
Visible-light promoted intramolecular carboamination of alkynes for the synthesis of oxazolidinone-fused isoquinolinones†
Qihang
Guo
abc,
Dongpo
Lu
a,
Yihui
Mao
a and
Zhan
Lu
 *ad
*ad
aCenter of Chemistry for Frontier Technologies, Department of Chemistry, Zhejiang University, Hangzhou, 310027, China. E-mail: luzhan@zju.edu.cn
bInstitute of Drug Discovery and Design, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, 310027, China
cZJU-Hangzhou Global Scientific and Technological Innovation Center, Hangzhou, 311200, China
dCollege of Chemistry, Zhengzhou University, Zhengzhou, 450001, China
First published on 19th January 2023
Abstract
An efficient method for the synthesis of isoquinolinone derivatives via photopromoted carboamination of alkynes is developed. Starting from the readily available propargyl alcohol derivatives, the polycyclic isoquinolinone derivatives could be obtained with good aryl and heterocycle tolerance. Both terminal and alkyl substituted alkynes could be employed. This protocol is operationally easy, and easily conducted on a gram-scale. A possible mechanism involving radical addition and cyclization following aromatization was proposed.
As an important nitrogen-containing heterocycle, isoquinolinone is widely found in natural products, pharmaceutical agents, and biologically active molecules.1 Therefore, the methods of efficient construction of isoquinolinones have attracted extensive attention. In recent years, the transition metals, including rhodium,2 ruthenium,3 palladium,4 nickel,5 cobalt,6 and so on, have been able to catalyze C–H bond activation inter-/intramolecular [4+2] cycloaddition reaction of benzamides and alkynes affording straight forward methods to construct substituted isoquinolinones (Scheme 1a).7 In addition, the research groups of Ackermann,8a,b Lei,8c and others developed a dual catalytic approach to synthesize isoquinolinones by combination of metal catalysis and electrosynthesis.8
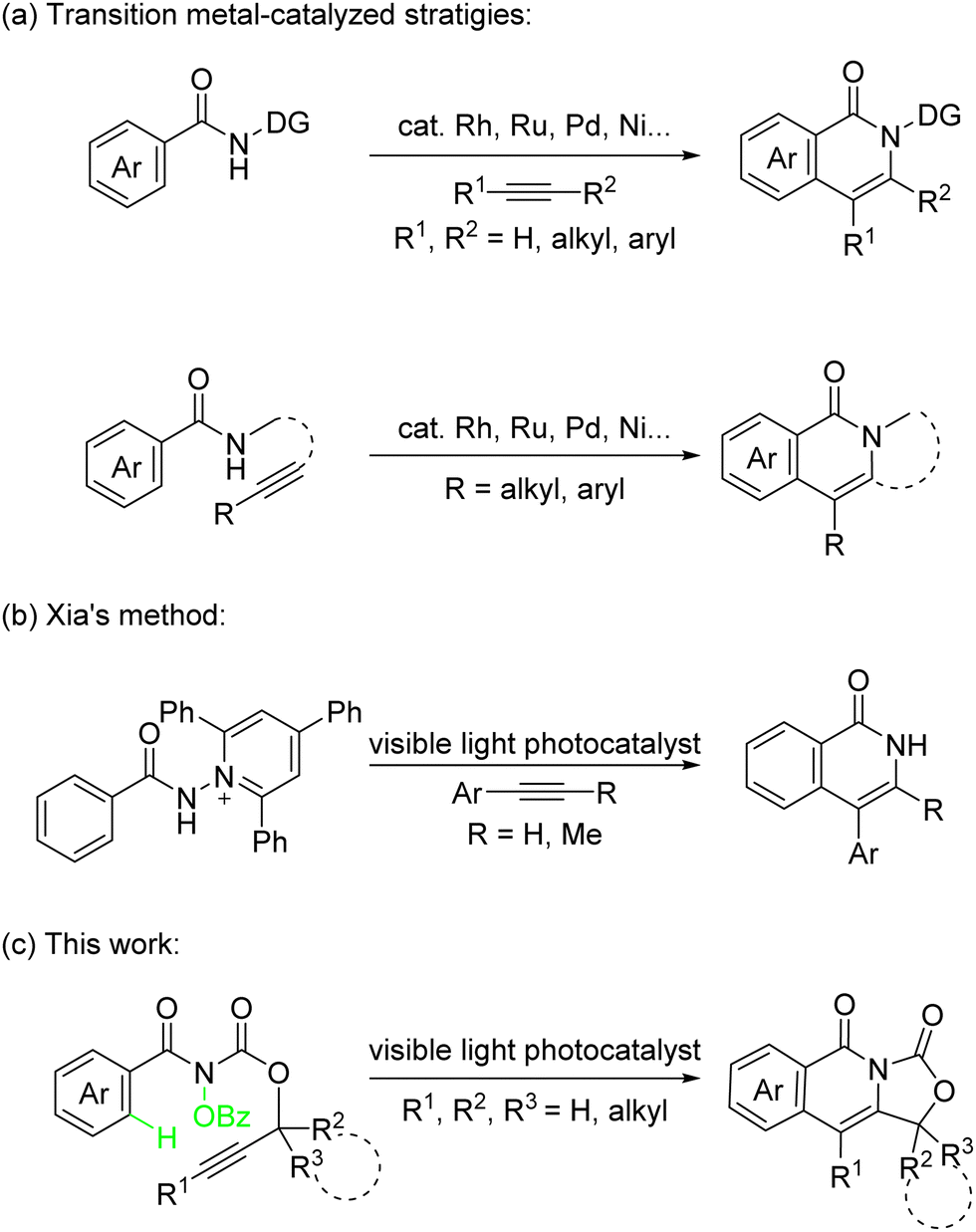 | ||
| Scheme 1 Synthesis of isoquinolinone derivatives via metal-catalysis and photocatalysis. | ||
As a safe, inexpensive, abundant, and nonpolluting energy source, visible light has attracted extensive attention.9 Recently, interests in visible-light photoredox catalysis have provided a number of approaches for C–N bond formation.10 The difunctionalization of olefins initiated by a nitrogen radical is a direct strategy to construct molecules containing a nitrogen heterocycle.11 However, because of the strong bond energy of alkynes and the instability of vinyl radicals derived from radical additions, there are few examples of the synthesis of nitrogen heterocycles using difunctionalization of alkynes initiated by a nitrogen radical.12 The development of methods for the difunctionalization of alkynes is still highly desirable.
In our previous work,13 an amidyl radical intermediate generated from hydroxylamines has been studied for visible light-catalyzed diastereodivergent intramolecular oxyamination of alkenes and intramolecular unactivated C(sp3)–H amination to construct nitrogen-containing compounds. Here, the difunctionalization of alkynes was proposed. The photoinduced synthesis of isoquinolinones from N-amidepyridinium and alkynes was reported by Xia and co-workers in 2020 (Scheme 1b).14 However, there are still some challenges: (1) it is difficult to control the regioselectivity based on unsymmetrical alkynes. (2) There is no construction of polycyclic structures, which are widely found in nature products.15 (3) The reaction is limited to aryl alkynes, and the results using terminal alkynes and alkyl alkynes have not been discussed. Alternatively, intramolecular [4+2] reactions can avoid the problem of regioselectivity for the construction of complex polycyclic structures. Here, we report a visible-light-promoted intramolecular radical-type cycloaddition of alkynes with benzamide to construct isoquinolinone skeletons (Scheme 1c).
At the beginning of our study by using 2-methylbut-3-yn-2-yl benzoyl(benzoyloxy)carbamate 1a as a model substrate, DIPEA as an electron sacrificial donor, and MeCN as a solvent at 50 °C under the irradiation of an 18 W cold fluorescent lamp (CFL), a variety of photocatalysts were screened (Table S1 in ESI†). The reaction using fac-Ir(ppy)3 as a photocatalyst without DIPEA could occur smoothly (entry 1, Table 1). After the optimization of the solvents (entries 1–12, Table 1), the reaction using DMSO as a solvent could give 2a in 64% yield (entry 12). Using 5 W blue LEDs at 60 °C, the reaction could deliver 2a in 88% yield (entries 13 and 14). Based on these results, the standard reaction conditions were determined. Specifically, benzoyl(benzoyloxy)carbamate 1 with 2 mol% of Ir(ppy)3 in DMSO (0.05 M) were stirred at 60 °C under 5 W blue LEDs for 12 h.
|
|
|||
|---|---|---|---|
| Entry | Solvent | 2a (%) | Recovery of 1a (%) |
| a Reaction conditions: 1a (1.0 equiv.) and photocatalyst (2 mol%) in solvent (0.05 M) were stirred at 50 °C under an 18 W compact fluorescent lamp (CFL) for 12 hours. b 5 W blue LEDs. c 60 °C. | |||
| 1 | MeCN | 20 | 70 |
| 2 | Toluene | 34 | 46 |
| 3 | PhCl | 30 | 59 |
| 4 | PhCF3 | 36 | 59 |
| 5 | THF | 17 | 5 |
| 6 | Dioxane | 42 | 3 |
| 7 | DME | 32 | 40 |
| 8 | DCM | 27 | 63 |
| 9 | DCE | 24 | 67 |
| 10 | DMF | 57 | — |
| 11 | DMA | 48 | — |
| 12 | DMSO | 64 | 16 |
| 13b | DMSO | 83 | — |
| 14bc | DMSO | 88 | — |
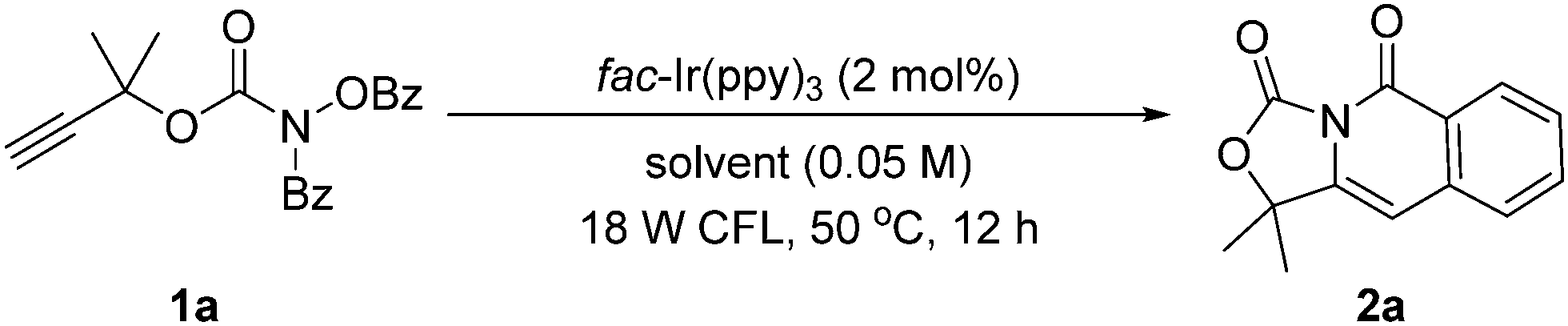
The scope of alkyne carboamination was investigated under the standard conditions (Scheme 2). The substrates (1b–1e) with electron-withdrawing and electron-donating substituents could be converted into alkyne carboamination products in 70–79% yields. The substrate containing naphthalen-1-yl could give 2f in 52% yield. Other heterocycles, such as indole (2g) and thiophene (2h), could give 62–64% yields. The dimethyl group in the substrate can be replaced by diethyl, cyclopentyl, cyclohexyl, and menthol derivatives (2i–2m). The reactions of the internal alkyne derivative 1n gave carbo-amination products in 77% yield. For aryl-terminal alkynes, the reaction only gave trace product. Secondary alcohol derivatives could be transformed to 2o and 2p in 26% and 33% yield, respectively.
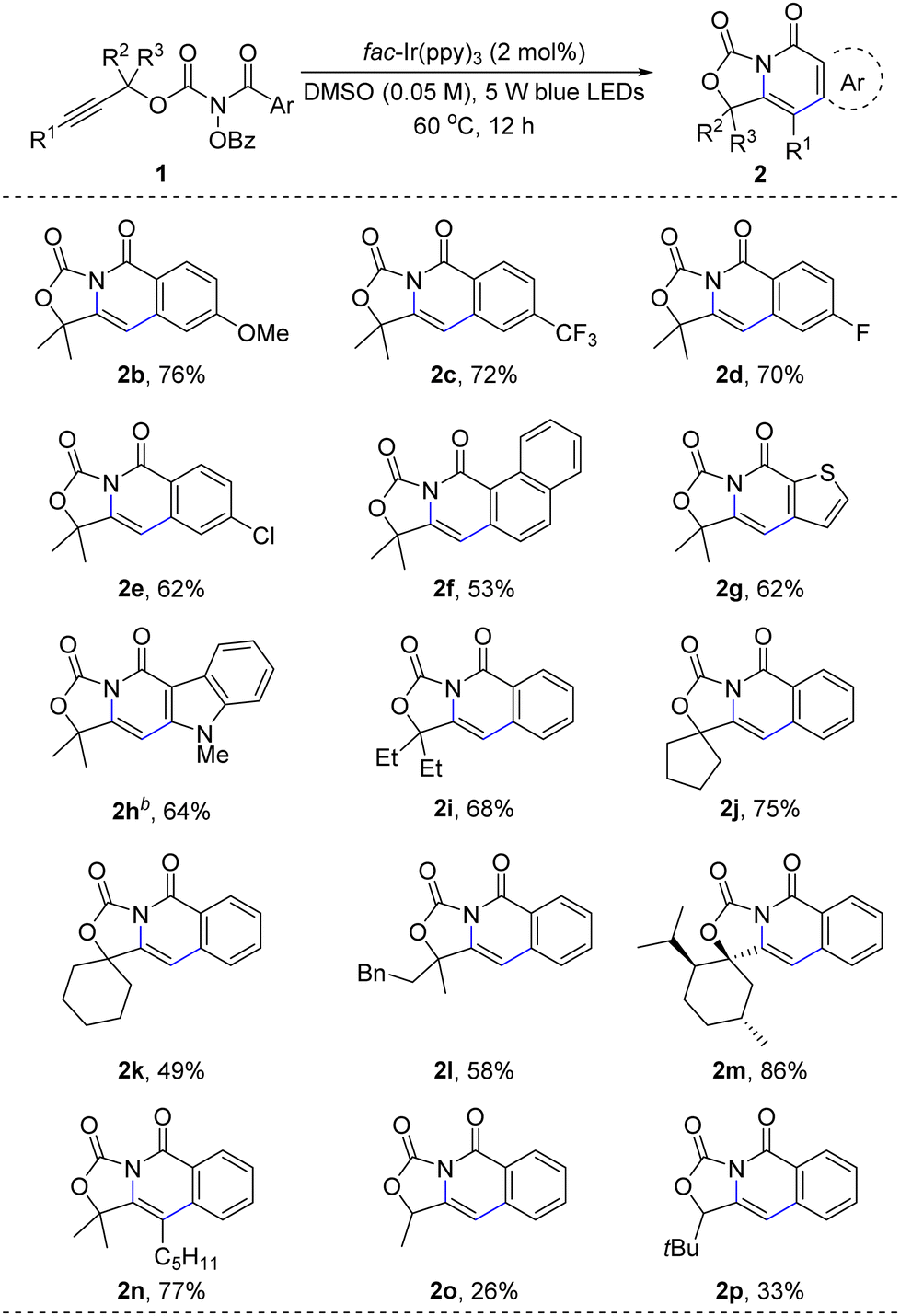 | ||
| Scheme 2 Photo-promoted alkyne difunctionalization. a Reaction conditions: 1 (1.0 equiv.) and fac-Ir(ppy)3 (2 mol%) in DMSO (0.05 M) were stirred at 60 °C under 5 W blue LEDs for 12 h. The yield of the isolated product after column chromatography on a silica gel. b 80 °C, 60 h. | ||
The 5 mmol-scale reaction of 1a could be conducted to afford 2a in 79% yield (Scheme 3a). To further investigate the mechanism of the reaction, a control experiment was conducted. When 3 equivalents of radical inhibitor TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl) were added under the standard reaction conditions, the reaction was almost inhibited (Scheme 3b). This result indicated that the reaction process might contain a radical-initiated process. Prop-2-yn-1-yl(benzoyloxy)carbamate 3 could give alkyne oxyamination product 4 in 15% NMR yield under the standard conditions (Scheme 3c). The product configuration was determined by the nuclear Overhauser effect (NOE). The hydrogen bond between the N–H in the oxazolidin-2-one and the benzoate might lead to this selectivity. The light-on-off experiment demonstrated that the radical chain mechanism was less likely involved (Scheme 3d).
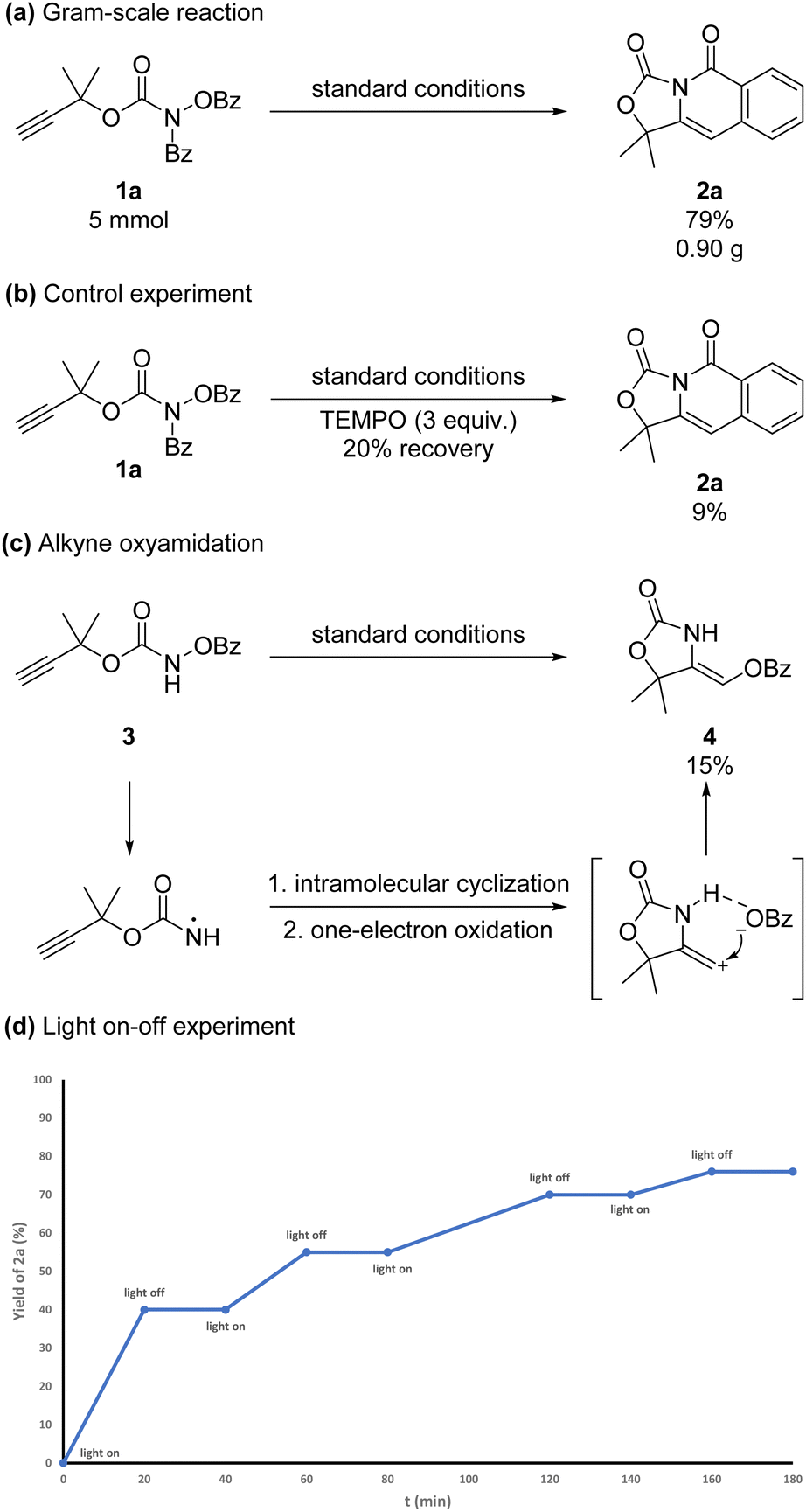 | ||
| Scheme 3 Scale-up reaction and mechanistic studies. | ||
The quantum yield of this reaction was 1 (see in ESI†), so the radical chain mechanism was less likely involved.
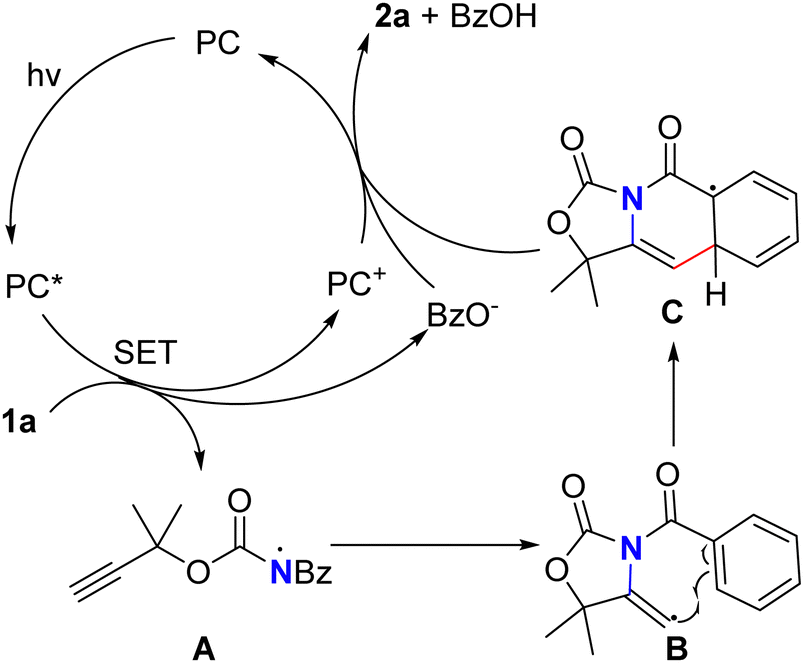
Based on the above experimental results and previously reported literature using photo-/electro-catalysis to construct azaheterocycles,11,16 a possible reaction mechanism was proposed (Scheme 4). Firstly, the substrate 1a could be reduced by PC* species generated by photoexcitation of the visible light photocatalyst to generate the high valence photocatalyst PC+ and afford the radical ion intermediate, which might undergo O–N bond cleavage to form a distonic radical ion A and benzoic acid anion. The intramolecular addition reaction between the nitrogen radical intermediate A and the alkyne gave the enyl radical intermediate B. Subsequently, the enyl radical intermediate B underwent intramolecular cyclization to give the aryl radical intermediate C. This intermediate could undergo one-electron oxidation by PC+ and dehydrogenation to afford 2a and regenerate PC simultaneously.
 | ||
| Scheme 4 Proposed mechanism. | ||
In summary, by using the visible-light-promoted intramolecular carboamination of alkynes, we developed an alternative way to construct aryl and heterocycle polycyclic isoquinolinone derivatives. Both terminal and alkyl substituted alkynes could be tolerated. This reaction could be easily scaled up to 5 mmol. Control experiments and light-on-off experiments indicated that the reaction process might contain a radical-initiated process and the radical chain mechanism was less likely involved.
Financial support was provided by NSFC (22271249); the Fundamental Research Funds for the Central Universities (226-2022-00224); Center of Chemistry for Frontier Technologies; and iChemFoundry, ZJU-Hangzhou Global Scientific and Technological Innovation Center.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) V. A. Glushkov and Y. V. Shklyaev, Chem. Heterocycl. Compd., 2001, 37, 663 CrossRef CAS; (b) G. R. Pettit, Y. Meng, D. L. Herald, K. A. N. Graham, R. K. Pettit and D. L. Doubek, J. Nat. Prod., 2003, 66, 1065 CrossRef CAS PubMed; (c) R. Hili and A. Yudin, Nat. Chem. Biol., 2006, 2, 284 CrossRef CAS PubMed; (d) S.-Q. Zhou and R.-B. Tong, Chem. – Eur. J., 2016, 22, 7084 CrossRef CAS PubMed; (e) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS PubMed.
- (a) T. K. Hyster and T. Rovis, J. Am. Chem. Soc., 2010, 132, 10565 CrossRef CAS PubMed; (b) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908 CrossRef CAS PubMed; (c) F. W. Patureau and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1977 CrossRef CAS PubMed; (d) D.-G. Yu, F. de Azambuja and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 2754 CrossRef CAS PubMed; (e) S. S. Gupta, D. Parmar, R. Kumar, D. Chandra and U. Sharma, Catal. Rev., 2022 DOI:10.1080/01614940.2022.2097640; (f) V. B. Kharitonov, D. V. Muratov and D. A. Loginov, Coord. Chem. Rev., 2022, 471, 214744 CrossRef CAS.
- (a) L. Ackermann, A. V. Lygin and N. Hofmann, Angew. Chem., Int. Ed., 2011, 50, 6379 CrossRef CAS PubMed; (b) B. Li, H. Feng, S. Xu and B. Wang, Chem. – Eur. J., 2011, 17, 12573 CrossRef CAS PubMed; (c) M. Deponti, S. I. Kozhushkov, D. S. Yufit and L. Ackermann, Org. Biomol. Chem., 2013, 11, 142 RSC; (d) C. Kornhaaß, C. Kuper and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1619 CrossRef; (e) K. Gollapelli, S. Kallepu, N. Govindappa, J. Nanubolu and R. Chegondi, Chem. Sci., 2016, 7, 4748 RSC; (f) G. Duarah, P. P. Kaishap, T. Begum and S. Gogoi, Adv. Synth. Catal., 2019, 361, 654 CrossRef CAS; (g) G. Duarah, P. P. Kaishap, T. Begum and S. Gogoi, Adv. Synth. Catal., 2019, 361, 654 CrossRef CAS; (h) L. Song, X. Zhang, X. Tang, L. Van Meervelt, J. Van der Eycken, J. N. Harvey and E. V. Van der Eycken, Chem. Sci., 2020, 11, 11562 RSC.
- (a) H. Zhong, D. Yang, S. Wang and J. Huang, Chem. Commun., 2012, 48, 3236 RSC; (b) N. Sharma, R. Saha, N. Parveen and G. Sekar, Adv. Synth. Catal., 2017, 359, 1947 CrossRef CAS; (c) M. Qiu, X. Fu, P. Fu and J. Huang, Org. Biomol. Chem., 2022, 20, 1339 RSC.
- (a) Y. Kajita, S. Matsubara and T. Kurahashi, J. Am. Chem. Soc., 2008, 130, 6058 CrossRef CAS PubMed; (b) T. Miura, M. Yamauchi and M. Murakami, Org. Lett., 2008, 10, 3085 CrossRef CAS PubMed; (c) H. Shiota, Y. Ano, Y. Aihara, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 14952 CrossRef CAS PubMed; (d) Z. Q. He and Y. Huang, ACS Catal., 2016, 6, 7814 CrossRef CAS; (e) A. Obata, Y. Ano and N. Chatani, Chem. Sci., 2017, 8, 6650 RSC; (f) W.-Z. Wang, J. Xie and B. Zhang, Org. Biomol. Chem., 2018, 16, 3983 RSC; (g) H. Xie, Q. Xing, Z. Shan, F. Xiao and G.-J. Deng, Adv. Synth. Catal., 2019, 361, 189 Search PubMed; (h) I. Nohira, S. Liu, R. Bai, Y. Lan and N. Chatani, J. Am. Chem. Soc., 2020, 142, 17306 CrossRef CAS PubMed.
- (a) L.-B. Zhang, X.-Q. Hao, Z.-J. Liu, X.-X. Zheng, S.-K. Zhang, J.-L. Niu and M.-P. Song, Angew. Chem., Int. Ed., 2015, 54, 10012 CrossRef CAS PubMed; (b) C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 2383 CrossRef CAS PubMed; (c) B.-J. Wang, G.-X. Xu, Z.-W. Huang, X. Wu, X. Hong, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2022, 61, e20220891 Search PubMed.
- (a) G. Y. Song, F. Wang and X. W. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (b) H. Huang, X. Ji, W. Wu and H. Jiang, Chem. Soc. Rev., 2015, 44, 1155 RSC; (c) V. P. Boyarskiy, D. S. Ryabukhin, N. A. Bokach and A. V. Vasilyev, Chem. Rev., 2016, 116, 5894 CrossRef CAS PubMed; (d) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS PubMed; (e) R. Gujjarappa, N. Vodnala and C. C. Malakar, Adv. Synth. Catal., 2020, 362, 4896 CrossRef CAS.
- (a) C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 2383 CrossRef CAS PubMed; (b) R. Mei, N. Sauermann, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2018, 140, 7913 CrossRef CAS PubMed; (c) S. Tang, D. Wang, Y. Liu, L. Zeng and A. Lei, Nat. Commun., 2018, 9, 798 CrossRef PubMed; (d) H. Mei, Z. Yin, J. Liu, H. Sun and J. Han, Chin. J. Chem., 2019, 37, 292 CAS; (e) J. Zhong, Y. Yu, D. Zhang and K. Ye, Chin. Chem. Lett., 2021, 32, 963 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- (a) T. Xiong and Q. Zhang, Chem. Soc. Rev., 2016, 45, 3069 RSC; (b) J. R. Chen, X. Q. Hu, L. Q. Lu and W. J. Xiao, Chem. Soc. Rev., 2016, 45, 2044 RSC.
- (a) M. D. Kärkäs, ACS Catal., 2017, 7, 4999 CrossRef; (b) Y. Zhao and W. Xia, Chem. Soc. Rev., 2018, 47, 2591 RSC.
- (a) M.-H. Huang, W.-J. Hao, G. Li, S.-J. Tu and B. Jiang, Chem. Commun., 2018, 54, 10791 RSC; (b) X. Ren and Z. Lu, Chin. J. Catal., 2019, 40, 1003 CrossRef CAS.
- (a) X. Ren, Q. Guo, J. Chen, H. Xie, Q. Xu and Z. Lu, Chem. – Eur. J., 2016, 22, 18695 CrossRef CAS PubMed; (b) Q. Guo, X. Ren and Z. Lu, Org. Lett., 2019, 21, 880 CrossRef CAS PubMed.
- Y. Zhao, C. Shi, X. Sua and W. Xia, Chem. Commun., 2020, 56, 5259 RSC.
- A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565 CrossRef CAS PubMed.
- (a) Z.-W. Hou, Z.-Y. Mao, H.-B. Zhao, Y. Y. Melcamu, X. Lu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 9168 CrossRef CAS PubMed; (b) Z.-W. Hou, Z.-Y. Mao, J. Song and H.-C. Xu, ACS Catal., 2017, 7, 5810 CrossRef CAS; (c) Z.-W. Hou, Z.-Y. Mao, Y. Y. Melcamu, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed., 2018, 57, 1636 CrossRef CAS PubMed; (d) Z. Ye and F. Zhang, Chin. J. Chem., 2019, 37, 513 CrossRef CAS; (e) J.-S. Li, P.-P. Yang, X.-Y. Xie, S. Jiang, L. Tao, Z.-W. Li, C.-H. Lu and W.-D. Liu, Adv. Synth. Catal., 2020, 362, 1977 CrossRef CAS; (f) P. Wang, Q. Zhao, W. Xiao and J. Chen, Green Synth. Catal., 2020, 1, 42 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc06542h |
| This journal is © The Royal Society of Chemistry 2023 |