DOI:
10.1039/D3MD00017F
(Research Article)
RSC Med. Chem., 2023,
14, 1165-1171
Synthesis of tryptanthrin appended dispiropyrrolidine oxindoles and their antibacterial evaluation†
Received
13th January 2023
, Accepted 1st May 2023
First published on 3rd May 2023
Abstract
The synthesis of sixteen tryptanthrin appended dispiropyrrolidine oxindoles, employing [3 + 2] cycloaddition of tryptanthrin-derived azomethine ylides with isatilidenes, and their detailed antibacterial evaluation is described. The in vitro antibacterial activities of the compounds were evaluated against ESKAPE pathogens and clinically relevant drug-resistant MRSA/VRSA strains, from which the bromo-substituted dispiropyrrolidine oxindole 5b (MIC = 0.125 μg mL−1) was found to be a potent molecule against S. aureus ATCC 29213 with a good selectivity index.
1. Introduction
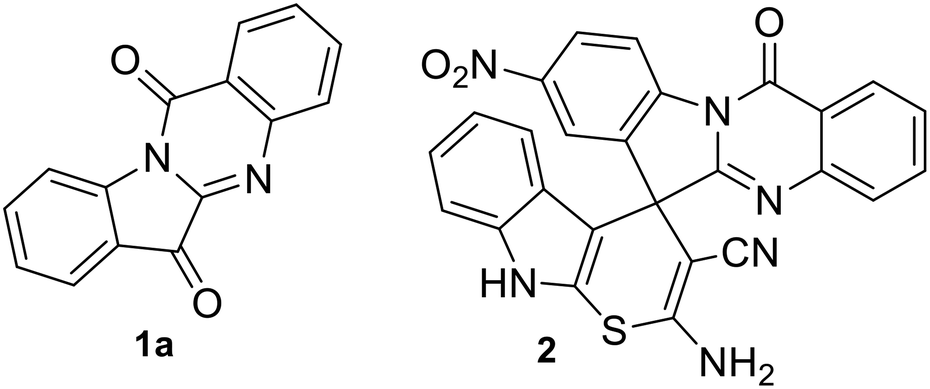
Molecular hybridization approach in medicinal chemistry has been used extensively for the synthesis of bioactive agents including antibacterials.1 The process involves linking two or more bioactive scaffolds in order to produce a new molecule with enhanced biological properties. The necessity to develop new generation antimicrobials, in order to combat multidrug resistance (MDR),2,3 further underpins the significance of molecular hybridization approaches for antimicrobial development. The current paper describes the synthesis of a molecular hybrid combining a quinazoline alkaloid, namely, tryptanthrin (indolo[2,1-b]quinazoline-6,12-dione 1a, Fig. 1) with spiropyrrolidine oxindoles. The former is known to possess a wide range of biological activity4 including anti-MRSA activity with low cytotoxicity5 and is therefore considered as a potential therapeutic agent.6 Our groups recently reported the green synthesis and detailed antibacterial evaluation of spirofused tryptanthrin-thiopyrano[2,3-b]indole hybrids and disclosed a potent compound (2, Fig. 1) which synergizes with linezolid against S. aureus ATCC 29213.7
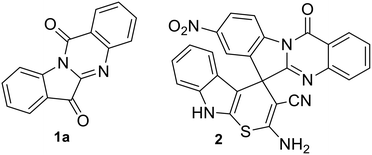 |
| | Fig. 1 Tryptanthrin (1a) and a biologically active spirofused tryptanthrin-thiopyrano[2,3-b]indole hybrid (2). | |
In this context, we synthesized tryptanthrin–spiropyrrolidine oxindole hybrid molecules and have evaluated their antibacterial properties in detail. The hybrid molecules were synthesized by 1,3-dipolar cycloaddition of azomethine ylides (AYs)8 generated from tryptanthrin to N-substituted isatilidenes. Except for very few reports,9 the generation of AYs from tryptanthrin and α-amino acids by decarboxylative routes is scarcely mentioned in literature and there has been no report on the synthesis of spiropyrrolidine oxindole cycloadducts from tryptanthrin. The spiropyrrolidine oxindoles constitute a privileged class of structural moieties on account of their enhanced biological properties.10 Therefore, hybridization of tryptanthrin with spiropyrrolidine oxindoles seems logical and intuitive.
2. Results and discussion
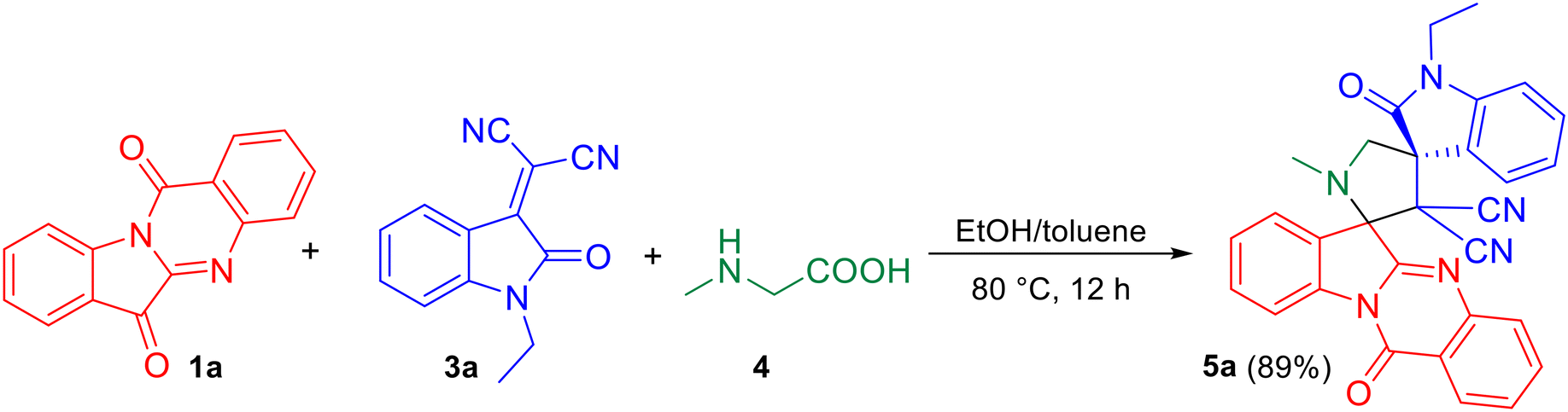
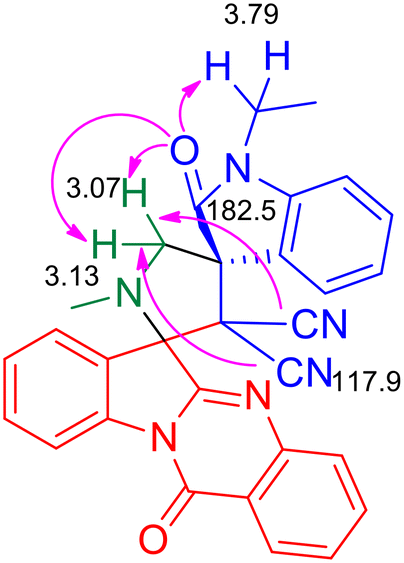
As the pilot reaction, tryptanthrin 1a, N-ethyl isatilidene 3a and sarcosine 4 (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1.2) were refluxed in ethanol until complete consumption of 1a was observed from TLC. Work-up followed by silica gel column chromatography using 75:25 hexane–ethyl acetate eluted the compound 5a which was isolated in 70% yield (Scheme 1). The structure of 5a was confirmed using spectroscopic analysis. In the IR spectrum, the stretching at 1722 cm−1 and 1685 cm−1 were due to the oxindole ring carbonyl group and the amide carbonyl group, respectively, while the peak at 2119 cm−1 corresponded to nitrile stretch. In the 1H NMR spectrum, –NCH3 protons appeared as a singlet at δ 2.04 ppm while the methylene protons adjacent to the N were seen as doublets at δ 3.06 and 3.13 ppm. In the 13C NMR spectrum, the oxindole carbonyl carbon was observed at 182.5 ppm while the quinazoline carbonyl carbon was discernible at 169 ppm. Fig. 2 represents the HMBC spectrum of compound 5a. From the figure, it is clear that the cyano carbon appearing at 117.9 ppm correlates with N–CH2 protons at 3.07 and 3.13 ppm, and the carbonyl carbon appearing at 182.5 ppm correlates with N–CH2 protons at 3.07, 3.13 and 3.79 ppm. Repeated attempts to crystallize 5a did not yield good crystals which indicate the amorphous nature of the compound and its stereochemistry was assigned in line with our previous finding.11 In order to optimize the conditions, the reaction was carried out in ethanol–DCM and ethanol–toluene mixtures, both of which led to an increase in product yield to 72% and 89% respectively, while the use of a polar aprotic solvent like acetonitrile decreased the yield to 65%. Thus, the 3:1 ethanol–toluene mixture was found to be the optimal solvent system for the reaction.
:1:1.2) were refluxed in ethanol until complete consumption of 1a was observed from TLC. Work-up followed by silica gel column chromatography using 75:25 hexane–ethyl acetate eluted the compound 5a which was isolated in 70% yield (Scheme 1). The structure of 5a was confirmed using spectroscopic analysis. In the IR spectrum, the stretching at 1722 cm−1 and 1685 cm−1 were due to the oxindole ring carbonyl group and the amide carbonyl group, respectively, while the peak at 2119 cm−1 corresponded to nitrile stretch. In the 1H NMR spectrum, –NCH3 protons appeared as a singlet at δ 2.04 ppm while the methylene protons adjacent to the N were seen as doublets at δ 3.06 and 3.13 ppm. In the 13C NMR spectrum, the oxindole carbonyl carbon was observed at 182.5 ppm while the quinazoline carbonyl carbon was discernible at 169 ppm. Fig. 2 represents the HMBC spectrum of compound 5a. From the figure, it is clear that the cyano carbon appearing at 117.9 ppm correlates with N–CH2 protons at 3.07 and 3.13 ppm, and the carbonyl carbon appearing at 182.5 ppm correlates with N–CH2 protons at 3.07, 3.13 and 3.79 ppm. Repeated attempts to crystallize 5a did not yield good crystals which indicate the amorphous nature of the compound and its stereochemistry was assigned in line with our previous finding.11 In order to optimize the conditions, the reaction was carried out in ethanol–DCM and ethanol–toluene mixtures, both of which led to an increase in product yield to 72% and 89% respectively, while the use of a polar aprotic solvent like acetonitrile decreased the yield to 65%. Thus, the 3:1 ethanol–toluene mixture was found to be the optimal solvent system for the reaction.
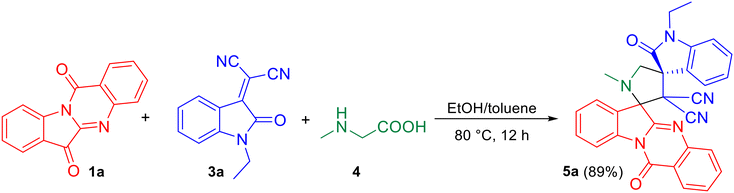 |
| | Scheme 1 One-pot, three-component reaction of tryptanthrin 1a, N-ethyl isatilidene 3a and sarcosine 4. | |
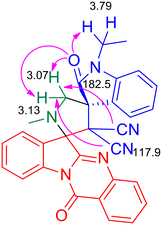 |
| | Fig. 2 Selected HMBC correlations of compound 5a. | |
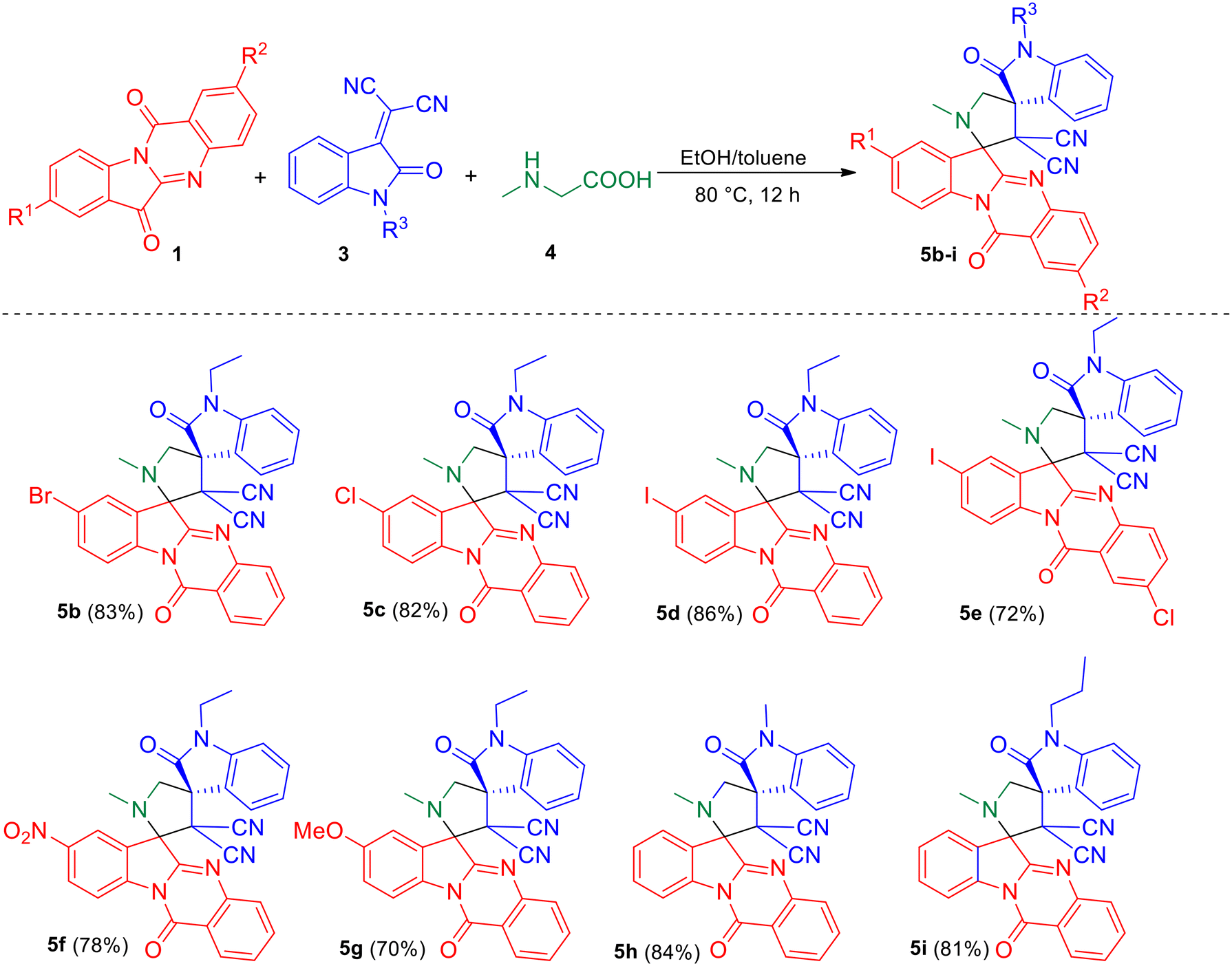
With the optimized reaction conditions, we tested the generality of the reaction. Initially, various substituted tryptanthrins 1b–g were employed for the reaction with N-alkyl isatilidene malononitriles 3a–c and sarcosine 4 (Scheme 2) to study the effect of different C8 substituents on tryptanthrin on the reaction. It was observed that when the C8 position is substituted with electron-withdrawing groups, like halo groups and the nitro group, the desired dispiro compounds 5b–5e and 5f were obtained in good yields, respectively. However, when electron-donating groups, such as the methoxy group, was employed in the 1,3-dipolar cycloaddition reaction, a slight decrease in the yield of the product (5g) was observed. No appreciable change in yield of the products 5h and 5i were observed using isatilidenes 3b (N-methyl) and 3c (N-propyl).
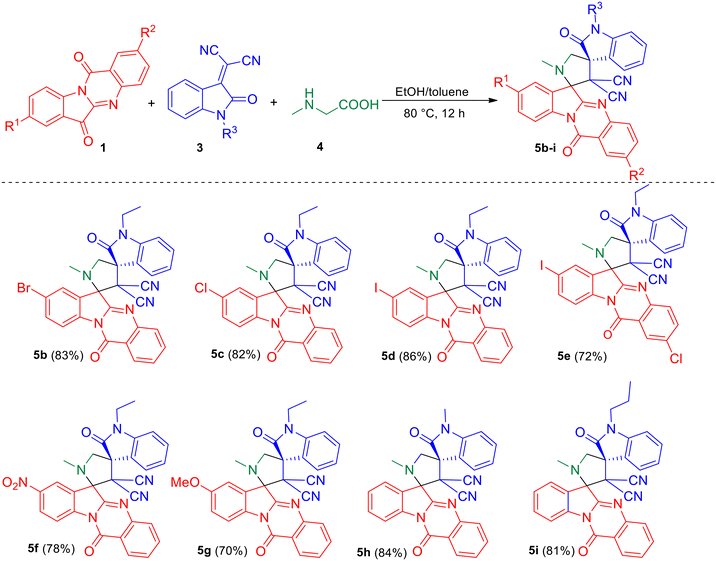 |
| | Scheme 2 Synthesis of dispirooxindole by a three-component reaction of substituted tryptanthrin 1, N-alkyl isatilidene malononitrile 3 and sarcosine 4. | |
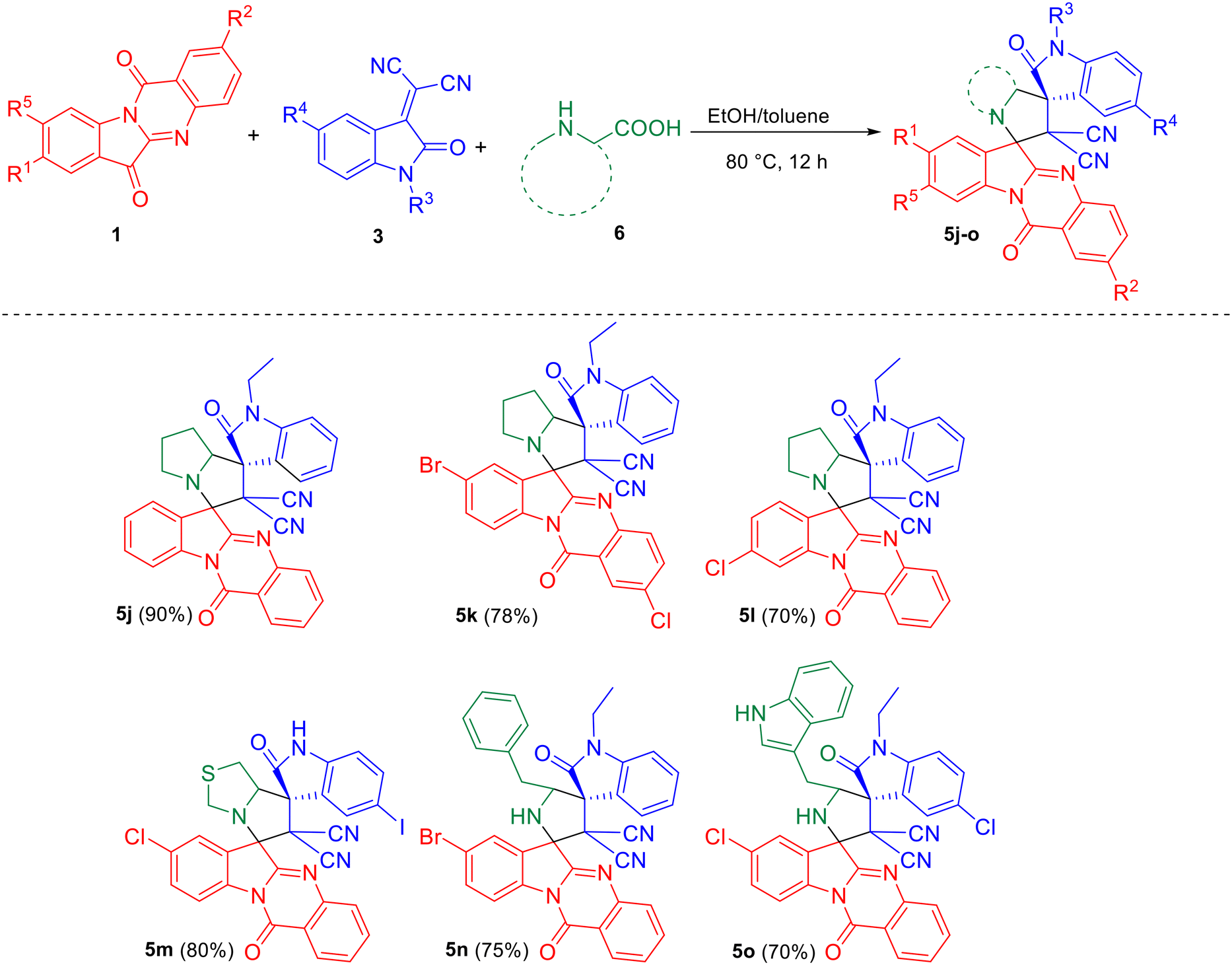
Scheme 3 depicts the reaction of azomethine ylide generated from tryptanthrin/ring-substituted tryptanthrins 1 and other amino acids such as proline/thioproline/tryptophan/phenylalanine 6 with 3 leading to the formation of the desired dispiro compounds 5j–o.
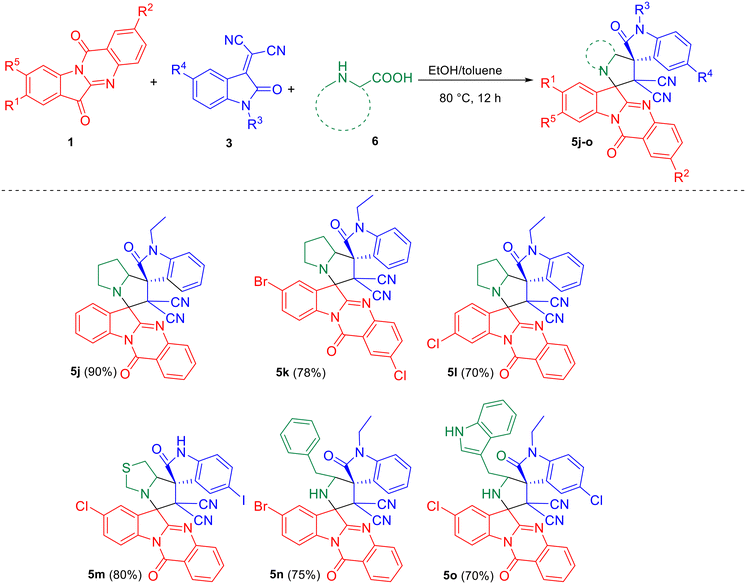 |
| | Scheme 3 Synthesis of dispirooxindoles by a three-component reaction of substituted tryptanthrin 1, substituted isatilidene malononitrile 3 and proline/thioproline/tryptophan/phenylalanine 6. | |
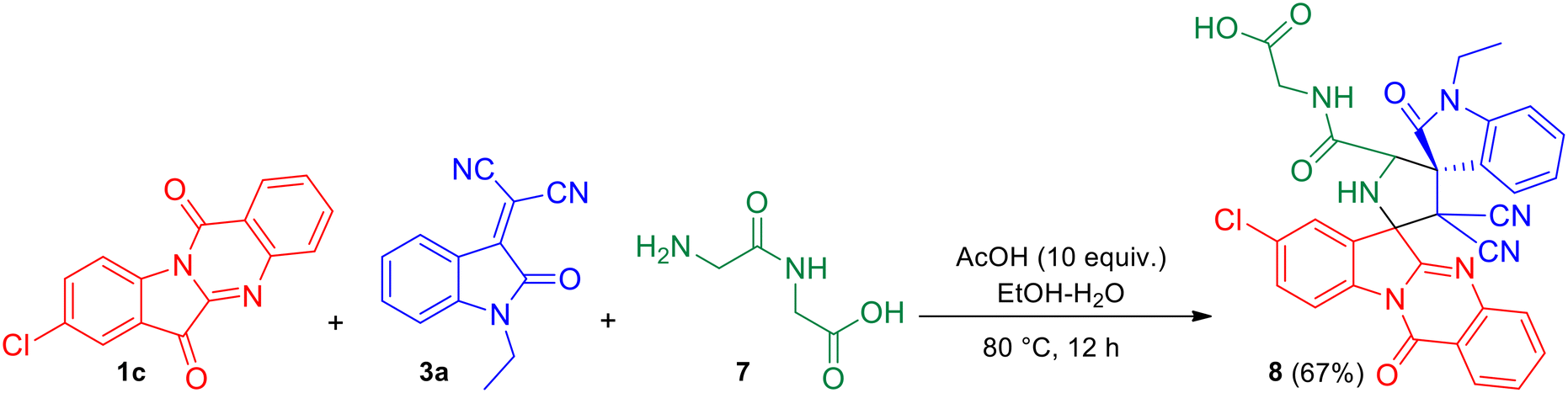
It was also observed that the azomethine ylide generated from 8-chloro tryptanthrin 1c (representative example) and glycylglycine (Gly–Gly) 7 underwent facile [3 + 2] cycloaddition with N-ethyl isatilidene 3a to yield (8′′-chloro-3′,3′-dicyano-1-ethyl-2,12′′-dioxo-12′′H-dispiro[indoline-3,4′-pyrrolidine-2′,6′′-indolo[2,1-b]quinazoline]-5′-carbonyl)glycine 8 in 67% yield (Scheme 4). The reaction was conducted in the presence of acetic acid (10 equiv.) in EtOH–H2O (3:1) under reflux for 12 h. In the 1H NMR spectrum of compound 8, signals at δ 13.45 and 8.44 ppm were attributed to the COOH and NH protons of the peptide fragment while the signal of the NH-proton of the pyrrolidine ring appeared at 3.92 ppm. In the 13C NMR spectrum, the four carbonyl carbons of compound 8 were discernible at δ 182.2, 176.1, 167.3 and 157.8 ppm.
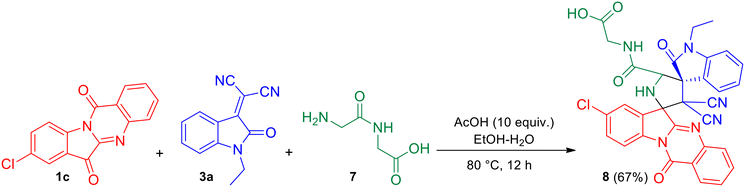 |
| | Scheme 4 One-pot, three-component reaction of 8-chloro tryptanthrin 1c, N-ethyl isatilidene 3a and glycylglycine (Gly–Gly) 7. | |
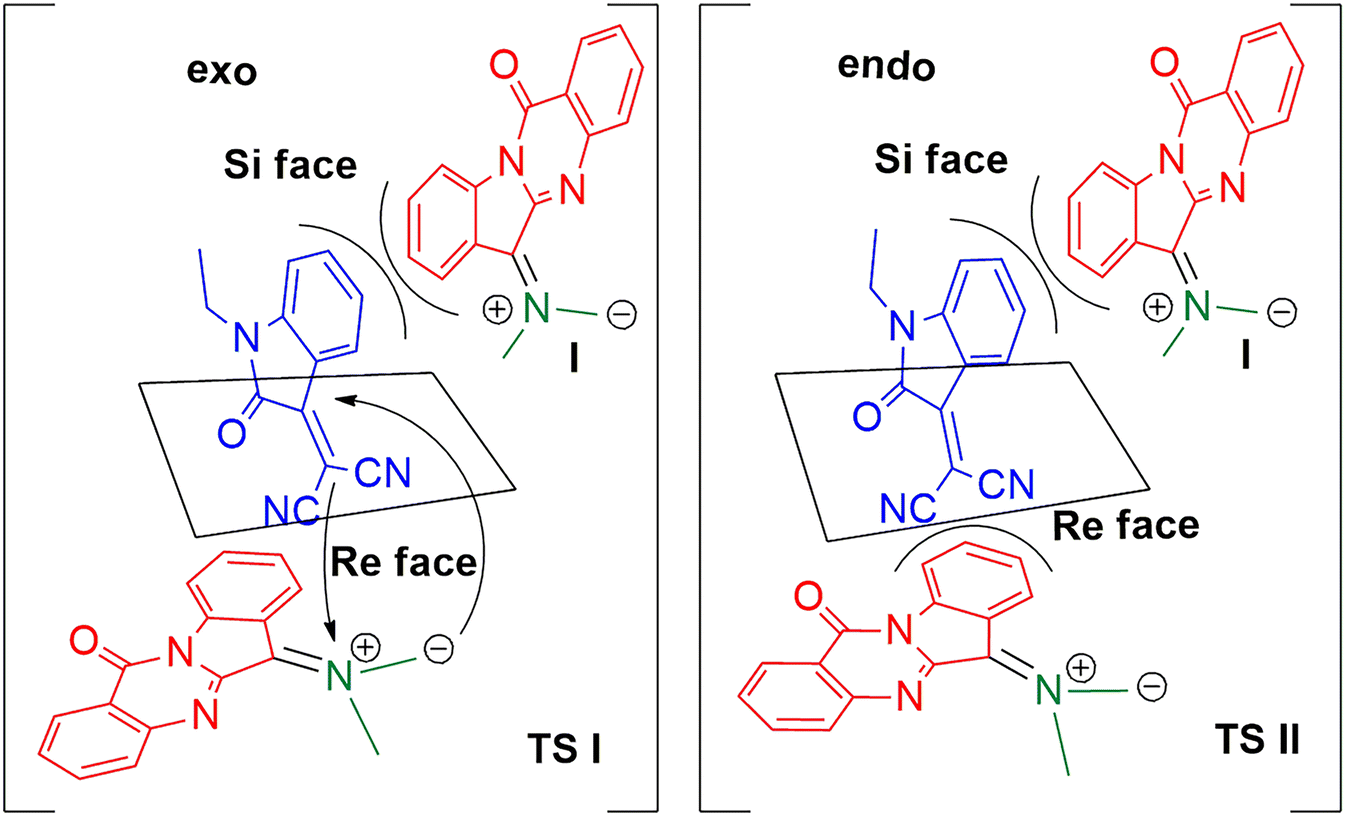
The possible stereochemical approach of ylide I to dipolarophile 3a is depicted in Scheme 5. The transition states TS I and TS II are formed by exo and endo modes of approach of the ylide to both the faces of the dipolarophile 3a. In TS I, the dipole probably experiences a steric repulsion due to the presence of the benzene ring present in 3a when approaching it from a Si face, while no such steric effects operate in the approach through the Re face; hence the exo Re face approach is more favourable for the diastereoselective formation of the product. The endo approach of the dipole is not favourable due to the steric repulsion of the dipole with benzene ring (at the Si face of 3a) or with the nitrile group (at the Re face of 3a). As the cycloaddition was proceeding through the exo mode only, a single diastereomer was only observed in all cases.
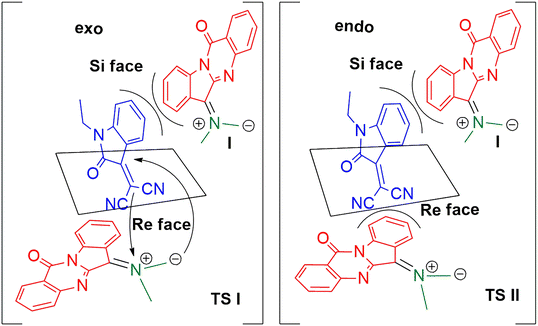 |
| | Scheme 5 Stereochemical approach to dipolarophile showing the exo Re face approach. | |
2.1
In vitro antibacterial activity
2.1.1 Antimicrobial activity of against bacterial pathogen panel.
All compounds 5a–h, 5j, 5n–o and 8 were evaluated for their antibacterial activity against an ESKAPE pathogen panel to determine the minimum inhibitory concentration (MIC). Levofloxacin was used as the positive control and results are depicted in Table 1. As can be seen, all compounds exhibited low MIC values against S. aureus ATCC 29213, among which 5b–c and 5g showed very potent activity (MIC 0.125 μg mL−1), which is lower than levofloxacin and is much better than vancomycin (MIC 2 μg mL−1), the standard of care for treatment of staphylococcal infections.
Table 1 MIC (μg mL−1) of 5a–h, 5j, 5n–o and 8 against bacterial pathogen panel along with comparators
| Compound |
MIC (μg mL−1) |
|
S. aureus ATCC 29213 |
A. baumannii BAA-1605 |
P. aeruginosa ATCC 27853 |
E. coli ATCC 25922 |
K. pneumoniae BAA 1705 |
| NT: not tested. |
|
5a
|
1 |
>64 |
>64 |
>64 |
>64 |
|
5b
|
0.125
|
>64 |
>64 |
>64 |
>64 |
|
5c
|
0.125
|
>64 |
>64 |
>64 |
>64 |
|
5d
|
0.25 |
>64 |
>64 |
>64 |
>64 |
|
5e
|
1 |
>64 |
>64 |
>64 |
>64 |
|
5f
|
0.25 |
>64 |
>64 |
>64 |
>64 |
|
5g
|
0.125
|
>64 |
>64 |
>64 |
>64 |
|
5h
|
8 |
>64 |
>64 |
>64 |
>64 |
|
5j
|
0.25 |
>64 |
>64 |
>64 |
>64 |
|
5n
|
0.25 |
>64 |
>64 |
>64 |
>64 |
|
5o
|
0.25 |
>64 |
>64 |
>64 |
>64 |
|
8
|
1 |
>64 |
>64 |
>64 |
>64 |
| Levofloxacin |
0.25 |
8 |
1 |
0.0156 |
64 |
| Vancomycin |
2 |
NT* |
NT* |
NT* |
NT* |
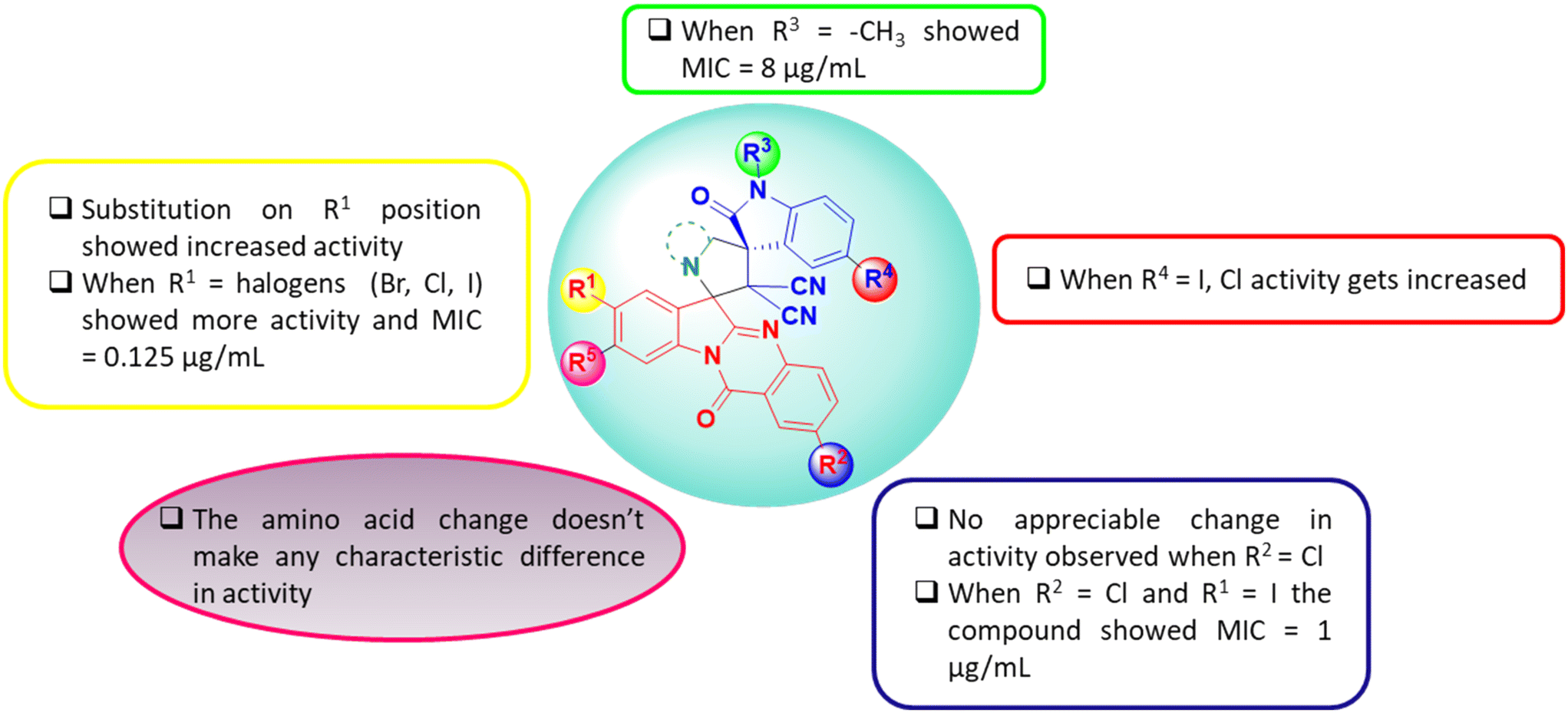
The structure activity relationship (SAR) of the molecular hybrids is depicted in Fig. 3. From the SAR studies, it was found that halogen substitutions at the R1 and R4 positions caused an enhancement in anti-bacterial activity while the same in R2 had a null effect. Further, anti-bacterial activity remained unaltered upon N-alkyl substitutions and on altering the pyrrolidine moiety (by using different amino acids for the reaction).
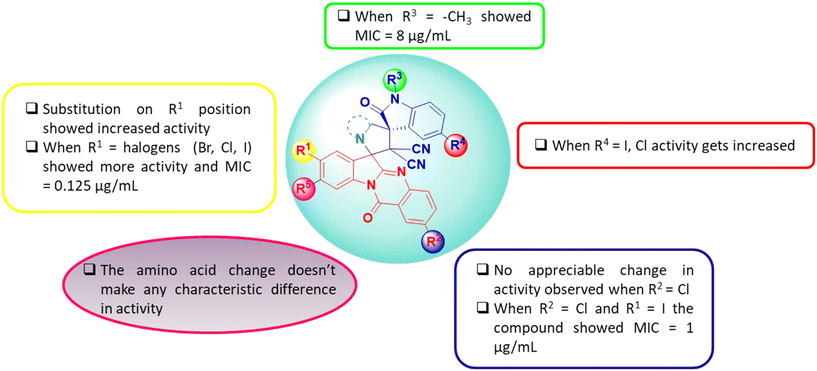 |
| | Fig. 3 Structure activity relationship (SAR) of tryptanthrin–dispiropyrrolidine oxindole molecular hybrids. | |
2.1.2 Cytotoxicity profile of 5b–c, 5f–g and 8 against Vero cells.
The cytotoxicity of 5b–c, 5f–g and 8 were determined against Vero cells by MTT assay, the selectivity index (SI = CC50/MIC) was calculated and results are shown in Table 2. As can be seen, majority of the compounds are non-toxic to Vero cells, especially compounds 5b, 5c and 5g which display high selectivity indices (SI 40 ≥ 80).
Table 2 Cytotoxicity profile and SI of compounds against Vero cells (ATCC CCL-81)
| Compound |
MIC (μg mL−1) |
SI (CC50/MIC) |
|
5b
|
0.125 |
40 |
|
5c
|
0.125 |
>80 |
|
5f
|
0.25 |
<10 |
|
5g
|
0.125 |
>80 |
|
8
|
1 |
10 |
2.1.3 Activity against clinical multidrug-resistant MRSA and VRSA isolates.
After confirming that compounds 5b, 5c, and 5g possessed strong antimicrobial activity against Staphylococcus aureus with high SI, we next assessed their activity against an array of clinically relevant MDR methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Staphylococcus aureus (VRSA) isolates and the results are shown in Table 3.
Table 3 MIC (μg mL−1) of compounds 5b, 5c and 5g against clinical MDR MRSA and VRSA isolates
|
S. aureus
|
MIC (μg mL−1) |
|
5b
|
5c
|
5g
|
Levofloxacin |
Meropenem |
Vancomycin |
|
MSSA
|
ATCC 29213 |
0.125 |
0.25 |
0.5 |
0.125 |
0.125 |
1 |
|
MRSA
|
NRS 119 |
0.125 |
0.125 |
0.25 |
16 |
64 |
1 |
| NRS 129 |
0.125 |
0.25 |
0.25 |
0.25 |
4 |
1 |
| NRS 186 |
0.25 |
0.125 |
0.5 |
4 |
8 |
1 |
| NRS 191 |
0.0625 |
0.0625 |
0.25 |
16 |
64 |
1 |
| NRS 192 |
0.125 |
0.25 |
0.5 |
4 |
8 |
1 |
| NRS 193 |
0.25 |
0.25 |
0.5 |
32 |
64 |
1 |
| NRS 194 |
0.5 |
0.5 |
0.5 |
0.125 |
1 |
1 |
| NRS 198 |
0.125 |
0.25 |
0.5 |
32 |
64 |
1 |
|
VRSA
|
VSR 4 |
0.5 |
0.25 |
0.5 |
>64 |
64 |
>64 |
| VSR 12 |
0.25 |
0.25 |
8 |
32 |
8 |
>64 |
As can be seen, 5b, 5c and 5g exhibited potent activities against various clinical isolates of MRSA and VRSA, although variations in MIC was observed with 5g against VRSA.12 The equipotent activity exhibited by 5b and 5c against MDR MRSA and VRSA indicates that these compounds possess a new mode of action and is able to bypass existing drug-resistance mechanisms, auguring well for their pre-clinical translation.
As 5b consistently exhibited more potent activity against clinical MDR MRSA VRSA isolates, it progressed further for time kill analysis.
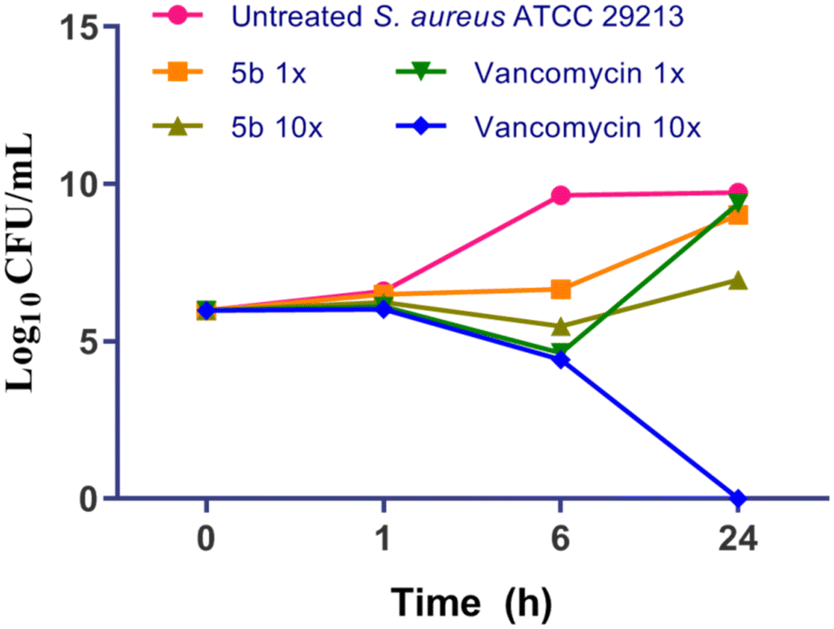
2.1.4 Time kill kinetics of 5b against S. aureus ATCC 29213.
Time kill analysis is conducted to determine the capacity of the compound, whether it is bacteriostatic or bactericidal in nature. Typically, bactericidal antibiotics are preferred since they may retard the generation of resistance. In this context, S. aureus ATCC 29213 was exposed to 1× and 10× MIC of 5b and vancomycin for 24 h and cells were plated at regular intervals and log10 CFU ml−1 was determined and plotted in Fig. 4. As can be seen, treatment with 10× MIC of 5b lead to a reduction of ∼2.7 log10 CFU ml−1 as compared to that of the untreated at 24 h whereas treatment with vancomycin at 10× MIC completely eliminated all culture. Thus, 5b exhibits concentration dependent bactericidal activity.
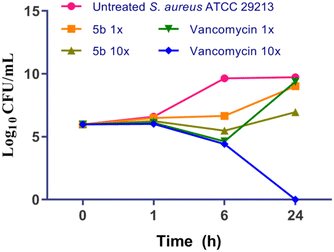 |
| | Fig. 4 Time kill kinetics of 5b and comparators against S. aureus ATCC 29213. | |
2.1.5 Determination of synergy of 5b with FDA approved drugs against S. aureus ATCC 29213.
Since drug combinations are especially recommended for treatment of serious staphylococcal infections, the ability of 5b to interact with various drugs utilized for the treatment of staphylococcal infections was tested using the checkerboard method and the results are depicted in Table 4. As can be seen, 5b does not synergize or interact with ceftazidime, daptomycin, gentamicin, levofloxacin, minocycline, rifampicin, linezolid, meropenem, ceftriaxone and vancomycin, and thus it can be progressed further in pre-clinical translation.
Table 4 Synergy interaction studies of 5b with antibiotics against S. aureus ATCC 29213
| Drug |
MIC (μg mL−1) against S. aureus ATCC 29213 |
MIC (μg mL−1) of 5b in presence of drug ‘A’ |
MIC (μg mL−1) of drug in presence of 5b ‘B’ |
FIC A |
FIC B |
∑FIC = FIC A + FIC B |
Inference |
|
5b
|
0.25 |
|
|
|
|
|
|
| Ceftazidime |
8 |
0.25 |
8 |
1 |
1 |
2 |
No interaction |
| Daptomycin |
1 |
0.25 |
1 |
1 |
1 |
2 |
No interaction |
| Gentamycin |
0.25 |
0.25 |
0.25 |
1 |
1 |
2 |
No interaction |
| Levofloxacin |
0.125 |
0.25 |
0.125 |
1 |
1 |
2 |
No interaction |
| Linezolid |
2 |
0.25 |
2 |
1 |
1 |
2 |
No interaction |
| Meropenem |
0.0625 |
0.25 |
0.0625 |
1 |
1 |
2 |
No interaction |
| Minocycline |
0.125 |
0.25 |
0.125 |
1 |
1 |
2 |
No interaction |
| Rifampicin |
0.0078 |
0.25 |
0.0078 |
1 |
1 |
2 |
No interaction |
| Vancomycin |
1 |
0.25 |
1 |
1 |
1 |
2 |
No interaction |
3. Conclusions
The stereoselective synthesis of sixteen tryptanthrin–dispiropyrrolidine oxindole molecular hybrids was achieved and their detailed antibacterial evaluation was carried out. It was found that 5b, 5c and 5g exhibited very low MIC, out of which 5b showed the best results in MRSA/VRSA panel studies and SI. Furthermore, time kill analysis of 5b showed concentration dependant bactericidal activity and did not interact with any FDA approved drug utilized for the treatment of serious staphylococcal infections. Thus, further studies positioning 5b as a serious therapeutic option for staphylococcal treatment are underway.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
SSL and AD thank the University of Kerala for research and instrumental facilities. SSL thanks the University of Kerala for research fellowship. AA and DS thank UGC for their fellowship while RM thanks DST-INSPIRE for his fellowship. This manuscript bears CSIR-CDRI communication number 10591.
References
- V. Ivasiv, C. Albertini, A. E. Gonçalves, M. Rossi and M. L. Bolognesi, Curr. Top. Med. Chem., 2019, 19(19), 1694–1711 CrossRef CAS PubMed.
- P. T. Alpert, Home Health Care Manag. Pract., 2016, 29(2), 130–133 CrossRef.
- High levels of antibiotic resistance found worldwide, WHO 2018, https://www.who.int/news/item/29-01-2018-high-levels-of-antibiotic-resistance-found-worldwide-new-data-shows.
-
(a) R. Kaur, S. K. Manjal, R. K. Rawal and K. Kumar, Bioorg. Med. Chem., 2017, 25, 4533–4552 CrossRef CAS PubMed;
(b) P. T. Parvatkar, P. S. Parameswaran and S. G. Tilve, Curr. Org. Chem., 2011, 15, 1036–1057 CrossRef CAS;
(c) K. Iwaki, E. Ohashi, N. Arai, K. Kohno, S. Ushio, M. Taniguchi and S. Fukuda, J. Ethnopharmacol., 2011, 134(2), 450–459 CrossRef CAS PubMed;
(d) Y. Takei, T. Kunikata, M. Aga, S. I. Inoue, S. Ushio, K. Iwaki and M. Kurimoto, Biol. Pharm. Bull., 2003, 26(3), 365–367 CrossRef CAS PubMed.
- D. C. M. Costa, M. M. B. D. Azevedo, D. O. E. Silva, M. T. V. Romanos, T. C. B. S. Souto-Padrón, C. S. Alviano and D. S. Alviano, Nat. Prod. Res., 2017, 31(17), 2077–2080 CrossRef CAS PubMed.
- Y. C. Tsai, C. L. Lee, H. R. Yen, Y. S. Chang, Y. P. Lin, S. H. Huang and C. W. Lin, Biomolecules, 2020, 10(3), 366 CrossRef CAS PubMed.
- S. S. Leena, G. Kaul, A. Akhir, D. Saxena, S. Chopra and A. Deepthi, Bioorg. Chem., 2022, 128, 106046 CrossRef PubMed.
-
(a) M. Singh, A. V. Amrutha Krishnan, R. Mandal, J. Samanta, V. Ravichandiran, R. Natarajan, P. Y. Bharitkar and A. Hazra, Mol. Diversity, 2020, 24(3), 627–639 CrossRef CAS PubMed;
(b) E. Sansinenea, E. F. Martínez and A. Ortiz, Eur. J. Org. Chem., 2020, 2020(32), 5101–5118 CrossRef CAS.
- A. S. Filatov, N. A. Knyazev, S. V. Shmakov, A. A. Bogdanov, M. N. Ryazantsev, A. A. Shtyrov and A. V. Stepakov, Synthesis, 2019, 51(03), 713–729 CrossRef CAS.
-
(a) A. Deepthi, N. V. Thomas and V. Sathi, Curr. Green Chem., 2019, 6(3), 210–225 CrossRef CAS and references cited therein;
(b) L. M. Zhou, R. Y. Qu and G. F. Yang, Expert Opin. Drug Discovery, 2020, 15(5), 603–625 CrossRef CAS PubMed.
- S. Vidya, K. Priya, D. Velayudhan Jayasree, A. Deepthi and P. G. Biju, Synth. Commun., 2019, 49(12), 1592–1602 CrossRef CAS.
- B. Yu, D.-Q. Yu and H.-M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *bc and
Ani
Deepthi
*bc and
Ani
Deepthi