
N-heterocyclic carbene-ligated metal complexes and clusters for photocatalytic CO2 reduction
Yilin
Jiang
and
Honghan
Fei
 *
*
Shanghai Key Laboratory of Chemical Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, 1239 Siping Rd., Shanghai 200092, P. R. China. E-mail: fei@tongji.edu.cn
First published on 31st May 2023
Abstract
Photocatalytic reduction of CO2 to high value-added products is a green and sustainable approach to reducing excessive CO2 emissions. N-heterocyclic carbenes (NHCs) are a class of structurally versatile ligands due to their strong σ-donor properties, forming covalent bonds with many metal centers, and ability to activate CO2 molecules. The resulting NHC-ligated metal centers and clusters have high stability, tunable π-conjugation and unique reactivity, making them excellent candidates for CO2 photoreduction. Herein, this frontier article provides a comprehensive review of the literatures on active NHC-stabilized metal complexes and clusters for photocatalytic CO2 reduction. We discuss the synthetic strategies of NHC-ligated metal species, including their incorporation into porous matrices such as metal–organic frameworks (MOFs). We also summarize their photocatalytic applications in CO2 reduction, providing guidance for the rational design and development of NHC-based MOF photocatalysts for efficient and sustainable CO2 photoreduction.
1 Introduction
Excessive carbon dioxide (CO2) emissions resulting from the combustion of fossil fuels are a global problem that causes serious environmental issues, including the greenhouse effect responsible for global warming.1,2 To address these issues, the photocatalytic conversion of CO2 into high-value products using solar energy has recently gained increasing attention since the discovery of the Fujishima–Honda effect in 1972.3 The ultimate goal is to couple the half-reaction of CO2 reduction with the half-reaction of water oxidation to mimic the natural photosynthesis.4,5Metal complexes (e.g., porphyrins,6–10 phthalocyanines,11,12 pyridine and amine derivatives13–15) and metal clusters16,17 have emerged to become a promising class of photocatalysts for CO2 reduction. These catalysts allow for precise structural modifications at the atomic level, facilitating the rational design and understanding of the structure–property relationship for CO2 photoreduction. The tunable activity and rich redox valence of metal species enable the modulation of their reduction potentials for photocatalysis. Importantly, the metal–ligand coordination motifs, especially the interface between metal clusters and surface ligands, play a critical role in improving the catalytic turnover numbers (TON) and turnover frequencies (TOF). Sufficient contact between the reaction substrates and the catalytic sites often leads to high TON and TOF. Additionally, the plasmonic nanosized metals (e.g., Au, Ag and Cu) are able to harvest visible light by the localized surface plasmon resonance (LSPR) effect to enhance photon absorption and photo-induced hot electron transport.18–20
In recent years, N-heterocyclic carbenes (NHCs) have become a well-established family of strong σ-donor ligands with diverse stereochemical and electronic features, capable of forming covalent bonds with many metal centers.21,22 More importantly, the reactive basic nature of NHCs finds them in potential applications for CO2 activation and catalytic conversion.23–25 Therefore, the unique properties and stability of metal–NHC catalysts make them highly desirable in photocatalytic CO2 reduction. For example, Chang and colleagues recently reported a basic homogeneous Ni2+–NHC catalyst for solar-driven catalytic conversion of CO2 into CO, with TON and TOF reaching 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 3.9 s−1, respectively.26 Although photosensitizers and electron donors are still required, this represents the first system combining visible-light excitation with first-row transition metal centers to achieve sustainable CO2 conversion with no detectable H2 formation from the proton reduction process. This finding demonstrated the potential of NHC motifs as molecular building blocks for application in solar-to-fuel transformation reactions.
000 and 3.9 s−1, respectively.26 Although photosensitizers and electron donors are still required, this represents the first system combining visible-light excitation with first-row transition metal centers to achieve sustainable CO2 conversion with no detectable H2 formation from the proton reduction process. This finding demonstrated the potential of NHC motifs as molecular building blocks for application in solar-to-fuel transformation reactions.
This frontier article provides a comprehensive summary of the literature on active NHC-stabilized metal complexes and clusters for photocatalytic CO2 reduction. The review article discusses the synthetic strategies of NHC-ligated metal complexes and clusters, as well as their incorporation into a porous matrix such as metal–organic frameworks (MOFs). Moreover, the article summarizes the catalytic applications of NHC-based photocatalysts in CO2 reduction. We hope the article will provide scientific insights into the rational design and development of NHC-based metal complexes and nanoclusters for efficient and selective CO2 photoreduction.
2 Synthesis of and photocatalytic CO2 reduction by metal–NHC complexes
2.1 Ni complexes
Rational modification of the NHC ligand structure shows great promise in developing new molecular photocatalysts for CO2 reduction with enhanced catalytic rates, selectivity and longevity. Two reliable strategies for designing CO2 reduction photocatalysts include strengthening the π-conjugation system and tuning the substituents of the NHC ligands.First, expanding π-conjugation can stabilize key intermediates by electron delocalization. For instance, Chang and coworkers reported that catalytic activities can be significantly improved by expanding the π-conjugated systems of tetradentate NHC–pyridine complexes.26 This was indeed the first type of metal–NHC complex for visible-light photocatalytic CO2 reduction. The typical synthesis involves the treatment of an imidazolium salt with Ag2O to form the AgI–NHC complex, followed by transmetalation with NiII. The resulting NiII–NHC–isoquinoline complex, [Ni(Prbimiq1)]2+ (Prbimiq1 = bis(3-(imidazolyl)isoquinolinyl)propane), was employed in photocatalytic CO2 reduction, in conjunction with Ir(ppy)3 (ppy = 2-phenylpyridine) as the photosensitizer and triethylamine (TEA) as the electron donor (Fig. 1). The photocatalytic system achieved the highest TON and TOF values of 98000 and 3.9 s−1, respectively, as determined by CO production and the [Ni(Prbimiq1)]2+ complex (Table 1, entry 1). These values may have been limited by the generation of an active nickel center and the CO2 conversion by the reduced nickel center. The high CO2 reduction selectivity over proton reduction was confirmed by no detectable H2 evolution. Compared to CdS semiconductive powders, this Ni–NHC molecular photocatalytic system exhibited two orders of magnitude higher solar-to-fuel efficiency.27,28 The limiting factor for long-term CO2 reduction is the photosensitizer degradation rather than catalyst deactivation. Despite the use of a noble-metal photosensitizer and a sacrificial agent, these results revealed the ability of the NHC–amine skeleton to form molecular building blocks for metal–NHC photocatalysis in solar-to-fuel transformations.
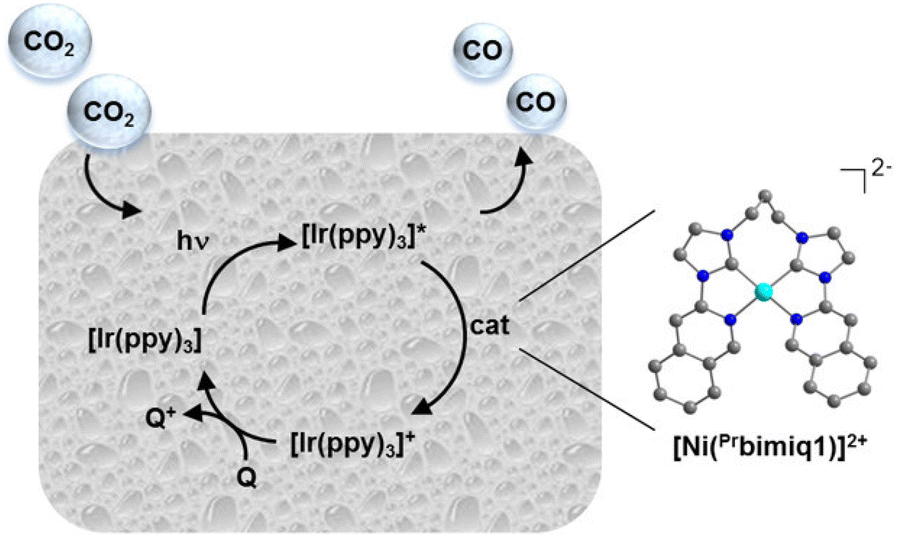 | ||
| Fig. 1 Photocatalytic CO2 reduction process by the [Ni(Prbimiq1)]2+ complex. Reprinted with permission from ref. 26. Copyright 2013 American Chemical Society. | ||
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Compound | Photosensitizer | Sacrificial reductant | QY (%) | Selectivityb (CS) (%) | TONCO | TONH2 | TONCH4 | Ref. |
| a QY, quantum yields; TON, turnover numbers; TEA, triethylamine; MeCN, acetonitrile; BIH, 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]-imidazole; DMF, N,N-dimethylformamide; Pheno, 3,7-di([1,1′-biphenyl]-4-yl)-10-(naphthalen-1-yl)-10H-phenoxazine; TEAB, tetraethylammonium bicarbonate. b CS (carbon-selective) reduction percentage is calculated as CS % = ((CO TON + CH4 TON)/total observed products TON) × 100. c TON after photocatalysis for 7 h with 2 nM catalyst, 0.2 mM Ir(ppy)3, 0.07 M TEA. d TON after photocatalysis for 6 h in an acetonitrile solution with catalyst, Ir(ppy)3, BIH and TEA. e TON after photocatalysis for 6 h in a DMF solution with catalyst, Ir(ppy)3, BIH and TEA. f TON after photocatalysis for 72 h with 2 nM catalyst, 0.1 mM Ir(ppy)3, 10 mM BIH and 5% v/v TEA. g TON after photocatalysis for 72 h with 2 nM catalyst, 0.1 mM Ir(ppy)3, 10 mM BIH, 5% v/v TEA and 2% v/v H2O. h TON after photocatalysis for 4 h with 0.1 mM catalyst, 0.1 mM Ir(ppy)3, 11 mM BIH and 5% v/v TEA. i TON after photocatalysis for 1 h with 0.05 mM catalyst, 0.1 mM Pheno, 11 mM BIH, 0.1 M TEAB and 2% v/v H2O. j TON after photocatalysis for 40 h in an acetonitrile solution with catalyst, Ir(ppy)3, BIH and TEA. k TON after photocatalysis with 1 nM catalyst, BIH and TEA. l TON after photocatalysis for 4 h with 0.1 mM catalyst, 10 mM BIH and 5% v/v TEA. m TON after photocatalysis for 72 h with 1 μM catalyst, 0.1 mM Ir(ppy)3, 20 mM BIH and 5% v/v TEA. | |||||||||
| 1 | 1 | Ir(ppy)3 | TEA | 0.01 | 100 | 98000c |
0 | Trace | 26 |
| 2 | 2 | Ir(ppy)3 | BIH, TEA | — | — | 10.6d | — | — | 29 |
| 3 | 2 | Ir(ppy)3 | BIH, TEA | — | — | 9e | — | — | 29 |
| 4 | 3 | Ir(ppy)3 | BIH, TEA | — | 90 | 310000f |
33000 |
0 | 30 |
| 5 | 3 | Ir(ppy)3 | BIH, TEA, H2O | 5 × 10−3 | 87 | 175000g |
29000 |
19000 |
30 |
| 6 | 4 | Ir(ppy)3 | BIH, TEA | — | 100 | 102.6h | 0 | 0 | 31 |
| 7 | 4 | Pheno | BIH, TEAB, H2O | 11.2 | 100 | 101i | 0 | 0 | 31 |
| 8 | 5 | Ir(ppy)3 | BIH, TEA | — | 100 | 250j | 0 | 0 | 32 |
| 9 | 6 | Self-sensitized | BIH, TEA | 5.4 × 10−5 | — | 33000k |
— | — | 33 |
| 10 | 7 | Self-sensitized | BIH, TEA | — | — | 32l | — | — | 34 |
| 11 | 8 | Ir(ppy)3 | BIH, TEA | — | > 97.8 | 45m | Trace | 0 | 36 |
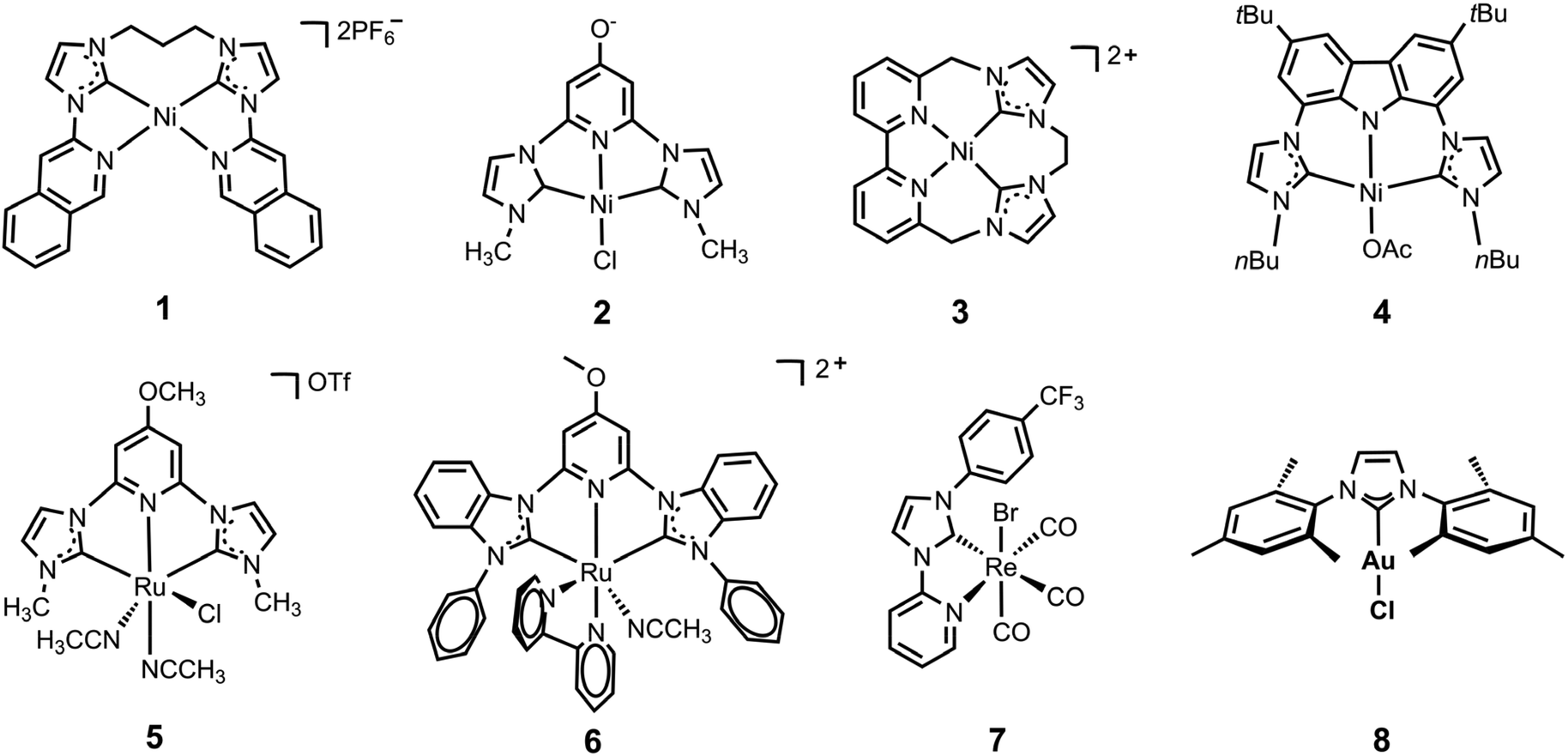
Second, the stronger electron-donating group often facilitates CO2 activation, which is usually considered the rate-limiting step for CO formation. Webster, Delcamp, Papish and co-workers revealed a surprising effect from a remote O-group on a pincer NiII complex, resulting in dramatically enhanced activity.29 They used a NiII–pyridinol–NHC-pincer complex in place of the Ru metal center to continue research on the electronic modifications at the metal center by introducing a protic donor group (–OH) at the para (to N) position of the pincers. The NiII–NHC–pyridinol–NHC-pincer catalyst can effectively catalyse the photoreduction of CO2 to CO in the presence of Ir(ppy)3 and 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]-imidazole (BIH), with a TON of 10.6 (Table 1, entry 2). However, the photocatalytic activity only lasts for 6 hours in the presence of a photosensitizer. In contrast, the pyridine–NHC complex without a remote O-substituent demonstrated negligible catalytic activity. Moreover, this catalyst exhibited reversible in situ protonation or deprotonation, which enabled the switching on or off of its photocatalytic performance. The incorporation of oxygen-bearing groups into CNHCNCNHC pincer ligands was found to be highly effective in NiII-catalyzed CO2 photoreduction, which effectively lowered the redox potentials and facilitated the electron transfer from the photosensitizer to the catalyst and from the catalyst to CO2 during the first reduction potential.
Delcamp and colleagues investigated the influence of macrocyclization and ligand planar geometry on catalytic reactivity of the redox-active NiII–NHC–bipyridyl complexes under aqueous conditions.30 Their study revealed that both macrocyclic structures exhibit high selectivity for CO2 reduction over proton reduction, while the less planar macrocycle structure demonstrates greater catalytic activity with a TON of 19000 for CO2 to CH4 conversion in the presence of water (Table 1, entries 4 and 5). The introduction of an additional aliquot of Ir(ppy)3 will again turn-on the photocatalysis process, suggesting the robust nature of the active nickel photocatalyst. This unique reactivity from a tunable, highly durable macrocyclic framework was studied via a series of photocatalytic and electrocatalytic reactions at different atmosphere compositions. The NiII–NHC–bipyridyl complex afforded a TON of 12000 for CH4 under a 1:1 CO2: H2 atmosphere, while a substantially increased TON of 570000 was achieved under a 1:1 CO:H2 atmosphere (CO in place of CO2) (Fig. 2). The isotope labeling experiments revealed that both H2 and CO were essential for increasing the CH4 TON value. These results demonstrate the potential of NHC and bipyridyl as ligand scaffolds for NiII-photocatalysis.
 | ||
| Fig. 2 Photocatalytic reduction of CO2/CO to CH4 by the NiII–NHC–bipyridyl complex. Reprinted with permission from ref. 30. Copyright 2019 American Chemical Society. | ||
Inspired by previous studies, Ke and coworkers were motivated to simultaneously apply these two strategies on one CO2-reduction catalyst to achieve enhanced catalytic performance by the introduction of a strong electron donor with large π-conjugation.31 They reported the development of a highly efficient NiII–bis(NHC)–carbazolide catalyst for selective CO2 photoreduction. The introduction of the carbazolide fragment into the scaffold significantly enhanced the photocatalytic activity, attributed to the strong characteristic σ/π donor and large π-conjugation of the carbazolide moieties. The NiII–bis(NHC)–carbazolide catalyst exhibited TON and TOF values that were 8- to 9-fold higher than those of the NiII–bis(NHC)–pyridinol catalyst at the same catalyst concentration using an identical photosensitizer (Table 1, entries 2 and 6).29 The ligand modifications altering the electron density at the metal center were proved again to be a viable strategy to improve the reactivity and selectivity of the photocatalysts. Notably, the authors employed an organic dye to replace the Ir photosensitizer and develop a noble-metal-free photocatalytic system with an outstanding quantum yield of 11.2% (Table 1, entry 7).
2.2 Ru complexes
CNC-pincer ligands, which combine the strong s-donor and moderate p-acceptor properties of the NHC and pyridine rings, have been shown to exhibit higher thermal stability and have thus garnered significant attention. Papish and coworkers reported the synthesis of (OMeCNC)RuII, a ruthenium complex coordinated by an NHC precursor, that was deprotonated with CsCO3 and treated with [(p-cymene)RuCl2]2 in acetonitrile.32 By affixing a pyridinol–NHC ligand to the ruthenium center, the authors evaluated the effects of modulating the electron density of the metal centers by introducing the methoxy group at the para (to N) position. The resulting 4-methoxy-pyridine derived RuII–NHC–pyridyl–NHC-pincer catalysts show greater durability in visible-light photocatalytic CO2 reduction, reaching a TON of 250 over 40 h with an initial TOF of 15 h−1 (Table 1, entry 8). The significant increase in durability is ascribed to the strongly chelating NHC and 4-methoxy-pyridine pincer ligand. The unsubstituted analog, with H in place of the methoxy group, was found to be inactive. These results suggest that the enhanced electron-donor properties of the 4-methoxy-pyridine substituent contribute to the increased catalyst stability through its strong chelation with metal centers.The same group continued to report a series of pincer RuII–NHC complexes for photocatalytic self-sensitized CO2 reduction to CO.33 The expanded ligand π-conjugated system of benzimidazole-derived NHC rings increases the light-harvesting ability of the photocatalysts, demonstrating that CO2 reduction without an additional photosensitizer is feasible and can produce quantities of CO (TON of 33000 and TOF of 250 h−1) comparable to the sensitized reactions (Table 1, entry 9).
2.3 Re complexes
A series of ReI–pyridyl–NHC (ReI–PyNHC) complexes with different substituents, including a resonance-donating –OC6H13 group, an electron-neutral –H group and an electron-deficient –CF3 group, have been synthesized by Delcamp and coworkers.34 Among them, Re(PyNHC-PhCF3)(CO)3Br with the electron-withdrawing substituent was found to exhibit the highest photocatalytic CO2 reduction activity, even outperforming the benchmark Re(bpy)(CO)3Br in the presence of BIH as electron donor and fac-Ir(ppy)3 as photosensitizer. Indeed, the Re complex was also found to function without a photosensitizer to afford a TON of 32, exceeding the TON of 14 for Re(bpy)(CO)3Br (Table 1, entry 10). However, all photocatalysts were found to be inactive after 4 h irradiation. The robustness of the ReI–pyridyl–NHC catalyst, Re(PyNHC–PhCF3)(CO)3X (X = Cl/Br), was investigated by the same group with respect to the introduction of water and oxygen during photocatalytic CO2 reduction.35 The NHC ligands afford enhanced stability under anhydrous conditions by retaining 67% of their peak performance, while the benchmark Re(bpy)(CO)3Cl retains 36% reactivity under anhydrous conditions. The introduction of ambient air into the reaction also affected the reaction. Under 40% ambient atmosphere, Re(PyNHC-PhCF3)(CO)3Cl retained 50% of the photocatalytic performance it had in a 100% CO2 atmosphere, while 91% reduction in TON was observed in the benchmark Re(bpy)(CO)3Cl. These results highlight the contribution of high robustness of ReI-based photocatalysts through NHC ligands.2.4 Au complexes
Nugegoda et al. reported the first examples of Au-based NHC-ligated molecular catalysts for CO2 photoreduction.36 They studied the photocatalytic performances of eleven Au–NHC complexes, which varied in their wingtip substituents, the saturated or unsaturated NHC backbone and the labile ligand (counterion) on the Au center. Among them, the mesitylene wingtip-bearing complex showed the highest TOF of 8.4 h−1 and TON of 45, ascribed to the small wingtip ligand, unsaturated backbone and easily dissociated chloride ligand (Table 1, entry 11). Although the Au–NHC photocatalysts studied are relatively stable for prolonged photocatalysis compared to benchmark rhenium–NHC complexes,34,35 the photoreaction lasts for 72 hours due to the decomposition of the photosensitizer.3 Synthesis and photocatalytic CO2 reduction by metal–NHC complexes in MOFs
Although homogeneous metal complexes are amenable for structural design and understanding the structure–property relationship,37 heterogeneous photocatalytic CO2 reduction catalysts offer a potentially robust platform to stabilize the metal–NHC species. Metal–organic frameworks (MOFs) are a unique class of porous crystalline materials constructed from metal–oxo nodes and tailorable organic struts, often possessing high surface area and well-defined porosity for gas absorption.38 Since many of them have CO2 capture abilities to increase local concentrations around catalytic sites, integration of photocatalytic active species into MOFs is highly desirable to realize efficient and stable CO2 photoreduction.39Inspired by synthetic strategies of homogeneous molecular catalysts, chemists have made great efforts to introduce metal–NHC species into MOFs for CO2 activation. Direct synthesis of MOFs containing activated, metal-free NHC or metal–NHC species is not straightforward due to the moisture/oxygen sensitivity and versatile metal-coordination nature of NHC. Traditional approaches involving post-modification of imidazolium-functionalized MOFs with strong alkaline reagents are often not suitable for NHC incorporation due to the potential decomposition of the frameworks. For example, a decrease in crystallinity was observed by treating the CoII-imidazolate framework with n-butyl lithium to generate NHC sites, which also demonstrated that most of the generated NHC species are located on the MOF surface.40 To overcome this issue, two viable strategies have been developed to access metal–NHC functional MOFs (Fig. 3). The first strategy employs a soluble metal source instead of conventional insoluble species, such as Ag2O, which is followed by transmetalation to afford M–NHC (M = PdII/IrI) single-site catalysts inside the MOFs (Fig. 3, route 1).41 The other strategy involves in situ generation of a metal-free NHC-functionalized MOF by post-synthetic ligand exchange, which generates NHC as functionalized backbone linkers of the MOF. The formation of activated, free NHC sites in MOFs has been evidenced by 1H NMR and 13C solid-state NMR spectroscopy (Fig. 3, route 2).42 The MOF has the ability to strongly coordinate with a variety of metal sites (e.g., ZnII, CuI and AuI), owing to the high σ-donor and low π-acceptor features of NHC.43,44
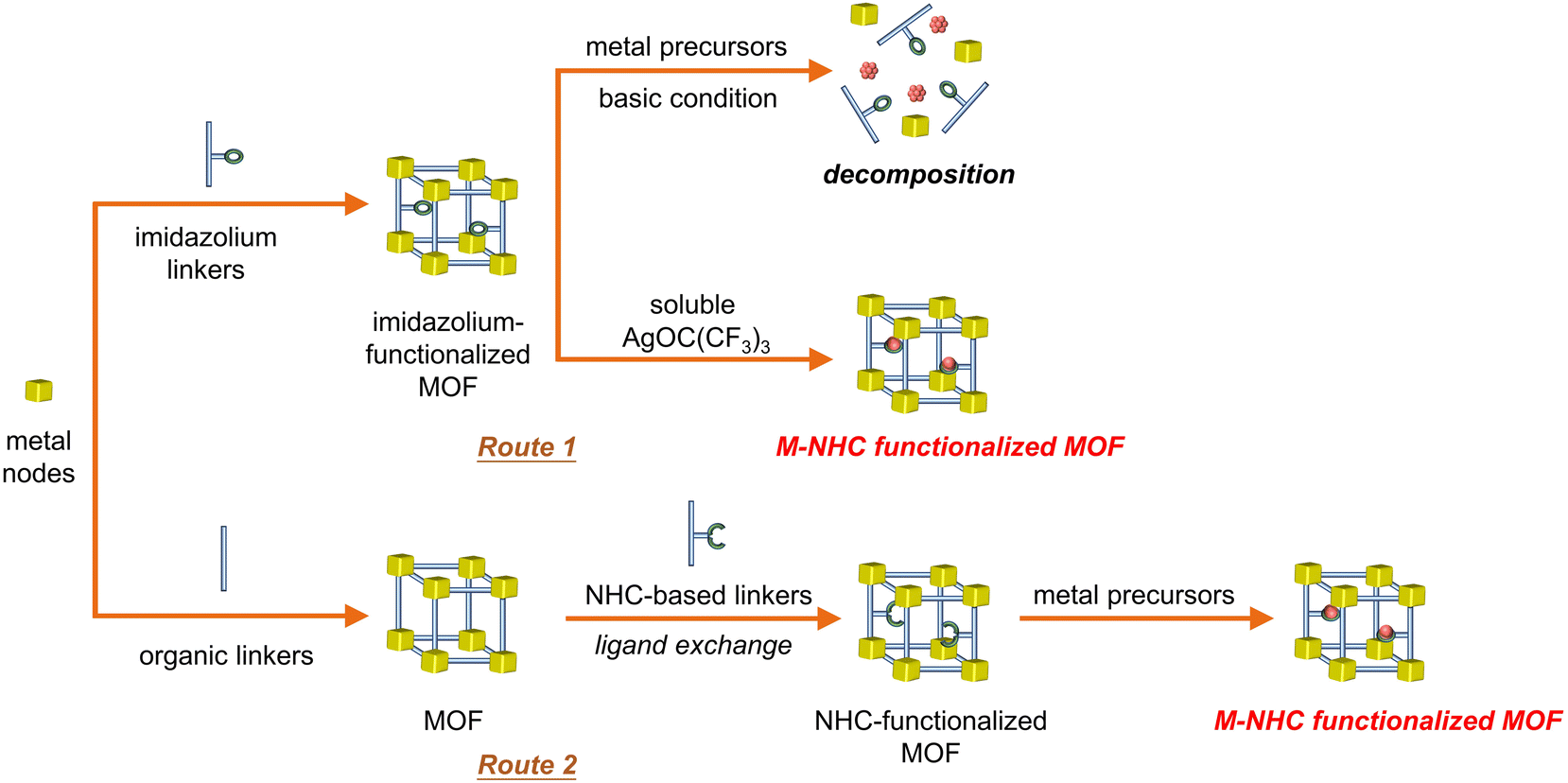 | ||
| Fig. 3 Schematic representation of the synthesis of metal–NHC-functionalized MOFs. | ||
Besides their single M–NHC sites, metal nanoclusters have also garnered increasing attention in photocatalysis over the last decades, owing to their LSPR effect to enhance light harvesting.45 Their formation requires strong covalent or coordination bonding by organic ligands, which is essential to prevent migration and aggregation. In recent years, NHC groups have emerged as a new type of ligand to functionalize metal nanoclusters, thanks to the pioneering work of ligand–surface coordination between 1-methyl-3-butyl imidazolylidene and Ir(0) nanoclusters by Finke et al.46 The synthetic approaches of NHC-stabilized metal nanoclusters in MOFs include chemical reduction of M–NHC precursors, the reduction of metal cations in the presence of imidazole-containing ionic liquids, ligand exchange between free NHC ligands and pre-made metal nanoclusters, and thermal decomposition of active metal precursors.47 By covalently loading NHC-protected plasmonic metal nanoclusters into a MOF platform, one is able to achieve the covalent interface between the MOF and metal nanoclusters for efficient carrier transport in photocatalysis. Considering the MOF advantages for CO2 adsorption and diffusion and the metal nanocluster advantages of strong light absorption, the NHC-stabilized metal nanoclusters in MOFs should be a promising platform for photocatalytic CO2 reduction.
Our group demonstrates a heterogeneous nucleation approach to accommodate ultrasmall and highly dispersed Au nanoclusters in an NHC-functionalized MOF (Fig. 4a).44 We employed the parent UiO-68-NHC with free activated NHC sites and used it directly for nanocluster nucleation in a methanolic solution of HAuCl4·3H2O. After sufficient diffusion of the Au species in the NHC-functionalized MOF, the reduction of AuIII to Au0 by L-glutathione affords ultrasmall nanoclusters with sizes in the range of 1.8 ± 0.2 nm. The formation of strong Au–carbene covalent bonds is confirmed by 13C solid-state NMR spectroscopy, which is crucial to stabilize Au nanoclusters. Control experiments using NH2 or imidazolium as the functional groups on MOFs form large Au aggregates due to the weak Au–support interactions, highlighting the key importance of NHC ligands. The resulting Au@UiO-68-NHC composite exhibited steady photoreduction of CO2 to CO at a rate of 57.6 μmol g−1 h−1 under mild CO2 reduction conditions (0.5 atm CO2) with MeOH as the electron donor (Fig. 4b and c). This value is over four times higher than the control Au/UiO mixtures without NHC stabilization. Notably, excellent photocatalytic performance was obtained using diluted CO2 (0.5 atm) as the gas source. The high photocatalytic activity is largely contributed to by the efficient charge transport between the plasmonic process of Au nanoclusters and the photocatalytically active UiO-68, evidenced by both experimental studies and density functional theory calculations. This is one of the few studies to introduce the strong covalent metal–NHC interface into a heterogeneous matrix, which is a proof of concept for employing a wide range of structurally diverse NHC ligands to stabilize different metal nanoclusters in porous solid-state materials.
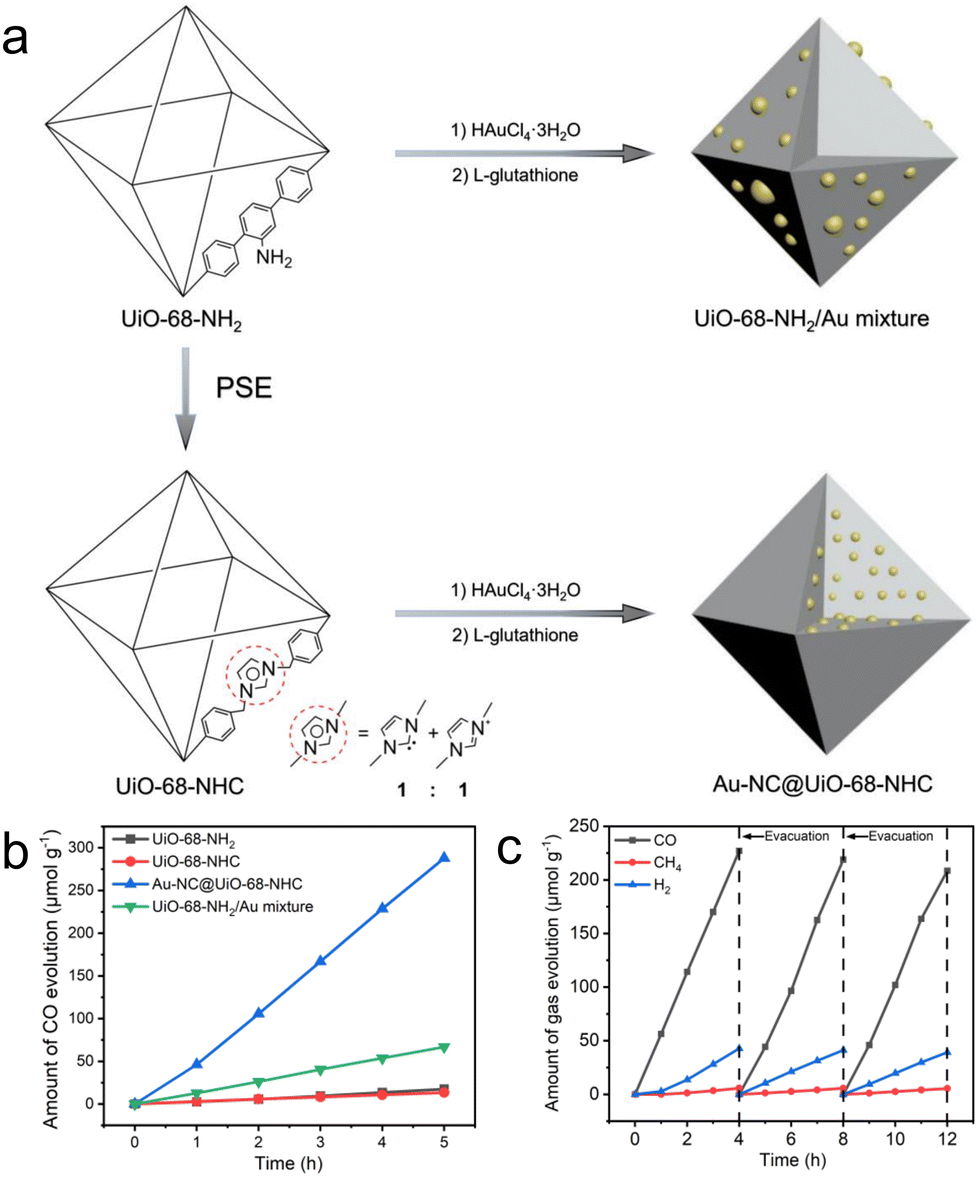 | ||
| Fig. 4 (a) Schematic representation of the synthesis of UiO-68-NHC, Au-NC@UiO-68-NHC and UiO-68-NH2/Au mixture. (b) Time courses of CO evolution by photocatalytic CO2 reduction using UiO-68-NH2 (black), UiO-68-NHC (red), Au-NC@UiO-68-NHC (blue) and UiO-68-NH2/Au mixture (green) as photocatalysts upon AM 1.5G irradiation. (c) Time courses of photocatalytic CO2 reduction on Au-NC@UiO-68-NHC under AM 1.5G irradiation for 12 h, with evacuation every 4 h (dashed line). Reprinted with permission from ref. 44. Copyright 2021 Wiley-VCH. | ||
4 Conclusions and future perspectives
This frontier article discusses recent developments of NHC-based photocatalysts for CO2 reduction. These photocatalysts, which include NHC-ligated metal complexes and clusters, demonstrate the potential of NHC scaffolds as molecular building blocks to form homogeneous molecular entities or heterogeneous MOFs for solar-to-fuel conversion. The robust M–NHC covalent bonds in these catalysts exhibit a variety of advantages including stable coordination, easy synthesis and modification and versatile steric/electronic modulation, therefore showing improved catalytic reactivity in CO2 conversion.Despite the significant advances of NHC ligands, the utilization of NHC-stabilized metal species for photocatalytic CO2 reduction is still in its early stages, especially in terms of NHC-functionalized MOFs.
First, the design of structurally diverse NHC ligands in the tailorable functionality of MOFs will be promising to enhance the performance of photocatalysts, but to date, the vast majority of NHC-MOFs have limited π-conjugation for photocatalysis owing to the synthetic challenge. Introducing a variety of substituents on the NHC species of MOF ligands has the ability to strengthen the π-conjugation effect and modify the σ-donation, thus tuning the spatial and electronic levels of the MOFs. For instance, Zhang and coworkers have synthesized a series of mesoporous porphyrin MOFs with increasing macrocyclic π-conjugated units to tune the light absorption range and photocatalytic performances.48
Second, the current M–NHC species in MOFs are largely focused on noble metals (e.g. Pd, Ru, Ir, Au).49 Therefore, the development of future NHC-ligated metal complexes should be expanded to a variety of Earth-abundant redox-active transition metal centers, such as Ni, Fe, Co and Mn, which have been described as power photocatalysts for visible-light-driven CO2 reduction. For example, Zhang et al. have reported two newly developed MOFs via incorporating unsaturated Co or Zn ions into the porphyrin rings with the efficient ability of capturing and photoreducing CO2 molecules.50 The presence of unsaturated metal sites can not only absorb and activate CO2 molecules but also boost electron–hole separation. Moreover, the type of metal cation can greatly influence the product selectivity for CO2 photoreduction.51,52 The covalent M–NHC coordination provides a great platform for charge transfer between metal species and the MOF skeleton in photocatalysis. At present, no C2 or C2+ production is observed in NHC photocatalysts. Developing bis-NHC photocatalysts with bimetallic sites is promising to achieve asymmetric charge distribution of the catalytic sites and promote C–C coupling.
Third, in addition to the aforementioned advantages of using NHC ligands, it is worth noting that most photocatalytic reactions in this field are conducted in organic solvents. However, it is more desirable to perform the CO2 photoreduction in aqueous media, which requires high aqueous stability for the MOF-based photocatalysts. The gas-phase reactions between CO2 and water vapor offer a new solution to reduce the competitiveness of the proton reduction process, as there is a higher potential barrier for proton reduction in the gas phase (5.39 eV, H2O to H2) compared to the liquid phase (1.25 eV, H+ to H2).53,54 Modulating the hydrophilic/hydrophobic nature of the catalyst may largely overcome the low solubility problem and slow diffusion kinetics of CO2 in the aqueous phase.55 Moreover, the confined MOF porosity is an ideal platform for the fast mass transport of gas-phase molecules, which is promising to achieve selective CO2 photoreduction by M–NHC-functionalized MOFs. In summary, it is highly desirable to immobilize the NHC-based photocatalysts in MOFs to achieve artificial photosynthesis, which not only has high potential to develop highly efficient photocatalysts but also provides insights into the structure–property relationship.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (22171214, 21971197), the Shanghai Rising-Star Program (20QA1409500), the Natural Science Foundation of Shanghai (22ZR1463200), the Xiaomi Young Talents Program, the Recruitment of Global Youth Experts by China and the Science and Technology Commission of Shanghai Municipality (19DZ2271500).References
- J. C. Zachos, G. R. Dickens and R. E. Zeebe, An early Cenozoic perspective on greenhouse warming and carbon-cycle dynamics, Nature, 2008, 451, 279–283 CrossRef CAS.
- C. A. Deutsch, J. J. Tewksbury, M. Tigchelaar, D. S. Battisti, S. C. Merrill, R. B. Huey and R. L. Naylor, Increase in crop losses to insect pests in a warming climate, Science, 2018, 361, 916–919 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders, Nature, 1979, 277, 637–638 CrossRef CAS.
- W. Tu, Y. Zhou and Z. Zou, Photocatalytic Conversion of CO2 into Renewable Hydrocarbon Fuels: State-of-the-Art Accomplishment, Challenges, and Prospects, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- J. Zhou, J. Li, L. Kan, L. Zhang, Q. Huang, Y. Yan, Y. Chen, J. Liu, S.-L. Li and Y.-Q. Lan, Linking oxidative and reductive clusters to prepare crystalline porous catalysts for photocatalytic CO2 reduction with H2O, Nat. Commun., 2022, 13, 4681 CrossRef CAS PubMed.
- J. Grodkowski, D. Behar, P. Neta and P. Hambright, Iron Porphyrin-Catalyzed Reduction of CO2. Photochemical and Radiation Chemical Studies, J. Phys. Chem. A, 1997, 101, 248–254 CrossRef CAS.
- D. Behar, T. Dhanasekaran, P. Neta, C. M. Hosten, D. Ejeh, P. Hambright and E. Fujita, Cobalt Porphyrin Catalyzed Reduction of CO2. Radiation Chemical, Photochemical, and Electrochemical Studies, J. Phys. Chem. A, 1998, 102, 2870–2877 CrossRef CAS.
- T. Dhanasekaran, J. Grodkowski, P. Neta, P. Hambright and E. Fujita, p-Terphenyl-Sensitized Photoreduction of CO2 with Cobalt and Iron Porphyrins. Interaction between CO and Reduced Metalloporphyrins, J. Phys. Chem. A, 1999, 103, 7742–7748 CrossRef CAS.
- J. Bonin, M. Robert and M. Routier, Selective and Efficient Photocatalytic CO2 Reduction to CO Using Visible Light and an Iron-Based Homogeneous Catalyst, J. Am. Chem. Soc., 2014, 136, 16768–16771 CrossRef CAS PubMed.
- H. Rao, J. Bonin and M. Robert, Non-sensitized selective photochemical reduction of CO2 to CO under visible light with an iron molecular catalyst, Chem. Commun., 2017, 53, 2830–2833 RSC.
- J. Grodkowski, T. Dhanasekaran, P. Neta, P. Hambright, B. S. Brunschwig, K. Shinozaki and E. Fujita, Reduction of Cobalt and Iron Phthalocyanines and the Role of the Reduced Species in Catalyzed Photoreduction of CO2, J. Phys. Chem. A, 2000, 104, 11332–11339 CrossRef CAS.
- E. Nikoloudakis, I. López-Duarte, G. Charalambidis, K. Ladomenou, M. Ince and A. G. Coutsolelos, Porphyrins and phthalocyanines as biomimetic tools for photocatalytic H2 production and CO2 reduction, Chem. Soc. Rev., 2022, 51, 6965–7045 RSC.
- K. Koike, N. Okoshi, H. Hori, K. Takeuchi, O. Ishitani, H. Tsubaki, I. P. Clark, M. W. George, F. P. A. Johnson and J. J. Turner, Mechanism of the photochemical ligand substitution reactions of fac-[Re(bpy)(CO)3(PR3)]+ complexes and the properties of their triplet ligand-field excited states, J. Am. Chem. Soc., 2002, 124, 11448–11455 CrossRef CAS PubMed.
- J. Agarwal, E. Fujita, H. F. Schaefer III and J. T. Muckerman, Mechanisms for CO Production from CO2 Using Reduced Rhenium Tricarbonyl Catalysts, J. Am. Chem. Soc., 2012, 134, 5180–5186 CrossRef CAS.
- H. Takeda, H. Koizumi, K. Okamoto and O. Ishitani, Photocatalytic CO2 reduction using a Mn complex as a catalyst, Chem. Commun., 2014, 50, 1491–1493 RSC.
- L. Liu, X. Zhang, L. Yang, L. Ren, D. Wang and J. Ye, Metal nanoparticles induced photocatalysis, Natl. Sci. Rev., 2017, 4, 761–780 CrossRef CAS.
- M. Nemiwal and D. Kumar, TiO2 and SiO2 encapsulated metal nanoparticles: Synthetic strategies, properties, and photocatalytic applications, Inorg. Chem. Commun., 2021, 128, 108602 CrossRef CAS.
- S. Linic, S. Chavez and R. Elias, Flow and extraction of energy and charge carriers in hybrid plasmonic nanostructures, Nat. Mater., 2021, 20, 916–924 CrossRef CAS PubMed.
- S. Luo, X. Ren, H. Lin, H. Song and J. Ye, Plasmonic photothermal catalysis for solar-to-fuel conversion: current status and prospects, Chem. Sci., 2021, 12, 5701–5719 RSC.
- S. Yu, A. J. Wilson, G. Kumari, X. Zhang and P. K. Jain, Opportunities and Challenges of Solar-Energy-Driven Carbon Dioxide to Fuel Conversion with Plasmonic Catalysts, ACS Energy Lett., 2017, 2, 2058–2070 CrossRef CAS.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An overview of N-heterocyclic carbenes, Nature, 2014, 510, 485–496 CrossRef CAS.
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, N-Heterocyclic Carbenes in Materials Chemistry, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS.
- S. N. Riduan, Y. Zhang and J. Y. Ying, Conversion of Carbon Dioxide into Methanol with Silanes over N-Heterocyclic Carbene Catalysts, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS.
- S. Das, F. D. Bobbink, G. Laurenczy and P. J. Dyson, Metal-Free Catalyst for the Chemoselective Methylation of Amines Using Carbon Dioxide as a Carbon Source, Angew. Chem., Int. Ed., 2014, 53, 12876–12879 CrossRef CAS PubMed.
- T. Ohishi, M. Nishiura and Z. Hou, Carboxylation of Organoboronic Esters Catalyzed by N-Heterocyclic Carbene Copper(I) Complexes, Angew. Chem., Int. Ed., 2008, 47, 5792–5795 CrossRef CAS PubMed.
- V. S. Thoi, N. Kornienko, C. G. Margarit, P. Yang and C. J. Chang, Visible-light photoredox catalysis: selective reduction of carbon dioxide to carbon monoxide by a nickel N-heterocyclic carbene-isoquinoline complex, J. Am. Chem. Soc., 2013, 135, 14413–14424 CrossRef CAS PubMed.
- J. T. S. Irvine, B. R. Eggins and J. Grimshaw, Solar energy fixation of carbon dioxide via cadmium sulphide and other semiconductor photocatalysts, Sol. Energy, 1990, 45, 27–33 CrossRef CAS.
- P. Johne and H. Kisch, Photoreduction of carbon dioxide catalysed by free and supported zinc and cadmium sulphide powders, J. Photochem. Photobiol., A, 1997, 111, 223–228 CrossRef CAS.
- D. B. Burks, S. Davis, R. W. Lamb, X. Liu, R. R. Rodrigues, N. P. Liyanage, Y. Sun, C. E. Webster, J. H. Delcamp and E. T. Papish, Nickel(II) pincer complexes demonstrate that the remote substituent controls catalytic carbon dioxide reduction, Chem. Commun., 2018, 54, 3819–3822 RSC.
- H. Shirley, X. Su, H. Sanjanwala, K. Talukdar, J. W. Jurss and J. H. Delcamp, Durable Solar-Powered Systems with Ni-Catalysts for Conversion of CO2 or CO to CH4, J. Am. Chem. Soc., 2019, 141, 6617–6622 CrossRef CAS PubMed.
- H. H. Huang, J. H. Zhang, M. Dai, L. Liu, Z. Ye, J. Liu, D. C. Zhong, J. W. Wang, C. Zhao and Z. Ke, Dual electronic effects achieving a high-performance Ni(II) pincer catalyst for CO2 photoreduction in a noble-metal-free system, Proc. Natl. Acad. Sci. USA, 2022, 119, e2119267119 CrossRef CAS.
- C. M. Boudreaux, N. P. Liyanage, H. Shirley, S. Siek, D. L. Gerlach, F. Qu, J. H. Delcamp and E. T. Papish, Ruthenium(II) complexes of pyridinol and N-heterocyclic carbene derived pincers as robust catalysts for selective carbon dioxide reduction, Chem. Commun., 2017, 53, 11217–11220 RSC.
- S. Das, R. R. Rodrigues, R. W. Lamb, F. Qu, E. Reinheimer, C. M. Boudreaux, C. E. Webster, J. H. Delcamp and E. T. Papish, Highly Active Ruthenium CNC Pincer Photocatalysts for Visible-Light-Driven Carbon Dioxide Reduction, Inorg. Chem., 2019, 58, 8012–8020 CrossRef CAS.
- A. J. Huckaba, E. A. Sharpe and J. H. Delcamp, Photocatalytic Reduction of CO2 with Re-Pyridyl-NHCs, Inorg. Chem., 2016, 55, 682–690 CrossRef CAS PubMed.
- C. A. Carpenter, P. Brogdon, L. E. McNamara, G. S. Tschumper, N. I. Hammer and J. H. Delcamp, A Robust Pyridyl-NHC-Ligated Rhenium Photocatalyst for CO2 Reduction in the Presence of Water and Oxygen, Inorganics, 2018, 6, 22 CrossRef.
- D. Nugegoda, N. V. Tzouras, S. P. Nolan and J. H. Delcamp, N-Heterocyclic Carbene Gold Complexes in a Photocatalytic CO2 Reduction Reaction, Inorg. Chem., 2022, 61, 18802–18809 CrossRef CAS PubMed.
- A. M. Voutchkova, L. N. Appelhans, A. R. Chianese and R. H. Crabtree, Disubstituted Imidazolium-2-Carboxylates as Efficient Precursors to N-Heterocyclic Carbene Complexes of Rh, Ru, Ir, and Pd, J. Am. Chem. Soc., 2005, 127, 17624–17625 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The Chemistry and Applications of Metal-Organic Frameworks, Science, 2013, 341, 1230444 CrossRef PubMed.
- Y.-H. Luo, L.-Z. Dong, J. Liu, S.-L. Li and Y.-Q. Lan, From molecular metal complex to metal-organic framework: The CO2 reduction photocatalysts with clear and tunable structure, Coord. Chem. Rev., 2019, 390, 86–126 CrossRef CAS.
- M. B. Lalonde, O. K. Farha, K. A. Scheidt and J. T. Hupp, N-Heterocyclic Carbene-Like Catalysis by a Metal-Organic Framework Material, ACS Catal., 2012, 2, 1550–1554 CrossRef CAS.
- C. He, J. Liang, Y. H. Zou, J. D. Yi, Y. B. Huang and R. Cao, Metal-organic frameworks bonded with metal N-heterocyclic carbenes for efficient catalysis, Natl. Sci. Rev., 2022, 9, nwab157 CrossRef CAS.
- X. Zhang, J. Sun, G. Wei, Z. Liu, H. Yang, K. Wang and H. Fei, In Situ Generation of an N-Heterocyclic Carbene Functionalized Metal-Organic Framework by Postsynthetic Ligand Exchange: Efficient and Selective Hydrosilylation of CO2, Angew. Chem., Int. Ed., 2019, 58, 2844–2849 CrossRef CAS PubMed.
- X. Zhang, Y. Jiang and H. Fei, UiO-type metal-organic frameworks with NHC or metal-NHC functionalities for N-methylation using CO2 as the carbon source, Chem. Commun., 2019, 55, 11928–11931 RSC.
- Y. Jiang, Y. Yu, X. Zhang, M. Weinert, X. Song, J. Ai, L. Han and H. Fei, N-Heterocyclic Carbene-Stabilized Ultrasmall Gold Nanoclusters in a Metal-Organic Framework for Photocatalytic CO2 Reduction, Angew. Chem., Int. Ed., 2021, 60, 17388–17393 CrossRef CAS PubMed.
- O. J. H. Chai, Z. Liu, T. Chen and J. Xie, Engineering ultrasmall metal nanoclusters for photocatalytic and electrocatalytic applications, Nanoscale, 2019, 11, 20437–20448 RSC.
- L. S. Ott, M. L. Cline, M. Deetlefs, K. R. Seddon and R. G. Finke, Nanoclusters in Ionic Liquids: Evidence for N-Heterocyclic Carbene Formation from Imidazolium-Based Ionic Liquids Detected by 2H NMR, J. Am. Chem. Soc., 2005, 127, 5758–5759 CrossRef CAS PubMed.
- H. Shen, G. Tian, Z. Xu, L. Wang, Q. Wu, Y. Zhang, B. K. Teo and N. Zheng, N-heterocyclic carbene coordinated metal nanoparticles and nanoclusters, Coord. Chem. Rev., 2022, 458, 214425 CrossRef CAS.
- J.-Y. Zeng, X.-S. Wang, B.-R. Xie, Q.-R. Li and X.-Z. Zhang, Large π-Conjugated Metal-Organic Frameworks for Infrared-Light-Driven CO2 Reduction, J. Am. Chem. Soc., 2022, 144, 1218–1231 CrossRef CAS PubMed.
- C. I. Ezugwu, N. A. Kabir, M. Yusubov and F. Verpoort, Metal-organic frameworks containing N-heterocyclic carbenes and their precursors, Coord. Chem. Rev., 2016, 307, 188–210 CrossRef CAS.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Efficient Visible-Light-Driven Carbon Dioxide Reduction by a Single-Atom Implanted Metal-Organic Framework, Angew. Chem., Int. Ed., 2016, 55, 14310–14314 CrossRef CAS PubMed.
- Y. Zhao, G. Chen, T. Bian, C. Zhou, G. I. Waterhouse, L. Z. Wu, C. H. Tung, L. J. Smith, D. O'Hare and T. Zhang, Defect-Rich Ultrathin ZnAl-Layered Double Hydroxide Nanosheets for Efficient Photoreduction of CO2 to CO with Water, Adv. Mater., 2015, 27, 7824–7831 CrossRef CAS PubMed.
- X. Xiong, Y. Zhao, R. Shi, W. Yin, Y. Zhao, G. I. N. Waterhouse and T. Zhang, Selective photocatalytic CO2 reduction over Zn-based layered double hydroxides containing tri or tetravalent metals, Sci. Bull., 2020, 65, 987–994 CrossRef CAS PubMed.
- S. Wang, M. Xu, T. Peng, C. Zhang, T. Li, I. Hussain, J. Wang and B. Tan, Porous hypercrosslinked polymer-TiO2-graphene composite photocatalysts for visible-light-driven CO2 conversion, Nat. Commun., 2019, 10, 676 CrossRef CAS PubMed.
- G. Jia, Z. Wang, M. Gong, Y. Wang, L. H. Li, Y. Dong, L. Liu, L. Zhang, J. Zhao, W. Zheng and X. Cui, Ultrathin origami accordion-like structure of vacancy-rich graphitized carbon nitride for enhancing CO2 photoreduction, Carbon Energy, 2023, 5, e270 CrossRef CAS.
- H. Huang, R. Shi, Z. Li, J. Zhao, C. Su and T. Zhang, Triphase Photocatalytic CO2 Reduction over Silver-Decorated Titanium Oxide at a Gas-Water Boundary, Angew. Chem., Int. Ed., 2022, 61, e202200802 CAS.
| This journal is © the Partner Organisations 2023 |