DOI:
10.1039/D3RA06715G
(Review Article)
RSC Adv., 2023,
13, 31047-31058
Dehydroabietane-type bifunctional organocatalysts in asymmetric synthesis: recent progress
Received
3rd October 2023
, Accepted 19th October 2023
First published on 24th October 2023
Abstract
Dehydroabietane-type bifunctional organocatalysts derived from rosane-type diterpenes of dehydroabietic acid (DHAA) and dehydroabietylamine (DA) have been utilized in a wide variety of highly enantioselective reactions. Since one well-documented review exclusively reported on the development of terpene-derived bifunctional thioureas in asymmetric organocatalysis in 2013, fragmentary progress on the dehydroabietane-type bifunctional thioureas and squaramides has been mentioned in other reviews. In this mini-review, we systematically analyze and reorganize the published literature on dehydroabietane-type bifunctional organocatalysts in the recent decade according to the type of catalysts. Our aim is for this review to provide helpful research information and serve as a foundation for further design and application of rosin-based organocatalysts.
1. Introduction
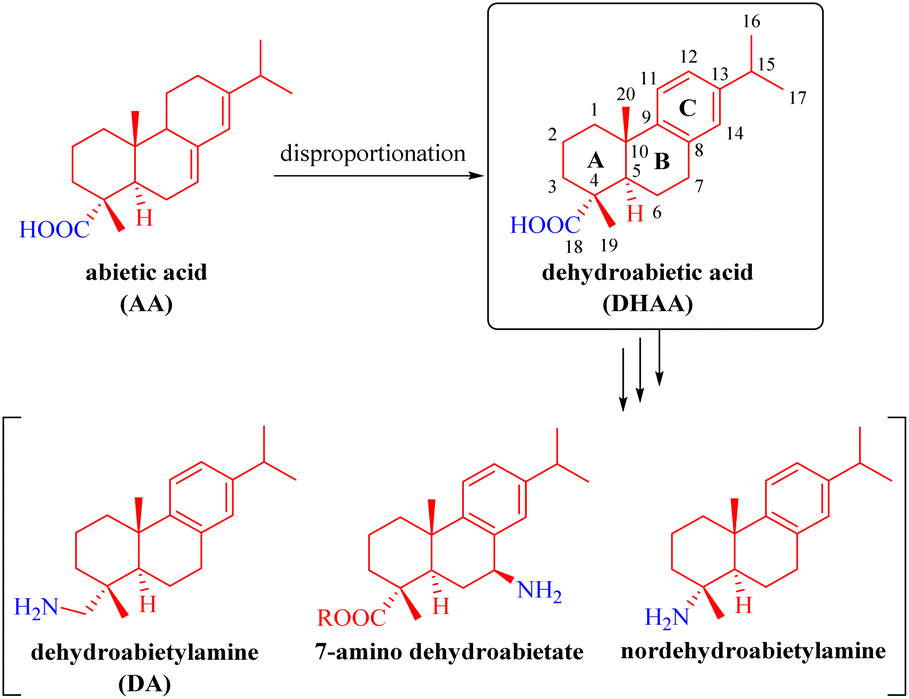
Rosin is an abundant, renewable and biodegradable natural resource.1,2 Rosane-type diterpenes, especially abietic acid (AA), which is the primary component of rosin, along with dehydroabietic acid (DHAA, a well-defined dehydroabietane-type diterpene) and dehydroabietylamine (DA) (Fig. 1), have been extensively utilized as chiral building blocks for the synthesis of numerous natural and synthetic molecules with diverse chemical catalytic or biological activities that find wide applications in pharmaceuticals, medicine, asymmetric synthetic chemistry, and various other fields.3–26 As depicted in Fig. 1, AA is a monocarboxylic acid diterpene possessing a unique chiral tricyclic rigid framework with two conjugated double bonds. It can be chemically converted into DHAA and others through disproportionation. After further modification of DHAA via various functional group transformations, several dehydroabietane-type derivatives such as commercial DA, nordehydroabietylamine,10 and 7-amino dehydroabietate11,12 can be fabricated.
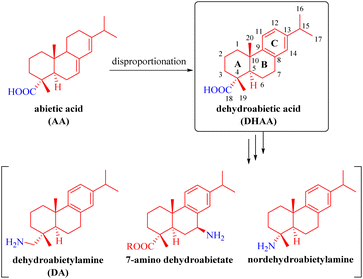 |
| | Fig. 1 Abietic and dehydroabietic acid derivatives. | |
Chiral thioureas and squaramides are representative organocatalysts in asymmetric synthesis that have been developed for enantioselective reactions.11–34 Owing to their specific strong hydrogen-bonding capabilities combined with the notable features of the dehydroabietane scaffold, a series of effective bifunctional organocatalysts based on the dehydroabietane structure have been employed in various catalytic asymmetric reactions so far.11–26
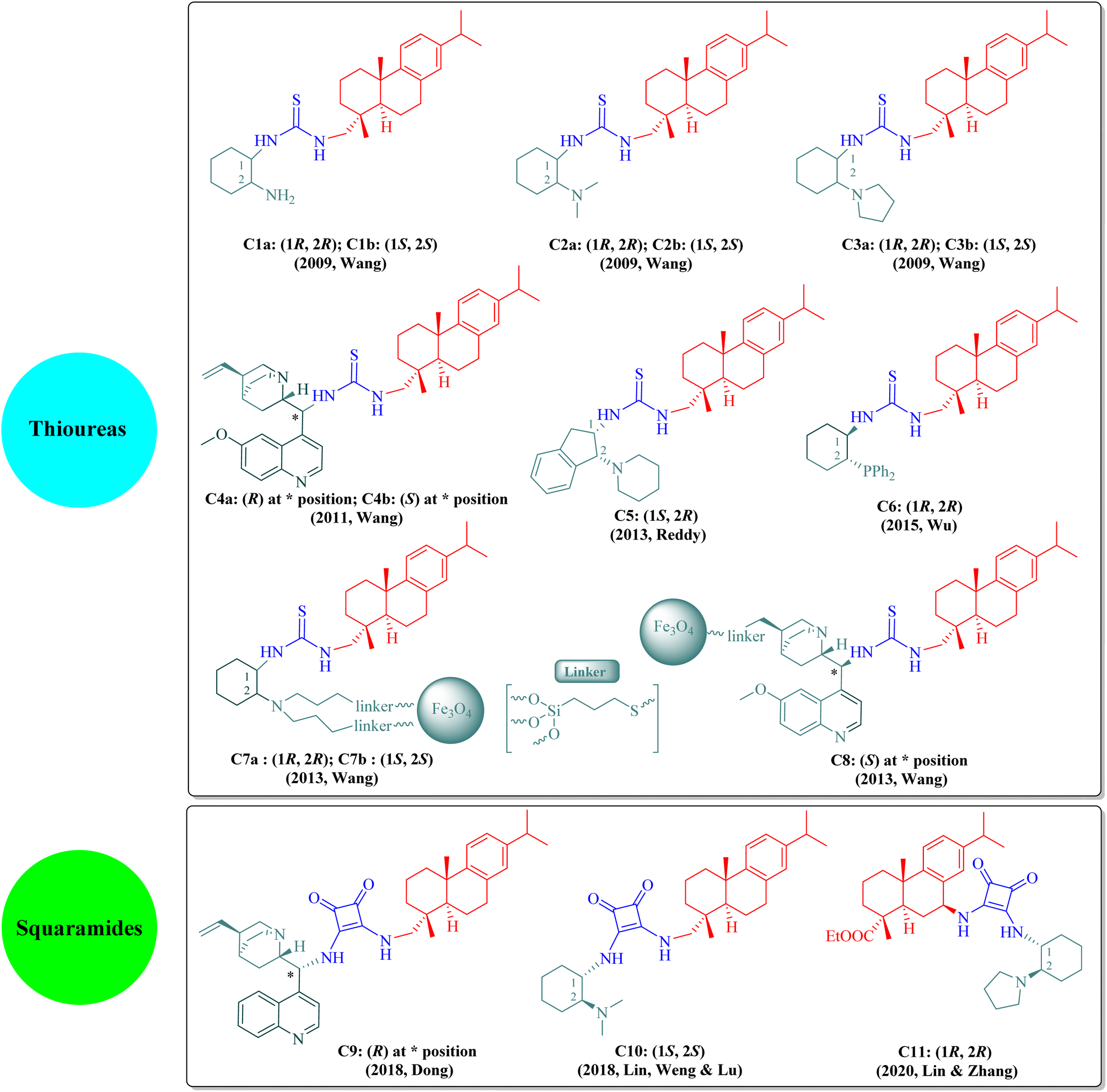
The dehydroabietane-type bifunctional organocatalysts can be broadly classified into two categories according to their hydrogen-bonding donor structures: thioureas (C1a–C8, including immobilized thioureas) and squaramides (C9–C11) as illustrated in Fig. 2.
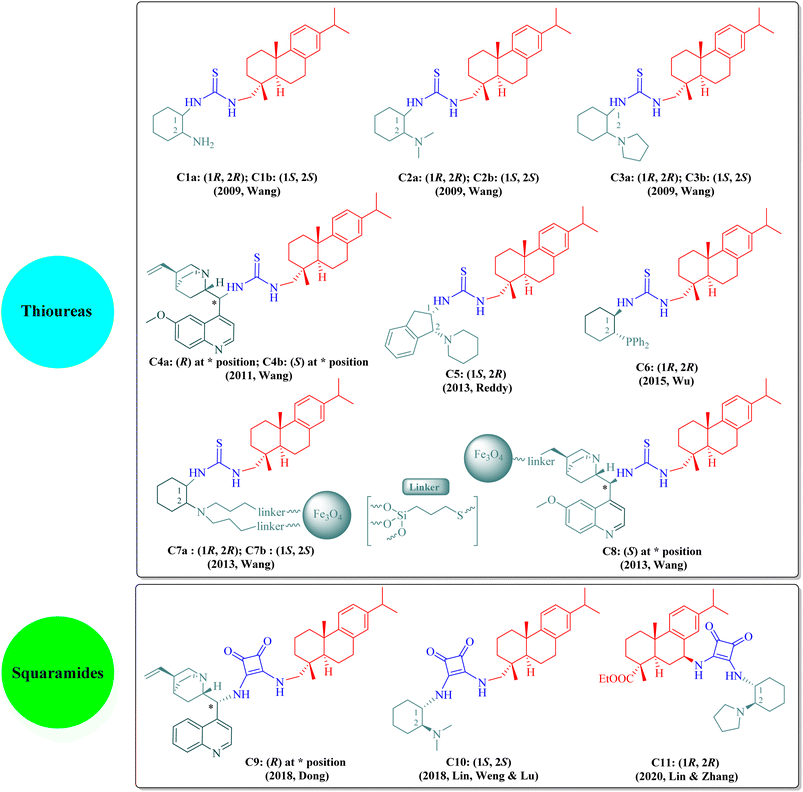 |
| | Fig. 2 Dehydroabietane-type bifunctional organocatalysts described in this review. | |
The literature has extensively investigated the significant achievements of dehydroabietane-type bifunctional thiourea and squaramide organocatalysts in various enantioselective reactions. In 2013, Rivera and Paixão et al. published a well-documented review paper exclusively on the bulky and rigid chiral terpene-based bifunctional thiourea organocatalysts (2009–2012).13 Since then, other review articles have sporadically introduced rosin-derived thiourea or squaramide organocatalysts from separate aspects such as asymmetric inverse-electron-demand Diels–Alder (IEDDA) reactions,14 synthesis of spirooxindoles via organocascade strategies,15 asymmetric cycloaddition reactions,16 enantioselective syntheses of 3,4-dihydropyran derivatives,17 supported hydrogen-bond organocatalysts,18 novel catalysts for asymmetric Michael reactions,19 sustainable organocatalysts from renewable resources,20 1,3-dipolar cycloadditions,21 organocatalytic synthesis of aza-spirocyclic compounds,22 asymmetric organocatalytic reactions for medicinal chemistry,23 enantioselective inverse-electron-demand hetero-Diels–Alder (IEDHDA) reactions,24 stereoselective synthesis and applications of spirocyclic oxindoles,25 and catalytic stereoselective multicomponent reactions.26 As shown in Fig. 2, these dehydroabietane-type organocatalysts generally contain two chiral moieties as the thiourea or squaramide substituents, namely, the dehydroabietane backbone and the other chiral skeleton such as trans-1,2-diaminocyclohexane, natural cinchona alkaloids, cis-1,2-diaminoindane, or trans-1-amino-2-diphenylphosphinocyclohexane, corporately reinforcing high reactivity and stereoselectivity. Wang's group has made outstanding contributions to the development of rosin-derived bifunctional thiourea organocatalysts (C1a–C4b and C7a–C8) for highly enantioselective reactions. Additionally, other research groups have also focused on rosin-based bifunctional thiourea (C5 and C6) and squaramide (C9–C11) organocatalysts employed in different asymmetric reactions. Nevertheless, to the best of our knowledge, all current dehydroabietane-type organocatalysts are derived from commercial DA, except the squaramide catalyst C11 based on 7-amino dehydroabietate. Given our persistent interest in rosin-based organocatalysts and the advancements over the past decade, an updated review is imperative to facilitate the sustained utilization of natural rosin.
Rosin-derived bifunctional primary or tertiary amine thioureas have been identified as effective promoters for specific catalytic asymmetric reactions, including Michael addition reactions of β-nitroalkenes (C1a/C1b,35 C2a/C2b,36 and C4a37), Aldol reactions of α-isothiocyanato imides (C3a/C3b38 and C3b39), Mannich reaction of α-isothiocyanato imides (C4a40), Michael/cyclization reactions involving α-isothiocyanato imides (C3a41 and C3b42) and α,α-dicyanoolefins (C4a43–45), Friedel–Crafts alkylation (C2b/C3a46), and IEDDA reaction of cyclic keto/enolate salts (C1b47) (the optimal catalyst(s) denoted in parentheses for each reaction). This mini-review aims to provide a comprehensive overview of enantioselective reactions catalyzed by dehydroabietane-type bifunctional thioureas and squaramides (2013–2022), including certain reports not covered by the existing review13 as supplementary content. We categorize and reorganize those reactions by different types of organocatalysts.
2. Dehydroabietane-type bifunctional thioureas
2.1. Asymmetric reactions catalyzed by dehydroabietane-type bifunctional thioureas with the trans-1,2-diaminocyclohexane scaffold
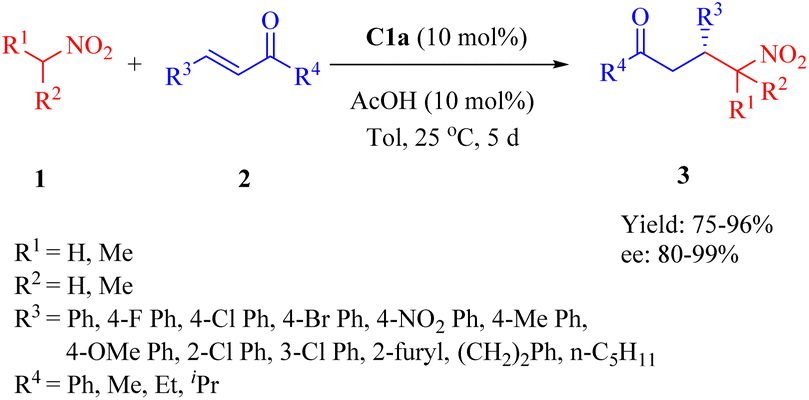
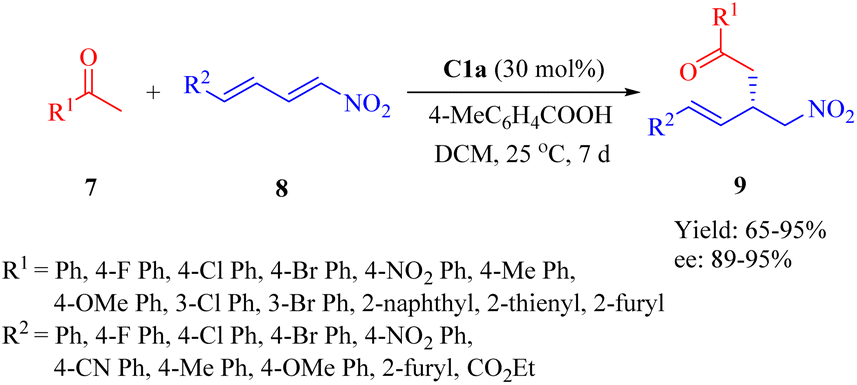
2.1.1. Asymmetric Michael addition reaction. In 2015, Wu et al. reported a highly enantioselective Michael addition of nitroalkanes 1 to α,β-unsaturated ketones 2 facilitated by primary amine thiourea C1a with acetic acid as additive, yielding γ-nitro ketones 3 with up to 99% ee (Scheme 1).48 The diastereomer C1b exhibited relatively lower enantioselectivity compared to C1a, indicating that the DA moiety demonstrated better compatibility with the (1R,2R)-1,2-diaminocyclohexane scaffold. This observation contrasts with those reported by Wang in the Michael reaction involving methyl ketones and nitroolefins.35 Additionally, the DA-derived thiourea containing a racemic 1,2-diaminocyclohexane backbone provided only a 4% ee value, which revealed that the DA moiety had negligible impact on stereocontrol. Notably, sterically hindered nitroalkanes such as nitrocyclopentane and 2-nitropropane did not affect the catalyst's efficiency and provided the corresponding adducts with excellent enantioselectivities.
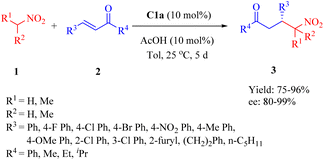 |
| | Scheme 1 Asymmetric Michael addition of nitroalkanes to α,β-unsaturated ketones. | |
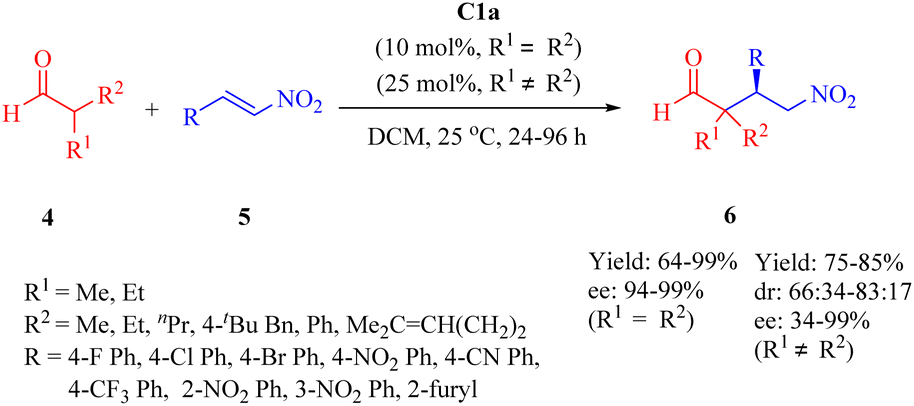
In 2016, Wu's group presented C1a for the asymmetric conjugate addition of α,α-disubstituted aldehydes 4 to β-nitroalkenes 5 to obtain γ-nitro aldehydes 6 (Scheme 2).49 Except 2-phenylpropanal with relatively poor enantioselectivity (34% ee), various α,α-disubstituted aldehydes, including cyclohexanecarbaldehyde and 2-ethylbutanal with a large hindrance, exhibited excellent reactivity with a range of (hetero)aromtic nitroolefins, whereas aliphatic substituted nitroolefins were not tolerated under the optimal reaction conditions.
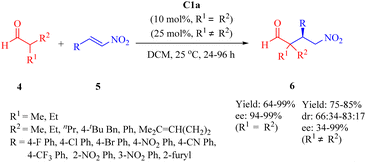 |
| | Scheme 2 Asymmetric Michael addition of α,α-disubstituted aldehydes and β-nitroalkenes. | |
In 2017, Wu et al. demonstrated the use of C1a for the asymmetric Michael addition of (hetero)aromatic ketones 14 to α,β,γ,δ-unsaturated nitro compounds 15 in the presence of 4-methylbenzoic acid as additive to afford the products 16 with high enantioselectivities (Scheme 3).50 The conjugate addition of aliphatic ketones such as acetone was more reactive than acetophenone even at lower catalyst loading (10 mol%), resulting in 85% yield and 87% ee (not shown in the scheme). Furthermore, this method enabled the total synthesis of (R)-3-(aminomethyl)-5-phenylpentanoic acid, which was not achieved previously.
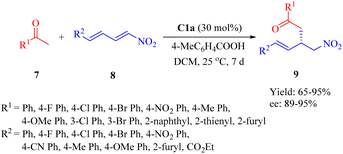 |
| | Scheme 3 Asymmetric Michael addition of (hetero)aromatic ketones to nitrodienes. | |
With respect to the investigated Michael reactions of nitroolefins by Wu's group, the diastereomer C1b efficiently promoted the enantioselective conjugation additions to furnish the corresponding products with opposite absolute configuration, showing that the DA moiety is compatible with both chiral 1,2-diaminocyclohexane with the configuration of (1R,2R) and (1S,2S). These observations were in agreement with Wang's previous report.35 However, in these two reactions, it was found that the DA-derived primary amine thiourea containing a racemic 1,2-diaminocyclohexane skeleton led to the corresponding racemic Michael adducts but with retained yields. For comparison, the Michael reactions catalyzed by other thioureas without a rosin scaffold proceeded with excellent enantioselectivity, but the catalytic activities were lower than DA-derived thioureas. These results showed that the stereoselectivity mainly depended on the chiral trans-1,2-diaminocyclohexane scaffold, and that the chiral dehydroabietane structure did not affect the enantioselectivity but remarkable influence on the reactivity.
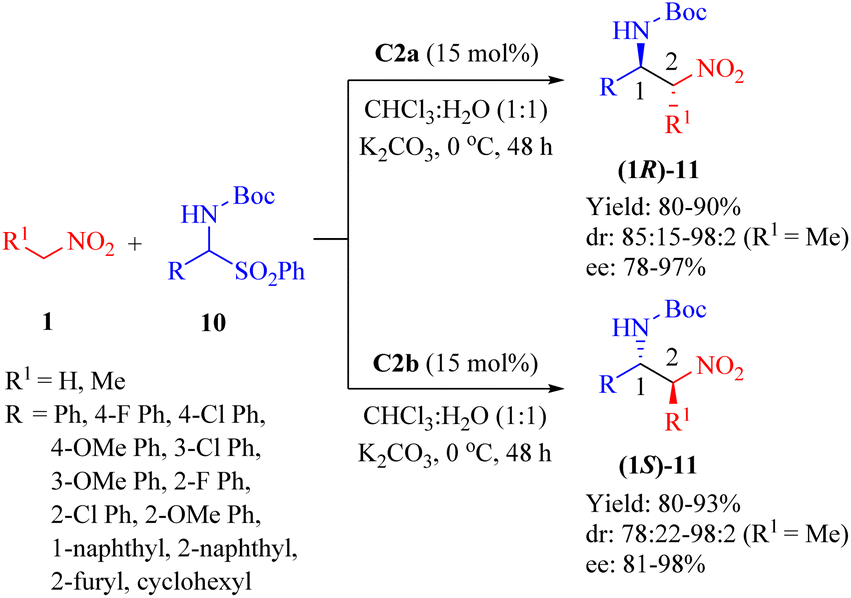
2.1.2. Asymmetric aza-Henry reaction. In 2009, Wang et al. employed the primary amine thioureas C2a and its diastereomer C2b for the first time to investigate the highly enantioselective aza-Henry reaction of nitroalkanes 1 and α-amidosulfones 10 (up to 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 anti/syn, and 98% ee) carried out in a doubly stereocontrolled manner (Scheme 4).51 In this heterogeneous procedure, all reactions proceeded efficiently apart from cyclohexyl α-amido sulfone as substrate (R1 = H, R = cyclohexyl).
:2 anti/syn, and 98% ee) carried out in a doubly stereocontrolled manner (Scheme 4).51 In this heterogeneous procedure, all reactions proceeded efficiently apart from cyclohexyl α-amido sulfone as substrate (R1 = H, R = cyclohexyl).
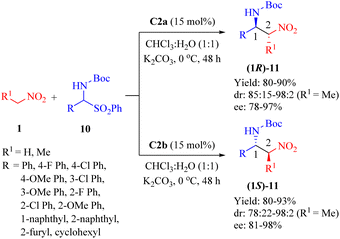 |
| | Scheme 4 Asymmetric aza-Henry reaction of nitroalkanes and α-amidosulfones. | |
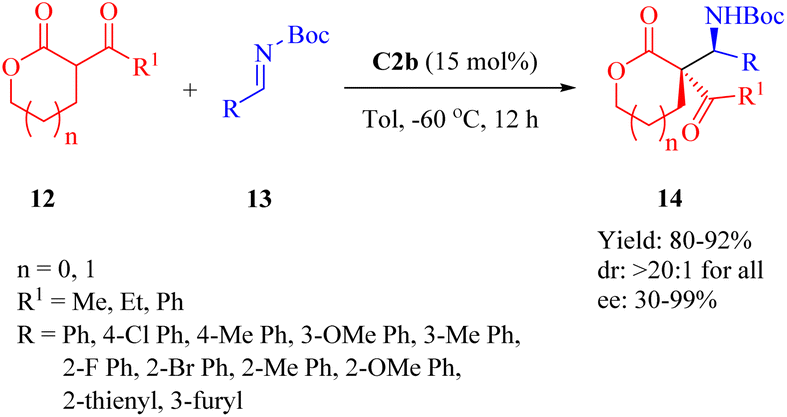
2.1.3. Asymmetric Mannich reaction. Wang's previous report in 2011 demonstrated the quinine-derived thiourea C4b for the efficient asymmetric Mannich/cyclization reaction of α-isothiocyanato imides and N-Ts-aldimines.40As early as 2010, Wang et al. revealed that C2b promoted the asymmetric Mannich reaction between lactones 12 and N-Boc-aldimines 13, leading to the formation of the adducts 14 with excellent stereoselectivities (>20:1 dr for all, and up to 99% ee) containing two adjacent carbon centers, including a quaternary carbon atom (Scheme 5).52 Although C2a provided high yield and excellent diastereoselectivity, it showed an obvious decrease in enantioselectivity for the major diastereomer. C3b gave excellent diastereoselectivity, but relatively lower enantioselectivity. Regarding the substrates, both the size of lactone ring and substituents on aromatic imine had no impact on diastereoseletivity (>20:1 dr for all), but enantioselectivities clearly depended on the lactone ring size and R1 substituent (91% ee for five cyclic lactone, n = 0, R1 = Me; 57% ee for five cyclic lactone, n = 0, R1 = Ph; 30% ee for six cyclic lactone, n = 1, R1 = Ph). Overall, various (hetero)aromatic N-Boc-aldimines along with five cyclic lactone (n = 0, R1 = Me) underwent this reaction to afford the corresponding products with high to excellent enantioselectivities (81–99% ee), although moderate enantioselectivity was observed when using p-chloro phenyl aldimines (75% ee).
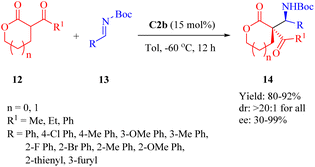 |
| | Scheme 5 Asymmetric Mannich reaction of lactones and N-Boc-aldimines. | |
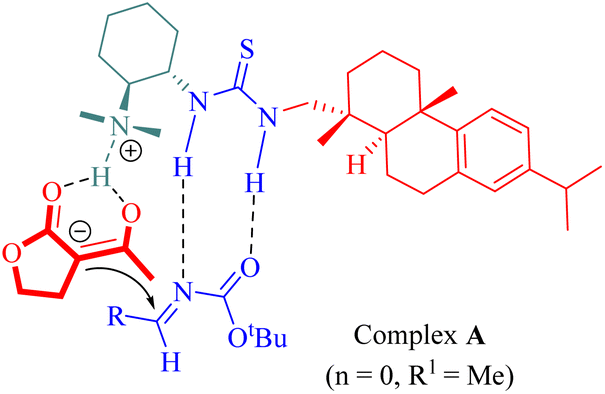
The proposed mechanism involved the generation of ternary complex A through activation by rosin-derived tertiary amine thiourea catalyst simultaneously targeting both lactone (n = 0, R1 = Me) and N-Boc-aldimines as illustrated in Fig. 3.
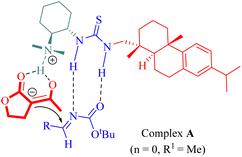 |
| | Fig. 3 Ternary complex A. | |
2.1.4. Asymmetric 1,3-dipolar cycloaddition reaction. In 2013, Wang et al. reported the first 1,3-dipolar cycloaddition reaction of homoserine lactone-derived cyclic imino esters 15 with methyleneindolinones 16 catalyzed by C3a to afford spiro[γ-butyrolactone-pyrrolidin-3,3′-oxindole] tricyclic skeletons 17 (Scheme 6).53 as extensively described in several reviews.15,16,21 Compared to the (1S,2S)-configuration of the diaminocyclohexane skeleton in the diastereomer C3b, the (1R,2R) configuration in C3a exhibited enhanced compatibility with the DA unit for this cycloaddition. In contrast, other DA-derived catalysts such as C2b and C4b displayed diminished levels of enantioselectivity.
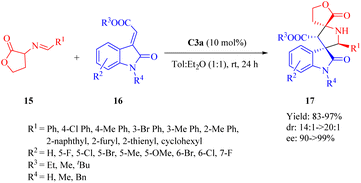 |
| | Scheme 6 Asymmetric 1,3-dipolar cycloaddition reaction of homoserine lactone derived cyclic imino esters and methyleneindolinones. | |
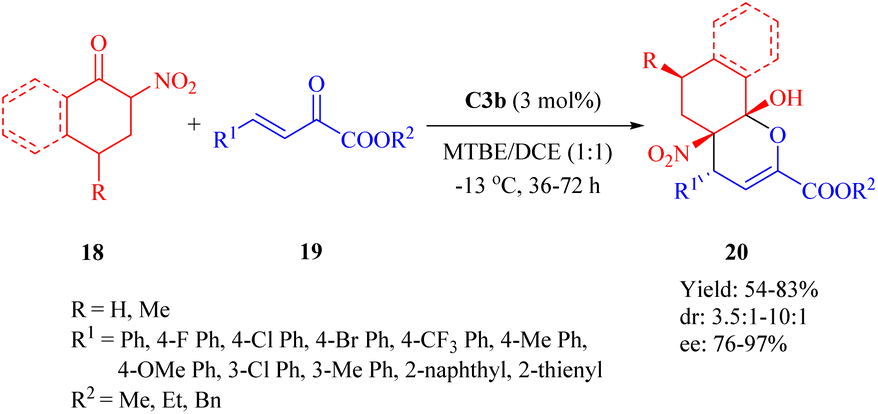
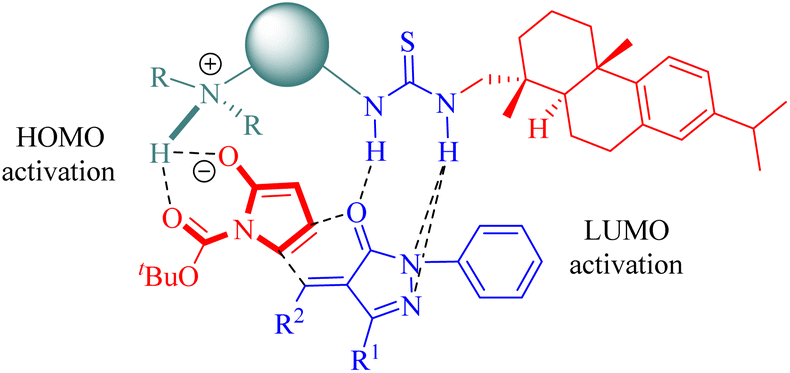
2.1.5. Asymmetric IEDDA and IEDHDA reaction. The catalytic asymmetric IEDDA14 and IEDHDA24 reactions have been explored using both metallic and organocatalytic methods to date, showcasing their immense potential. In 2003, Jøgensen pioneered the first organocatalytic asymmetric IEDHDA reaction employing enamine catalysis to elevate the energy of the HOMOdienophile.54 In 2012, Wang published an elegant asymmetric IEDHDA reaction facilitated by a single catalyst through a hydrogen-bonding dual activation strategy that simultaneously activates both the HOMOdienophile and the LUMOdiene.47In 2012, Wang and colleagues reported another highly enantioselective IEDHDA reaction of 2-nitrocycloketones 18 and β,γ-unsaturated α-ketone esters 19 for the first time to synthesize functionalized dicyclic hemiketals 20 via a hydrogen-bonding dual HOMOdienophile and LUMOdiene-activation pathway (Scheme 7).55 Evaluation of thiourea catalysts (C1b, C2b, C3a, C3b and C4b) revealed that C3b exhibited superior performance. Subsequently, different solvents were screened in combination with C3b to investigate their impact on the reaction outcome. Altering the solvent composition significantly influenced both yield and stereoselectivity, ultimately leading to the identification of a mixed solvent system with methyl tert-butyl ether (MTBE) and 1,2-dichloroethane (DCE) in a ratio of 1:1 as optimal.
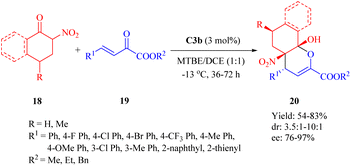 |
| | Scheme 7 Asymmetric IEDDA reaction of 2-nitrocycloketones and β,γ-unsaturated α-ketone esters. | |
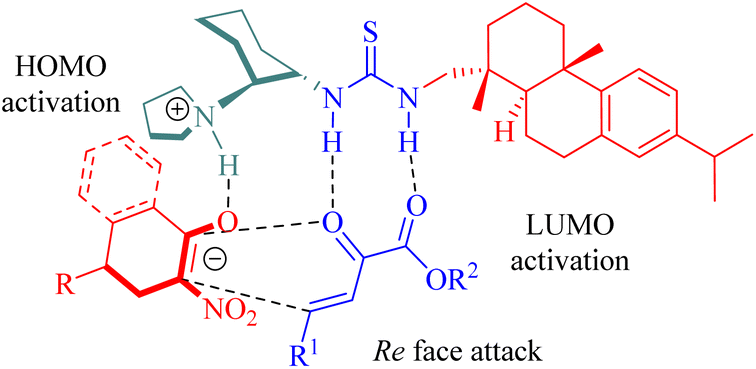
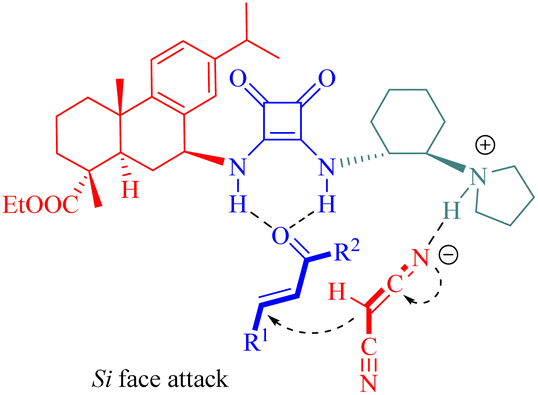
The stereochemistry was elucidated by the proposed transition state as depicted in Fig. 4. The enones were activated by the thiourea moiety, while the chiral tertiary amine moiety influenced the enolization of 2-nitrocycloketone. The endo-selective reaction mode was achieved via the stereochemical control of 1,2-diaminocyclohexane and steric hindrance from the dehydroabietane moiety of the thiourea. Activation of the enone occurred through attack from its Re face, whereas exo-selectivity was suppressed due to shielding effects on the Si face of the enol caused by the (1S,2S)-configuration of diaminocyclohexane and a bulky group.
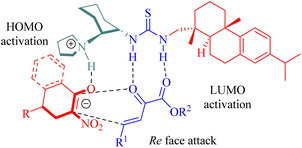 |
| | Fig. 4 A possible transition-state model. | |
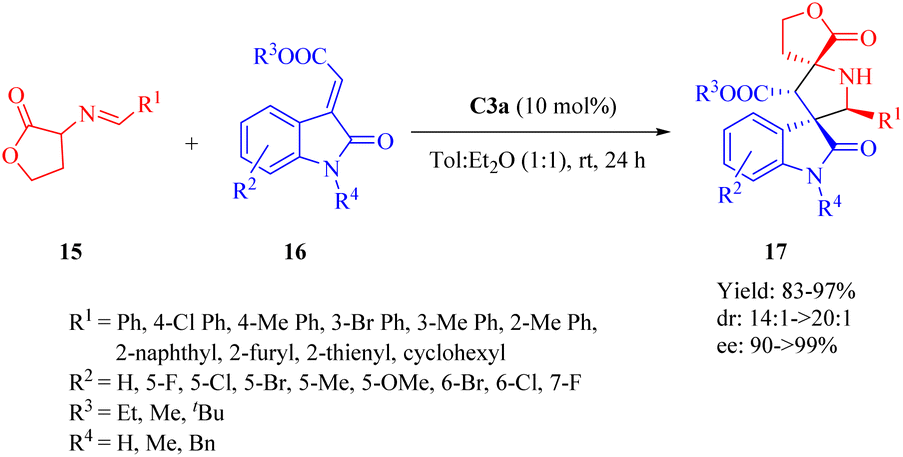
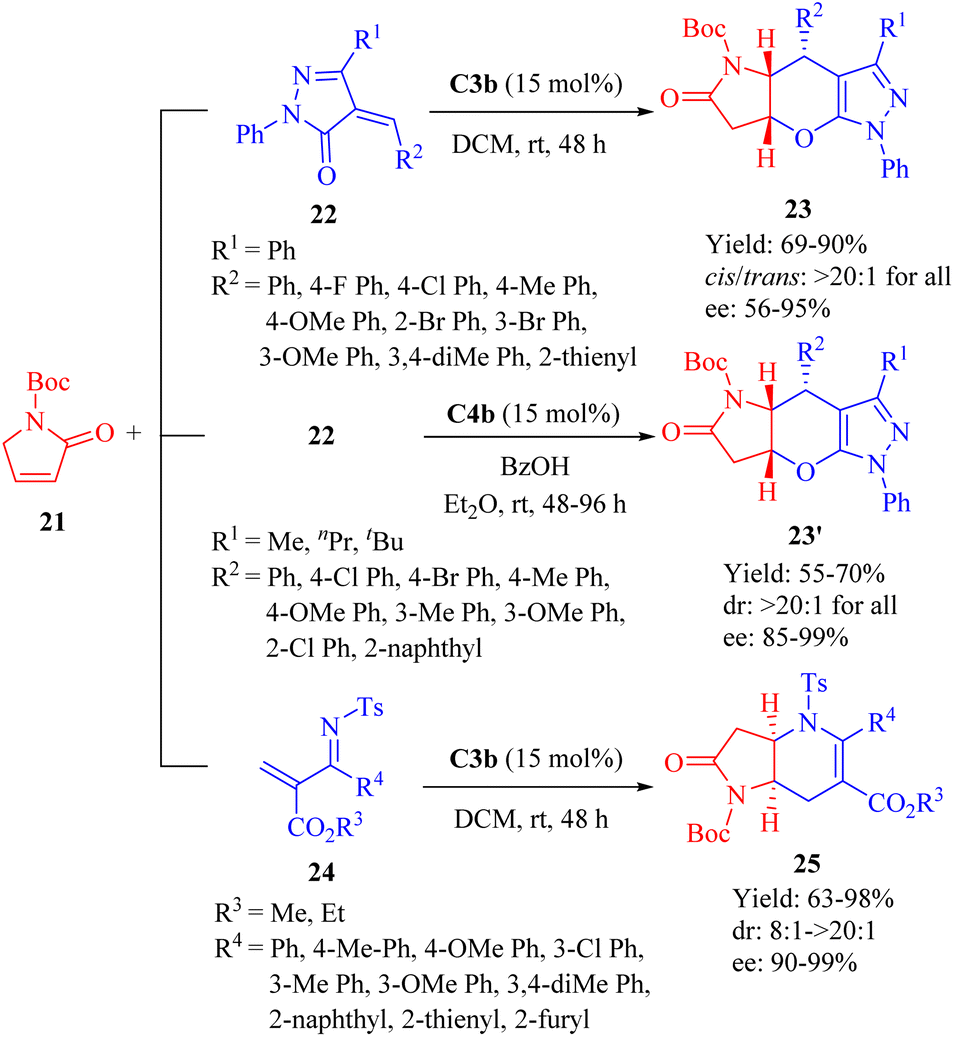
In 2013, Wang et al.disclosed the highly enantioselective IEDHDA [4 + 2] annulation involving α,β-unsaturated γ-butyrolactams 21 to efficiently assemble bridged tricyclic or bicyclic compounds in a single step through direct β,γ-selective activation (Scheme 8).56 C3b effectively facilitated the reaction of 21 with unsaturated pyrazolones 22 (R1 = Ph), affording β,γ-functionalized bridged tricyclic dihydropyranopyrrolidin-2-ones 23 with moderate to excellent enantioselectivities. In comparison to 21, the α,β-unsaturated lactone exhibited negligible reactivity for this annulation (<5% yield). Furthermore, the cinchona thiourea C4b was also found suitable to this [4 + 2] annulation when the R1 substituent varied. The reaction between 21 and 22 (R1 = Me, nPr or tBu) outcome another set of β,γ-functionalized bridged tricyclic dihydropyranopyrrolidin-2-ones 23′ with good to excellent enantioselectivities. In addition, the catalytic system of C3b promoted the annulation of 21 with substituted N-tosyl-2-methylenebut-3-enoates 24 to furnish β,γ-functionalized bridged bicyclic dihydropyranopyrrolidin-2-ones 25 (Scheme 8).
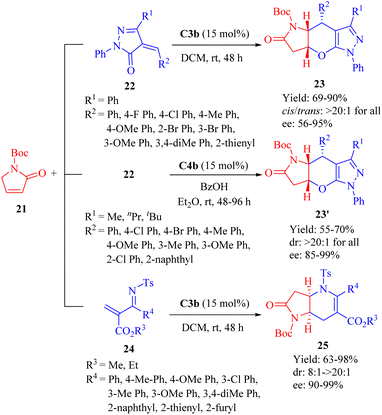 |
| | Scheme 8 Asymmetric IEDDA reaction of α,β-unsaturated γ-butyrolactams. | |
Based on the experimental results, a plausible transition-state model was proposed to explain the stereochemistry of the β,γ-selective IEDDA reaction of α,β-unsaturated γ-butyrolactams (Fig. 5). The activation of both the β and γ positions in α,β-unsaturated γ-butyrolactams was achieved by simultaneous in situ formation of a 1,4-unsaturated enolate via the tertiary amine moiety of the catalyst, leading to an increase in HOMO energy, while the activation of the unsaturated pyrazolone occurred through weak hydrogen bonding with two thiourea hydrogen atoms resulting in a decrease in LUMO energy. This combination facilitated high Re face and endo-β,γ selectivity for obtaining the desired chiral product. These findings are consistent with the previous reports highlighting the primary stereochemical control exerted by the dehydroabietane moiety of the thiourea.35,39–41,46,52
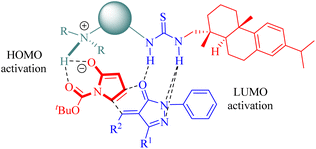 |
| | Fig. 5 A possible transition-state model of the β,γ-selective IEDDA reaction. | |
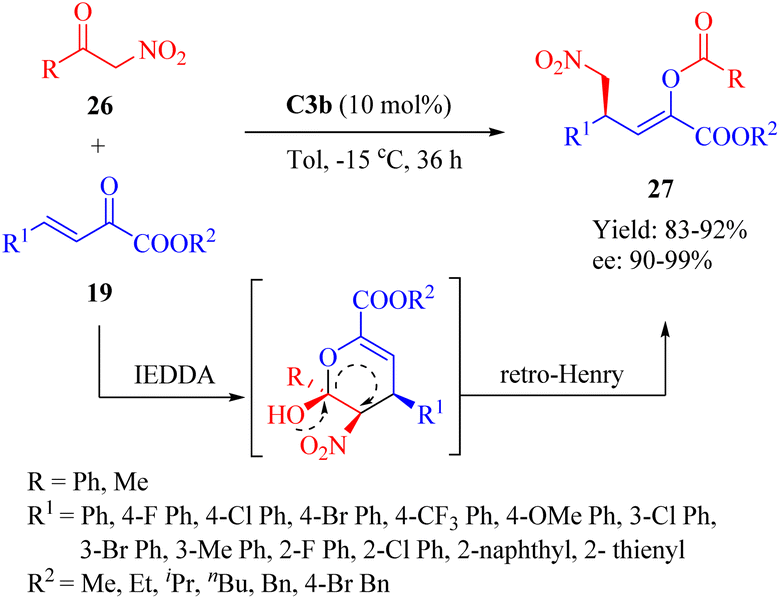
In 2014, Zhang et al. reported the highly enantioselective Diels–Alder/inverse Henry reaction of α-nitroketones 26 and β,γ-unsaturated α-ketone esters 19 (Scheme 9).57 C2a–C3b exhibited comparable yields, while C3b gave the best enantioselectivity. Conversely, C4b led to the least favorable result. The configuration of product 27 was determined by the chiral center in the diaminocyclohexane moiety, and both chiral moieties in C3b contributed to the observed enantioselectivities. A novel mechanism involving an unstable dihydropyran intermediate was proposed for this IEDDA and inverse Henry reaction.17,20
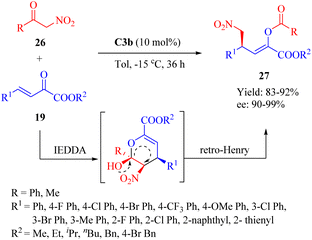 |
| | Scheme 9 Asymmetric IEDDA/Henry inverse reaction of α-nitroketones and β,γ-unsaturated α-ketone esters. | |
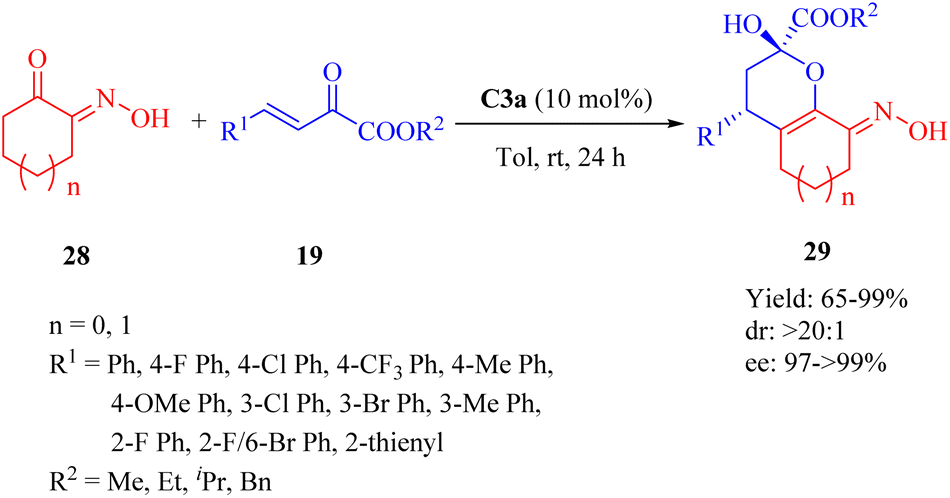
2.1.6. Asymmetric Michael addition/cyclization reaction. In 2013, Wang et al. conducted the catalytic 1,5(6)-selective Michael/cyclization reaction of α-hydroxyimino cyclic ketones 28 with β,γ-unsaturated α-keto esters 19 in the presence of 10 mol% C3a to synthesize ring-fused dihydropynan compounds 29 with moderate to high yields and excellent diastereo- and enantioselectivities (>20:1 dr for all, and up to >99% ee) (Scheme 10).58 C3b afforded the product with the opposite configuration in excellent diastereo- and enantioselectivity but lower yield. C1a, C2a and C4a exhibited excellent diastereoselectivity but lower yield and enantioselectivity. Remarkably, the electronic properties of substituents on β,γ-unsaturated α-ketone esters did not affect the diastereomeric ratio.
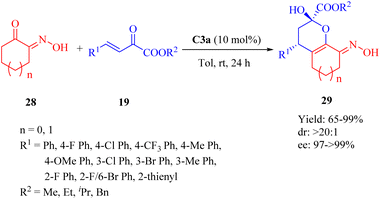 |
| | Scheme 10 Asymmetric Michael/cyclization of α-hydroxyimino cyclic ketones to β,γ-unsaturated α-keto esters. | |
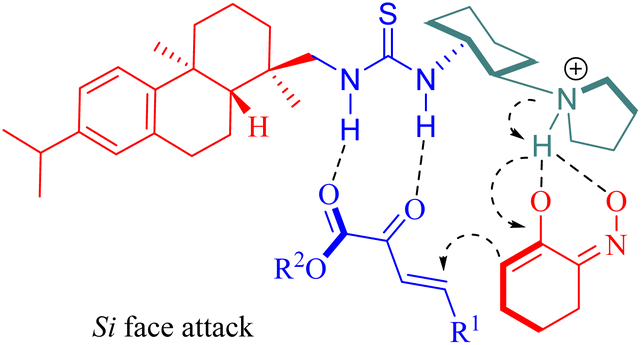
A plausible mechanism was proposed wherein tertiary amine activated α-hydroxyimino cyclic ketones while two hydrogen bonds from the chiral thiourea catalyst activated β,γ-unsaturated α-ketone esters, and subsequently, the nucleophilic attack occurred on the Si face of β,γ-unsaturated α-ketone ester(Fig. 6).
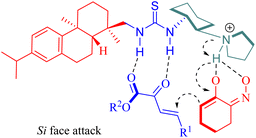 |
| | Fig. 6 A possible transition-state model of the asymmetric Michael/cyclization. | |
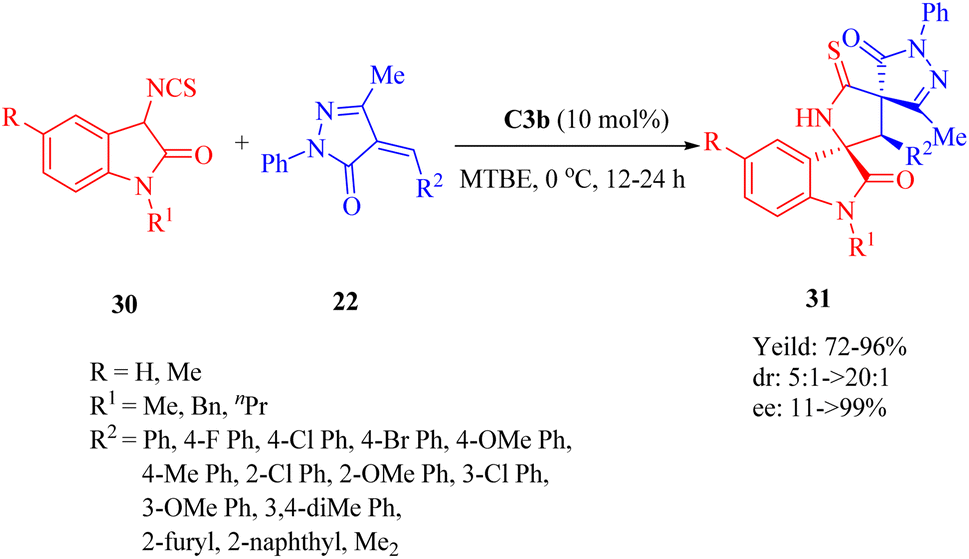
In 2013, Wang et al. discovered the enantioselective Michael-cyclization sequence of 3-isothiocyanato oxindoles 30 and unsaturated pyrazolones 22 utilizing C3b to construct the multicyclic spiro[oxindole/thiobutyrolactam/pyrazolone] core structures 31 with three consecutive stereocenters including two chiral spiro-quaternary carbon atoms (Scheme 11).59 C3a produced the cycloadduct with the opposite configuration in excellent diastereoselectivity, but lower yield and enantioselectivity. C4b exhibited poor results in terms of yield, diastereo- and enantioselectivity. Remarkably, an unsaturated heteroaromatic pyrazolone (R2 = 2-furyl) was effective for this reaction with >20:1 dr and 96% ee. However, poor enantioselectivity (11% ee) was observed for an unsaturated pyrazolone with dimethyl groups (R2 = Me2), in spite of good yield and excellent diastereoselectivity. Encouragingly, there was no obvious impact on both reactivity and enantioselectivity when a relatively large-scale reaction was conducted even under only 0.2 mol% ligand loading.
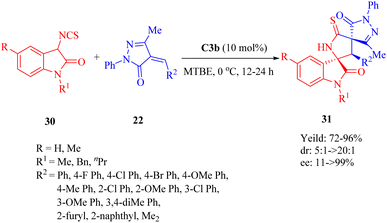 |
| | Scheme 11 Asymmetric Michael/cyclization of 3-isothiocyanato oxindoles to unsaturated pyrazolones. | |
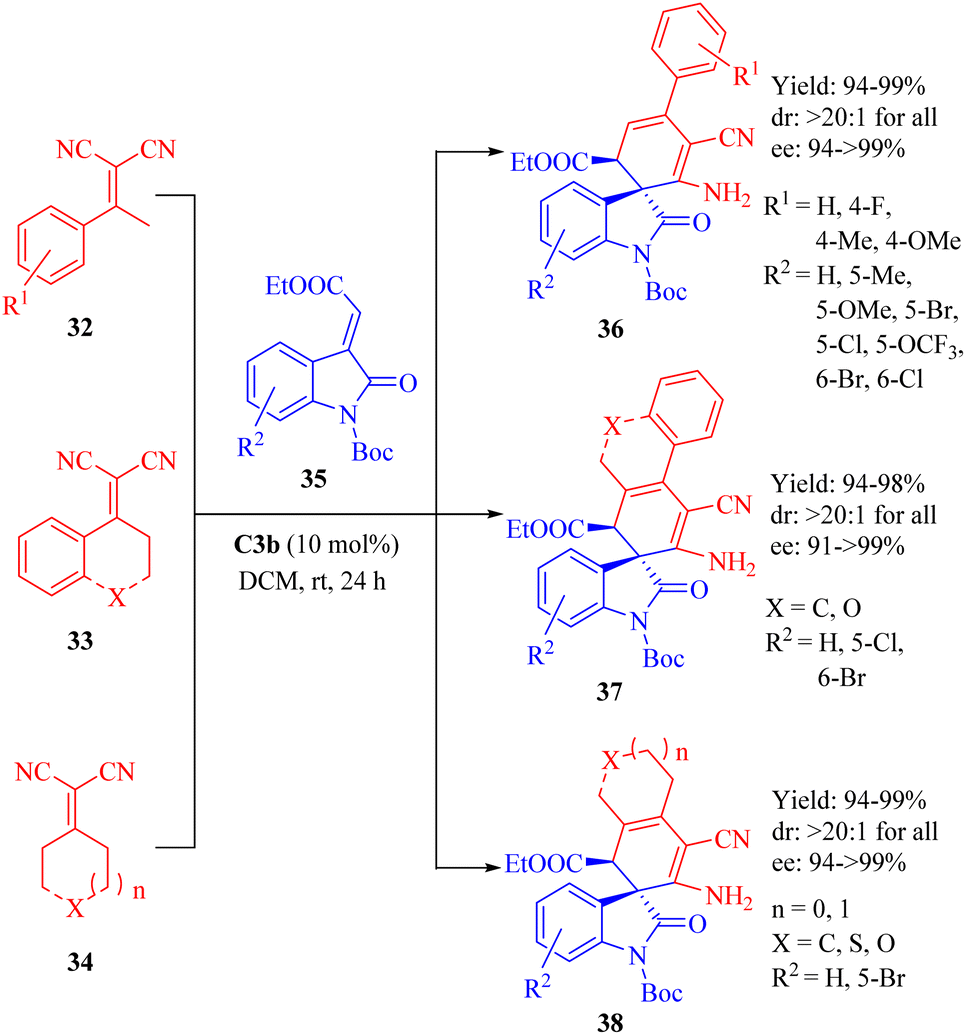
In 2013, Wang et al. reported the C3b-catalyzed asymmetric vinylogous Michael addition/cyclization cascade reaction between ketone-derived electron-deficient α,α-dicyanoalkenes 32–34 and 3-alkylideneoxindoles 35 (Scheme 12).60 This resulted in the formation of spirooxindole derivatives 36–38 with one all-carbon spiro-quaternary carbon atom in excellent stereoselectivities (up to >20:1 dr and 99% ee). In this cyclization, the diastereomer C3a provided the product with the opposite configuration in excellent diastereoselectivity but lower enantioselectivity.
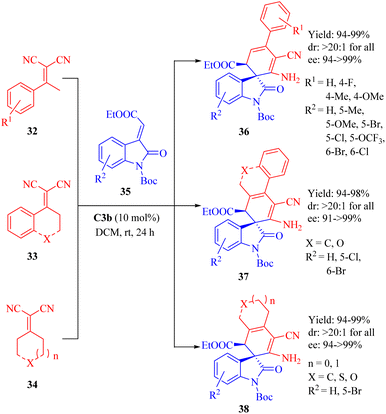 |
| | Scheme 12 Asymmetric Michael/cyclization of α,α-dicyanoalkenes to 3-alkylideneoxindoles. | |
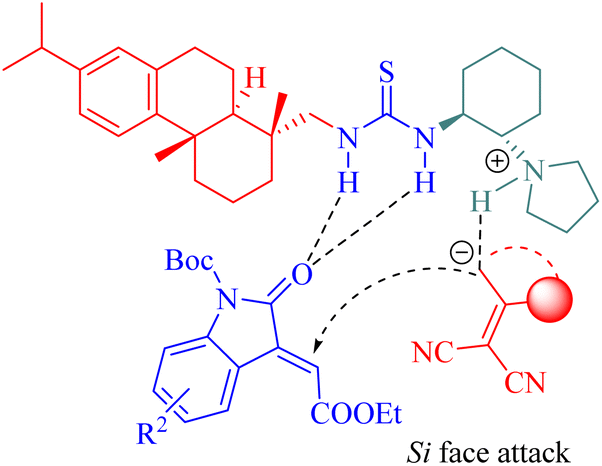
Substrates were activated by tertiary amine thioureas as described in Fig. 7, and the generated vinylogous carbanion subsequently attacked from the Si face of activated 3-alkylideneoxindoles to form the corresponding intermediate leading to the final products through a cyclization-tautomerization sequence.
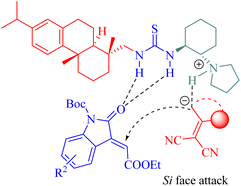 |
| | Fig. 7 A possible transition-state model of the asymmetric Michael/cyclization. | |
2.2. Asymmetric reactions catalyzed by dehydroabietane-type bifunctional thioureas with the cinchona alkaloid scaffold
The dehydroabietane-type cinchona thiourea C4a was found to be suitable for the asymmetric Mannich/cyclization reaction,40 while its diastereomer C4b was employed in the enantioselective Diels–Alder [4 + 2] annulation (see Section 2.1.5).
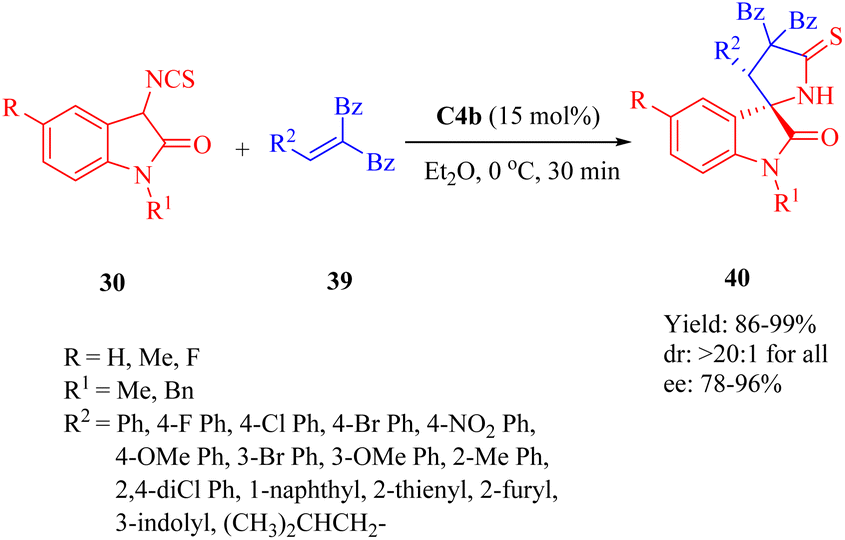
In 2013, Wang et al. presented C4b for the first highly efficient asymmetric Michael addition/cyclization of compound 30 toward electron-deficient olefins 39, leading to the diverse functionalized 3,2′-pyrrolidinyl spirooxindoles 40 with a chiral spiro-quaternary carbon center (Scheme 13).61 This method demonstrated practicality for both aromatic- and aliphatic-substituted alkenes (up to 99% yield, >20:1 dr and 96% ee). Electron-withdrawing and electron-donating groups at different positions on the aromatic ring (R2) of olefins were well-tolerated. Moreover, reactions with the substrates possessing heteroaromatic rings and a larger hindrance 1-naphthyl worked well, except the substrate containing a 2-furyl group which gave only a moderate enantioselectivity (78% ee).
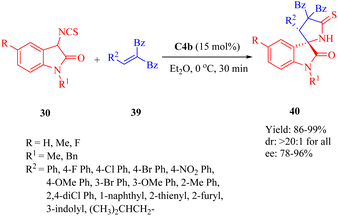 |
| | Scheme 13 Asymmetric Michael/cyclization of 3-isothiocyanato oxindoles to electron-deficient olefins. | |
2.3. Asymmetric reactions catalyzed by dehydroabietane-type bifunctional thioureas with the cis-1,2-diaminoindane scaffold
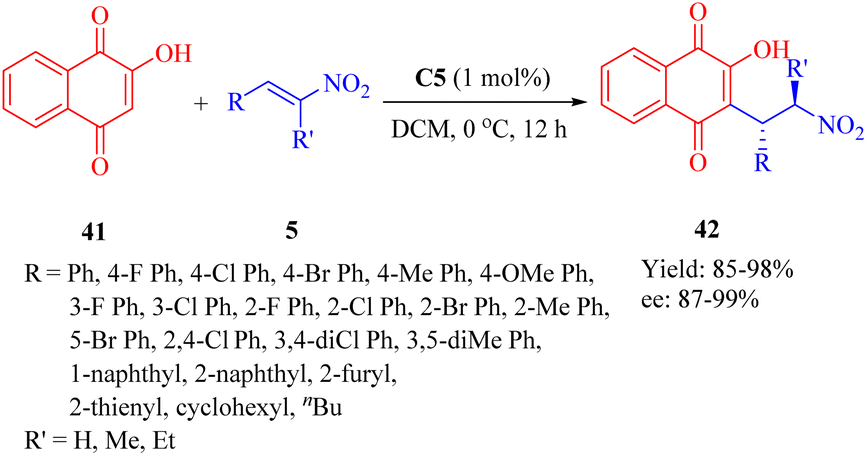
In 2013, Reddy et al. developed a series of bifunctional rosin- and sugar-indane tertiary amine thiourea catalysts as well as indane-based squaramides, inspired by the success of natural rosin- and sugar-derived thiourea organocatalysts.62 Initially, the asymmetric Michael addition of 2-hydroxy-1,4-naphthoquinone 41 to β-nitroalkenes 5 was conducted using indane-based tertiary amine thioureas or squaramides. Remarkably, the DA-indane thiourea C5 exhibited high efficiency in this conjugate addition even at low catalyst loading (1 mol%), resulting in excellent enantioselectivities (up to 99% ee, R = nBu, and R′ = H) (Scheme 14). Furthermore, this same catalyst demonstrated positive effects on the reactions between compound 41 and α-branched β-nitrostyrenes (R = Ph, and R′ = Me, Et), leading to admirable diastereoselectivities (only one single diastereomer detected for each) and remarkable enantioselectivities (95% and 98% ee values, respectively).
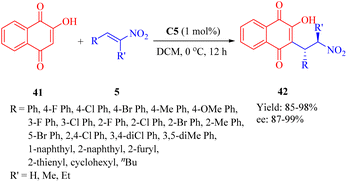 |
| | Scheme 14 Asymmetric Michael addition of 2-hydroxy-1,4-naphthoquinone to β-nitroalkenes. | |
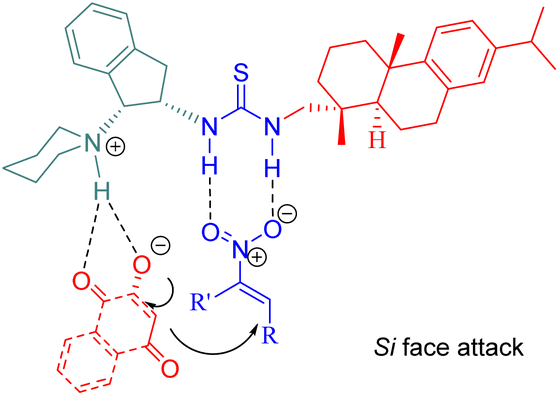
A possible transition state model was proposed in Fig. 8. C5 activated the nitroalkenes through doubly hydrogen bonding with its thiourea groups while simultaneously activating substrate 41 via its indane tertiary amine moiety. The nucleophile attacked from the Si face of the activated nitroalkene.
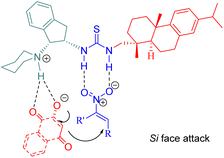 |
| | Fig. 8 A possible transition-state model of the asymmetric Michael reaction. | |
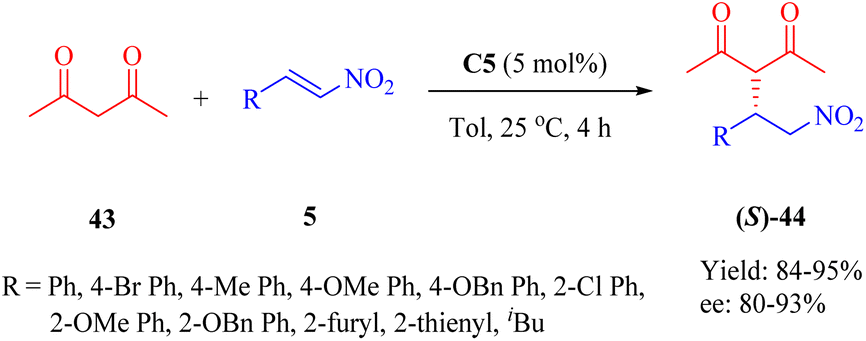
Moreover, the conjugate additions of 1,3-dicarbonyl compounds to β-nitroolefins 5 were also investigated (Scheme 15). Indeed, the utilization of catalyst C5 enhanced the reaction of 2,4-pentanedione 43 and compound 5 with high reactivities and enantioselectivities (up to 95% yield and 93% ee), albeit with a higher catalyst loading. In addition, efficient Michael reaction of other 1,3-dicarbonyl compounds such as malonate esters with β-nitrostyrene was observed under similar reaction conditions, however, the enantioselectivity did not exceed 93% ee (not shown in the scheme).
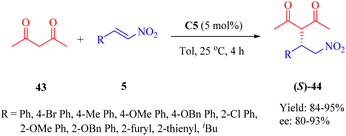 |
| | Scheme 15 Asymmetric Michael addition of 1,3-dicarbonyl compounds to β-nitroalkenes. | |
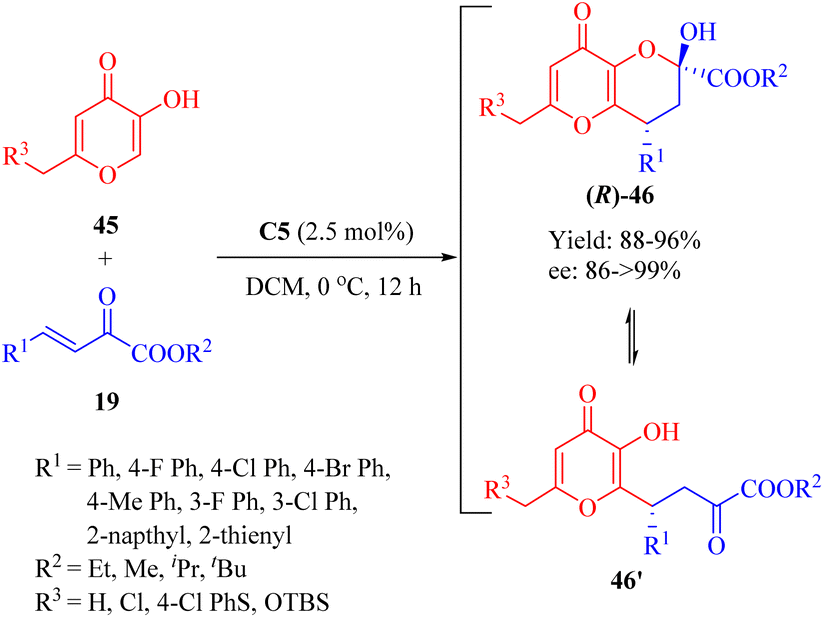
Subsequently in 2014, Reddy and coworkers discovered the enantioselective Michael/hemiketalization reaction (a type of cascade reaction) of various derivatives 45 from kojic acid (R3 = OH, a fungal metabolite with diverse biological activities) and β,γ-unsaturated α-ketone esters 19 promoted by the tertiary amine thiourea C5 with a catalyst loading of 2.5 mol% (Scheme 16).63 This reaction efficiently constructed chiral tetrahydro pyrano[3,2-b]pyran scaffolds 46 with high enantioselectivities (up to 99% ee). A rapid equilibrium was observed between the cyclic hemiketal 46 and the Michael-type adduct 46′, similarly to the equilibriums previously studied.64
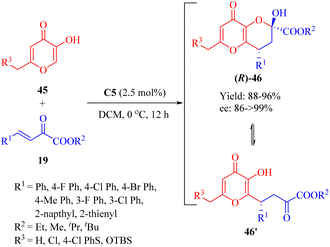 |
| | Scheme 16 Asymmetric Michael/hemiketalization of kojic acid derivatives to β,γ-unsaturated α-ketone esters. | |
2.4. Asymmetric reactions catalyzed by dehydroabietane-type bifunctional thioureas with the trans-1-amino-2-diphenylphosphinocyclohexane scaffold
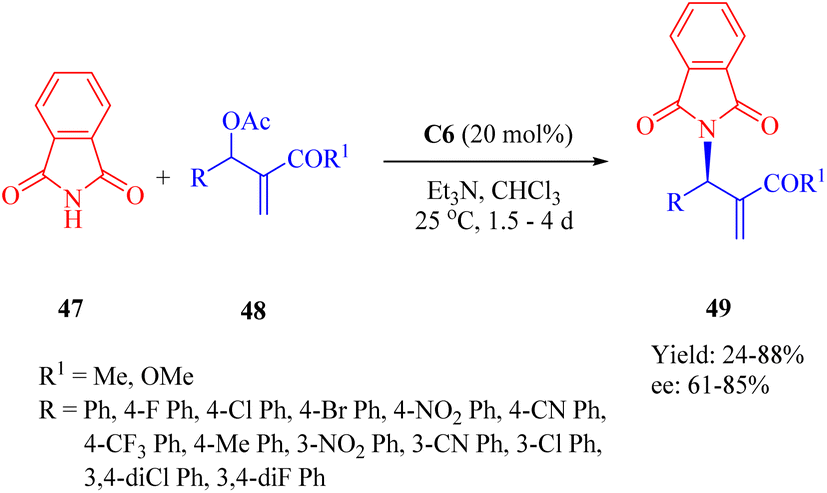
In 2015, Wu and Sha et al. investigated the allylic amination reaction of phthalimide 47 and Morita–Baylis–Hillman (MBH) acetates 48 using the thiourea C6 (Scheme 17).65
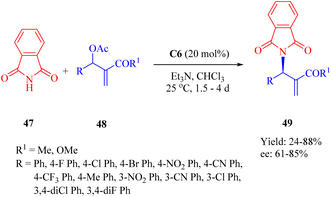 |
| | Scheme 17 Asymmetric allylic amination of phthalimide and Morita–Baylis–Hillman acetates. | |
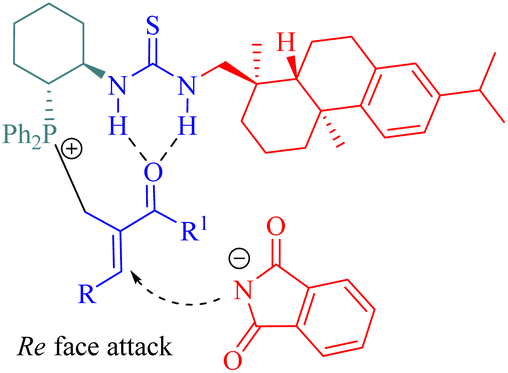
In the catalytic process, nucleophilic addition of the phosphine C6 to MBH acetates occurred, followed by the acetoxy group departure to form a cationic (E)-enone intermediate. This intermediate was activated through the hydrogen-bonding interaction. A nucleophile generated from deprotonated phthalimide by triethylamine attacked the Re face of cationic enone to produce chiral amines 49 (Fig. 9). Catalyst screening revealed that the dehydroabietane part matched well with the (1R,2R)-cyclohexyl backbone to reach a high level of enantioselectivity.
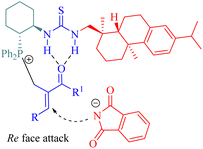 |
| | Fig. 9 A possible transition-state model of the asymmetric MBH reaction. | |
3. Immobilized dehydroabietane-type bifunctional thioureas
The immobilization of hydrogen-bond organocatalysts offers heterogeneous systems that provide various advantages for a sustainable approach in green chemistry, including chemical stability, facile work-up, and/or potential reusability. Several kinds of supports have been employed while preserving the benefits of the homogeneous catalysts.19 Among these support material, magnetic nanoparticles (MNPs) are considered ideal due to their special magnetic behavior, high specific surface area, chemical durability, and low toxicity.66 Therefore, MNPs hold great promise as versatile catalyst supports for catalytic synthetic transformations under environmentally friendly reaction conditions.
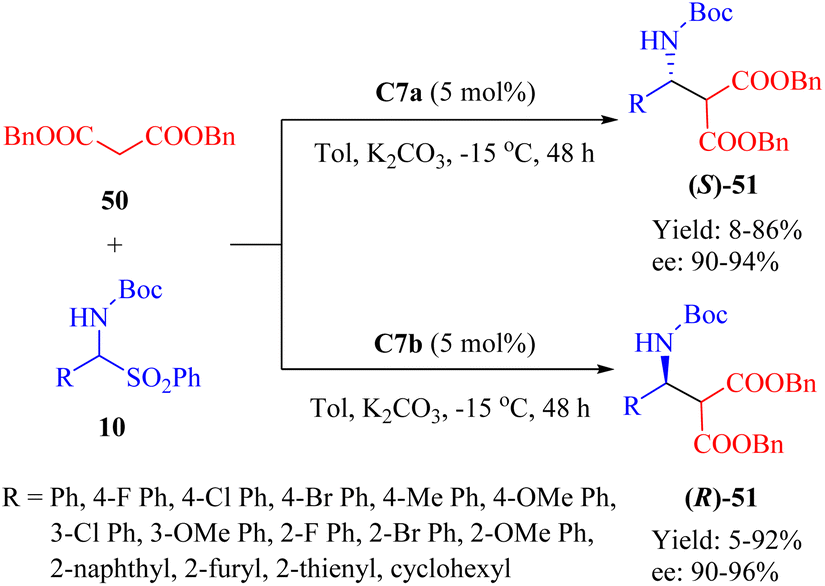
In 2013, Wang et al. developed a new class of MNPs-supported tertiary amine thioureas, namely C7a and its diastereomer C7b, to facilitate the asymmetric Mannich reaction between dibenzyl malonate 50 and α-amidosulfones 10 (Scheme 18).67 Under identical conditions, these immobilized thioureas exhibited higher catalytic activities compared to the free thioureas C2a and C2b, whose ligand loading must be 15 mol% for achieving high yield and enantioselectivity. With the exception of an aliphatic substrate (yielding only 5% and 8%, R = cyclohexyl, ee not determined), a range of aromatic amido sulfones with various substituents successfully underwent the reaction. Notably, these two catalysts were reproducible up to 15 times with a minimal impact on enantioselectivity.
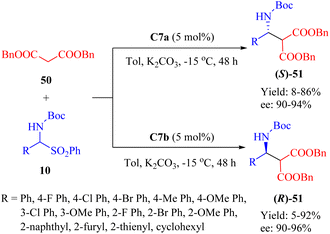 |
| | Scheme 18 Asymmetric heterogeneous Mannich reaction of dibenzyl malonate and α-amidosulfones. | |
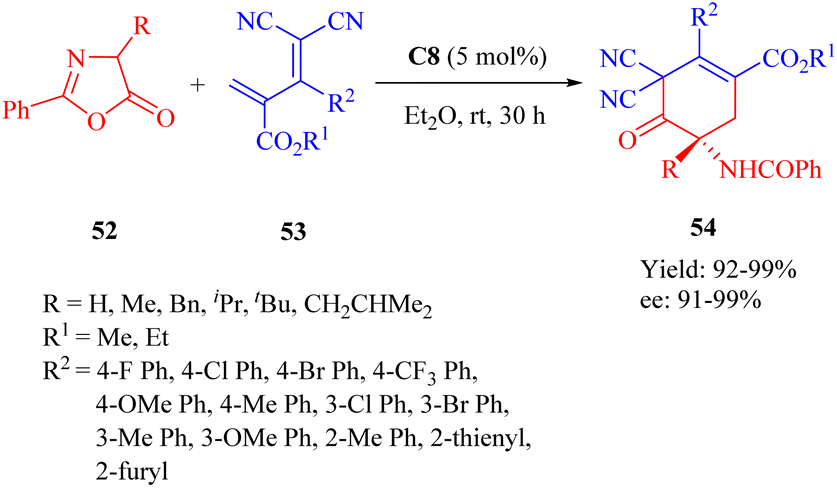
In the same year, Wang et al. developed the MNPs-supported cinchona thiourea C8 for the highly enantioselective heterogeneous IEDDA reaction of azlactones 44 with multisubstituted dicyano-2-methylenebut-3-enoates 45 through a double HOMOdienophiles and LUMOdienes activation manner (Scheme 19).68 Additionally, the immobilized catalyst exhibited excellent stability and recyclability over 10 cycles without any noticeable loss of activity.
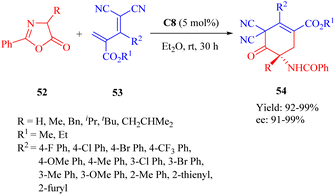 |
| | Scheme 19 Asymmetric heterogeneous IEDDA reaction of azlactones and multisubstituted dicyano-2-methylenebut-3-enoates. | |
4. Dehydroabietane-type bifunctional squaramides
Over and above thioureas, squaramides have emerged as highly efficient hydrogen-bonding donors. Drawing inspiration from the extensive utilization of squaramide organocatalysts in asymmetric transformations,12,14,15,17–21 several research groups have endeavored to develop novel dehydroabietane-type bifunctional squaramides, although successful examples remain limited.
4.1. Asymmetric reactions catalyzed by dehydroabietane-type bifunctional squaramides with the cinchona alkaloid scaffold
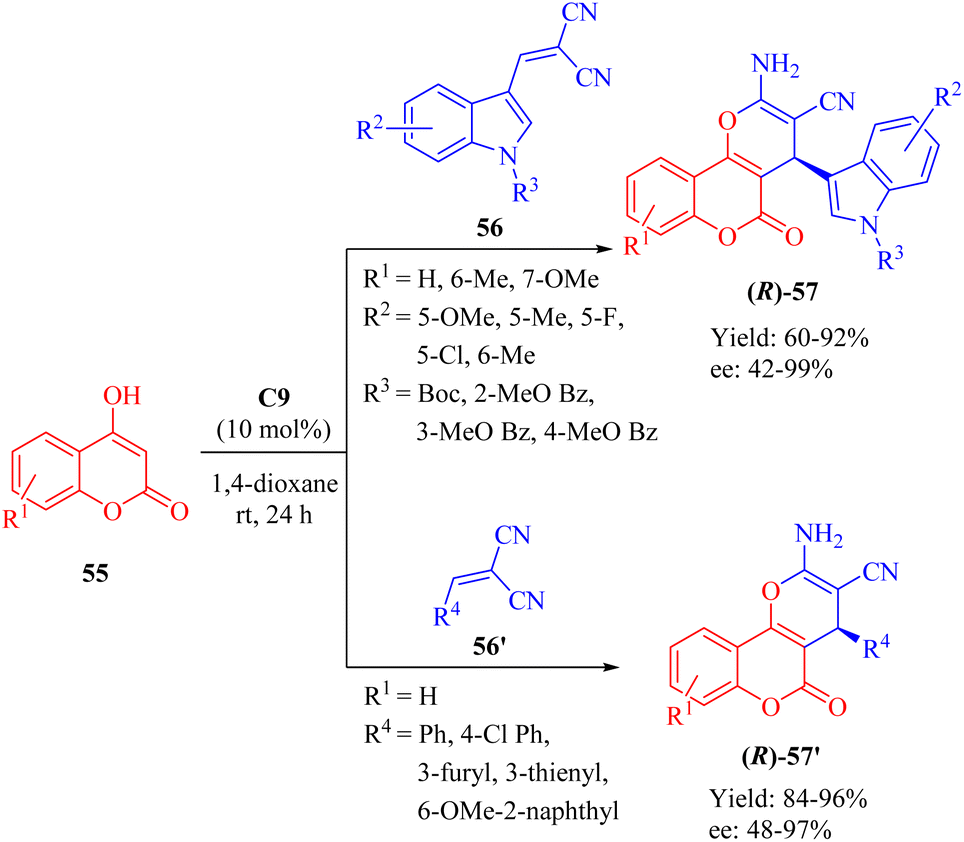
In 2018, Dong et al. developed a DA-cinchonine-derived squaramide C9 for the asymmetric domino reaction of 4-hydroxy-2H-chromen-2-ones 55 and substituted methylene malononitriles 56/56′ (Scheme 20).69 This methodology successfully afforded a range of optically active pyrano[3,2-c]chromene derivatives 57/57′ with moderate to excellent enantioselectivities. The resulting cyclic products were evaluated for their inhibitory activities against acetylcholinesterase (AChE) in vitro. Quite notably, it was observed that the (S)-configured product exhibited higher bioactivity than the (R)-configured counterpart, except for the significantly potent product 57′ containing 6-OMe-2-naphthyl group where both enantiomers displayed similar activities.
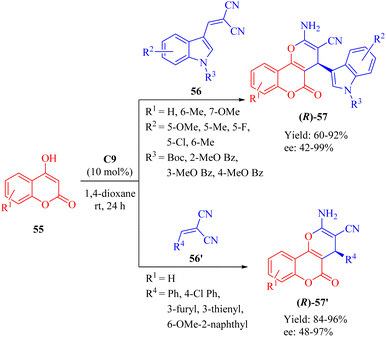 |
| | Scheme 20 Asymmetric domino reaction of 4-hydroxy-2H-chromen-2-ones and methylene malononitriles. | |
4.2. Asymmetric reactions catalyzed by dehydroabietane-type bifunctional squaramides with the trans-1,2-diaminocyclohaxane scaffold
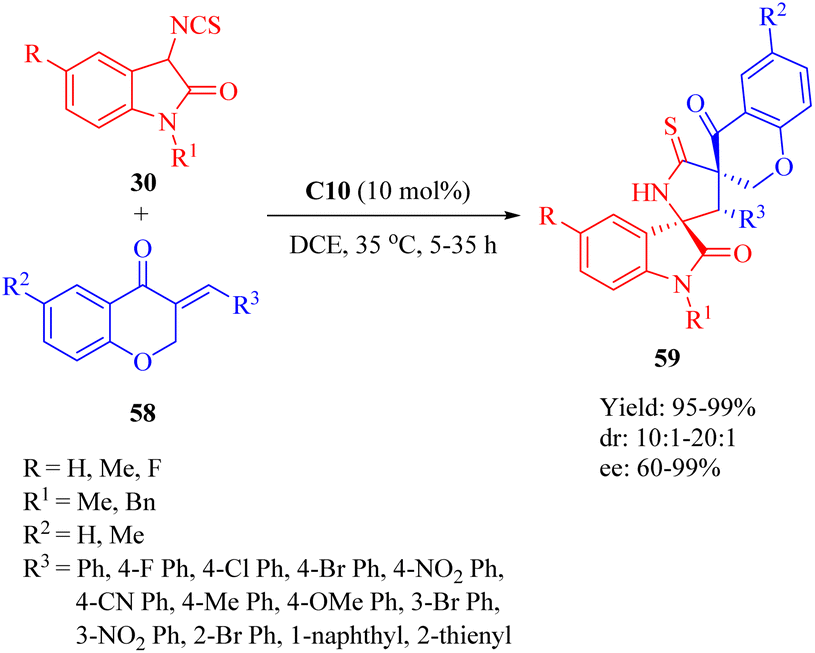
In 2018, our group developed a DA-derived tertiary amine squaramide C10 for the asymmetric Michael/cyclization cascade reaction of 3-isothiocyanato oxindoles 30 with (E)-3-arylidenechroman-4-ones 58 to successfully construct a series of spirocyclic compounds 59 with three continuous stereocenters, including two spiro-quaternary carbon centers, in high yields and stereoselectivities (Scheme 21).11 The gram-scale preparation of this reaction did not compromise its stereoinduction and reactivity. Moreover, through substitution and oxidation reactions, these spirocyclic compounds would be transformed into other chiral spirocyclic skeletons with considerable yield and stereoselectivities.
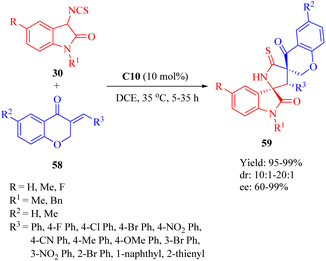 |
| | Scheme 21 Asymmetric Michael/cyclization of 3-isothiocyanato oxindoles to (E)-3-arylidenechroman-4-ones. | |
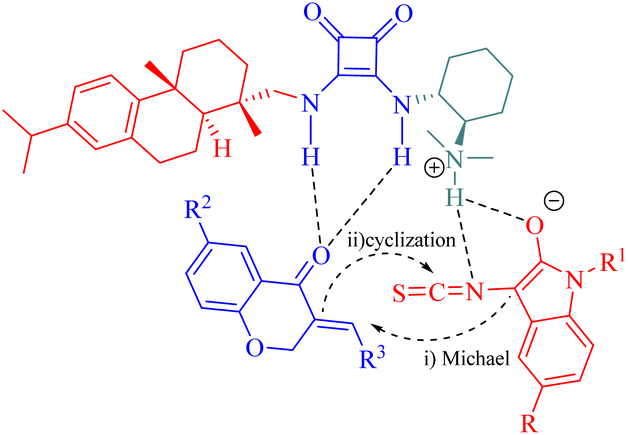
In addition, to rationalize the stereochemical outcome of this cycloaddition, a catalytic transition state model was proposed as shown in Fig. 10.
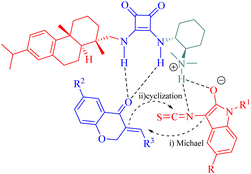 |
| | Fig. 10 A possible transition-state model. | |
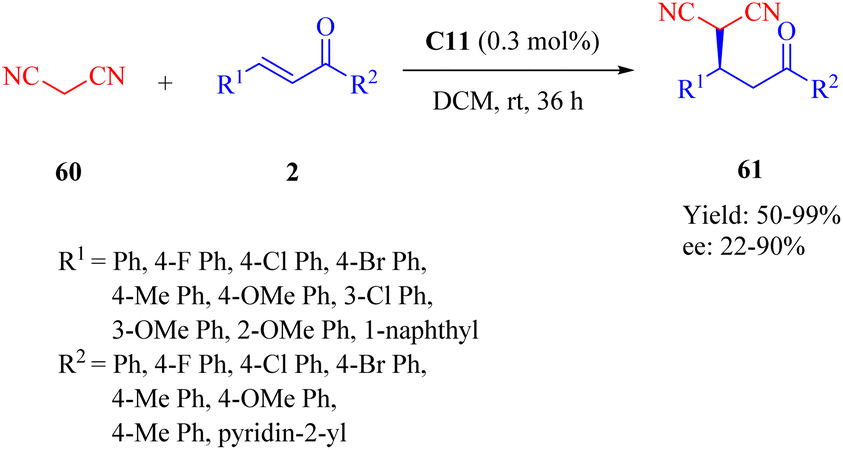
The novel dehydroabietane-type tertiary amine squaramide C11 was designed and synthesized by our group in 2020 and successfully applied for the asymmetric Michael reaction of malononitrile 60 to α,β-unsaturated ketones 2, leading to chiral γ-cyano carbonyl compounds 61 under significantly low catalyst loading (0.3 mol%) (Scheme 22).12
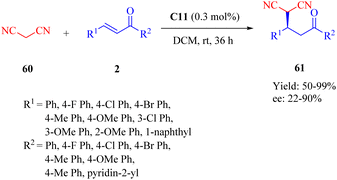 |
| | Scheme 22 Asymmetric Michael addition of malononitrile to α,β-unsaturated ketones. | |
In the transition state formation, both malononitrile and α,β-unsaturated ketones interacted via hydrogen bonding with the squaramide moiety and the tertiary amine moiety of C11, respectively. Subsequent nucleophilic attack of malononitrile on the Si face of activated α,β-unsaturated ketones offered the (R)-configured products (Fig. 11).
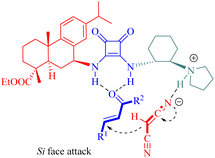 |
| | Fig. 11 Proposed model of nucleophilic attack. | |
5. Conclusions
Rosin-based bifunctional organocatalysts, particularly, dehydroabietane-type bifunctional thioureas and squaramides, have demonstrated remarkable efficacy in facilitating extensive enantioselective reactions over the past decade, as summarized in this concise review. Despite their significant contributions to this captivating research field of asymmetric organocatalysis, there remain several topics worthy of discussion concerning the development of rosin-based organocatalysts. Firstly, in those successful asymmetric reactions, bifunctional thiourea and squaramide organocatalysts generally feature the dehydroabietane backbone together with a secondary chiral scaffold such as trans-1,2-diaminocyclohexane, cinchona alkaloids, cis-1,2-diaminoindane, and 1-amino-2-diphenylphosphinecyclohexane. This combination of two chiral moieties appears an additive effect. Therefore, it is anticipated that the second chiral scaffold can be diversified by incorporating various chiral amino acids or alternative chiral skeletons. Secondly, most dehydroabietane-type organocatalysts have undergone structural modifications at the C-4 position on the A ring of the DHAA skeleton, except one example for B-ring modification at the C-7 position.12 In particular, derivatization of the amino moiety at the C-18 position has been performed, resulting in the activation site appears distant from the steric atmosphere of rosin. Consequently, the stereoselectivity and absolute configuration of the products primarily rely on the secondary chiral unit, such as the chiral trans-1,2-diaminocyclohexane scaffold. It is worth noting that while the chiral dehydroabietane structure does not significantly impact enantioselectivity, it exerts a remarkable influence on reactivity. However, no catalyst resulting from simultaneous modifications on both A and B rings has been reported yet. To fully exploit and elucidate the potential of the chiral tricyclic rigid skeleton found in dehydroabietane-type diterpenes, a rational design approach for novel organocatalysts by modifying both A and B rings to achieve desired catalytic activities represents an intriguing avenue for future research. These insights are expected to significantly contribute to the further refinement and practical implementation of rosin-based organocatalysts.
Author contributions
Conceptualization, Z.-W. Zhang, Y.-Q. Deng and N. Lin; funding acquisition, Z.-W. Zhang, Y.-Q. Deng and N. Lin; writing—original draft preparation, S.-W. Liu, H.-P. Huang, Y.-H. Xie and R.-C. Huang; writing—review and editing, Z.-W. Zhang, Y.-Q. Deng and N. Lin.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Guangxi Natural Science Foundation (2020GXNSFAA297215, 2021GXNSFDA075016, and AD19245138); Gui Style Xinglin Top Talent Funding Project of Guangxi University of Chinese Medicine (2022C005); Thousands of Young and Middle-aged Backbone Teachers Training Project of Guangxi Colleges and Universities (Gui-Jiao 2019–81); Guangxi First-class Discipline Construction Project (Gui Education-Research 2022-1); Guangxi University of Chinese Medicine Science of Chinese Materia Medica Platform Construction (First-class Discipline); Discipline and Platform Construction Funds (Science of Chinese Materia Medica Platform Construction); Guangxi University of Chinese Medicine General Program (2021MS006); Guangxi University of Chinese Medicine Graduate Education Innovation Program (YCSW2023394); and Guangxi Graduate Joint Training Base (Gui Degree 2021-6).
Notes and references
- A. J. D. Silvestre and A. Gandini, Monomers, Polymers and Composites from Renewable Resources, M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, The Netherlands, 2008, ch. 4, pp. 67–88 Search PubMed.
- B. K. Yadav, B. Gidwani and A. Vyas, J. Bioact. Compat. Polym., 2016, 31, 111–126 CrossRef CAS.
- C. Faustino, Í. Neto, P. Fonte and A. Macedo, Curr. Pharm. Design, 2018, 24, 4362–4375 CrossRef CAS PubMed.
- J. Wiemann, A. Al-Harrasi and R. Csuk, Anti-Cancer Agents Med. Chem., 2020, 20, 1756–1767 CrossRef CAS PubMed.
- S. Kugler, P. Ossowicz, K. Malarczyk-Matusiak and E. Wierzbicka, Molecules, 2019, 24, 1651 CrossRef PubMed.
- F. Steppeler, D. Iwan, E. Wojaczyńska and J. Wojaczyński, Molecules, 2020, 25, 401 CrossRef PubMed.
- R. J. Rafferty, R. W. Hicklin, K. A. Maloof and P. J. Hergenrother, Angew. Chem., Int. Ed., 2014, 53, 220–224 CrossRef CAS PubMed.
- R. Tapia, H. Bouanou, E. Alvarez, R. Alvarez-Manzaneda, R. Chahboun and E. Alvarez-Manzaneda, J. Org. Chem., 2014, 79, 4405–4413 CrossRef CAS PubMed.
- S. Bhowmick, S. S. Kunte and K. C. Bhowmick, Tetrahedron: Asymmetry, 2014, 25, 1292–1297 CrossRef CAS.
- W.-G. Huang, H.-S. Wang, G.-B. Huang, Y.-M. Wu and Y.-M. Pan, Eur. J. Org Chem., 2012, 2012, 5839–5843 CrossRef CAS.
- N. Lin, X.-W. Long, Q. Chen, W.-R. Zhu, B.-C. Wang, K.-B. Chen, C.-W. Jiang, J. Weng and G. Lu, Tetrahedron, 2018, 74, 3734–3741 CrossRef CAS.
- N. Lin, Q.-X. Wei, L.-H. Jiang, Y.-Q. Deng, Z.-W. Zhang and Q. Chen, Catalysts, 2020, 10, 14 CrossRef.
- S. Narayanaperumal, D. G. Rivera, R. C. Silva and M. W. Paixão, ChemCatChem, 2013, 5, 2756–2773 CrossRef CAS.
- X. Jiang and R. Wang, Chem. Rev., 2013, 113, 5515–5546 CrossRef CAS PubMed.
- D. Cheng, Y. Ishihara, B. Tan and C. F. Barbas, ACS Catal., 2014, 4, 743–762 CrossRef CAS.
- F. E. Held and S. B. Tsogoeva, Catal. Sci., 2016, 6, 645–667 RSC.
- G. Desimoni, G. Faita and P. Quadrelli, Chem. Rev., 2018, 118, 2080–2248 CrossRef CAS PubMed.
- A. Franconetti and G. de Gonzalo, ChemCatChem, 2018, 10, 5554–5572 CrossRef CAS.
- S. Kamal, A. F. Zahoor, S. Ahmad, R. Akhtar, I. Khaliq, W. Qurban and A. Mehreen, Curr. Org. Chem., 2020, 24, 1397–1458 CrossRef CAS.
- S. Meninno, ChemSusChem, 2020, 13, 439–468 CrossRef CAS PubMed.
- G. Molteni and A. Silvani, Eur. J. Org Chem., 2021, 2021, 1653–1675 CrossRef CAS.
- A. Laviós, A. Sanz-Marco, C. Vila, G. Blay and J. R. Pedro, Eur. J. Org Chem., 2021, 2021, 2268–2284 CrossRef.
- B. Han, X.-H. He, Y.-Q. Liu, G. He, C. Peng and J.-L. Li, Chem. Soc. Rev., 2021, 50, 1522–1586 RSC.
- V. Laina-Martín, J. A. Fernández-Salas and J. Alemán, Chem.–Eur. J., 2021, 27, 12509–12520 CrossRef PubMed.
- A. J. Boddy and J. A. Bull, Org. Chem. Front., 2021, 8, 1026–1084 RSC.
- D. M. Patel, P. J. Patel and H. M. Patel, Eur. J. Org Chem., 2022, 2022, e202201119 CrossRef CAS.
- N. Melnyk, I. Iribarren, E. Mates-Torres and C. Trujillo, Chem.–Eur. J., 2022, 28, e202201570 CrossRef CAS PubMed.
- P. Chauhan, S. Mahajan, U. Kaya, D. Hack and D. Enders, Adv. Synth. Catal., 2015, 357, 253–281 CrossRef CAS.
- X. Han, H.-B. Zhou and C. Dong, Chem. Rec., 2016, 16, 897–906 CrossRef CAS PubMed.
- B. L. Zhao, J. H. Li and D. M. Du, Chem. Rec., 2017, 17, 994–1018 CrossRef CAS PubMed.
- Y. Lin and D. Du, Chinese J. Org. Chem., 2020, 40, 3214–3236 CrossRef CAS.
- D. Krištofíková, V. Modrocká, M. Mečiarová and R. Šebesta, ChemSusChem, 2020, 13, 2828–2858 CrossRef PubMed.
- A. Biswas, A. Ghosh, R. Shankhdhar and I. Chatterjee, Asian J. Org. Chem., 2021, 10, 1345–1376 CrossRef CAS.
- S. Vera, A. García-Urricelqui, A. Mielgo and M. Oiarbide, Eur. J. Org Chem., 2023, 26, e202201254 CrossRef CAS.
- X. Jiang, Y. Zhang, A. S. C. Chan and R. Wang, Org. Lett., 2009, 11, 153–156 CrossRef CAS PubMed.
- X. Jiang, Y. Zhang, X. Liu, G. Zhang, L. Lai, L. Wu, J. Zhang and R. Wang, J. Org. Chem., 2009, 74, 5562–5567 CrossRef CAS PubMed.
- X. Liu, H. Song, Q. Chen, W. Li, W. Yin, M. Kai and R. Wang, Eur. J. Org Chem., 2012, 2012, 6647–6655 CrossRef CAS.
- X. Jiang, G. Zhang, D. Fu, Y. Cao, F. Shen and R. Wang, Org. Lett., 2010, 12, 1544–1547 CrossRef CAS PubMed.
- X. Jiang, Y. Cao, Y. Wang, L. Liu, F. Shen and R. Wang, J. Am. Chem. Soc., 2010, 132, 15328–15333 CrossRef CAS PubMed.
- X. Jiang, Y. Wang, G. Zhang, D. Fu, F. Zhang, M. Kai and R. Wang, Adv. Synth. Catal., 2011, 353, 1787–1796 CrossRef CAS.
- Y. Cao, X. Jiang, L. Liu, F. Shen, F. Zhang and R. Wang, Angew. Chem., 2011, 123, 9290–9293 (Angew. Chem., Int. Ed., 2011, 50, 9124–9127) CrossRef.
- L. Liu, Y. Zhong, P. Zhang, X. Jiang and R. Wang, J. Org. Chem., 2012, 77, 10228–10234 CrossRef CAS PubMed.
- G. Zhang, Y. Zhang, X. Jiang, W. Yan and R. Wang, Org. Lett., 2011, 13, 3806–3809 CrossRef CAS PubMed.
- X. Jiang, Y. Sun, J. Yao, Y. Cao, M. Kai, N. He, X. Zhang, Y. Wang and R. Wang, Adv. Synth. Catal., 2012, 354, 917–925 CrossRef CAS.
- G. Zhang, Y. Zhang, J. Yan, R. Chen, S. Wang, Y. Ma and R. Wang, J. Org. Chem., 2012, 77, 878–888 CrossRef CAS PubMed.
- X. Jiang, L. Wu, Y. Xing, L. Wang, S. Wang, Z. Chen and R. Wang, Chem. Commun., 2012, 48, 446–448 RSC.
- X. Jiang, X. Shi, S. Wang, T. Sun, Y. Cao and R. Wang, Angew. Chem., 2012, 124, 2126–2129 (Angew. Chem., Int. Ed., 2012, 51, 2084–2087) CrossRef.
- X.-Y. Wu, F. Sha, X.-T. Guo and J. Shen, Synthesis, 2015, 47, 2063–2072 CrossRef.
- X.-T. Guo, F. Sha and X.-Y. Wu, Res. Chem. Intermed., 2016, 42, 6373–6380 CrossRef CAS.
- X.-Y. Wu, X.-T. Guo and F. Sha, Synthesis, 2017, 49, 647–656 CrossRef.
- X. Jiang, Y. Zhang, L. Wu, G. Zhang, X. Liu, H. Zhang, D. Fu and R. Wang, Adv. Synth. Catal., 2009, 351, 2096–2100 CrossRef CAS.
- X. Jiang, D. Fu, G. Zhang, Y. Cao, L. Liu, J. Song and R. Wang, Chem. Commun., 2010, 46, 4294–4296 RSC.
- L. Wang, X.-M. Shi, W.-P. Dong, L.-P. Zhu and R. Wang, Chem. Commun., 2013, 49, 3458–3460 CAS.
- K. Juhl and K. A. Jørgensen, Angew. Chem., 2003, 115, 1536–1539 (Angew.Chem. Int. Ed., 2003, 42, 1498–1501) Search PubMed.
- X. Jiang, L. Wang, M. Kai, L. Zhu, X. Yao and R. Wang, Chem.–Eur. J., 2012, 18, 11465–11473 CrossRef CAS PubMed.
- X. Jiang, L. Liu, P. Zhang, Y. Zhong and R. Wang, Angew. Chem., 2013, 125, 11539–11543 (Angew. Chem., Int. Ed., 2013, 52, 11329–11333) CrossRef.
- H.-R. Zhang, J.-J. Xue, R. Chen, Y. Tang and Y. Li, Chin. Chem. Lett., 2014, 25, 710–714 CrossRef.
- L. Liu, D. Zhang, P. Zhang, X. Jiang and R. Wang, Org. Biomol. Chem., 2013, 11, 5222–5225 RSC.
- Q. Chen, J. Liang, S. Wang, D. Wang and R. Wang, Chem. Commun., 2013, 49, 1657–1659 RSC.
- X.-M. Shi, W.-P. Dong, L.-P. Zhu, X.-X. Jiang and R. Wang, Adv. Synth. Catal., 2013, 355, 3119–3123 CrossRef CAS.
- Y.-M. Cao, F.-F. Shen, F.-T. Zhang and R. Wang, Chem.–Eur. J., 2013, 19, 1184–1188 CrossRef CAS PubMed.
- B. V. S. Reddy, M. Swain, S. M. Reddy and J. S. Yadav, RSC Adv., 2013, 3, 8756–8765 RSC.
- B. V. S. Reddy, M. Swain, S. M. Reddy, J. S. Yadav and B. Sridhar, RSC Adv., 2014, 4, 42299–42307 RSC.
- S.-L. Zhao, C.-W. Zheng, H.-F. Wang and G. Zhao, Adv. Synth. Catal., 2009, 351, 2811–2816 CrossRef CAS.
- X. Zhao, T. Kang, J. Shen, F. Sha and X. Wu, Chin. J. Chem., 2015, 33, 1333–1337 CrossRef CAS.
- Z. Zeng, Y. Chen, X. Zhu and L. Yu, Chin. Chem. Lett., 2022, 34, 107728 CrossRef.
- H. Zhu, X. Jiang, X. Li, C. Hou, Y. Jiang, K. Hou, R. Wang and Y. Li, ChemCatChem, 2013, 5, 2187–2190 CrossRef CAS.
- X. Jiang, H. Zhu, X. Shi, Y. Zhong, Y. Li and R. Wang, Adv. Synth. Catal., 2013, 355, 308–314 CrossRef CAS.
- J. Zheng, M. He, B. Xie, L. Yang, Z. Hu, H.-B. Zhou and C. Dong, Org. Biomol. Chem., 2018, 16, 472–479 RSC.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Shao-Wu Liu†
a,
Hong-Ping Huang†a,
Yu-Hang Xiea,
Ruo-Chen Huanga,
Yan-Qiu Deng*ab and
Ning Lin*a
*ab,
Shao-Wu Liu†
a,
Hong-Ping Huang†a,
Yu-Hang Xiea,
Ruo-Chen Huanga,
Yan-Qiu Deng*ab and
Ning Lin*a