
Theoretical investigation on hydrogenation of dinitrogen triggered by singly dispersed bimetallic sites†
Xue-Lu
Ma
 *a,
Yue
Yang
a,
Le-Min
Xu
a,
Hai
Xiao
b,
Wen-Zhi
Yao
c and
Jun
Li
*b
*a,
Yue
Yang
a,
Le-Min
Xu
a,
Hai
Xiao
b,
Wen-Zhi
Yao
c and
Jun
Li
*b
aSchool of Chemical & Environmental Engineering, China University of Mining & Technology, Beijing 100083, China. E-mail: maxl@cumtb.edu.cn
bDepartment of Chemistry and Key Laboratory of Organic Optoelectronics & Molecular Engineering of Ministry of Education, Tsinghua University, Beijing 100084, China. E-mail: junli@tsinghua.edu.cn
cDepartment of Environmental and Municipal Engineering, North China University of Water Conservancy and Electric Power, Zhengzhou 450011, China
First published on 16th November 2021
Abstract
The conversion of molecular dinitrogen into ammonia is one of the most important chemical processes, which involves the inert N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond activation and multi-step hydrogenation. To meet the requirements of complicated catalytic processes, single-cluster catalysts (SCCs) have emerged as a promising platform to trigger synergistic effects. Herein, we present a systematic investigation on the stability and activity of a series of singly dispersed bimetallic catalysts, M1Con/CoOx SCCs, for the hydrogenation of dinitrogen by first-principles calculations. Our results indicate that late transition metals are prone to form stable singly dispersed bimetallic clusters by doping the O vacancy on CoO surfaces. Due to the low oxidation state of the doped metals in the stable M1Con/CoOx SCCs, the chemisorption of N2 is enhanced by the multiple “pull–push effect” of the isolated bimetallic sites. Subsequently, an improved electron descriptor (μ) is proposed to effectively estimate the correlation between the favorable bridging N2 adsorption energies and the essential electronic characteristics of M1Con/CoOx SCCs, in which the influence of the metal d orbital electrons and the intrinsic 1st ionization energy are considered. We focus on the first hydrogenation in the associative mechanism on the multicenter of M1Con/CoOx SCCs, and the Pd1Co4/CoOx SCC is proposed to exhibit superior charge buffer capacity towards thermal dinitrogen hydrogenation. Furthermore, a superimposed evaluation strategy associates the activity of nitrogen fixation on SCCs with certain intrinsic features of multi-active sites at both static and kinetic states. The present work not only provides a potential candidate for the thermal N2-to-NH3 conversion, but also sheds insights into the rational design of heterogeneous catalysts for thermal nitrogen fixation.
N bond activation and multi-step hydrogenation. To meet the requirements of complicated catalytic processes, single-cluster catalysts (SCCs) have emerged as a promising platform to trigger synergistic effects. Herein, we present a systematic investigation on the stability and activity of a series of singly dispersed bimetallic catalysts, M1Con/CoOx SCCs, for the hydrogenation of dinitrogen by first-principles calculations. Our results indicate that late transition metals are prone to form stable singly dispersed bimetallic clusters by doping the O vacancy on CoO surfaces. Due to the low oxidation state of the doped metals in the stable M1Con/CoOx SCCs, the chemisorption of N2 is enhanced by the multiple “pull–push effect” of the isolated bimetallic sites. Subsequently, an improved electron descriptor (μ) is proposed to effectively estimate the correlation between the favorable bridging N2 adsorption energies and the essential electronic characteristics of M1Con/CoOx SCCs, in which the influence of the metal d orbital electrons and the intrinsic 1st ionization energy are considered. We focus on the first hydrogenation in the associative mechanism on the multicenter of M1Con/CoOx SCCs, and the Pd1Co4/CoOx SCC is proposed to exhibit superior charge buffer capacity towards thermal dinitrogen hydrogenation. Furthermore, a superimposed evaluation strategy associates the activity of nitrogen fixation on SCCs with certain intrinsic features of multi-active sites at both static and kinetic states. The present work not only provides a potential candidate for the thermal N2-to-NH3 conversion, but also sheds insights into the rational design of heterogeneous catalysts for thermal nitrogen fixation.
1. Introduction
Ammonia (NH3) is a vital chemical for agriculture and industry and it can be directly synthesized on a large scale through the Haber–Bosch process (N2 + 3H2 → 2NH3) under harsh conditions, in which the nitrogen and hydrogen atoms do not react until the strong NN and H–H bonds have been broken (the dissociative mechanism).1 However, the reaction mechanism in enzymes is quite different, in which N2 molecules are hydrogenated initially before N–N cleavage (the associative mechanism).2 The principal obstacle of the thermal N2-to-NH3 conversion essentially lies in the inert NN bond activation and multi-step hydrogenation. Inspired by biological nitrogen fixation to improve the Haber–Bosch process, extensive efforts have been made to search for sustainable and clean methods for ammonia production, as well as effective catalysts with high performance.3
As the bridge of homogeneous catalysts and heterogeneous catalysts, single-atom catalysts (SACs) with well-defined single-atom dispersion on the surfaces4,5 exhibit high selectivity and activity, as well as maximizing the utilization efficiency of supported metal atoms.6,7 To adapt to the requirements of complicated catalytic reactions and break the limitation of a single adsorption site, single-cluster catalysts (SCCs), singly dispersed metal clusters anchored on nonmetallic supports with natural pores or defects, have emerged as promising candidates in the field of heterogeneous catalysis.8–14 Singly dispersed bimetallic clusters anchored on the oxide surface (M1An/AOx) can be regarded as a typical category of SCCs, in which an oxygen atom on the metal oxide (AOx) surface is substituted by a guest metal atom (M).15
It is noteworthy that the guest metal M in M1An/AOx shows a strong charge rearrangement.16 For example, the oxygen vacancy on the CeO2 surface results in very stable anchoring sites for the Au adatom, which binds at the O vacancy site with two Ce nearest neighbors, forming AuCe2 clusters.17 The charge transfer occurs from the reduced oxide surface to the supported metal atom, which results in a negatively charged Auδ− adatom and leads to the formation of Au–Ce bonds. Benefiting from the presence of metal–metal bonds and the special electronic state of M1An, which could trigger synergistic effects, singly dispersed bimetallic sites have been considered as a promising platform for complicated catalytic reactions, such as the reduction of NO to N2 on the Pd1Con/Co3O4 catalyst,8 and methanol partial oxidation (MPO) on the Ir1Znn/ZnO catalyst.9 In our previous work,18 a high degree of NN triple bond activation occurs in a bridging manner over the Co–Rh bond of the bimetallic SCC Rh1Co3/CoO, and the co-adsorption of reactants is favorable on isolated Rh1Co3 sites, which are proposed to be directly hydrogenated to form the NNH intermediate in the thermal N2-to-NH3 conversion.12 The catalytic ability of M1An SCCs arises from both the electronic effect of doped low-valent metal M that serves as a charge buffer, and the complementary role of synergic metal A in catalysis, which offer more possibilities for bond cleavage/formation and contribute to achieving unique catalytic properties different from SACs. Additionally, active clusters in SCCs strongly prefer to bind adsorbates with certain sites, and such adsorbate-induced reconstructions can dynamically generate active sites during catalytic reactions, which is known as the cluster fluxionality and also partially responsible for the high catalytic activity of SCCs.19,20
Due to the Brønsted–Evans–Polanyi (BEP) relation, the dissociation barrier of N2 and the desorption energies of NHx are limited by the adsorption energy of the N atom.21–23 Rather than the N2 dissociative adsorption, the NNH associative mechanism on the multicenter of SCCs can bypass the BEP relation.14 The electrochemical N2 reduction reactions on SCCs have been proposed as one of the most attractive alternatives for the production of NH3.1,10,13,24–33 However, the energy variation changes significantly when the chemical potential is increased by applying a negative electric potential because hydrogen is not entering as H2 but as protons and electrons, H+ and e−.34 The mechanism of thermal N2-to-NH3 conversion is totally different because of the definite activation energy of the reaction, in which H transfer, M–H bond cleavage and N–H bond formation should be considered intensively.
Using first-principles calculation, we present a systematic investigation on the stability and activity of a series of M1Con/CoOx SCCs for dinitrogen activation. Here, the electron descriptor of singly dispersed bimetallic sites is adopted to analyze the correlation between the favorable bridging N2 adsorption energies and the intrinsic properties of M1Con/CoOx SCCs. We focus on the first hydrogenation in the associative mechanism of dinitrogen fixation and the correlation factors of dinitrogen hydrogenation triggered by bimetallic SCCs are discussed. Our results demonstrated that the Pd1Co4/CoOx SCC exhibits superior charge buffer capacity towards thermal dinitrogen hydrogenation. The correlation of the activation energy in this work sheds insights into the rational design of heterogeneous catalysts for thermal nitrogen fixation.
2. Computational details
The calculations were based on spin-polarized density functional theory methods, as implemented in the Vienna Ab initio Simulation Package (VASP version 5.4) with projector-augmented wave pseudopotential.35,36 The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional was used to describe the exchange–correlation interaction of valence electrons.37 In this work, we have not included the vdW correction, because the commonly used approach for including the vdW interaction, such as the D3 correction, may lead to overestimation of chemisorption energies.38 The cutoff energy for the plane wave basis set was taken as 400 eV. A Monkhorst-Pack grid of size 2 × 2 × 1 was used to sample the surface Brillouin zone. To partially correct the strong electron-correlation property of cobalt monoxide, DFT+U calculations were carried out with the parameter of Ueff = 3.0 eV for Co.39 The convergence criterion of electronic iteration was set at 10−5 eV for energy and 0.03 eV Å−1 for the maximal force. The transition states were obtained by relaxing the force <0.02 eV Å−1 by using the dimer method. Atomic charges were calculated using the atom-in-molecule (AIM) theory proposed by Bader.40The CoO (011) surface was represented by a periodic slab model with vacuum layers of 12 Å constructed using the cubic CoO with cell parameters of a = b = c = 4.2667 Å. Since CoO is antiferromagnetic and has atomic moment on cobalt atoms, the primitive cubic unit cell of CoO was used to build the surface slab, which was previously proved to be the most energetically stable configuration for CoO.18 The slab model contains five layers of 60 atoms, and the three top-layer slabs of the surface were allowed to relax while the other layers were frozen during the geometry optimization.
3. Results and discussion
3.1 Stability and electronic structures of M1Con/CoOx SCCs
Different vacancies on the surface exhibit quite different binding capacities with the same doped metal.41 To evaluate the thermodynamic stabilities of SACs (doping the Co vacancy) and SCCs (doping the O vacancy) on the CoO surface as shown in Fig. 1, binding energies between 24 transition metals and the CoO surface with different vacancy defects were taken into consideration.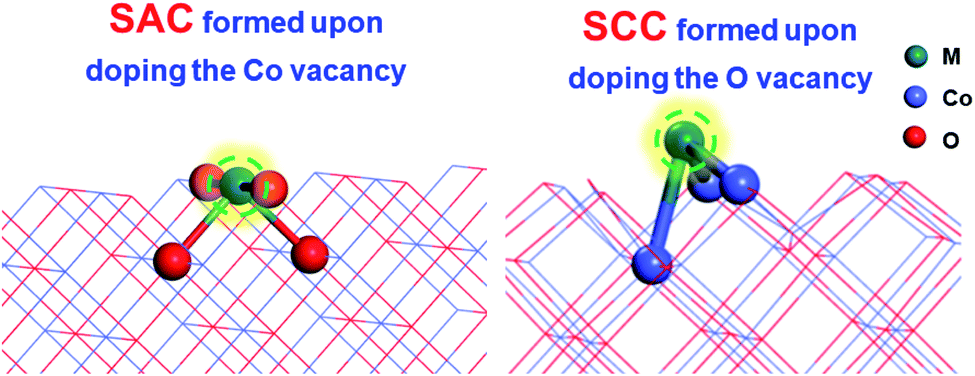 | ||
| Fig. 1 The model structures of SACs (doping the Co vacancy) and SCCs (doping the O vacancy) on the CoO surface. | ||
The binding energy Eb(SCC) of M1 anchored on the O vacancy is calculated as,
| Eb(SCC) = ESCC − EM1 − EOv |
| Eb(SAC) = ESAC − EM1 − ECov |
Based on the stronger binding energies as shown in Fig. 2, it was suggested that early transition metals are prone to form SACs with doping the Co vacancy on the CoO surface, while late transition metals (Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, Au) can more easily generate SCCs with doping the O vacancy. In addition, the previous calculations suggest that the host metal oxide surfaces CoO(011) could stabilize most of the guest metal atoms (Ni, Ru, Ir, etc.) due to large energy barriers, which proves that the doped metal atoms are kinetically stable and not prone to migrate out of the surface O vacancy.42 The stable configurations of M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, Au) and corresponding computed electron density difference maps are displayed in Fig. S1.† It is noteworthy that pure late transition metal surfaces are limited by the adsorption of molecular nitrogen and the first proton transfer step (N2 + H+ + e− → *NNH) on the volcano plots for ammonia formation.2 For this reason, the following discussion focuses on the first hydrogenation of dinitrogen to reveal the activity of M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, Au) in dinitrogen fixation.
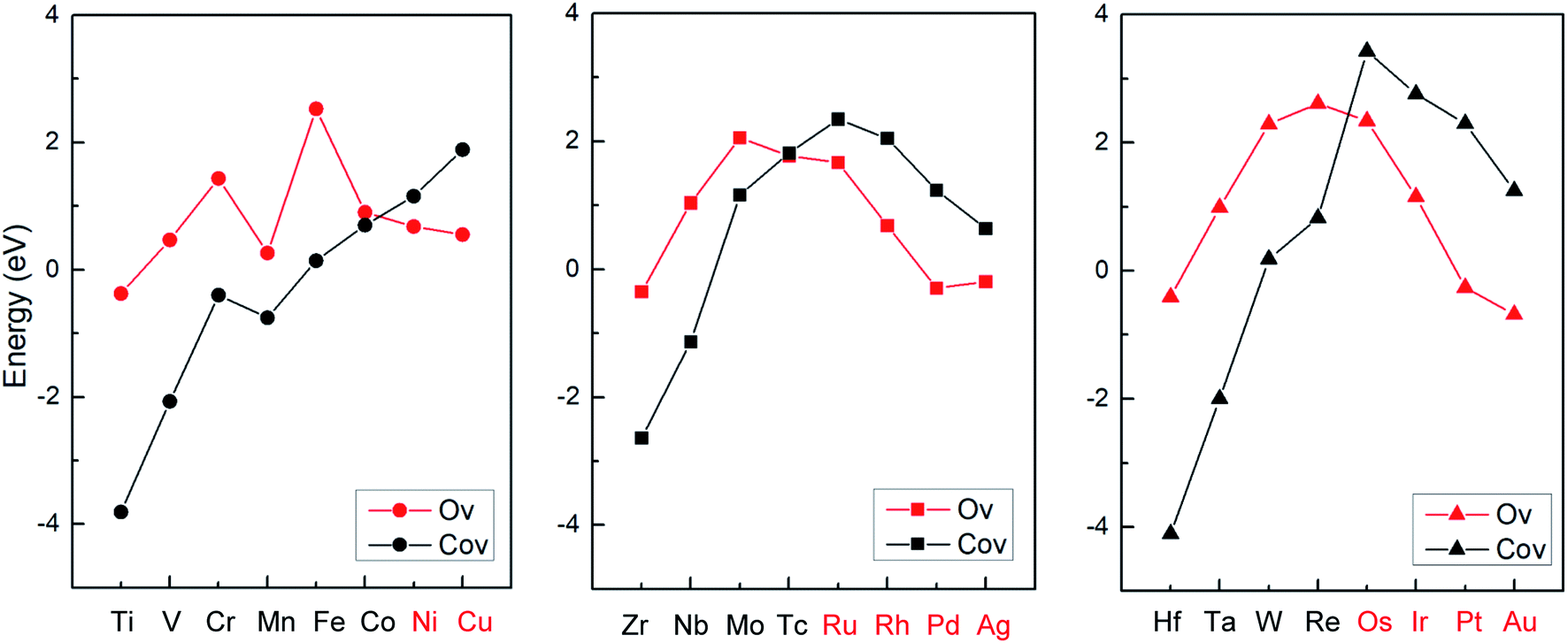 | ||
| Fig. 2 Binding energies between 24 transition metals and the CoO surface with different vacancy defects. | ||
From Bader charge analysis, it is shown that the doped metals are negatively charged in the stable SCCs (M = Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, Au) as shown in Fig. 3. With the most negative charge in the period, the Bader charge on doped Pd and Pt is −0.56|e| and −0.81|e|, respectively. The results indicate that late transition metals (Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, Au) are prone to be further reduced to obtain electrons and form singly dispersed bimetallic clusters with neighboring Co atoms in the process of metal doping to the O vacancy on the CoO surface. The phenomenon is related to the intrinsic electron characteristics of the doped transition metals, which can be reflected by the first ionization energy and the electron affinity energy as displayed in Fig. S2.† In the same period, the smaller the first ionization energy is, the more easily electrons are lost; the larger the electron affinity energy is, the more easily electrons are obtained. Therefore, early transition metals with smaller first ionization energy and electron affinity are more prone to lose electrons; while late transition metals with larger first ionization energy and electron affinity are more likely to gain electrons during doping with the O vacancy on the CoO surface, leading to the relatively stable singly dispersed bimetallic clusters, M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, Au).
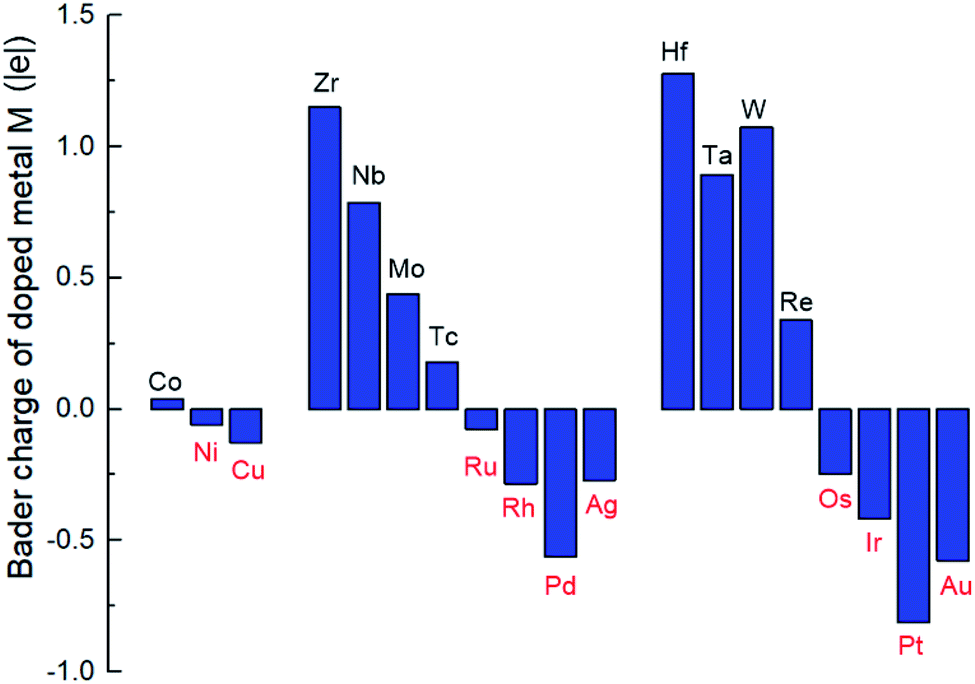 | ||
| Fig. 3 Bader charge of the doped metal M in M1Con/CoOx SCCs. | ||
The composition of the singly dispersed bimetallic cluster, M1Con, can be referenced to the Co–M distance between the doped metal M and neighboring Co atoms. The key bond lengths of Co–M in M1Con/CoOx SCCs are listed in Table S1.† It is noteworthy that the representation of composition is related to the distance criterion to some extent. The strong metal–metal interaction of the supported bimetallic cluster can be further confirmed by the overlap of the projected d-orbitals of the doped M and the neighboring Co atoms as illustrated in Fig. S3.† The present work can help to understand the formation of SCCs synthesized in the experiments so far, such as the Pd1Con/Co3O4 catalyst8 and Ir1Znn/ZnO catalyst,9 as well as give more insights into the development of SCCs into singly dispersed bimetallic clusters anchored on the oxide surface (M1An/AOx).
3.2 Adsorption of N2 and H2 on M1Con/CoOx SCCs
The adsorption of the N2 molecule on SCCs is vital to activate the inert NN triple bond, and also meaningful for subsequent hydrogenation.43 Owing to the low oxidation state of the doped metals in the stable M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, Au), the isolated bimetallic sites have the potential to enhance the chemisorption of N2 to effectively activate the NN triple bond.
For these stable M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, Au), the adsorption configurations of dinitrogen with the end-on and the bridging modes are considered. The corresponding adsorption energies and the N–N bond lengths are listed in Table S2.† The N–N bond lengths in the bridging mode are in the range from 1.171 Å (Ir) to 1.205 Å (Pt), and the N–N bond lengths in the end-on mode are in the range from 1.118 Å (Au) to 1.151 Å (Os). The results show that the N2 adsorption species with the bridging mode have a longer N–N bond length than those in the end-on mode on all M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, Au), which implies the stronger activation of the bridging N2 and the stronger π-backdonation from the bimetallic sites of M1Con/CoOx SCCs to N2. In addition, the bridging adsorption configurations of N2 on M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, Au) are more stable than the corresponding end-on adsorption configurations. The adsorption energies of the bridging mode range from −3.18 eV(Os) to −0.04 eV(Au), and the adsorption energies of the end-on mode range from −1.91 eV(Ir) to 0.29 eV(Au). For N2 adsorption on M1Con/CoOx SCCs (M = Ag, Au), the end-on configuration of N2 is physisorption, which indicates that it is difficult for the adsorption and activation of N2 to take place.
Furthermore, the electronic properties and the charge transfer induced by N2 adsorption are discussed. As convinced by the projected density of states (PDOS) in Fig. S4,† for the bridging adsorption configurations of N2 on M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, Au), there is strong d–π* orbital coupling above or below the Fermi energy level, as well as obvious overlap between the d orbitals of M1Con and the occupied orbitals of adsorbed N2, contributing the multiple “pull–push effect”,11 which could be considered as the reservoir for storing or supplying d electrons when needed. The strong interaction between the dinitrogen unit and bimetallic sites of M1Con/CoOx SCCs is mainly caused by the “acceptance–donation” of electrons,44 which is mostly associated with the unoccupied and occupied d orbitals of the doped metal and neighboring cobalt. As shown in Fig. S5 and S6,† the charge density difference of adsorbed N2 on SCCs further confirms the electron transfer between M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, Au) and the adsorbed N2, which shows that more electrons transfer from M1Con/CoOx SCCs to the bridging N2 than to the end-on N2 at the isovalue of 0.05 e Å−3. Obviously, the electron enrichment and decrease mainly occur between M1Con/CoOx SCCs and N2, implying that M1Con/CoOx SCCs act as both electron donors and acceptors, which is well consistent with the PDOS analysis above.
The cooperative effect of hydrogen adsorption is also an important issue of active site stability for dinitrogen activation.45 For most doped metals of M1Con/CoOx SCCs, dissociative H2 adsorption configurations are available, and corresponding adsorption energies are in the range from −1.95 eV(Ir) to −0.05 eV(Cu) as listed in Table S2.† However, M1Con/CoOx SCCs (M = Ag, Au) have no interaction with dihydrogen due to the full π-states of the doped metal. Nørskov and co-workers drew the volcano curves to describe the catalytic performance for hydrogen evolution on different metal surfaces,46 and also proposed that Au and Ag are unreactive metals for the HER. As shown in Fig. 4, it was also found that there is a linear correlation with an R2 of 0.964 between the H2 adsorption energy and the end-on N2 adsorption energy on M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Os, Ir, Pt), which indicates the active limitation of single sites for the adsorbed species. In contrast, the linear relationship between the energies of bridging N2 adsorption and H2 adsorption is rough with an R2 of 0.730, which means that the bridging adsorption on the bimetallic active site is beneficial to break through the restriction of catalytic nature of the single active metal.
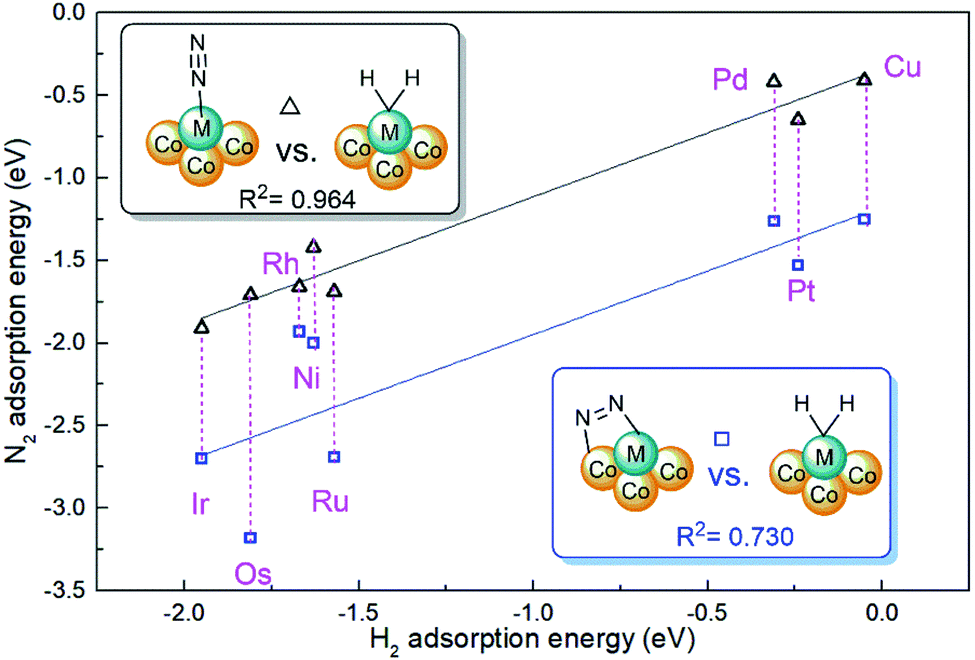 | ||
| Fig. 4 Relationship between N2 adsorption and H2 adsorption on M1Con/CoOx SCCs. | ||
In an attempt to gain further insight into the relationship between the properties of the active site and the stability of bridging N2-adsorbed species on M1Con/CoOx SCCs, an improved electronic descriptor (μ) was introduced, in which the influence of the metal d orbital electrons and the intrinsic 1st ionization energy were considered comprehensively.11 The electron descriptor (μ) of the SCC is calculated using the following formula,
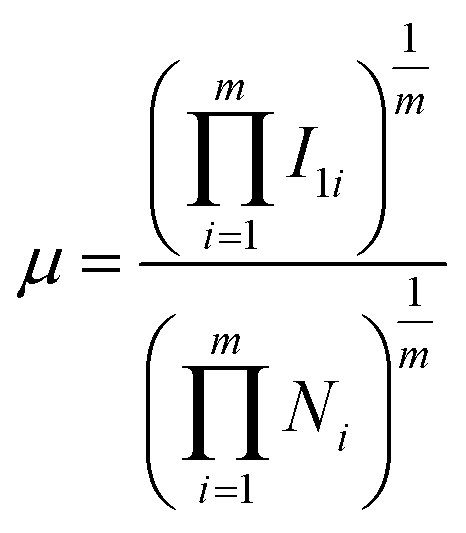
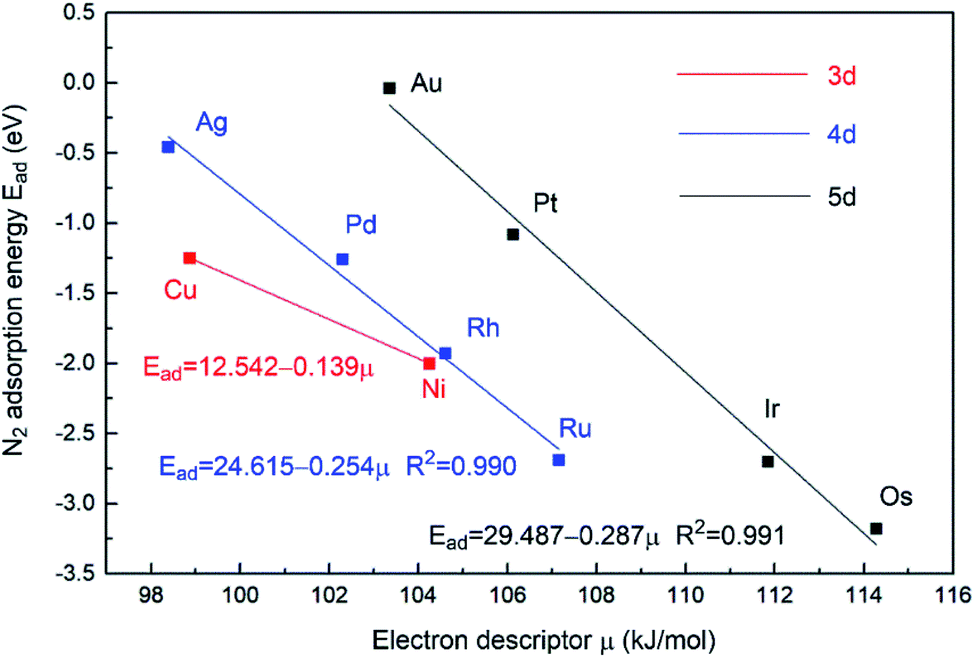 | ||
| Fig. 5 Relationship between bridging N2 adsorption energies at bimetallic sties and the electron descriptor μ of M1Con/CoOx SCCs. | ||
3.3 Hydrogenation of N2 on M1Con/CoOx SCCs
Catalytic activities of SCCs originate from their ability for the bridging N2 activation and dissociative H2 activation, which is related to the intrinsic electronic properties of SCCs as discussed above. Followed by the favorable N2 bridging adsorption, hydrogen is added in the form of H2 molecules. The thermodynamically advantageous configurations for the co-adsorption of N2 and H2 on M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Os, Ir, Pt) are shown in Fig. S7,† and they play an important role in the associative mechanism. In the thermal N2-to-NH3 conversion, the bridging N2 at the bimetallic site in co-adsorption provides more possible ways for subsequent hydrogenation, either the alternating pathway or the evolutionary distal pathway.12 From this viewpoint, multi-active sites on SCCs have certain advantages to overcome the limitation of SACs when complicated multi-steps are involved.The energy of favorable co-adsorption (Eco) is linear to the energy of bridging N2 adsorption with an R2 of 0.885 as shown in Fig. S8,† indicating that N2 bridging adsorption plays a leading role in the stability of the co-adsorption configuration. Due to the lower co-adsorption energies compared to the corresponding dinitrogen adsorption energies, it is suggested that the dissociative adsorption of H2 on SCCs has an enhanced influence on the stability of the co-adsorption on SCCs. Therefore, the electronic descriptor of singly dispersed bimetallic sites, μ of SCCs, also correlates with the adsorption energy of co-adsorption in the same period.
The adsorbed bridging dinitrogen unit on SCCs can be further activated by H transferring to form NNH* from I to II as shown in Fig. 6a, which plays a critical role in the subsequent steps to generate ammonia. To evaluate the reactivity of the first step in dinitrogen hydrogenation on M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Os, Ir, Pt), the activation energy barriers (Ea) and the reaction energies (Er) are calculated using the following formulas, Ea = ETSI-II − EI and Er = EII-I − EI, respectively. The activation energy barriers are in the range of 1.65 eV(Pd) to 3.09 eV(Ir) and the reaction energies range from −0.05 eV(Pd) to 0.87 eV(Ir) as listed in Table S4.†Fig. 6b shows the linear proportional relationship between the energy barrier and the reaction energy in the same period. In addition to the lowest activation energy barrier from I to II, the reaction energy of Pd1Co4/CoOx SCC is negative, which suggests that the H transferred NNH* intermediate is more stable than the co-adsorption of N2 and H2. The complete reaction pathway for the N2-to-NH3 thermal conversion on the Pd1Co4/CoOx SCC follows the alternating hydrogenation mechanism as shown in Fig. S9,† in which the N–N bond cleavage is not the rate-determining step.
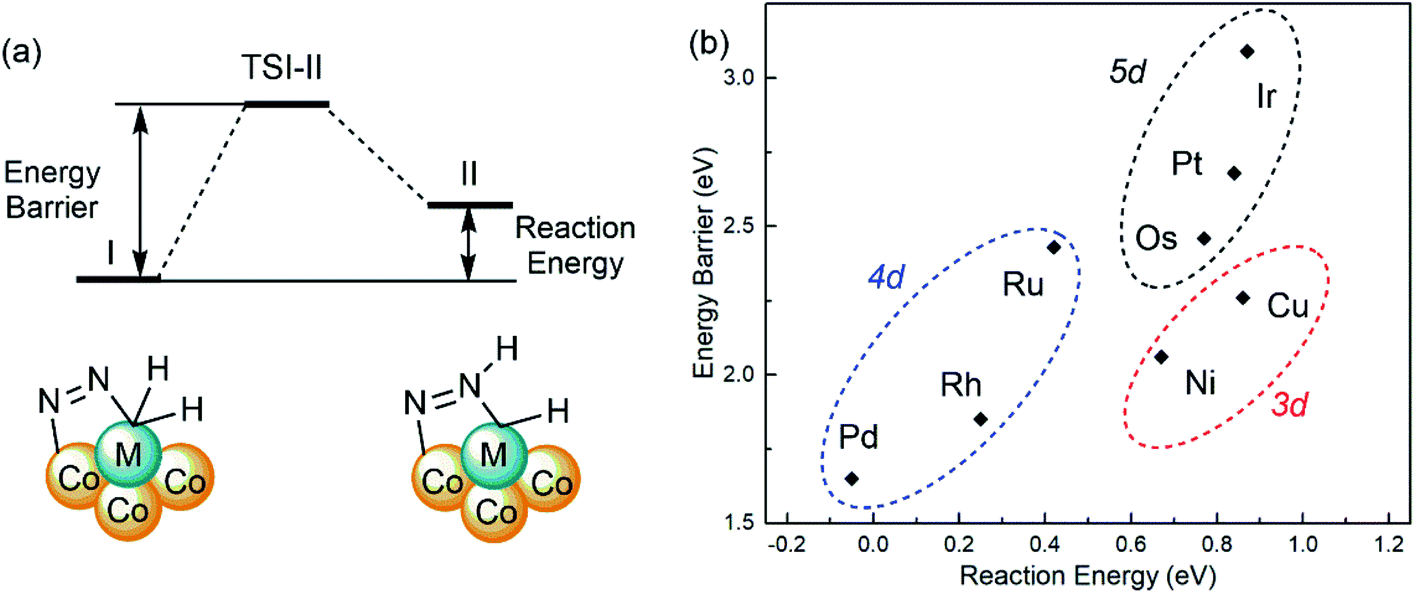 | ||
| Fig. 6 (a) Schematic diagram for H transferring on SCCs from I to II. (b) The linear proportional relationship between the energy barrier and the reaction energy. | ||
As we proposed previously, the doped metal with large charge buffer capacity and low N2 reduction activation energy is the ideal candidate for ammonia synthesis.12 The negatively charged doped metals on M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Os, Ir, Pt) could be regarded as electron reservoirs, which have the potential to regulate the charge variation in the dinitrogen hydrogenation. According to the Bader charge difference of doped M (ΔξM) between I and II, the doped Pd exhibits minimal charge fluctuations, and ΔξM is correlated with Er with an R2 of 0.821 as shown in Fig. S10.† However, the correlation of ΔξM and Ea is poor with an R2 of 0.457. To gain improved insight into hydrogenation of dinitrogen triggered by singly dispersed bimetallic sites, the essential electronic characteristics of SCCs, the co-adsorption energies, and the changes of geometric structures should be combined to explore the correlation of the activation energies.
When the bridging dinitrogen is hydrogenated to *NNH on M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Os, Ir, Pt), occupation of the π* orbitals in *NNH facilitates the increase of N–N bond length. Besides the N–N bond activation, the process also involves M–H bond cleavage and N–H bond formation. These bond length variations from I to II are listed in Table S5.† The root mean square deviation (RMSD) of these key bonds between I and II could be used to measure the variations of two geometric structures on the same bimetallic sites, which should also be related to the activation energy barrier. That is, the activation energy should be considered as the contribution of resisting the combined effects of geometric changes and the charge rearrangement, which is defined as the descriptor (α) calculated using the following formula, α = RMSDbond × ΔξM. Based on the descriptor α, the correlation with Ea is improved with an R2 of 0.570. If the essential electronic characteristics of SCCs (μ) are stacked up to define the descriptor (β) with the formula of β = RMSDbond × ΔξM × μ, the R2 of the linear relationship between β and Ea is 0.684. If the co-adsorption energy (Eco) is continued to stack, the descriptor (γ) is defined with the formula of γ = RMSDbond × ΔξM × μ × Eco, and the R2 of the linear relationship between γ and Ea is 0.719. Therefore, the improved descriptors from α to γ with informative data could associate the activity of nitrogen fixation on SCCs with some intrinsic features of multi-active sites at static and kinetic states. Similar to the previously used descriptors, the complexity of the model increases exponentially with the number of the considered features.47
4. Conclusion
In summary, we performed DFT calculations to systematically investigate the stability and activity of a series of singly dispersed bimetallic catalysts, M1Con/CoOx SCCs, in the hydrogenation of dinitrogen. Firstly, it is demonstrated that early transition metals with smaller first ionization energy and electron affinity are more prone to lose electrons, and are preferred to form relatively stable M1/Cox-1Ox SACs by doping with the Co vacancy on the CoO surface; while late transition metals with larger first ionization energy and electron affinity are more likely to gain electrons during doping with the O vacancy, leading to the relatively stable singly dispersed bimetallic clusters, M1Con/CoOx SCCs (M = Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, Au). Secondly, the strong interaction between the bridging dinitrogen unit and bimetallic sites of M1Con/CoOx SCCs is mainly caused by the “acceptance–donation” of electrons, which is mostly associated with the unoccupied and occupied d orbitals of the doped metal with negative charge and neighboring cobalt. Based on the descriptor identification, the correlation between the favorable bridging N2 adsorption energies and the integrated electronic capability of M1Con/CoOx SCCs is effectively estimated. Furthermore, it is suggested that the activation energy barrier of dinitrogen hydronation should be related to the overlay of the essential electronic characteristics of SCCs and the N2/H2 co-adsorption energy at the static state, as well as the changes of geometric structures and charge arrangements in the kinetic process. We finally concluded that the Pd1Co4/CoOx SCC is the most promising M1Con/CoOx SCC for the thermal dinitrogen hydrogenation due to the superior charge buffer capacity. Overall, the present work can help to understand the formation of SCCs synthesized in the experiments so far, as well as shed insights into the development of SCCs into singly dispersed bimetallic clusters anchored on the oxide surface (M1An/AOx) for the thermal N2-to-NH3 conversion.Author contributions
X.-L. Ma, and J. Li conceived and designed the project. X.-L. Ma, Y. Yang and W.-Z. Yao carried out the calculations. X.-L. Ma, Y. Yang, L.-M. Xu and H. Xiao discussed the results and prepared the manuscript. All the authors reviewed and contributed to this paper.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21902182), the Fundamental Research Funds for the Central Universities (2021YQHH04) and the Training Program of Innovation and Entrepreneurship for Undergraduates (C202003262).References
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C. P. Chen, M. R. Smith III and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS.
- E. Skulason, T. Bligaard, S. Gudmundsdottir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jonsson and J. K. Norskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- Q. Wang, J. Guo and P. Chen, J. Energy Chem., 2019, 36, 25–36 CrossRef.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- J.-C. Liu, Y. Tang, Y.-G. Wang, T. Zhang and J. Li, Natl. Sci. Rev., 2018, 5, 638–641 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- J. Liu, ACS Catal., 2016, 7, 34–59 CrossRef.
- L. Nguyen, S. Zhang, L. Wang, Y. Li, H. Yoshida, A. Patlolla, S. Takeda, A. I. Frenkel and F. Tao, ACS Catal., 2016, 6, 840–850 CrossRef CAS.
- L. Nguyen, S. Zhang, L. Tan, Y. Tang, J. Liu and F. F. Tao, ACS Sustainable Chem. Eng., 2019, 7, 18793–18800 CrossRef.
- B. Han, H. Meng, F. Li and J. Zhao, Catalysts, 2020, 10, 974 CrossRef CAS.
- X. Zheng, Y. Liu and Y. Yao, Chem. Eng. J., 2021, 426, 130745 CrossRef CAS.
- X. L. Ma, J. C. Liu, H. Xiao and J. Li, J. Am. Chem. Soc., 2018, 140, 46–49 CrossRef CAS.
- G. Zheng, L. Li, Z. Tian, X. Zhang and L. Chen, J. Energy Chem., 2021, 54, 612–619 CrossRef.
- J.-C. Liu, X.-L. Ma, Y. Li, Y.-G. Wang, H. Xiao and J. Li, Nat. Commun., 2018, 9, 1–9 CrossRef.
- N. López and F. Illas, J. Phys. Chem. B, 1998, 102, 1430–1436 CrossRef.
- R. Rousseau, V.-A. Glezakou and A. Selloni, Nat. Rev. Mater., 2020, 5, 460–475 CrossRef CAS.
- M. F. Camellone and S. Fabris, J. Am. Chem. Soc., 2009, 131, 10473–10483 CrossRef PubMed.
- S. Zhang, L. Nguyen, J. X. Liang, J. Shan, J. J. Liu, A. I. Frenkel, A. Patlolla, W. Huang, J. Li and F. F. Tao, Nat. Commun., 2015, 6, 7938 CrossRef CAS PubMed.
- B. W. J. Chen, L. Xu and M. Mavrikakis, Chem. Rev., 2021, 121, 1007–1048 CrossRef CAS.
- B. Zandkarimi and A. N. Alexandrova, J. Phys. Chem. Lett., 2019, 10, 460–467 CrossRef PubMed.
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, J. Catal., 2015, 328, 36–42 CrossRef CAS.
- T. Bligaard, J. K. Nørskov, S. Dahl, J. Matthiesen, C. H. Christensen and J. Sehested, J. Catal., 2004, 224, 206–217 CrossRef CAS.
- S. Dahl, A. Logadottir, C. J. H. Jacobsen and J. K. Nørskov, Appl. Catal., A, 2001, 222, 19–29 CrossRef CAS.
- Y. Gambo, S. Adamu, A. A. Abdulrasheed, R. A. Lucky, M. S. Ba-Shammakh and M. M. Hossain, Appl. Catal., A, 2021, 609, 117914 CrossRef CAS.
- H. Zhang, C. Cui and Z. Luo, J. Phys. Chem. C, 2020, 124, 6260–6266 CrossRef CAS.
- C. Yao, N. Guo, S. Xi, C. Q. Xu, W. Liu, X. Zhao, J. Li, H. Fang, J. Su, Z. Chen, H. Yan, Z. Qiu, P. Lyu, C. Chen, H. Xu, X. Peng, X. Li, B. Liu, C. Su, S. J. Pennycook, C. J. Sun, J. Li, C. Zhang, Y. Du and J. Lu, Nat. Commun., 2020, 11, 4389 CrossRef CAS PubMed.
- M. Li, Y. Cui, X. Zhang, Y. Luo, Y. Dai and Y. Huang, J. Phys. Chem. Lett., 2020, 11, 8128–8137 CrossRef CAS PubMed.
- C. Cui, H. Zhang and Z. Luo, Nano Res., 2020, 13, 2280–2288 CrossRef CAS.
- Z. W. Chen, L. X. Chen, M. Jiang, D. Chen, Z. L. Wang, X. Yao, C. V. Singh and Q. Jiang, J. Mater. Chem. A, 2020, 8, 15086–15093 RSC.
- D. Ma, Z. Zeng, L. Liu, X. Huang and Y. Jia, J. Phys. Chem. C, 2019, 123, 19066–19076 CrossRef CAS.
- X. Zhang, A. Chen, Z. Zhang and Z. Zhou, J. Mater. Chem. A, 2018, 6, 18599–18604 RSC.
- Z. W. Chen, J. M. Yan and Q. Jiang, Small Methods, 2018, 3, 1800291 CrossRef.
- J.-C. Chen, H. Cao, J.-W. Chen, S.-J. Qian, G.-J. Xia, Y.-G. Wang and J. Li, J. Phys. Chem. C, 2021, 125, 19821–19830 CrossRef CAS.
- M. Boudart, J. Am. Chem. Soc., 1952, 74, 1531–1535 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- B. Liu, L. Cheng, L. Curtiss and J. Greeley, Surf. Sci., 2014, 622, 51–59 CrossRef CAS.
- L. Wang, T. Maxisch and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195107 CrossRef.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- M. Ha, D. Y. Kim, M. Umer, V. Gladkikh, C. W. Myung and K. S. Kim, Energy Environ. Sci., 2021, 14, 3455–3468 RSC.
- N. Liu, X.-L. Ma, J. Li and H. Xiao, J. Phys. Chem. C, 2021 DOI:10.1021/acs.jpcc.1c07706.
- M. Zafari, A. S. Nissimagoudar, M. Umer, G. Lee and K. S. Kim, J. Mater. Chem. A, 2021, 9, 9203–9213 RSC.
- W. Zhao, L. Chen, W. Zhang and J. Yang, J. Mater. Chem. A, 2021, 9, 6547–6554 RSC.
- C. Ling, Y. Zhang, Q. Li, X. Bai, L. Shi and J. Wang, J. Am. Chem. Soc., 2019, 141, 18264–18270 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- M. Zafari, D. Kumar, M. Umer and K. S. Kim, J. Mater. Chem. A, 2020, 8, 5209–5216 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08350c |
| This journal is © The Royal Society of Chemistry 2022 |