
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom liquid to solid-state, solvent-free oxidative ammonolysis of lignins – an easy, alternative approach to generate “N-lignins”†
Gerhild K. Wurzer a,
Markus Bachera,
Oliver Muslab,
Nadine Kohlhubera,
Irina Sulaevaac,
Theres Kelza,
Karin Facklerd,
Robert H. Bischofd,
Hubert Hetteggera,
Antje Potthasta and
Thomas Rosenau*ae
a,
Markus Bachera,
Oliver Muslab,
Nadine Kohlhubera,
Irina Sulaevaac,
Theres Kelza,
Karin Facklerd,
Robert H. Bischofd,
Hubert Hetteggera,
Antje Potthasta and
Thomas Rosenau*ae
aDepartment of Chemistry, Institute of Chemistry of Renewable Resources, University of Natural Resources and Life Sciences, Vienna (BOKU), Konrad-Lorenz-Strasse 24, A-3430 Tulln, Austria. E-mail: thomas.rosenau@boku.ac.at
bDepartment of Chemical and Biological Engineering, Biobased Colloids and Materials, UBC University of British Columbia, Vancouver, 2385 East Mall, Vancouver, Canada
cCore Facility Analysis of Lignocellulosics (ALICE), University of Natural Resources and Life Sciences, Vienna (BOKU), Konrad-Lorenz-Straße 24, A-3430 Tulln, Austria
dLenzing AG, Research & Development, A-4860 Lenzing, Austria
eJohan Gadolin Process Chemistry Centre, Åbo Akademi University, Porthansgatan 3, FI-20500 Åbo, Finland
First published on 23rd March 2023
Abstract
A new chemical modification protocol to generate N-lignins is presented, based on Indulin AT and Mg2+-lignosulfonate. The already known ammonoxidation reaction in liquid phase was used as a starting point and stepwise optimised towards a full solid-state approach. The “classical” liquid ammonoxidation products, the transition products from the optimization trials, as well as the “solid-state” products were comprehensively analysed and compared to the literature. The N-lignins obtained with the conventional ammonoxidation protocol showed the same properties as reported. Their molar mass distributions and the hydroxy group contents, hitherto not accessible due to solubility problems, were measured according to a recently reported protocol. N-Indulin showed an N-content up to 11 wt% and N-lignosulfonate up to 16 wt%. The transition experiments from liquid to solid-state gave insights into the influence of chemical components and reaction conditions. The use of a single chemical, the urea-hydrogen peroxide complex (UHP, “carbamide peroxide”), was sufficient to generate N-lignins with satisfying N-content. This chemical acts both as an N-source and as the oxidant. Following the optimization, a series of solid-state ammonoxidation tests were carried out. High N-contents of 10% in the case of Indulin and 11% in the case of lignosulfonate were obtained. By varying the ratio of UHP to lignin, the N-content can be controlled. Structural analysis showed that the N is organically bound to the lignin, similar to the “classical” ammonoxidation products obtained under homogeneous conditions. Overall, a new ammonoxidation protocol was developed which does not require an external gas supply nor liquids or dissolved reactants. This opens the possibility for carrying out the lignin modification in closed continuous reactor systems, such as extruders. The new, facile solid-state protocol will hopefully help N-lignins to find more consideration as a fertilizing material and in soil-improving materials.
1 Introduction
Food supply is one of the biggest challenges of mankind. Population growth and changes in dietary habits increase the demands for agricultural production. In addition, extreme weather events are becoming more and more frequent due to climate change, which threatens the stable production of agricultural goods. Plants, especially crops, are required to be highly adaptable, not only to produce high yields under changing conditions, but also to meet our food demands.1–4 Besides phosphorus (P) and potassium (K), nitrogen (N) is one of the most important macronutrients for crops, which usually has to be supplied “externally” in the form of synthetic fertilisers. Mainly inorganic/mineral N fertiliser have been applied in the form of ammonium and nitrate salts, later by urea and the use of nitrogen-phosphorus-potassium (NPK) mixtures.1,5,6 However, their use has also some grave disadvantages.The efficiency of the N-uptake of synthetic N fertilisers is typically rather low. Only about 50% of the applied N is taken up by the plant, a minor quantity stays in the soil, while the rest is lost.1–3,7,8 The major mechanisms for N loss are nitrate leaching, runoff, erosion and gaseous loss through denitrification and ammonia volatilisation.1,10 The environmental pollution caused by conventional inorganic N-fertilisers as well as their contribution to the climate crisis is immense. Paired with the urgent necessity to meet the global food supply, on the other hand, their use has been and remains a double-edged sword.
Organic fertilisers, e.g. animal manure, have also been used in larger organic farming systems. Although organic farming scores with valorising waste organic matter, recycling of nutrients and improving the soil quality,1 the use of manure has a negative impact on soil, water and air.4,10
The challenge is to increase the efficiency of nitrogen uptake and to achieve the high yields needed to optimise natural resource efficiency and feed the growing population, while at the same time minimising nitrogen losses from agro-ecosystems.4,6,8,9 Modern N-fertilisers require specific properties: they are expected to be plant-compatible, offer high N-availability for the plant, which should be tuneable over time (short-, medium- and long-term availability).11 From an economical point of view, fertilizers are bulk products: thus their availability in huge quantities at a competitive low price is very important. In terms of cost, also the long-term fertilising activity and humus formation should be considered when comparing them.12
Wood scientists and biorefinery experts have proposed the use of lignin for fertilising purposes as well as soil conditioning.11,13,14 Lignin, the most plentiful araliphatic biopolymer on earth, plays an important role in agriculture soil quality improvement due to its natural role in humification and its similarity to humic substances.11,13,14 The lignin structure is recalcitrant to fast (microbial) degradation, and its humification products represent an integral part of the organic matter in the soil. The improvement of the humus situation in terms of supply and quality is one of the most significant goals of soil rehabilitation measures. By the use of lignin instead of the plant matter as a whole, humus formation can be accelerated.11 The use of lignin instead of harmful substances in agriculture, extends not only the life of a carbon source, but would also lead to a lower global warming potential.15 As lignin is still mainly seen as a by-product of the classical pulp and paper biorefinery and mostly burned for energy generation, its use in agriculture would mean carbon sequestration and have a positive sustainable implication.16 The structurally highly diverse biopolymer is obtained through different chemical processes, such as kraft, sulphite, or organosolv pulping,17–21 which cause significant structural changes, eventually yielding so-called “technical lignins”, e.g., kraft lignin, lignosulfonates or organosolv lignins.22 Changes in molecular weight, lignin depolymerization and repolymerization, condensation reactions, the introduction of functional groups (especially sulphur-containing moieties during kraft and sulfite pulping), and oxidative structural changes occur. In the case of lignosulfonate, strongly acidic sulfonate groups are introduced on the lignin backbone, which impose solubility in water.17 None of these modifications of natural lignins is per se incompatible with the use of the technical lignins as fertilisers or fertiliser precursors. Moreover, the structural changes occurring upon pulping and during natural humification even have many similarities.
The chemical modification of technical lignins to implement nitrogen and thus to simulate natural humification and its products, the humic matter, more closely is a very promising approach. This process, invented in 1930, is synonymously referred to as oxidative ammonolysis, ammonoxidation or ammoxidation (AO). The products, lignin-based N fertilisers, are often denoted as “N-lignins” or artificial humic matter.11 Note that IUPAC has highlighted artificial humus formation as one of the top ten emerging technologies in chemistry for a circular and climate neutral future.23 It is stated that artificial humic matter synthesis requires control over the process and leads to greener and safer fertiliser solutions.23
AO is usually carried out in alkaline ammonia solution or suspension, under elevated pressure and temperature in an oxygen atmosphere, whereby nitrogen from ammonia is incorporated into lignin. The amount of nitrogen bound to lignin varies greatly (up to 20 wt% absolute N-content), depending on the conditions used. Also the type of chemical binding of nitrogen differs widely.11,24,25 Fischer and Science (2002) divided the N-containing functional groups into ionically bound ammonia (ammonium, NH4+), hydrolysable N, such as nitriles (R–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), amines (R–NH2, R2NH, R3N) or amides (R–CO–NR2), and strong organically bound N (sob N, in heterocyclic and/or aromatic structures).11 Due to the different binding modes of nitrogen both immediate availability (ammonium) and a slow-release and long-term availability of nitrogen is achieved. NH4+–N is supposed to act as readily available N, amide-N as medium-term fertiliser, and sob N as long-term fertiliser. A low C/N ratio, around 25 or even lower, has been shown to favour humus formation.13 During oxidative ammonolysis, the technical lignin undergoes additional severe structural changes, for example, O-demethylation, demethoxylation, chain cleavage/degradation, condensation, aromatization and dearomatization, or desulfonation in the case of lignosulfonate.
N), amines (R–NH2, R2NH, R3N) or amides (R–CO–NR2), and strong organically bound N (sob N, in heterocyclic and/or aromatic structures).11 Due to the different binding modes of nitrogen both immediate availability (ammonium) and a slow-release and long-term availability of nitrogen is achieved. NH4+–N is supposed to act as readily available N, amide-N as medium-term fertiliser, and sob N as long-term fertiliser. A low C/N ratio, around 25 or even lower, has been shown to favour humus formation.13 During oxidative ammonolysis, the technical lignin undergoes additional severe structural changes, for example, O-demethylation, demethoxylation, chain cleavage/degradation, condensation, aromatization and dearomatization, or desulfonation in the case of lignosulfonate.
AO has been extensively studied in the past, especially before the turn of the millennium. Different parameters, optimum process conditions, and the applicability of the N-lignin products as eco-friendly fertilisers to soil have been investigated. Gonzales et al. studied the reactivity of kraft lignin and reactions during the AO process.26 Meier et al. reported that higher temperatures lead to a higher degradation of the biopolymer and that the bound N-content strongly depends on the lignin raw material used: lignosulfonate showed a higher reactivity compared to soda or kraft lignin. The optimum conditions were found to be 150 °C at an O2 pressure of 15 bar to bind at least 10 wt% of N.25 Capanema et al. studied the influence of reaction parameters (temperature, pressure, ammonia concentration, time) on N-incorporation with organosolv lignins.27–30 Also structural details of N-lignins, based on FTIR and 15N NMR experiments, have been addressed in several studies.24,25,31 Commercialization and large-scale use of N-lignins has been hampered mainly by their higher costs compared to the very low prices of bulk inorganic N-fertilizers. A main contributor to the higher expenses is the energy demand for water evaporation (to obtain a solid, powdery product), or the transportation costs when using the aqueous suspension/solution (= black liquor) as a starting material.
Only a few studies address the influence of N-lignins in soil.32–34 Ramirez et al. tested ammonoxidized kraft lignin in pot experiments with sorghum and compared it to a conventional N-fertiliser (ammonium sulfate).12 The fertilising activity of the modified lignin was satisfactory and yielded similar grain and biomass yields. Also, very positively, nitrate lixiviation was considerably reduced. In a follow-up project, the ammonoxidized kraft lignin was compared to a conventional urea-type fertiliser in sorghum field experiments. N-lignin performed weaker in the first growth cycle but yielded higher biomass in the second cycle.32 It showed long-term availability and functioned as a fertiliser for more than one growth period. In another study, the crop yield obtained with N-lignin was 82% relative to ammonium sulphate.25 Despite the lower yield, less fertiliser had to be used, as leaching was prevented, and humus formation was stimulated. De la Rosa et al. stated that only a small part of the N was used for plant biomass production and a large part was remaining in the soil matrix where it enhanced the soil organic matter content.33 Impurities of technical lignins, especially monosaccharides and oligosaccharides and their degradation products, were shown to be transformable into N-heterocyclic compounds or other potentially phytotoxic compounds during AO in model compound experiments.13,35,36 However, sufficiently mild conditions avoid or at least significantly decrease formation of (phyto)toxic substances.13,36
In our study, we report a novel procedure to introduce N into lignins, more precisely a solvent-free solid-state ammonoxidation. The use of an aqueous ammonium hydroxide solution and an external gas supply is eliminated, which renders the process attractive for extrusion, kneading or ball milling, and might have the potential to significantly lower production costs. The transition from liquid- to solid-state AO is described, including a comprehensive analytical characterization of the obtained N-lignin materials.
2 Materials and methods
2.1 Materials
Indulin® AT was obtained from MeadWestvaco, North Charleston, SC, USA. Lignosulfonate was purchased from Lenzing AG, Austria. The LS used, was purified according to a protocol by Sumerskii et al.37 with a ratio of 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (mass of Amberlite® XAD-7 to mass of sulphite liquor). NH4HCO3, hydrogen peroxide urea complex, NaOH, NH4OH (28.0–30.0% based on NH3), H2O2 and 15N urea (99%) were purchased from Sigma Aldrich, Steinheim, Germany. A dialysis membrane, Spectra/Por® Biotech CE MWCO 100–500, 16 mm was purchased from Carl Roth GmbH, Karlsruhe, Germany.
:1 (mass of Amberlite® XAD-7 to mass of sulphite liquor). NH4HCO3, hydrogen peroxide urea complex, NaOH, NH4OH (28.0–30.0% based on NH3), H2O2 and 15N urea (99%) were purchased from Sigma Aldrich, Steinheim, Germany. A dialysis membrane, Spectra/Por® Biotech CE MWCO 100–500, 16 mm was purchased from Carl Roth GmbH, Karlsruhe, Germany.
2.2 Analytical methods
:2:1 w/v/v) in case of LS or 400 μL of DMF and 200 μL of pyridine in case of Indulin were injected. Then 100 μL of a stock solution containing the internal standard N-hydroxy-5-norbornene-2,3-dicarboxylic acid imide (e-HNDI, 50 mg mL−1) and 100 μL of the relaxation agent Cr(acac)3 dissolved in IL/DMF/pyridine (IL = 1-ethyl-3-methylimidazolium chloride [emim]Cl, 5 mg mL−1) in case of LS or pure DMF (5 mg mL−1) for the Indulin samples were added. After thorough mixing, the derivatization agent 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP) was added (100 μL) and the mixture was shaken for 1 h before NMR analysis. Before measuring, CDCl3 (100 μL) was added as a locking agent. All samples were measured within 12 h. For the blank sample prepared in the ternary solvent system, the preparation was carried out without LS addition.2.3 Ammonoxidation
| Sample | Indulin (g) | H2O (mL) | N-Source | O-Source |
|---|---|---|---|---|
| Tr.-N-Ind. 1 | 0.55 | 18 | 2.4 g NH4HCO3 | O2, 10 bar |
| Tr.-N-Ind. 2 | 0.55 | — | 2.4 g NH4HCO3 | O2, 10 bar |
| Tr.-N-Ind. 3 | 0.55 | 18 | 2 mL NH4OH | 2.6 g UHP |
| Tr.-N-Ind. 4 | 0.55 | — | 2 mL NH4OH | 2.6 g UHP |
| Tr.-N-Ind. 5 | 0.55 | — | 2.4 g NH4HCO3 | 2.6 g UHP |
Additional experiments were carried out with ammonium bicarbonate and urea hydrogen peroxide in different ratios to lignin. In some tests, powdered NaOH was additionally added. For purification, the reaction mixture was flushed from the reactor with water (reaction solutions had a pH around 8), acidified with aqueous HCl (1 M) to a pH slightly below 4, centrifuged and washed with water once and freeze-dried.
2.3.4.1 Indulin. Samples were prepared based on the complete substitution of N- and O-source. Temperature and time were kept the same as above. Purification was carried out as already described above. Briefly, Indulin was mixed with urea peroxide and ammonium bicarbonate at different ratios. To some tests, powdered NaOH was added. The mixture was heated up in the tightly closed reactor for 30 min to reach 130 °C. The mixture was stirred by magnetic stirring. The temperature was held at 130 °C for 2 h. After that, the mixture was cooled down to r.t. The mixture was removed from the reactor with water and purified. The N-lignin was precipitated with an aqueous HCl solution (1 M) to a pH around 4. After that, the mixture was centrifuged at 5000 rpm for 10 min, the supernatant was discarded and the precipitated N-lignin was washed with water and again centrifuged for 10 min at 5000 rpm. The N-lignin was transferred into a beaker with the help of water and freeze-dried. For analysis, the N-lignin was additionally dried in a vacuum oven set to 40 °C for 24 h.
2.3.4.2 Lignosulfonate. Based on the results obtained from the SS-AO of Indulin samples, a series of different ratios between LS and UHP was carried out. In more detail, LS was mixed with UHP in the reactor vessel. Then the mixture was heated up to 130 °C for 30 min while it was stirred by magnetic stirring. The temperature was held for 2 h at 130 °C and then the vessel was cooled down to room temperature. The reaction mixture was transferred to a beaker with the help of water. For purification, the dissolved reaction mixture was put in a dialysis membrane (MWCO 100–500 Da). After dialysis for one week against water, the remaining N-LS was freeze dried. For analysis, the N-LS was additionally dried in a vacuum oven at 40 °C overnight.
3 Results and discussion
In this study, the crossover from the “classical” ammonoxidation in liquid-state towards a new ammonoxidation protocol in solid-state is presented. Initial experiments were carried out using the classical ammonoxidation parameters, followed by a stepwise transition to solid-state conditions. The stepwise change involved varying one parameter at a time to gain insights into the influence of each factor. As a last part, a series of experiments completely in solid-state mode was performed for parameter optimization, in particular the ratio of lignin to reaction chemicals. All experiments were carried out in a pressurized reactor with a softwood kraft lignin, Indulin AT, and a hardwood resin-purified37 magnesium lignosulfonate; the transition steps only with Indulin AT, in a pressurizable reactor. The purity of the resin-purified lignosulfonate is according to the method over 98%. Comprehensive analysis of the respective products ensured that the parameter changes did not negatively affect the product parameters, such as N-content. Mechanistic considerations were supported by kinetic studies and isotopic labelling (15N) experiments.3.1 “Classical” liquid-state ammonoxidation
The reaction parameters for the classical liquid-state ammonoxidation were chosen according to established protocols.24,27 The reaction temperature was 130 °C, following a heating-up period of 30 minutes. The reaction time was set to 2 h at an oxygen pressure of 10 bar. A molarity of 1.48 M NH4OH was used for 0.55 g of lignin, this gives a ratio of 1.1 g NH4OH for 1 g lignin. This value was chosen based on a series of preliminary experiments, in which only the ammonium hydroxide concentration was varied to determine the conditions for the highest N-input. Temperature and pressure for optimum N-incorporation were taken from previous studies.27–30 A higher pressure does not only affect the oxygen uptake but also nitrogen incorporation, CO2 formation and the loss of carbon and methoxy groups.28 The same is true for the ammonium hydroxide concentration, with a high pH also increasing lignin dissolution.30 At higher temperatures, more N is incorporated into the lignin. Additionally, the quality and quantity of N functional groups are influenced, because the lignin undergoes more structural changes and therefore N is incorporated by stronger bonds, such as in nitriles.11,29 In lignosulfonates, also desulfonation occurs apart from demethoxylation.11 In triplicate experiments we determined the reproducibility of our experimental conditions based on the N-incorporation (wt%), which yielded a relative standard deviation of 0.4 to 0.6%. The recovery yield was in all experiments around 95%. In Table 2, the results of the elemental analysis before and after AO are shown. An N-content of about 11 wt% for Indulin and 16 wt% for lignosulfonate was obtained. These values agree well with the N-contents obtained by Meier et al.,25 Potthast et al.,24 and Capanema et al.27 As discussed by Meier et al.,25 lignosulfonates show a higher reactivity compared to kraft lignins in terms of higher N uptake upon ammonoxidation, which was explained through the higher sensitivity towards oxidation of this material. With the parameters chosen, we obtained a quite satisfactory C/N ratio below 5 for both of the industrial lignins. A low C/N ratio speeds up humus formation, as more N in the organic material becomes available to microorganisms.25| Sample | C% | H% | N% | S% | O% |
|---|---|---|---|---|---|
| Indulin | 61.17 | 5.61 | 1.15 | 1.88 | 28.12 |
| N-Indulin | 46.01 | 4.99 | 11.09 | 0.64 | 24.89 |
| LS | 49.67 | 5.53 | 0.43 | 4.38 | 37.04 |
| N-LS | 30.59 | 5.37 | 16.38 | 3.94 | 39.65 |
Fig. 1 presents the FT-IR spectra of the original lignins and the N-lignins. Upon AO, new bands appear between 2750 and 3600 cm−1, which are assigned to amides, a new maximum at around 3200 cm−1 occurs, which indicates the presence of ammonium-N and amides. Further, an increase between 1550 and 1750 cm−1 can be seen, which is assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–N and C–H–N stretching. A new absorbance band at 1400 cm−1 represents the C–N stretching. A decrease at 1512 cm−1 and 1270 cm−1 corresponds to a loss in aromatic structures and ether bonds. These FT-IR data are in full agreement with previously published spectra.11,24,25
O, C–N and C–H–N stretching. A new absorbance band at 1400 cm−1 represents the C–N stretching. A decrease at 1512 cm−1 and 1270 cm−1 corresponds to a loss in aromatic structures and ether bonds. These FT-IR data are in full agreement with previously published spectra.11,24,25
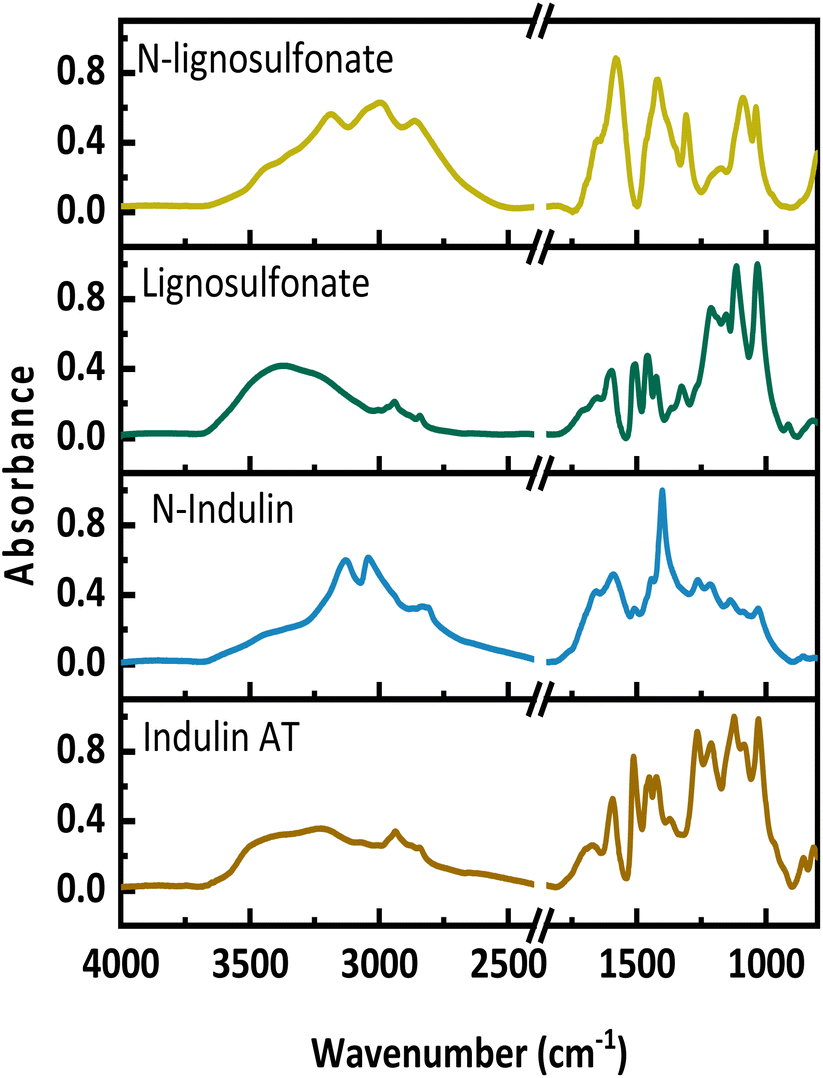 | ||
| Fig. 1 FT-IR spectra of Indulin and N-Indulin (below) as well as lignosulfonate and N-lignosulfonate (above). | ||
Gel permeation chromatography (GPC) was performed to determine the molar mass distribution of the starting materials and the obtained N-modified products (Fig. 2). The calculated statistical moments were based on multi-angle laser light scattering (MALS) detection at 785 nm with correction factors according to Zinovyev et al. to minimize fluorescence and UV influences.38 Data of the statistical moments are presented in the ESI.† In the case of Indulin AT, the molecular mass and molecular mass distribution increased significantly upon AO, see Fig. 2. The MMD curve is shifted towards higher molar masses and becomes multimodal. For lignosulfonate, the MMD curve was shifted towards lower molar masses, indicating the presence of smaller fractions of N-lignosulfonates. In case of N-Indulin, all calculated statistical moments differed significantly from the starting material, which points to significant structural changes (repolymerization or condensation) towards high molecular mass. The Mn, representing the low molecular weight fraction, as well as the Mz, representing the high molecular weight fraction, are at least doubled compared to the starting material. The high dispersity Mw/Mn shows additionally that a broader range of different molar mass molecules is present. For lignosulfonates, the change in MMD does not change significantly compared to the starting material. As the Mz value is most different, much more smaller molecular mass fractions seem to be present in the modified sample. Thus, when Indulin was used as starting material, condensation/polymerization and aggregation reactions dominated, whereas in the case of lignosulfonate, molar mass changes were minor, with a slight dominance of degradation reactions.
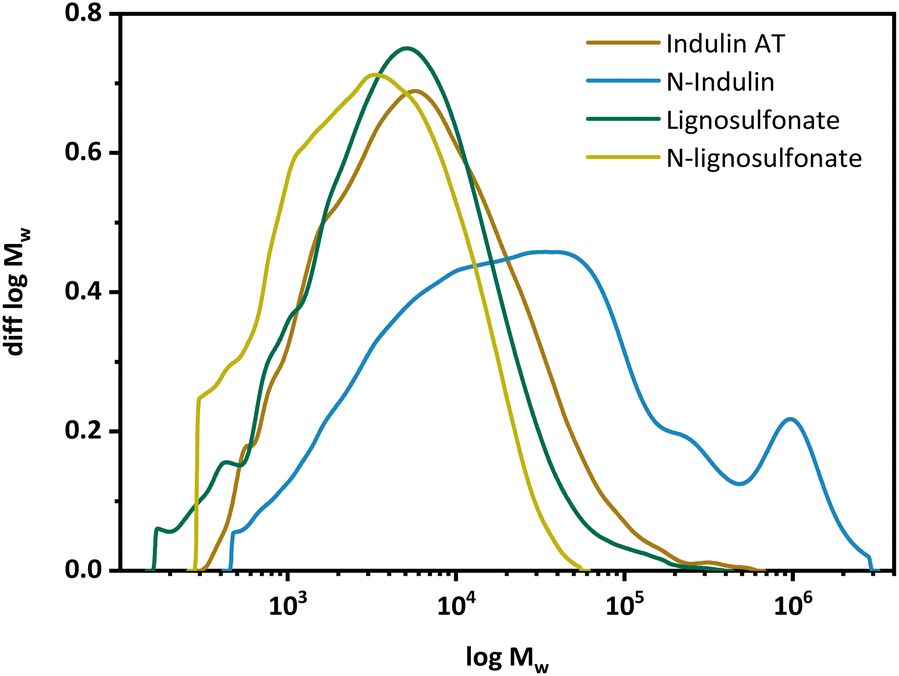 | ||
| Fig. 2 Molecular mass distributions of Indulin AT and lignosulfonate and the respective N-lignins. | ||
15N NMR spectra were recorded to give insights into which structures the N is incorporated into the lignin. For this, isotopic labelling with 15N was used and half of the ammonium hydroxide solution was replaced by 15NH4OH to obtain 50% isotopic enrichment. The spectra can be seen in Fig. 3. Reference data were taken from Potthast et al.24 and Fischer and Science.11 In the case of Indulin, the prominent resonances of ammonium ions at −358 ppm and urea at −304 ppm besides different types of amides between −240 ppm and −300 ppm were observed. Formamides gave a signal at −266 ppm, acetamides at −270 ppm, propionamides at −271 ppm and substituted benzamides at −280 ppm. Surprisingly, no peak at −358 ppm (ammonium) was observed for N-lignosulfonate, only the signals at −304 ppm and in the region of amides between −300 and −240 ppm were present. One possible reason for the absence of ammonium ions in case of the N-lignosulfonate could be the purification procedure. During evaporation, ammonia from ammonium salts is most likely volatised. In case of N-Indulin, in which precipitation was chosen as the purification procedure, ammonium ions remained in the sample. Below −240 ppm, no signals were present. Formation of nitriles, which would appear in the 15N NMR spectrum around −126 ppm, occurs only under harsher reaction conditions. No formation of nitriles was expected and seen under the conditions used.
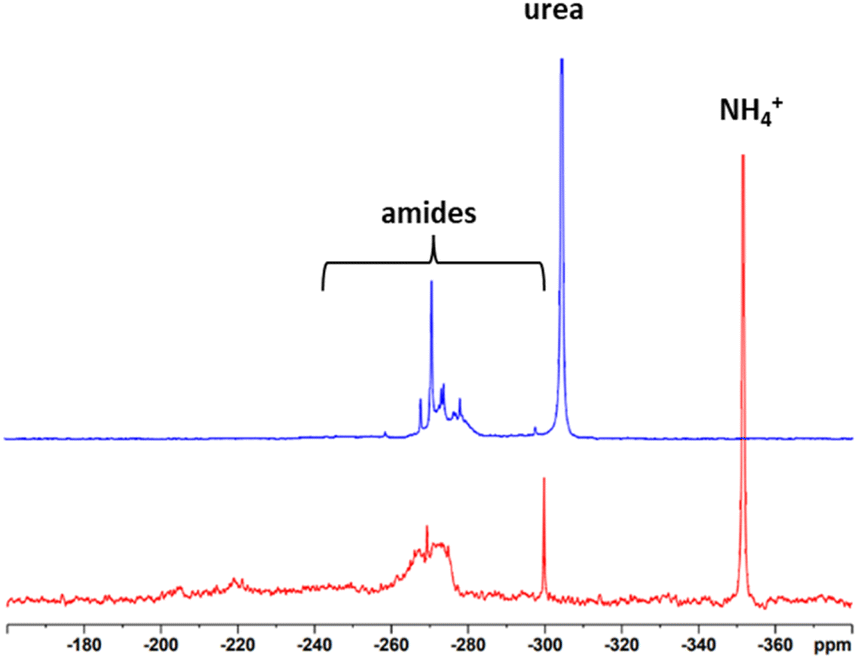 | ||
| Fig. 3 15N NMR spectrum of N-Indulin (red) and N-lignosulfonate (blue). | ||
1H/13C HSQC spectra of the ammonoxidized samples showed changes in the β-aryl ether, phenylcoumaran and resinol motifs, which were generall more drastic for the solid-state samples (Fig. 4C and F) than for the samples from conventional liquid-state ammonoxidation (Fig. 4B and E). The 1H/15N HMBC spectra did not provide much information beyond the interesting fact that some N was directly bound to (or in) aromatic ring structures (see the ESI† for an example spectrum). A detailed NMR analysis of the structural changes upon liquid-state and solid-state AO and of the differences between the two protocol variants will be the topic of an upcoming account.
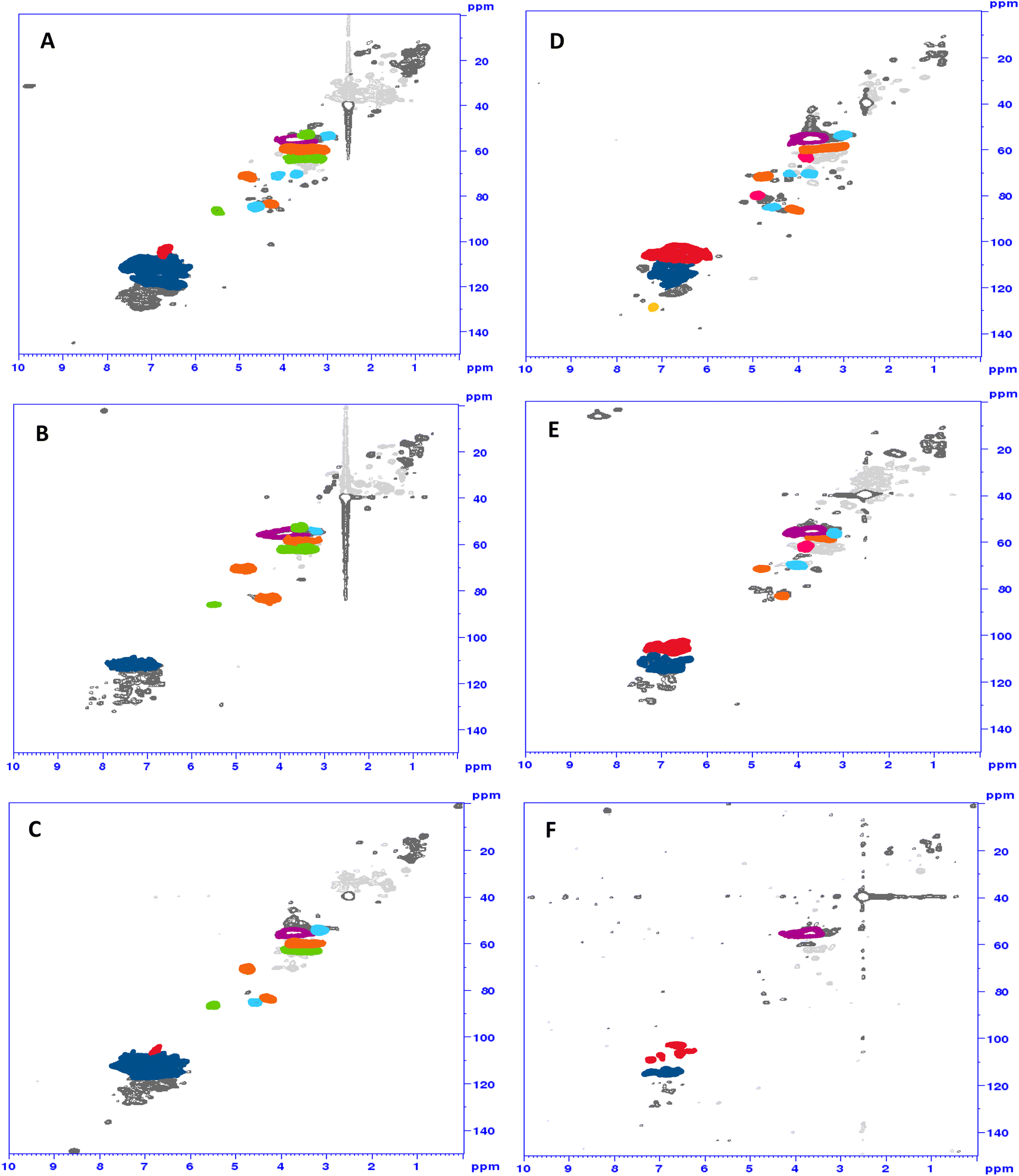 | ||
| Fig. 4 HSQC spectra of Indulin (A–C) and lignosulfonate (D–F): starting material (top), “classical” N-lignin (middle) and solid-state N-lignin (bottom). Red: S-units; dark blue: G-units; yellow: H-units; lila: methoxy groups; orange: β-aryl-ether; green: phenylcoumaran; light blue: resinol; pink: sulfonic acid group in α-position formaldehyde (formed through β,γ-C–C cleavage of the CH2–OH moiety) have already been identified by lignin model compounds.11,24 | ||
31P NMR spectra were recorded to determine the OH group contents before and after ammonoxidation, by reaction with a P-containing probe.42 To the best of our knowledge, no reference data for ammonoxidized N-lignin samples have been published yet. Due to the poor solubility of the modified lignosulfonate in the conventionally used solvents for 31P NMR of lignins (CDCl3/pyridine, CDCl3/DMF/pyridine and DMF/pyridine), a new solvent system had been developed as previously reported.39 This solvent system, containing the ionic liquid [emim]Cl additionally to the DMF/pyridine components, made solubilisation of the modified lignosulfonate and quantitation of their hydroxyl group contents via 31P NMR possible.
All types of aliphatic and aromatic hydroxyl groups in N-Indulin and N-lignosulfonate were significantly decreasing after modification (see ESI†). The total OH groups dropped to approx. one third for Indulin and to approx. one quarter in the lignosulfonate case. On the other hand, the COOH content increased, only moderately for Indulin, but rather drastically (six-fold) for lignosulfonate. The main mechanism decreasing aromatic hydroxyl groups is the oxidation of the lignins' aromatic rings to ortho-quinones, which is followed by several subsequent processes, such as direct oxidation to muconic acids under ring opening, the reaction with ammonia to quinone imines and oxidative ring opening to muconamides, or Michael-type addition of N-nucleophiles. Alternatively, oxidation of aromatic lignin rings with free 4-OH to para-quinone methide structures causes elimination of the C-α substituent (hydroxyl or alkoxyl) and, when followed by reaction with an N-nucleophile at C-α, N-incorporation under re-aromatisation. Direct substitution of hydroxy or alkoxy groups in the C3-aliphatic side chain are less likely to occur with ammonia than with primary or secondary amine structures already formed, because the latter are considerably better nucleophiles. Phenomena such as nitrogen being bound to the aromatic ring via 1,2-addition of ammonia to o-quinone structures, whereby quinone imines are formed and reduced by formaldehyde (formed through β,γ-C–C cleavage of the CH2–OH moiety) have already been identified by lignin model compounds.11,24
3.2 Transition: from liquid- towards solid-state ammonoxidation
Based on the results of the liquid-state ammonoxidation, the transition to a new process, which can be carried out completely in solid-state, was attempted. Eventually, this should create the possibility to carry out the lignin modification in an extruder or in a ball mill, without pressurized gas supply and liquid (= diluted) phases. The aqueous ammonium hydroxide solution was replaced by solid ammonium hydrogencarbonate, a harmless solid that is commonly used as baking salt, applied in the same molarity as NH4OH before. Under elevated temperatures, ammonium carbonate decomposes into equimolar amounts of ammonia, carbon dioxide and water. For the substitution of O2 as the oxidant, the urea hydrogen peroxide (UHP) complex, a white, crystalline solid, was used. This compound is used as a mild disinfectant in daily life. UHP starts to decompose at 60 °C,43 forming urea (which is further decomposed to ammonia and CO2) and H2O2, followed by the release of O2 at higher temperatures through the decay of H2O2.In the first experiment (Tr.-N-Ind. 1, see Table 3), NH4OH was replaced by NH4HCO3 while all other parameters from the liquid-state ammonoxidation were maintained. Despite the high temperature in the system and the high pressure from the degradation of the ammonium salt, comparatively little N was incorporated. The medium was neutral, thus the sample was only partially dissolved in the system, which could explain this result. Nevertheless, an N-content of about 4 wt% demonstrated that also under these conditions ammonoxidized lignin was generated. In the next experiment (Tr.-N-Ind. 2), no water was added, which yielded almost the same result. This run provided the proof that an ammonia hydroxide solution is not necessarily needed to implement N into lignins, although no high N-content was reached.
| Sample | NH4OH | NH4HCO3 | O2 | UHP | H2O | C% | H% | N% | S% | O% |
|---|---|---|---|---|---|---|---|---|---|---|
| Tr.-N-Ind. 1 | ✗ | ✓ | ✓ | ✗ | ✓ | 60.87 | 5.12 | 4.04 | 0.84 | 26.90 |
| Tr.-N-Ind. 2 | ✗ | ✓ | ✓ | ✗ | ✗ | 60.78 | 5.48 | 4.46 | 1.00 | 27.12 |
| Tr.-N-Ind. 3 | ✓ | ✗ | ✗ | ✓ | ✓ | 60.83 | 5.30 | 4.25 | 0.92 | 27.01 |
| Tr.-N-Ind. 4 | ✓ | ✗ | ✗ | ✓ | ✗ | 53.55 | 5.33 | 10.11 | 0.69 | 27.09 |
| Tr.-N-Ind. 5 | ✗ | ✓ | ✗ | ✓ | ✗ | 58.09 | 5.34 | 7.14 | 0.80 | 27.04 |
In the third run (Tr.-N-Ind. 3, see Table 3), only the gaseous oxygen was replaced by UHP. The pressure build-up in the vessel was solely obtained through the high temperature and the degradation of the reagents. In the system, a maximum pressure of 7 bar was reached. At a pH of around 12 and through the resulting good solubility of the sample, an N-content of around 4% was obtained, indicating that also at a lower pressure N can be incorporated into the lignin. Run Tr.-N-Ind. 4 equalled run Tr.-N-Ind. 3, but excluded the use of water. Interestingly, under these conditions, we reached a higher N-content of over 10%. Apparently, the higher effective concentration of hydrogen peroxide, without the diluting effect of additional water, caused an efficient oxidation providing enough functionalities for reaction with ammonia (and urea) with concomitant N-incorporation. Further, the pH in the system was high (pH ∼ 12), the overall pressure in the system was around 5–6 bar, but the lignin was rarely dissolved. In the final experiment (Tr.-N-Ind. 5, cf. Table 3), both the gaseous oxygen and the ammonium hydroxide solution were replaced by the solid alternatives, without water addition. Satisfyingly, an N-content >7% was reached, clearly demonstrating the general viability of the solid-state approach. It is important to note that the thermal degradation of UHP does not only generate O2 but also urea which, in turn, gets decomposed at elevated temperatures to a product mixture of cyanuric acid, biuret, carbon dioxide, and gaseous ammonia. This provides additional N-sources and reactive ammonia derivatives.44 The formed ammonia also increases the pH in the system to an alkaline range. These “transition” experiments proved that ammonoxidation of lignins with solid chemicals is possible and that sufficiently high amounts of nitrogen can be implemented into the lignins.
The transition samples were analytically characterized by FT-IR, GPC and 31P NMR. As in the case of the liquid-state modified samples, FT-IR spectra (Fig. 5) showed new bands occurring between 2750 and 3600 cm−1 and a new maximum at 3300 cm−1, which indicated the presences of amide N-bands and ammonium ions. These bands are especially pronounced in the Tr.-N-Ind. 4 and 5 samples. In all the five transition samples, new intensive bands between 1750 and 1550 cm−1 occur, which are assigned to CO, C–N and C–H–N stretching modes. Decreases at 1512 cm−1 and 1270 cm−1, which corresponds to a loss in aromatic structures and ether bonds, are rather minor.
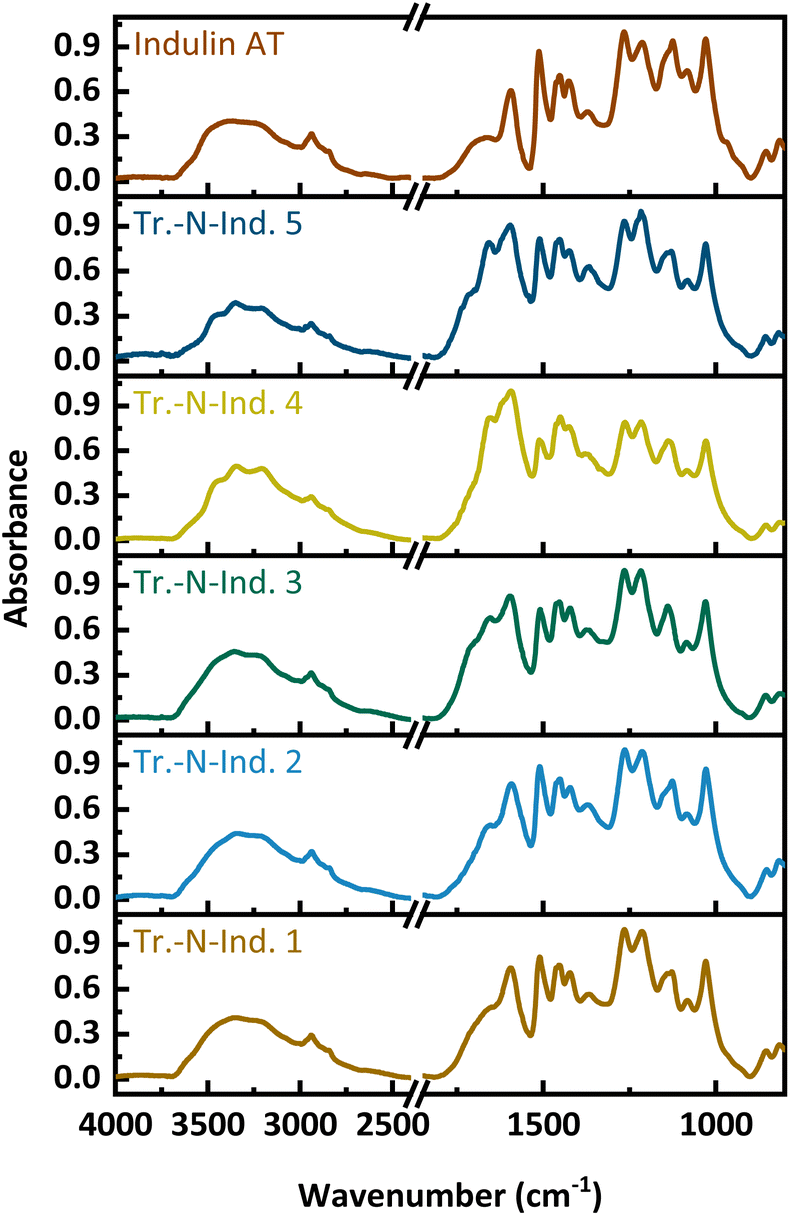 | ||
| Fig. 5 FT-IR spectra of the transition samples. | ||
GPC measurements (Fig. 6) showed the molar mass distribution to shift generally towards higher molar mass fractions. Also under varying ammonoxidation conditions, Indulin gets heavily condensed and the dispersity (diversity of low and high molecular weight molecules) increases significantly (see the ESI† for the statistical moments). The hydroxyl group content, as seen by 31P NMR after derivatization, generally decreases after modification, due to the mechanistic reasons presented above.
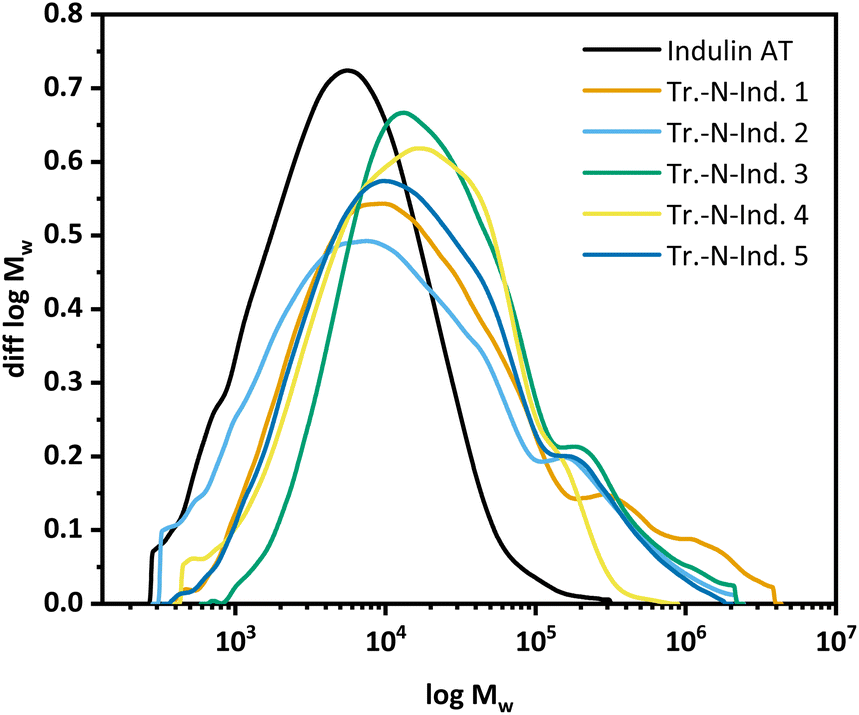 | ||
| Fig. 6 Molar mass distributions of the “transition” samples. | ||
In a series of follow-up experiments, carried out only with Indulin, the ratio of the reactants was optimised and the influence of the pH was tested. A summary of the reaction conditions and elemental analysis results is given in Table 4. In the samples SS-N-Ind. 1 to SS-N-Ind. 4, the amount of Indulin was increased while the number of reagents stayed the same. As expected, the higher the mass of Indulin, the less N was incorporated, although without a direct (reciprocal) proportionality. Importantly, even without ammonium carbonate as the ammonia source, a high amount of N was incorporated (SS-N-Ind. 5), indicating that UHP was able to function both as the N-source and the oxidant at the same time. Thus, only a single chemical was employed to modify the lignin in a solid-state ammonoxidation. Because of the good outcome, SS-N-Ind. 5 was used as the basis of the later solid-state experiments.
| Sample | Indulin (g) | NH4HCO3 (g) | NaOH | UHP (g) | C% | H% | N% | S% |
|---|---|---|---|---|---|---|---|---|
| SS-N-Ind. 1 | 0.55 | 2.4 | — | 2.6 | 52.20 | 5.07 | 7.75 | 0.66 |
| SS-N-Ind. 2 | 1.10 | 2.4 | — | 2.6 | 57.69 | 5.57 | 6.61 | 0.76 |
| SS-N-Ind. 3 | 2.20 | 2.4 | — | 2.6 | 59.78 | 5.59 | 4.18 | 0.87 |
| SS-N-Ind. 4 | 6.60 | 2.4 | — | 2.6 | 61.72 | 5.84 | 3.18 | 0.94 |
| SS-N-Ind. 5 | 0.55 | — | — | 2.6 | 50.17 | 4.95 | 9.55 | 0.58 |
| SS-N-Ind. 6 | 0.55 | — | — | 5.2 | 51.24 | 4.10 | 15.81 | 0.43 |
| SS-N-Ind. 6 dialysis | 0.55 | — | — | 5.2 | 50.15 | 3.80 | 15.62 | 0.49 |
| SS-N-Ind. 6 repeated | 0.55 | — | — | 5.2 | 51.40 | 4.09 | 14.84 | 0.84 |
| SS-N-Ind. 7 | 0.55 | — | ✓ | 2.6 | 55.22 | 4.81 | 8.17 | 0.54 |
| SS-N-Ind. 8 | 0.55 | 2.4 | — | 5.2 | 49.03 | 4.97 | 12.57 | 0.51 |
| SS-N-Ind. 9 | 0.55 | 2.4 | ✓ | 2.6 | 54.45 | 4.98 | 7.14 | 0.65 |
| SS-N-Ind. 10 | 6.60 | 2.4 | ✓ | 2.6 | 61.73 | 5.68 | 2.42 | 0.95 |
In the run SS-N-Ind. 6, the amount of UHP was doubled, and as the result a very high N-content was reached. Moreover, the N was shown to be strongly bound, as it was still present after dialysis to remove salts and low-molecular weight components. An independent experiment (SS-N-Ind. 6 rep.) confirmed the very high N-content of around 15%. Such high N-contents bound to Indulin are nearly out of reach and only attainable under rather severe and energy-intensive conditions according to the “classical” liquid-state ammonoxidation protocol. To determine the influence of the pH, powered NaOH was added in three runs (SS-N-Ind. 7, 9 and 10). The start pH for these experiments was therefore similar as in the “classical” ammonoxidation protocol (pH around 12). However, no higher N incorporation in the presence of NaOH was observed.
Based on these results, we concluded that the presence of ammonium hydrogencarbonate and NaOH was not needed and that the solid-state ammonoxidation can be carried out solely with UHP as the only solid reactant added. This renders the procedure both more facile and economical.
3.3 Solid-state ammonoxidation
Based on the optimized conditions (cf. sample SS-N-Ind. 5), a series of solid-state ammonoxidation experiments was carried out with Indulin and lignosulfonate. The amount of UHP was fixed while the amount of lignin was increased stepwise. For scaled-up procedures, the amount of used derivatization chemicals can be crucial in terms of cost, purification efforts and overall sustainability of the process. The detailed elemental analysis results from the solid-state ammonoxidation trials of Indulin and lignosulfonate are detailed in the ESI.† The N-content reached more than 9% in the case of Indulin and more than 11% in the case of lignosulfonate. With increasing UHP/lignin ratio, more N was incorporated. At higher ratios, the N-intake was higher for lignosulfonate, agreeing with its higher reactivity compared to Indulin as stated by Meier et al.25 The recovery yield was about 80–90% in case of Indulin and 60–70% for lignosulfonate. Losses occurred due to transfer, purification for analytical purposes and sample handling of the sticky samples and would be considerably higher for an upscaled, optimized procedure.It must be considered that with higher amounts of the respective lignin the miscibility in the system decreases. Especially in the solid-state approach, efficient and powerful mixing is crucial; in our case, magnetic stirring was used. The resulting pressure during the solid-state ammonoxidation was usually between 4 - 5 bar. Despite this pressure being comparatively low compared to the conventional solution state approach, high amounts of N (around 10%) were incorporated into the lignin. This indicated that with the new reaction system, neither a high pressure nor a high pH was needed to reach almost the same N-content as with the “classical” liquid-state ammonoxidation. The results provide clear proof that with the use of UHP as a single, solid co-reactant and a closed reactor without additional gas/pressure supply, a sufficiently efficient solid-state ammonoxidation with high N-incorporation was possible.
N incorporation was not reciprocally proportional to the amount of lignin used at a constant UHP amount. Between UHP/lignin ratios of 0.13 and 0.35 g UHP/g lignin, nearly the same N-content was achieved (Fig. 7), probably due to insufficient mixing. The amount of nitrogen incorporated can be controlled through the lignin-to-UHP ratio.
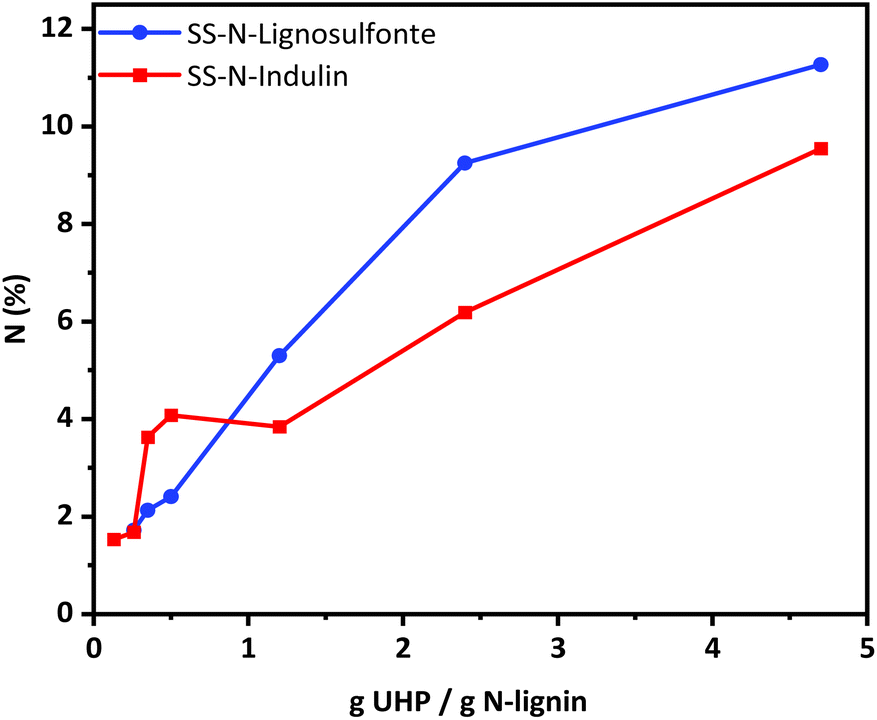 | ||
| Fig. 7 N-contents incorporated based on g UHP/g lignin. Red: Indulin, blue: lignosulfonate. | ||
Example FT-IR spectra of a solid-state ammonoxidized Indulin and lignosulfonate are shown in Fig. 8 for the samples with the highest N-content, SS-N-Ind. 5 and SS-N-LS 1, and the starting lignins; spectra of the other samples are in the supplemental info. In both spectra, the amide/amine band between 3300 and 3500 cm−1 is less pronounced than in the spectra of samples from the “classical” ammonoxidation (Fig. 1). The absorbance increased between 1550 and 1750 cm−1, which is again assigned to CO, C–N and C–H–N stretching bands, while the new maximum at 1400 cm−1 corresponds to the C–N stretching vibration. The decrease at 1512 cm−1 and 1270 cm−1, which corresponded to the loss in aromatic structures and ether bonds, was weaker compared to the “classical” ammonoxidation samples.
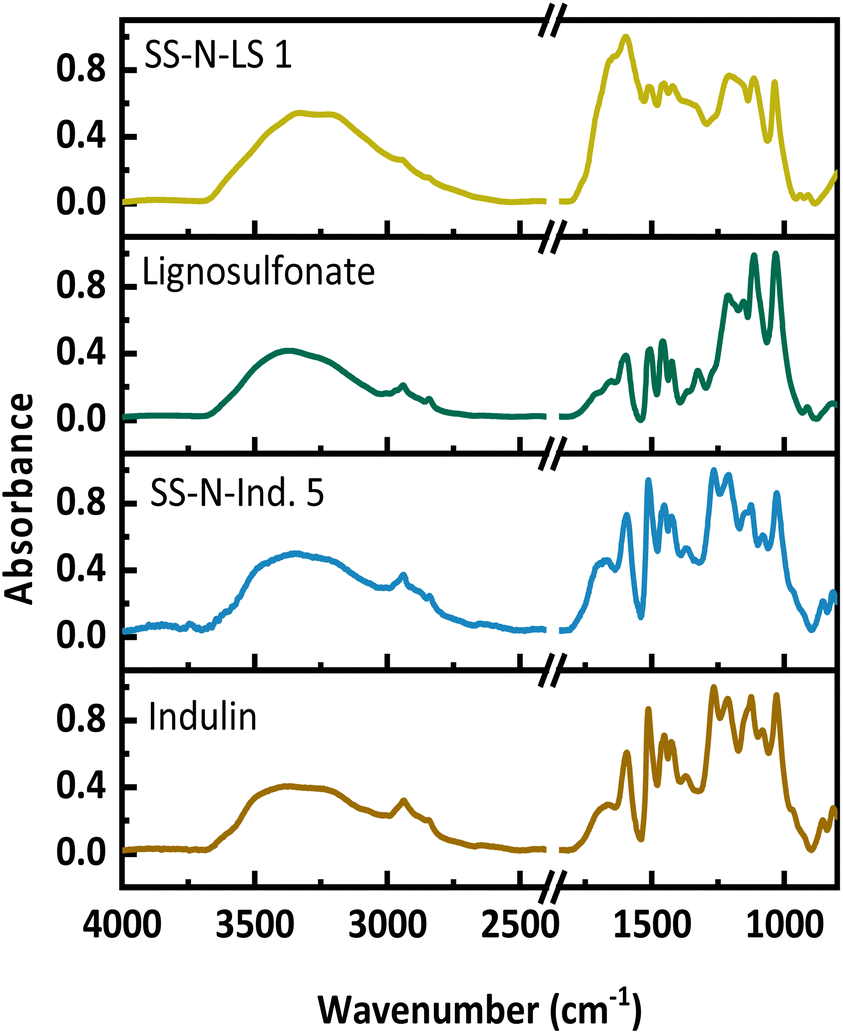 | ||
| Fig. 8 FT-IR spectra of the starting materials and the solid-state ammonoxidized Indulin and lignosulfonate samples SS-N-Ind. 5 and SS-N-LS 1. | ||
The analysis of the molar mass distribution of the solid-state N-Indulin samples was again performed by means of multi-angle light scattering (MALS) at 785 nm according to the protocol by Zinovyev et al. (2018),38 for the spectra see the ESI (page 11†). In all cases, the molar mass distribution was significantly shifted towards higher molar masses. This was mainly due to an increase of the high-molecular weight region, as emphasized by Mz, while the change in the low-molecular weight region (Mn) was rather minor. The dispersity index (Ð) was significantly increased for most of the samples compared to the starting material, which indicated an even broader molar mass distribution for the modified samples. In general, there was no clear correlation between the N-content and the statistical moments of the molar mass distributions.
The solid-state N-lignosulfonates were processed differently in GPC, because the samples caused high fluorescence interference, which would yield overestimated molar mass statistical moments with MALS detection. N-Modification caused a non-filterable fluorescence even at 785 nm. Therefore, the classical calibration approach was used: the molar mass fit of the reference sample (lignosulfonate) was used as a calibration curve for processing of the data of derivatized (ammonoxidized) samples. It is recommended to use the calibration approach for this set of samples as it provides more reliable (although not absolute) molar mass data. Surprisingly, the samples with the higher N-content had a molar mass profile more similar to the starting material than those with a lower N-content, for spectra see the ESI.† There was a general increase in the molar mass, which was stronger for samples with a lower N-content, and weaker for those with a high N-content.
To record meaningful 15N NMR spectra, we once more resorted to isotopic labelling. 15N UHP was synthesized from 15N urea and 30% H2O2 according to the procedure of Lu et al.45 and mixed with unlabelled UHP to achieve an isotopic enrichment of about 50%. Due to the fact that only half of the amount of the reaction chemical and lignin was used because of economic reasons (1.3 g of UHP and 0.28 g of lignin), the maximum pressure in the system reached only around 3 bar. Nevertheless, both samples had an N-content of about 7%. The 15N spectra of the SS-N-lignins are presented in Fig. 9. Both modified lignins show a sharp resonance at −358 ppm which is assigned to ammonium ions and another sharp peak at −304 ppm coming from urea. Additionally, the broad signals between −300 and −260 ppm originate from different amide structures. Intensive signals can be observed at −298 ppm and −260 ppm. These signals did not occur to such extent upon the “classical” AO, indicating that the amides formed in the solid-state process are more diverse. Assignment of these signals to a specific amide was not possible. Also previous studies with model compounds could not assign resonances in the amide range to specific compounds.24,27–30 Nevertheless, as with the “classical” ammonoxidised samples, the different binding types of N present are a definite advantage in terms of slow-release and long-term fertilization.
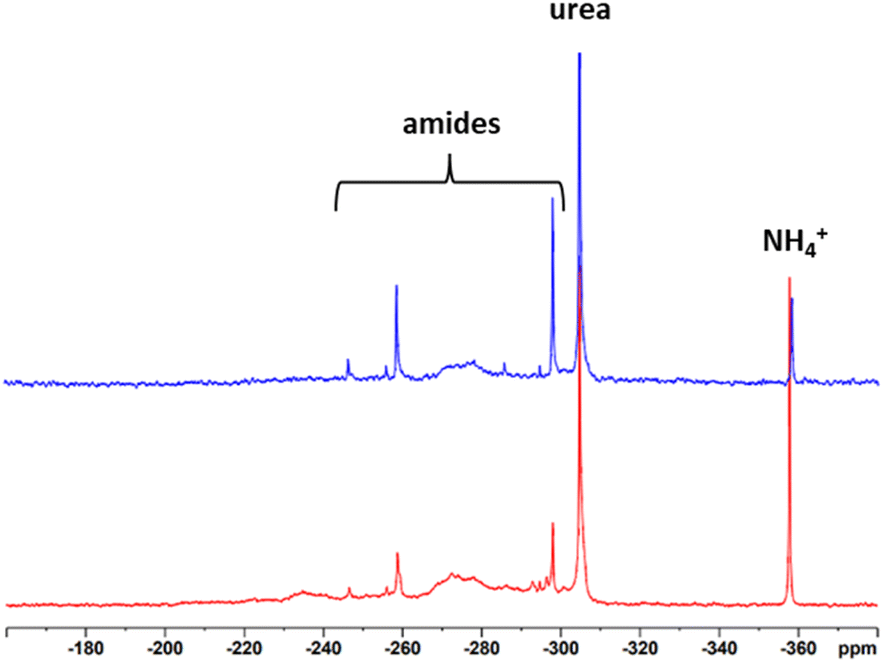 | ||
| Fig. 9 15N NMR spectra of SS-N-Indulin (red) and SS-N-LS (blue). | ||
The content of different hydroxyl groups was once more determined by 31P NMR. As in the case of the “classical” ammonoxidized samples, the DMF/pyridine solvent system was used for N-Indulin and the IL/DMF/pyridine solvent system for the lignosulfonate samples (see Tables 9 and 10 in the ESI†). Similar to the liquid-state ammonoxidized samples, a huge difference to the starting lignins was observed. For both lignins, the aliphatic and aromatic OH groups decreased significantly with increasing N-content, while the carboxylic OH groups increased. This points to the occurrence of similar processes as upon the classical ammonoxidation (oxidative ring opening, Michael-type additions to the products, carboxylic acid and subsequent amide formation, nucleophilic substitutions). This proves additionally that a significant part of the incorporated N in the samples is organically bound and not just present in the form of ammonium ions. It should be noted that the solid-state conditions and the absence of water should generally favour amide formation, condensation reactions and processes under water elimination.
4 Conclusions
A new protocol for the preparation of N-lignins was developed. With this new procedure the use of a single, non-hazardous and economical chemical, urea hydrogen peroxide complex, is sufficient to obtain N-lignins with an N-content of around 10% in the case of Indulin and 11% in the case of lignosulfonate. No additional water and gas supply are necessary. Further, the N is bound organically in a variety of different functional groups (ammonium, amide, amines, organically bound) similar to classical solution-state ammonoxidation samples. This opens the possibility to fabricate those N-lignins in other closed reactor system or in extruders.Lignin is an important precursor in the formation of humic substances. Naturally, their C/N ratio is quite high, which slows down the biological degradation. The more N is present, the better the substance becomes available for microorganisms. Therefore, the modification of lignin by ammonoxidation facilitates and mimics humus generation. The form of N present in the soil is another important key factor for soil quality. We proved that in the solid-state N-lignins nitrogen is present in many different forms. These binding types further influence the availability of N over time: NH4+ is readily available and covalently bound N (amides, amines, other organically bound N) shows a slower hydrolysis and thus a retarded release effect.
The use of lignin for agricultural purposes, in the form of N-lignins, does not only mean valorization of a waste product. More than that, it could replace potentially harmful chemicals, and improve the soil quality. Eventually, it helps mitigating the climate crisis: its action as a carbon sink to sequester CO2 and store it in the soil is an important favourable quality of artificial humic matter.
According to the new protocol, purification of the generated N-lignin can be avoided, depending on the respective application. This shortens the overall preparation procedure. With any residual ammonium salts or urea from the UHP coreactant, an additional short- and mid-term fertilizing effect can be achieved, while the N bound to lignin covers the longer-term fertilizing period. The fertilising properties of the solid-state ammonoxidized materials are currently tested in different systems. We hope that our improved protocol might help to induce a revival of ammonoxidized lignins as fertilizers and soil conditioners with many advantages.
Author contributions
G. K. Wurzer: data curation, investigation, methodology, visualization, writing – original draft. M. Bacher and O. Musl: data curation, investigation, writing – original draft. N. Kohlhuber, I. Sulaeva: data curation and visualization. T. Kelz: data curation. K. Fackler and R. H. Bischof: writing – review and editing. H. Hettegger: methodology, supervision, writing – original draft. A. Potthast: data curation, methodology, supervision. T. Rosenau: supervision, conceptualization, methodology, writing – original draft and revision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the University of Natural Resources and Life Sciences, Vienna (BOKU), the County of Lower Austria, and Lenzing AG for their financial support in the framework of the Austrian Biorefinery Center Tulln (ABCT) and the BOKU doctoral school “Advanced Biorefineries – Chemistry & Materials” (ABC&M). Further, we gratefully acknowledge Mag. J. Theiner at the Faculty of Chemistry of the University of Vienna for elemental analysis.Notes and references
- P. E. Fixen and F. B. West, AMBIO A J. Hum. Environ., 2002, 31, 169–176 CrossRef PubMed.
- P. P. Motavalli, K. W. Goyne and R. P. Udawatta, Crop Manag., 2008, 7, 1–15 CrossRef.
- D. Stuart, R. L. Schewe and M. McDermott, Land Use Pol., 2014, 36, 210–218 CrossRef.
- K. C. Cameron, H. J. Di and J. L. Moir, Ann. Appl. Biol., 2013, 162, 145–173 CrossRef CAS.
- A. M. Manschadi and A. Soltani, Field Crop. Res., 2021, 270, 108211 CrossRef.
- D. Stuart, R. L. Schewe and M. McDermott, Land Use Pol., 2014, 36, 210–218 CrossRef.
- P. M. Glibert, J. Harrison, C. Heil and S. Seitzinger, Biogeochemistry, 2006, 77, 441–463 CrossRef CAS.
- K. Chojnacka, K. Moustakas and A. Witek-Krowiak, Bioresour. Technol., 2020, 295, 122223 CrossRef CAS PubMed.
- J. Martínez-Dalmau, J. Berbel and R. Ordóñez-Fernández, Sustainability, 2021, 13, 5625 CrossRef.
- B. H. Byrnes, Fert. Res., 1990, 26, 209–215 CrossRef CAS.
- K. Fischer and R. Schiene, in Chemical Modification, Properties, and Usage of Lignin, ed. T. Q. Hu, Springer US, Boston, MA, 2002, pp. 167–198 Search PubMed.
- F. Ramírez, V. González, M. Crespo, D. Meier, O. Faix and V. Zúñiga, Bioresour. Technol., 1997, 61, 43–46 CrossRef.
- A. Abaecherli and V. I. Popa, Environ. Eng. Manag. J., 2005, 4, 273–292 CrossRef CAS.
- G. K. Wurzer, H. Hettegger, R. H. Bischof, K. Fackler, A. Potthast and T. Rosenau, Holzforschung, 2022, 76, 155–168 CrossRef CAS.
- C. Moretti, B. Corona, R. Hoefnagels, I. Vural-Gürsel, R. Gosselink and M. Junginger, Sci. Total Environ., 2021, 770, 144656 CrossRef CAS PubMed.
- M. Balakshin, E. A. Capanema, I. Sulaeva, P. Schlee, Z. Huang, M. Feng, M. Borghei, O. Rojas, A. Potthast and T. Rosenau, ChemSusChem, 2021, 14(4), 1016–1036 CrossRef CAS PubMed.
- T. Aro and P. Fatehi, ChemSusChem, 2017, 10, 1861–1877 CrossRef CAS PubMed.
- M. Ek, G. Gellerstedt and G. Hendriksson, Pulping chemistry and technology, De Gruyter, Berlin, 2009 Search PubMed.
- M. Ek, G. Gellerstedt and G. Hendriksson, Wood chemistry and biotechnology, De Gruyter, Berlin, 2009 Search PubMed.
- M. Ek, G. Gellerstedt and G. Hendriksson. Paper products physics and technology, De Gruyter, Berlin, 2009 Search PubMed.
- M. Ek, G. Gellerstedt and G. Hendriksson. Paper chemistry and technology, De Gruyter, Berlin, 2009 Search PubMed.
- A. Berlin and M. Balakshin, in Bioenergy Research: Advances and Applications, Elsevier, 2014, pp. 315–336 Search PubMed.
- F. Gomollón-Bel, Chem. Int., 2021, 43(4), 13–20 Search PubMed.
- A. Potthast, R. Schiene and K. Fischer, Holzforschung, 1996, 50, 554–562 CrossRef CAS.
- D. Meier, V. Zúñiga-Partida, F. Ramírez-Cano, N.-C. Hahn and O. Faix, Bioresour. Technol., 1994, 49, 121–128 CrossRef CAS.
- C. González, R. Alvarez and J. Coca, Water, Air, Soil Pollut., 1992, 61, 191–199 CrossRef.
- E. A. Capanema, M. Yu. Balakshin, C.-L. Chen, J. S. Gratzl and A. G. Kirkman, Holzforschung, 2001, 55, 397–404 CAS.
- E. A. Capanema, M. Yu. Balakshin, C.-L. Chen, J. S. Gratzl and A. G. Kirkman, Holzforschung, 2001, 55, 405–412 CAS.
- E. A. Capanema, M. Y. Balakshin, C.-L. Chen, J. S. Gratzl and A. G. Kirkman, Holzforschung, 2002, 56, 402–415 CAS.
- E. A. Capanema, M. Y. Balakshin, C.-L. Chen and J. S. Gratzl, J. Wood Chem. Technol., 2006, 26, 95–109 CrossRef CAS.
- C. Lapierre, B. Monties, D. Meier and O. Faix, Holzforschung, 2009, 48, 63–68 CrossRef.
- F. Ramírez-Cano, A. Ramos-Quirarte, O. Faix, D. Meier, V. González-Alvarez and V. Zúñiga-Partida, Bioresour. Technol., 2001, 76, 71–73 CrossRef PubMed.
- J. M. De la Rosa, F. Liebner, G. Pour and H. Knicker, Soil Biol. Biochem., 2013, 60, 125–133 CrossRef CAS.
- F. Liebner, G. Pour, J. la R. A. De, A. Hilscher, T. Rosenau and H. Knicker, Angew Chem. Int. Ed., 2011, 50, A34–A39 CrossRef PubMed.
- K. M. Klinger, F. Liebner, T. Hosoya, A. Potthast and T. Rosenau, J. Agric. Food Chem., 2013, 61, 9015–9026 CrossRef CAS PubMed.
- K. M. Klinger, F. Liebner, I. Fritz, A. Potthast and T. Rosenau, J. Agric. Food Chem., 2013, 61, 9004–9014 CrossRef CAS PubMed.
- I. Sumerskii, P. Korntner, G. Zinovyev, T. Rosenau and A. Potthast, RSC Adv., 2015, 5, 92732–92742 RSC.
- G. Zinovyev, I. Sulaeva, S. Podzimek, D. Rössner, I. Kilpeläinen, I. Sumerskii, T. Rosenau and A. Potthast, ChemSusChem, 2018, 11, 3259–3268 CrossRef CAS PubMed.
- G. K. Wurzer, M. Bacher, H. Hettegger, I. Sumerskii, O. Musl, K. Fackler, R. H. Bischof, A. Potthast and T. Rosenau, Anal. Methods, 2021, 13, 5502–5508 RSC.
- M. Balakshin and E. Capanema, J. Wood Chem. Technol., 2015, 35, 220–237 CrossRef CAS.
- A. W. T. King, L. Zoia, I. Filpponen, A. Olszewska, H. Xie, I. Kilpeläinen and D. S. Argyropoulos, J. Agric. Food Chem., 2009, 57, 8236–8243 CrossRef CAS PubMed.
- P. Korntner, I. Sumerskii, M. Bacher, T. Rosenau and A. Potthast, Holzforschung, 2015, 69, 807–814 CrossRef CAS.
- R. Matyáš, J. Selesovsky, V. Pelikán, M. Szala, S. Cudziło, W. A. Trzciński and M. Gozin, Propellants Explos. Pyrotech., 2017, 42, 198–203 CrossRef.
- S. Tischer, M. Börnhorst, J. Amsler, G. Schoch and O. Deutschmann, Phys. Chem. Chem. Phys., 2019, 21, 16785–16797 RSC.
- C.-S. Lu, E. W. Hughes and P. A. Giguère, J. Am. Chem. Soc., 1941, 63, 1507–1513 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00691c |
| This journal is © The Royal Society of Chemistry 2023 |