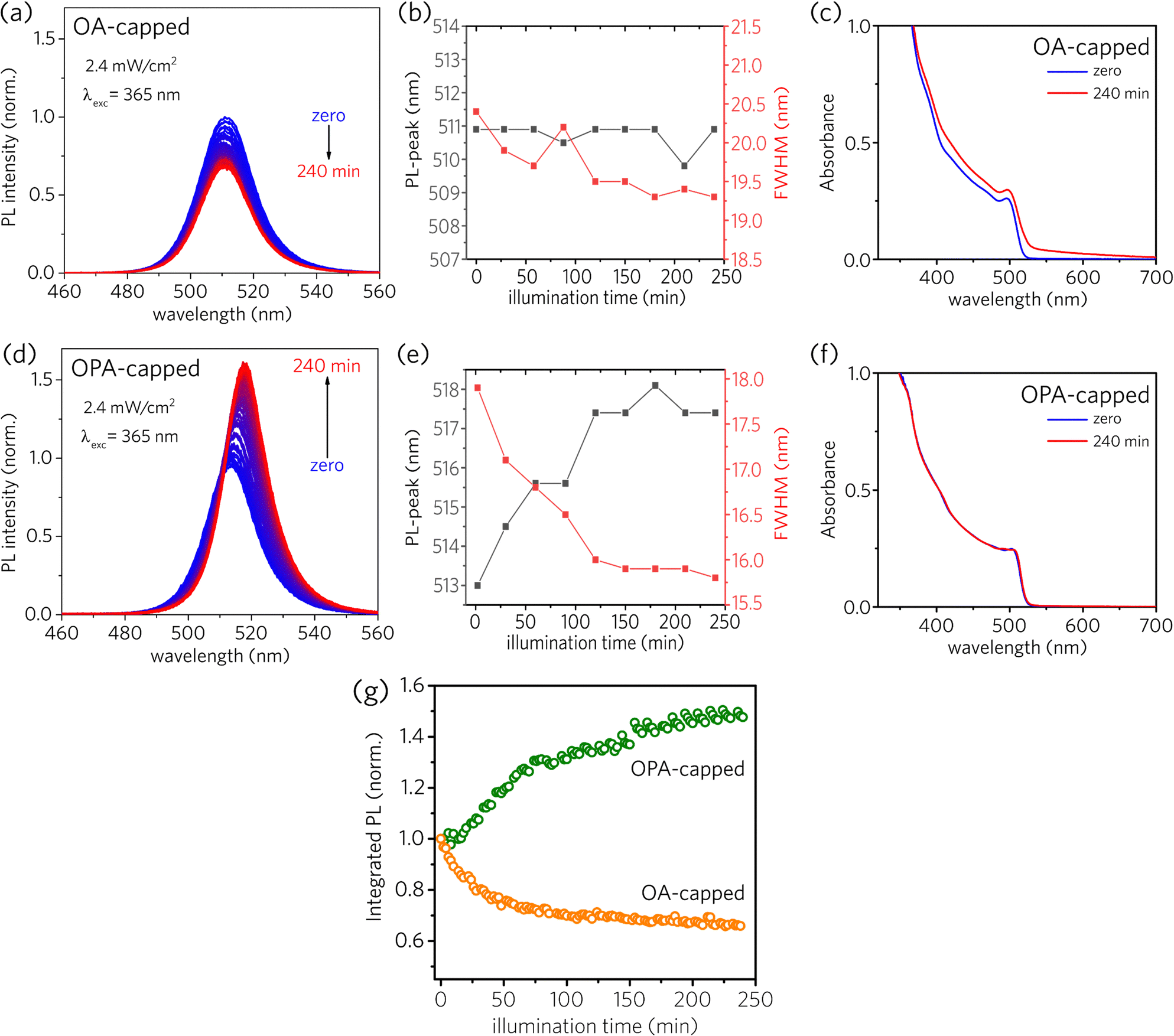 | ||
| Fig. 2 PL emission spectra as a function of illumination time for (a) OA-capped and (d) OPA-capped CsPbBr3 PNCs. Each PL spectra was registered every 2 min at continuous illumination and stirring, using excitation of 365 nm with irradiation of 2.6 mW cm−2. Analysis of FWHM and PL-peak for (b) OA- and (e) OPA-capped PNCs over the data depicted in (a) and (d). Absorbance profiles before and after the light soaking period for (c) OA- and (f) OPA-capped PNCs. (g) Integrated PL as a function of illumination time over the data depicted in (a) and (d). Both plots were normalized at the first PL spectra for each sample. | ||
For better visualization of the photostability behavior presented by PNCs, we plotted the integrated PL as a function of illumination time (Fig. 2g), where the first registered spectrum of each sample was used as the normalization factor for each set of spectra. As, for both samples, the changes in the absorption spectra near the excitation wavelength are small, variations in the integrated PL can be approximately linked to the variations in the photoluminescence quantum yield (PLQY). As a result, in Fig. 2g it is possible to observe two distinct behaviors of the PLQY as a function of the illumination time. For OA-capped PNCs, PLQY monotonically decreases over 240 minutes of continuous illumination, reaching about 66% of the initial PLQY (i.e., PLQYinitial = 0.66, PLQYfinal = 0.44). Thus, such decrease in the PLQY across the illumination time indicates the formation of additional surface defects due to the light-induced ligand loss. Differently, OPA-capped PNCs exhibited a monotonic increase in PLQY throughout the entire illumination time, reaching about 148% of the initial PLQY after 240 minutes of exposure to the UV light (i.e., PLQYinitial = 0.44, PLQYfinal = 0.65). Therefore, opposing what is observed for OA-capped PNCs, the increase in the PLQY and the narrowing in the PL linewidth for OPA-capped PNCs indicate a redistribution of surface defects across the surface of the NCs ensemble induced by the UV light exposure during the analyzed time window, without the formation of additional surface defect states.
Based on the PL evolution as a function of illumination time (Fig. 2), we propose a mechanism for the light-induced loss of surface ligands. Due to the low dielectric constant of hexane, we consider that the species desorbed from the NCs surfaces must be neutral. Consequently, the loss of ligands for OA-capped NCs should occur through the formation of lead oleate species (Pb(OA)2), as illustrated in Fig. 3a, whereas the loss of ligands for OPA-capped NCs should form lead oleylphosphate (Pb(OPA)), as depicted in Fig. 3b. The loss of surface ligands, for both samples, must occur through the formation of surface defects, which directly impacts the charge carrier recombination, and consequently the PLQY. Hence, the nature and the density of the surface traps are important to understand the variations in PL intensity as a function of UV illumination time.
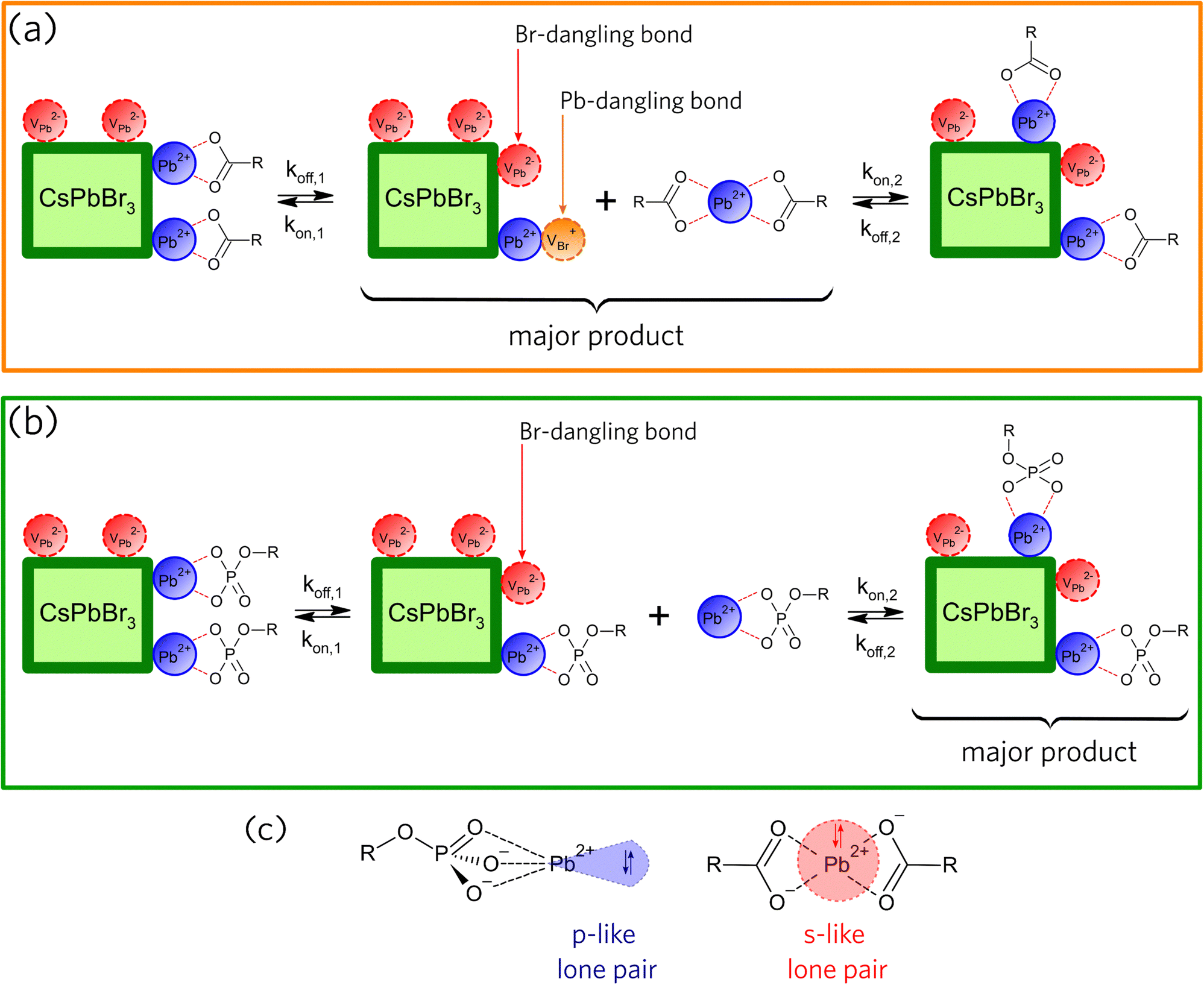 | ||
| Fig. 3 Proposed mechanisms for the light-induced loss of surface components in hexane for (a) OA- and (b) OPA-capped CsPbBr3 PNCs (kon: adsorption rate constant; koff: desorption rate constant). (c) Molecular structures of components lost from the surface during light exposure, showing the hemidirected geometry and the p-like lone-pairs for Pb(OPA), as well as the holodirected geometry and the s-like lone-pairs for Pb(OA)2. | ||
The interaction of defect sites with charge carriers and their relative energy positions depends on the type of point defect formed. According to theoretical calculations performed by ten Brinck et al.,29 considering cubic CsPbBr3 perovskite nanocrystals as archetypal to perform the calculations, only interstitial defects are energetically favorable to form spontaneously, whereas vacancy and anti-site defects are mostly endothermic in nature. Due to energetic reasons and the high surface-to-volume ratio in these nanostructures, it is expected that the trap states-forming defects are located at the surface of the nanocrystals. As a general observation in different theoretical works,29–34 point defects that yield mid-gap trap states have higher formation energies, while point defects that result in shallower traps have low formation energies. For instance, calculations performed by Li et al.33 in α-CsPbI3 have shown that the defects that present the lowest formation energies are VPb (Pb vacancy), CsX (antisite Cs), Pbi (interstitial Pb), PbX (antisite Pb), Csi (interstitial Cs), and VX (halide vacancy), respectively (considering Pb-rich conditions and the Fermi energy located at valence band maximum). Similar trends were observed for the defect energy formations in CsPbBr3, but not for CsPbCl3, as this last one forms primarily deep traps.35
To further discuss the role of the light-induced ligand loss in the formation of surface defects in CsPbBr3 PNCs, we consider in Fig. 3a and b that koff is a constant rate assigned to the desorption of species from the NC surface, whereas kon is a constant rate assigned to the adsorption of species on the NC surface. According to the schemes proposed in Fig. 3a and b, the loss of ligands through the formation of neutral compounds for both OA- and OPA-capped PNCs leads to the formation of two types of point defects. The detachment of Pb(OA)2 generates bromide vacancy (VBr+) and lead vacancy (VPb2−), while the detachment of Pb(OPA) generates only VPb2−. VBr+ point defects, which correspond to the Pb-dangling bond, act like electron traps, as under-coordinated Pb2+cations lead to the formation of shallow trap states near the conduction band minimum (CBM).28,29,36 On the other hand, VPb2− point defects, which corresponds to the Br-dangling bond, act like hole traps. However, the relative position of such defect levels generated by undercoordinated Br− anions is still under debate.28,29,36
In a recent work by du Fossé et al.,34 calculations using density-functional theory depict that the nonbonding Br− orbitals can be pushed into the bandgap depending on the local electrostatic potential. Thus, dangling bonds from surface Br− sites can be destabilized by different local electrostatic potentials, creating deep-trap states. In a nutshell, theoretical models suggest that the way the density of trap-states on the NCs' surface is changed under UV illumination is dependent on the type of the surface ligand. For OA-capped PNCs (Fig. 2g), PLQY decreases with a time constant of about 40 min, which is assigned to an increase in the density of surface trap-states (Fig. 3a, kon,1 ≪ koff,1). The density of trap-states saturates as some previously formed surface defects start being passivated by Pb(OA)2 species (Fig. 3a, kon,2 > koff,2) and the system reaches an equilibrium regime. This trend suggests a concentration-dependent light-induced resurfacing for OA-capped PNCs.
The reason for such concentration-dependent light-induced resurfacing can be explained in terms of the steric barrier presented by the holodirected geometry of Pb(OA)2 species (Fig. 3c).37–39 In Pb(OA)2 species, bonds of the ligand atoms are isotropically distributed around Pb atom, which hinder the effective approach of Pb2+ to Br-dangling bonds (VPb2−) and of oleate to Pb-dangling bonds (VBr+) when the concentration of the lead oleate is low. Hence, high concentration of Pb(OA)2 is requested to overcome the steric barrier and favor the passivation of some fraction of surface defects. As a result, one can see a reduction in the decrease rate of the PLQY due to a resurfacing of the OA-capped PNCs.37 On the other hand, Pb(OPA) ligands, which are desorbed from the surface of the OPA-capped PNCs, presents a molecular geometry called hemidirected (Fig. 3c),37–39 in which bonds of the ligand atoms are directed throughout only a part of the space around Pb2+. Consequently, in those complexes Pb-site is less sterically hindered. Thus, as soon as Pb(OPA) desorbs the NC surface, they can passivate the Br-dangling bonds (VPb2−) without the formation of Pb-dangling bonds (VBr+) (Fig. 3b, kon,1 < koff,1). Also, the red-shift of the PL peak (Fig. 2d) indicates that larger NCs become brighter, suggesting that the ligands desorbed from smaller NCs adsorb on the surface of the largest ones. So, VPb2− defects are homogeneously distributed through the NCs surfaces ensemble, resulting in an increased PLQY during the entire experiment (Fig. 3b, kon,2 > koff,2). Therefore, the OPA-capped CsPbBr3 PNCs exhibit a non-concentration-dependent light-induced resurfacing, which acts like a self-healing effect under the UV illumination.
It is important to point that the hemi- and holodirected geometry presented by Pb(II) complexes have a dependence on the nature of the ligands. According to Shimoni-Livny et al.,37 the formation of holodirected Pb(II) complexes is favored by higher coordination numbers and soft ligands, yielding more covalent metal–ligand bonds and s-character of the Pb(II) lone-pair. Contrarily, the formation of hemidirected Pb(II) complexes is favored by lower coordination numbers and hard ligands, yeilding more ionic metal–ligand bonds and an increase in the p-character of the Pb(II) lone-pair.
To correlate the nature of the ligand-NC interaction with each proposed mechanism we performed various NMR spectroscopy techniques, focusing on the organic moieties of the PNCs (for further details, see Fig. S5–S12 in ESI†). Briefly, diffusion-ordered spectroscopy (DOSY) experiments performed for both OA- and OPA-capped NC dispersions, as well as for neat-OA and neat-OPA (see Fig. S7–S10), showed that the diffusion coefficients, assigned as free- and bound-ligands, cannot be related to an equilibrium involving the adsorption/desorption of the ligands on the surface of the NCs.40 Our findings are analogous to those reported by Bonilla et al.,41 where they observed a large decrease in the diffusion coefficient for thioglycolic acid (TGA) ligands when they are attached to the surface of CdTe quantum dots (QD), in comparison with free TGA molecules (both in deuterated water). Therefore, there is strong evidence that oleate and oleylphosphate are tightly bound to the CsPbBr3 NC surface,26,40,42 but the equilibrium between bound (adsorbed) and unbound (desorbed) forms is too slow in comparison with NMR time-scale (typically seconds).
It is important to notice, however, that the NMR measurements were carried out in the absence of light and, therefore, do not reflect the conditions of the light exposure experiments summarized in Fig. 2. Our findings on surface-ligand dynamics for amine-free CsPbBr3 PNCs are analogous to those observed for oleate ligands tightly bound to the surface of PbSe NCs, as well as trioctylphosphine oxide ligands tightly bound to the surface of InP NCs, both deeply detailed by Moreels et al.43,44 A detailed description of the bimolecular exchange process used to define the dynamics of the ligands on the amine-free PNC surface is presented in the ESI† (see Section 4).
Differently from the dark conditions employed on the 1H NMR measurements, we developed a spectrophotochemical cell and performed in situ FTIR experiments under continuous illumination to access the light-induced surface modification (for further details, see Experimental section and Fig. S2 in the ESI†). Fig. 4a shows the standard spectra for OA, Pb(OA)2, and OA-capped PNC, all of them suspended in hexane. The spectrum of OA-capped PNC shows three distinct bands (1750, 1370, and 1240 cm−1) that are not observed on the spectra of OA and Pb(OA)2. As the uncapped perovskite (macroscopic CsPbBr3 crystals) does not demonstrate a response to infrared irradiation, and these bands are not present in the oleic acid and lead oleate spectra, we concluded that they are related to the organic capping bounded to the perovskite surface.45–47 Subsequently, Fig. 4c shows spectra of OA-capped PNC obtained under irradiation, collected across the experiment time. Two upwards (1370 and 1240 cm−1) and two downward bands (1120 and 1010 cm−1) emerge during the experiment. The upwards bands indicate that the concentration of a given species is reducing in the suspension. As those bands match the ones related to the OA attached to the PNC surface (Fig. 4a), they indicate that the irradiation promotes the detachment of the organic capping from the PNC structure. The downward bands suggest an increase in the concentration of a certain species in the suspension. However, we were not able to clearly identify which species are being formed and generating positive bands. Using literature data to identify those frequencies is less than ideal as most of the textbooks and handbook tables contain the frequency of specific vibrations in solid state (KBr pallet) or for the neat molecule (ATR acquisition); therefore, these standard frequencies may change completely in our system because of the interactions of the molecules with the NC core and solvation with hexane. Nevertheless, the downward band at 1120 cm−1 in Fig. 4c agrees with the weak signal observed in Fig. 4a for OA and Pb(OA)2.
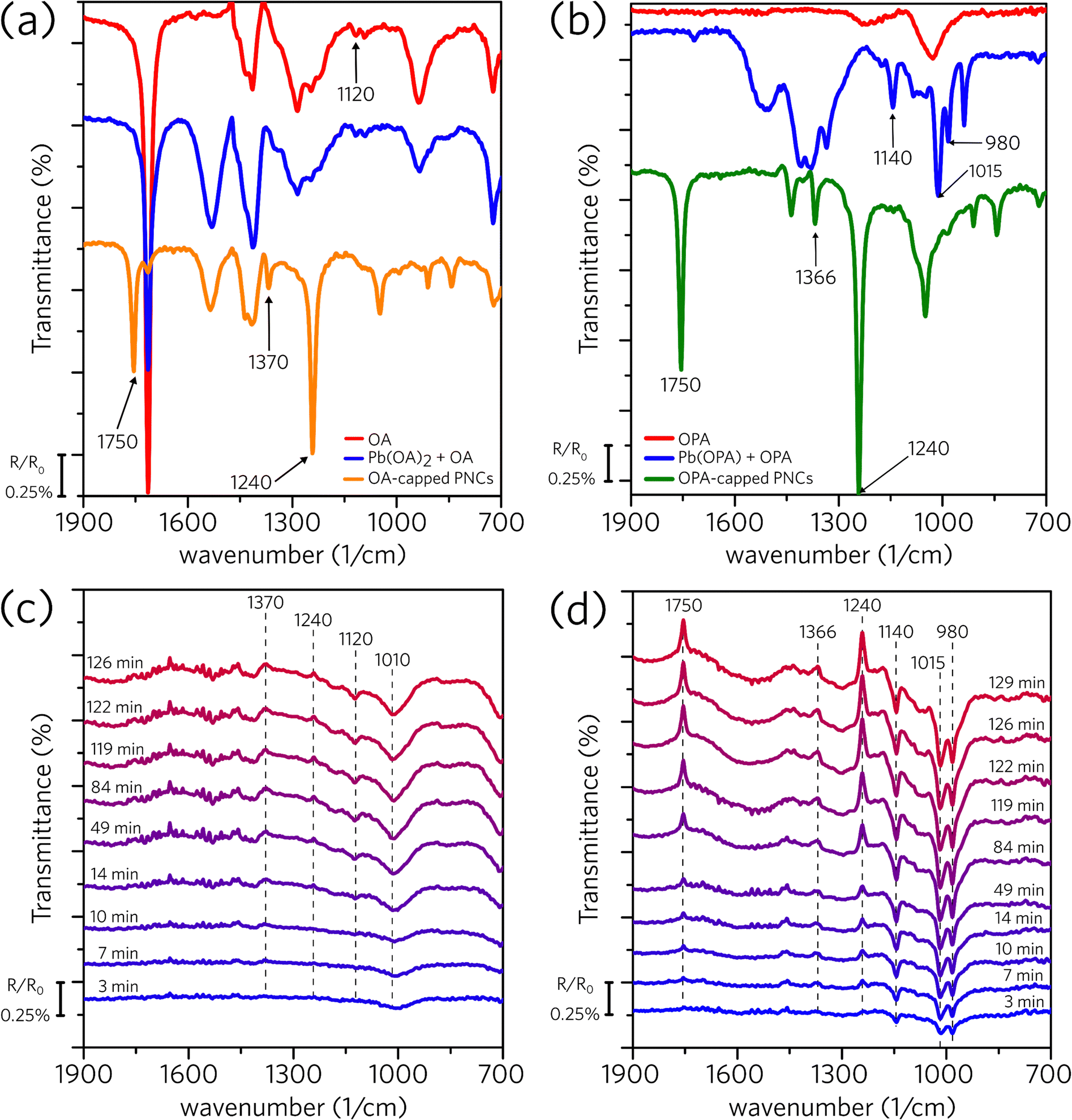 | ||
| Fig. 4 ATR-FTIR spectra of (a) oleic acid, lead oleate, and OA-capped CsPbBr3 PNC dispersion in hexane; (b) oleylphosphoric acid, lead oleylphosphate, and OPA-capped CsPbBr3 PNC dispersion in hexane. In situ ATR-FTIR of (c) OA-capped and (d) OPA-capped perovskite dispersions in hexane. In situ measurements were performed for around 120 minutes under continuous illumination at 365 nm, with an irradiance of 2.6 mW cm−2. R is the spectra obtained at a given time and R0 is the spectra obtained at the beginning of the experiment (time zero). | ||
Similarly, Fig. 4b shows the standard spectra for OPA, Pb(OPA), and OPA-capped PNC, whereas Fig. 4d shows the results obtained under irradiation for OPA-capped PNC dispersion, all of them dispersed in hexane. The upward bands presented in Fig. 4d match those for the capped perovskite (1750, 1366, and 1240 cm−1, Fig. 4b), meaning that the organic capping leaves the nanocrystal.48,49 The positive bands (1140, 1015, and 980 cm−1) match those of the complex lead oleylphosphate, indicating that the oleylphosphate leaves the perovskite surface carrying a Pb2+ ion. In summary, the in situ FTIR experiments indicate that UV irradiation promotes the detachment of the organic ligands from the perovskite surface. While in the OA-capped we were not able to identify which are the formed species, the results with the OPA-capped material clearly show the formation of a complex between the oleylphosphate and Pb2+.
To understand the trap sites formation (passivation) as the ligands are desorbed (adsorbed) to the PNCs surface, we performed time-resolved photoluminescence (TRPL) measurements as a function of illumination time (for further details, see Experimental section and Fig. S1 in the ESI†). According to An et al.,25 recombination of the photo-excited charge carriers in PNCs follows a multi-exponential decay, which considers fast defect-assisted recombination (i.e., hole-trap and electron-trap states), a slower component related to excitonic-state decay, and, for nanoparticles larger than the exciton Bohr radius, an even slower component due to free-carrier recombination. Unbounded free carriers are expected to emit at higher energies as compared to exciton recombination, as they are not subjected to the exciton binding energy, which is above tens of meV for CsPbBr3 PNCs.1 This shift is not observed in the spectrally resolved PL dynamics data as shown in the ESI† (Fig. S13). Consequently, free carrier recombination is neglected here, and all PL decay data were fitted considering a 2-channel decay as described by Equation 1:
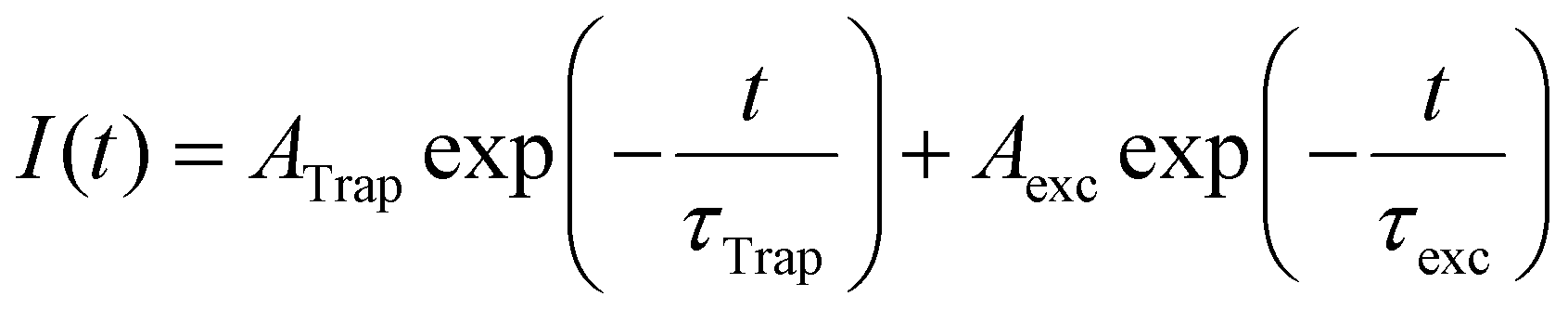 | (1) |
Fig. 5a and b illustrate that the PL dynamics change through the illumination time for both OA- and OPA-capped PNCs samples (the complete set of PL-decay spectra as a function of illumination time for both samples are presented in Fig. S14a and b, in the ESI†). Normalized PL dynamics for OA-capped PNCs exhibit an increase in the short lifetime decay channels, assigned to trap-assisted recombination, as the sample is kept under illumination (Fig. 5a), whereas an opposite trend is exhibited by OPA-capped PNCs (Fig. 5b). Indeed, this is in qualitative agreement with the changes on the integrated PL intensity shown in Fig. 2. To analyze properly the contribution of each recombination channel during the illumination time and obtain a quantitative relationship between the changes on the dynamics and on the PLQY, we fit all the PL decay plots (Fig. S14a and b) using eqn (1). For the OA-capped sample the involved decay lifetimes, τTrap and τexc do not change over the course of the experiment. Consequently, a global fitting considering τTrap and τexc as constants and allowing the free variation of amplitudes, Ai, could be applied. Note that this is in agreement with the results shown in Fig. 2a, which indicates no spectral changes on the PL for the OA-capped samples. The best fit has resulted in τTrap = 5 ± 1 ns and τexc = 13 ± 2 ns in good agreement with previously reported values for similar PNCs.25 Conversely, for OPA-capped PNCs, a global fitting, which considers constant lifetimes, is not possible, as we observe that both τTrap and τexc become longer during the course of UV irradiation. Fig. 5c shows how the decay lifetimes evolve under UV illumination. It shows that τexc doubles within the first ∼120 min of irradiation and then it is nearly constant through the rest of the exposure time. It is interesting to point out that similar trend is observed on the evolution of the PL spectra for the OPA-capped PNCs, as shown in Fig. 2e. In that case, the PL peak red shifts and the linewidth become narrow in the first 120 min of irradiation and no change is observed afterwards. This indicate that the change in PL spectra and PL lifetimes are directly connected phenomena and they suggest that the irradiation favors the passivation of larger nanoparticles, as in those the delocalization of the exciton wavefunction results on slower dynamics. Note, however, that the observed changes in PL dynamics and spectra do not indicate the formation of aggregation as the linear absorption is nearly unchanged upon illumination (Fig. 2f).
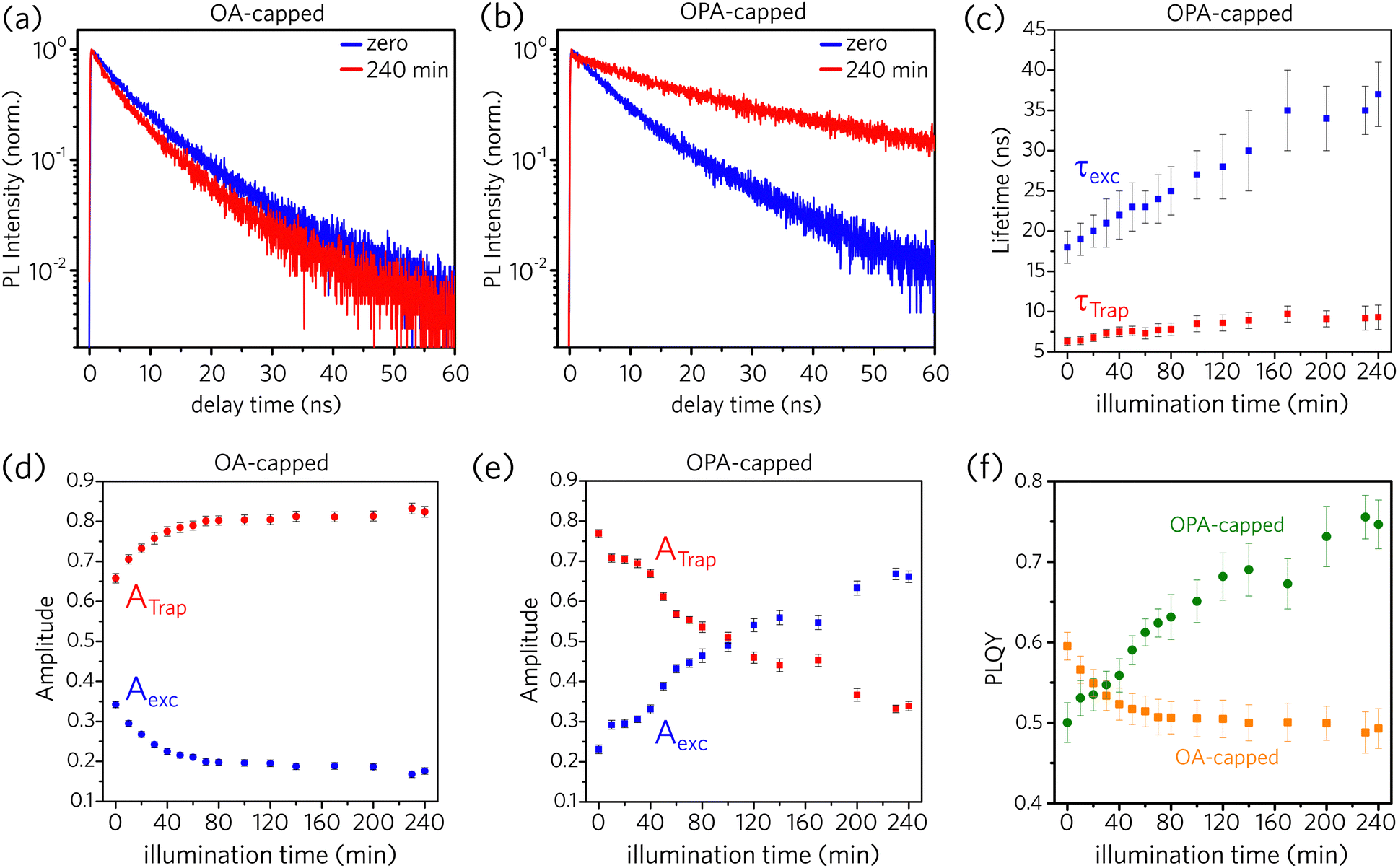 | ||
| Fig. 5 PL dynamics as a function of illumination time for (a) OA-capped and (b) OPA-capped CsPbBr3 PNCs. (c) Calculated lifetimes (τ) from PL decay data for OPA-capped PNCs. Calculated amplitudes (A) from PL decay data for (d) OA-capped and (e) OPA-capped PNCs (Trap: trap-assisted recombination; exc: excitonic recombination). (f) Calculated photoluminescence quantum yield (PLQY) from PL decay data for OA- and OPA-capped PNCs. | ||
According to the data from Fig. 5c, τTrap varies from 6.3 ± 0.5 ns to 9.3 ± 0.7 ns during the UV exposure. It is possible that the redistribution of ligands results on a lower density of defects per nanocrystal, reducing the trapping rate. From the theoretical work by du Fossé et al.,34 trap assisted PL quenching in PNCs is more likely when PbBr2 is stripped from the PNCs surface, leading to the creation of hole-trap states, in agreement with the proposed model in Fig. 3. For CdSe NCs, decreasing in the PLQY has been also attributed to the presence of surface hole traps.50,51
The values obtained for ATrap and Aexc as a function of illumination time, for both OA- and OPA-capped PNCs, are depicted in Fig. 5d and e, respectively. One can note that initially both samples are dominated by PNCs with active trap sites. Upon illumination, the redistribution of the ligands increases the fraction of OA-capped PNCs with active trap sites, which reaches up to 80% of those particles. On the other hand, for OPA-capped PNCs, the fraction of passivated particles increases, and it becomes the dominating species after 100 min of UV irradiation.
Finally, we use the decay data to estimate how the PLQY changes upon the UV exposure time. Following the picture described in Equation 1, the values for PLQY are calculated by:
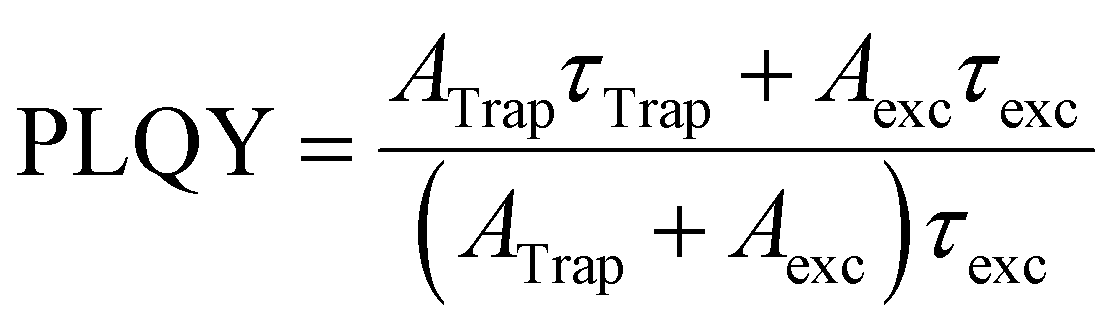 | (2) |
The results are shown in Fig. 5f for both samples. The changes in PLQY are in agreement with the one obtained by the evolution of the integrated PL (Fig. 2g), confirming that the UV-irradiance-assisted ligand redistribution in the OPA-capped PNCs works towards passivating the nanocrystals. Indeed, after 200 min of UV-irradiance, the PLQY reach values as high as 75%, compared to the 50% from the not-irradiated sample.
Although the mechanism proposed for OPA-capped PNCs (Fig. 3) suggests that the total number of hole-trap sites does not vary during the illumination time, the increase of the ensemble PLQY and of τTrap can occur due to passivation of specific defect sites over the PNC surface, reducing the number of active hole-trap sites. According to du Fossé et al.,34 defects formed at the edges and at the corners of the PNCs are more likely to generate deep-trap states than those formed at the middle of the facets. Hence, our data for OPA-capped PNCs suggest that light promotes the passivation of hole traps at the edges and at the corners, which makes the larger NCs brighter even at the ligand loss under UV illumination.
Conclusions
In conclusion, we demonstrated that oleylphosphate-capped (OPA) CsPbBr3 PNCs present improved photostability, under UV radiation, when compared to oleate-capped (OA) ones. We proposed a mechanism to explain this improvement by considering the light-mediated process of ligand attaching and detaching to the PNC surface. While the sterically hindered lead oleate species favor the formation of surface defects that are concentration-dependent, the oleylphosphate species allow the reconstruction of the surface already at early times of illumination. So, our set of data indicates that the organic coverage becomes dynamic under UV illumination, which can directly impact the PLQY. Our findings give insights into the impact of the light-induced trap-states formation in amine-free perovskite nanocrystals, as well as provide strategies for the suppression of such defects toward the increase in the PLQY through the utilization of hemidirected Pb-alkylphosphates as passivating ligands.Author contributions
L. G. B., L. A. P. and A. F. N. designed the experiments, conceived the presented idea and crucial parts of the interpretation. L. G. B. and R. F. M. synthesized the perovskite nanocrystals. R. F. M. carried out the X-ray diffraction measurements and characterization. L. G. B., B. R. C. V., and J. C. G. carried out the absorbance, PL, and TCSPC experiments. D. B. A. was the responsible for the HR-TEM images. The NMR spectroscopy experiments was carried out and analysed by G. D. P. and C. F. T. In Situ FTIR spectroscopy was conducted and analysed by P. S., R. F. M., and P. S. F. All authors provided critical feedback and helped writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the São Paulo Research Foundation (FAPESP), National Council for Scientific Research (CNPq), and the Coordination for the Improvement of Higher Education Personnel (CAPES) for the financial support. L. G. B. (grant no. 2021/13262-0), B. R. C. V. (grant no. 2020/16077-6), and D. B. A (grant no. 2019/22576-8) acknowledge FAPESP for their fellowship. R. F. M. acknowledges FAPESP (process no. 2019/25765-6) for him scholarship. J. C. G. acknowledges FCT/i3N/UA for (UIDB/50025/2020 & UIDP/50025/2020) and for the CEEC-ind grant (2021.02056.CEECIND). G. D. P. and C. F. T. thanks the support from FAPESP (grant no. 2020/10246-0). P. S. F. acknowledges for the support from FAPESP (grant no. 2020/04431-0). L. A. P. thanks FAPESP grant no. 2018/15574-6. A. F. N. gratefully acknowledges support from FAPESP (Grant no. 2017/11986-5) and Shell and the strategic importance of the support given by ANP (Brazil's National Oil, Natural Gas, and Biofuels Agency) through the R&D levy regulation. All authors acknowledge LNNano at CNPEM for the permission to acquire HR-TEM images.References
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- E. Yassitepe, Z. Yang, O. Voznyy, Y. Kim, G. Walters, J. A. Castañeda, P. Kanjanaboos, M. Yuan, X. Gong, F. Fan, J. Pan, S. Hoogland, R. Comin, O. M. Bakr, L. A. Padilha, A. F. Nogueira and E. H. Sargent, Adv. Funct. Mater., 2016, 26, 8757–8763 CrossRef CAS.
- G. Li, F. W. R. Rivarola, N. J. L. K. Davis, S. Bai, T. C. Jellicoe, F. De La Peña, S. Hou, C. Ducati, F. Gao, R. H. Friend, N. C. Greenham and Z. K. Tan, Adv. Mater., 2016, 28, 3528–3534 CrossRef CAS PubMed.
- G. Nagamine, J. O. Rocha, L. G. Bonato, A. F. Nogueira, Z. Zaharieva, A. A. R. Watt, C. H. De Brito Cruz and L. A. Padilha, J. Phys. Chem. Lett., 2018, 9, 3478–3484 CrossRef CAS PubMed.
- C. Y. Huang, C. Zou, C. Mao, K. L. Corp, Y. C. Yao, Y. J. Lee, C. W. Schlenker, A. K. Y. Jen and L. Y. Lin, ACS Photonics, 2017, 4, 2281–2289 CrossRef CAS.
- Y. Xu, Q. Chen, C. Zhang, R. Wang, H. Wu, X. Zhang, G. Xing, W. W. Yu, X. Wang, Y. Zhang and M. Xiao, J. Am. Chem. Soc., 2016, 138, 3761–3768 CrossRef CAS PubMed.
- K. J. Singh, X. Fan, A. S. Sadhu, C.-H. Lin, F.-J. Liou, T. Wu, Y.-J. Lu, J.-H. He, Z. Chen, T. Wu and H.-C. Kuo, Photonics Res., 2021, 9, 2341 CrossRef.
- Y. Zhang, R. Sun, X. Ou, K. Fu, Q. Chen, Y. Ding, L. J. Xu, L. Liu, Y. Han, A. V. Malko, X. Liu, H. Yang, O. M. Bakr, H. Liu and O. F. Mohammed, ACS Nano, 2019, 13, 2520–2525 CrossRef CAS PubMed.
- A. K. Srivastava, W. Zhang, J. Schneider, J. E. Halpert and A. L. Rogach, Adv. Sci., 2019, 6, 1–20 Search PubMed.
- J. A. Castañeda, G. Nagamine, E. Yassitepe, L. G. Bonato, O. Voznyy, S. Hoogland, A. F. Nogueira, E. H. Sargent, C. H. B. Cruz and L. A. Padilha, ACS Nano, 2016, 10, 8603–8609 CrossRef PubMed.
- R. F. Moral, L. G. Bonato, J. C. Germino, W. X. Coelho Oliveira, R. Kamat, J. Xu, C. J. Tassone, S. D. Stranks, M. F. Toney and A. F. Nogueira, Chem. Mater., 2019, 31, 9472–9479 CrossRef CAS.
- R. F. Moral, J. C. Germino, L. G. Bonato, D. B. Almeida, E. M. Therézio, T. D. Z. Atvars, S. D. Stranks, R. A. Nome and A. F. Nogueira, Adv. Opt. Mater., 2020, 8, 1–8 Search PubMed.
- A. Liu, L. G. Bonato, F. Sessa, D. B. Almeida, E. Isele, G. Nagamine, L. F. Zagonel, A. F. Nogueira, L. A. Padilha and S. T. Cundiff, J. Chem. Phys., 2019, 151, 1–6 Search PubMed.
- A. Liu, D. B. Almeida, L. G. Bonato, G. Nagamine, L. F. Zagonel, A. F. Nogueira, L. A. Padilha and S. T. Cundiff, Sci. Adv., 2021, 7, 1–7 Search PubMed.
- B. W. Boote, H. P. Andaraarachchi, B. A. Rosales, R. Blome-Fernández, F. Zhu, M. D. Reichert, K. Santra, J. Li, J. W. Petrich, J. Vela and E. A. Smith, ChemPhysChem, 2019, 20, 2647–2656 CrossRef CAS PubMed.
- G. Nagamine, B. G. Jeong, T. A. C. Ferreira, J. H. Chang, K. Park, D. C. Lee, W. K. Bae and L. A. Padilha, ACS Photonics, 2020, 7, 2252–2264 CrossRef CAS.
- L. Duan, L. Hu, X. Guan, C. H. Lin, D. Chu, S. Huang, X. Liu, J. Yuan and T. Wu, Adv. Energy Mater., 2021, 11, 1–23 Search PubMed.
- H. Huang, M. I. Bodnarchuk, S. V. Kershaw, M. V. Kovalenko and A. L. Rogach, ACS Energy Lett., 2017, 2, 2071–2083 CrossRef CAS PubMed.
- P. Ijaz, M. Imran, M. M. Soares, H. C. N. Tolentino, B. Martín-García, C. Giannini, I. Moreels, L. Manna and R. Krahne, J. Phys. Chem. Lett., 2020, 11, 2079–2085 CrossRef CAS PubMed.
- T. H. Le, S. Lee, H. Jo, G. Jeong, M. Chang and H. Yoon, J. Phys. Chem. Lett., 2021, 12, 5631–5638 CrossRef CAS PubMed.
- B. T. Diroll, G. Nedelcu, M. V. Kovalenko and R. D. Schaller, Adv. Funct. Mater., 2017, 27, 1606750 CrossRef.
- W. Nie, J. C. Blancon, A. J. Neukirch, K. Appavoo, H. Tsai, M. Chhowalla, M. A. Alam, M. Y. Sfeir, C. Katan, J. Even, S. Tretiak, J. J. Crochet, G. Gupta and A. D. Mohite, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- Y. Tian, M. Peter, E. Unger, M. Abdellah, K. Zheng, T. Pullerits, A. Yartsev, V. Sundström and I. G. Scheblykin, Phys. Chem. Chem. Phys., 2015, 17, 24978–24987 RSC.
- J. Chen, D. Liu, M. J. Al-Marri, L. Nuuttila, H. Lehtivuori and K. Zheng, Sci. China: Mater., 2016, 59, 719–727 CrossRef CAS.
- R. An, F. Zhang, X. Zou, Y. Tang, M. Liang, I. Oshchapovskyy, Y. Liu, A. Honarfar, Y. Zhong, C. Li, H. Geng, J. Chen, S. E. Canton, T. Pullerits and K. Zheng, ACS Appl. Mater. Interfaces, 2018, 10, 39222–39227 CrossRef CAS PubMed.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- P. Cottingham and R. L. Brutchey, Chem. Commun., 2016, 52, 5246–5249 RSC.
- Y. J. Yoon, Y. S. Shin, C. B. Park, J. G. Son, J. W. Kim, H. S. Kim, W. Lee, J. Heo, G. H. Kim and J. Y. Kim, Nanoscale, 2020, 12, 21695–21702 RSC.
- S. Ten Brinck, F. Zaccaria and I. Infante, ACS Energy Lett., 2019, 4, 2739–2747 CrossRef CAS.
- W. Yin, T. Shi, Y. Yan, W. Yin, T. Shi and Y. Yan, Appl. Phys. Lett., 2014, 104, 063903 CrossRef.
- J. Kang and L. W. Wang, J. Phys. Chem. Lett., 2017, 8, 489–493 CrossRef CAS PubMed.
- Y. Huang, W. J. Yin and Y. He, J. Phys. Chem. C, 2018, 122, 1345–1350 CrossRef CAS.
- Y. Li, C. Zhang, X. Zhang, D. Huang, Q. Shen, Y. Cheng and W. Huang, Appl. Phys. Lett., 2017, 111, 1–6 Search PubMed.
- I. du Fossé, J. T. Mulder, G. Almeida, A. G. M. Spruit, I. Infante, F. C. Grozema and A. J. Houtepen, J. Am. Chem. Soc., 2022, 144(25), 11059–11063 CrossRef PubMed.
- D. P. Nenon, K. Pressler, J. Kang, B. A. Koscher, J. H. Olshansky, W. T. Osowiecki, M. A. Koc, L. W. Wang and A. P. Alivisatos, J. Am. Chem. Soc., 2018, 140, 17760–17772 CrossRef CAS PubMed.
- R. E. Brandt, J. R. Poindexter, P. Gorai, R. C. Kurchin, R. L. Z. Hoye, L. Nienhaus, M. W. B. Wilson, J. A. Polizzotti, R. Sereika, R. Žaltauskas, L. C. Lee, J. L. Macmanus-Driscoll, M. Bawendi, V. Stevanović and T. Buonassisi, Chem. Mater., 2017, 29, 4667–4674 CrossRef CAS.
- L. Shimoni-Livny, J. P. Glusker and C. W. Bock, Inorg. Chem., 1998, 37, 1853–1867 CrossRef CAS.
- M. Thamim, K. Senthilkumar, N. Job, N. Palanisami and K. Thirumoorthy, Appl. Organomet. Chem., 2020, 34, 12 CrossRef.
- M. Imran, A. Mix, B. Neumann, H. G. Stammler, U. Monkowius, P. Gründlinger and N. W. Mitzel, Dalton Trans., 2014, 44, 924–937 RSC.
- A. Hassinen, I. Moreels, C. De Mello Donegá, J. C. Martins and Z. Hens, J. Phys. Chem. Lett., 2010, 1, 2577–2581 CrossRef CAS.
- C. A. M. Bonilla, M. H. T. Flórez, D. R. Molina Velasco and V. V. Kouznetsov, New J. Chem., 2019, 43, 8452–8458 RSC.
- I. Moreels, Y. Justo, B. De Geyter, K. Haustraete, J. C. Martins and Z. Hens, ACS Nano, 2011, 5, 2004–2012 CrossRef CAS PubMed.
- I. Moreels, B. Fritzinger, J. C. Martins and Z. Hens, J. Am. Chem. Soc., 2008, 130, 15081–15086 CrossRef CAS PubMed.
- Z. Hens, I. Moreels and J. C. Martins, ChemPhysChem, 2005, 6, 2578–2584 CrossRef CAS PubMed.
- Y. Lu and J. D. Miller, J. Colloid Interface Sci., 2002, 256, 41–52 CrossRef CAS.
- L. M. Bronstein, X. Huang, J. Retrum, A. Schmucker, M. Pink, B. D. Stein and B. Dragnea, Chem. Mater., 2007, 19, 3624–3632 CrossRef CAS.
- F. Söderlind, H. Pedersen, R. M. Petoral, P. Käll and K. Uvdal, J. Colloid Interface Sci., 2005, 288, 140–148 CrossRef PubMed.
- Y. Zhang, T. Zhang, G. Lv, G. Zhang, Y. Liu and W. Zhang, Hydrometallurgy, 2016, 166, 87–93 CrossRef CAS.
- A. A. Ahmed, S. Gypser, P. Leinweber, D. Freese and O. Kühn, Phys. Chem. Chem. Phys., 2019, 21, 4421–4434 RSC.
- A. Veamatahau, B. Jiang, T. Seifert, S. Makuta, K. Latham, M. Kanehara, T. Teranishi and Y. Tachibana, Phys. Chem. Chem. Phys., 2015, 17, 2850–2858 RSC.
- D. P. Morgan and D. F. Kelley, J. Phys. Chem. C, 2018, 122, 25661–25667 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc04953h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |