
π-Adsorption promoted electrocatalytic acetylene semihydrogenation on single-atom Ni dispersed N-doped carbon†
Wenxiu
Ma‡
a,
Zhe
Chen‡
 b,
Jun
Bu
a,
Zhenpeng
Liu
a,
Jinjin
Li
a,
Chen
Yan
a,
Lin
Cheng
d,
Lei
Zhang
a,
Hepeng
Zhang
a,
Jichao
Zhang
*c,
Tao
Wang
*b and
Jian
Zhang
*a
b,
Jun
Bu
a,
Zhenpeng
Liu
a,
Jinjin
Li
a,
Chen
Yan
a,
Lin
Cheng
d,
Lei
Zhang
a,
Hepeng
Zhang
a,
Jichao
Zhang
*c,
Tao
Wang
*b and
Jian
Zhang
*a
aKey Laboratory of Special Functional and Smart Polymer Materials of Ministry of Industry and Information Technology, School of Chemistry and Chemical Engineering, Northwestern Polytechnical University, Xi'an, 710129, P. R. China. E-mail: zhangjian@nwpu.edu.cn
bCenter of Artificial Photosynthesis for Solar Fuels, School of Science, Westlake University, Hangzhou 310024, P. R. China. E-mail: twang@westlake.edu.cn
cShanghai Synchrotron Radiation Facility, Zhangjiang Laboratory, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai, 201204, P. R. China. E-mail: zhangjichao@zjlab.org.cn
dSchool of Science, Xi'an Polytechnic University, Xi'an, 710048, P. R. China
First published on 15th November 2021
Abstract
In pursuit of environmental friendliness and high economic efficiency, renewable energy-driven selective acetylene semihydrogenation under ambient conditions is urgently required but remains a grand challenge. Herein, we explore single-atom nickel (Ni) dispersed N-doped carbon (SA-Ni-NC) as a novel electrocatalyst for catalyzing selective acetylene semihydrogenation. In situ electrochemical Raman and theoretical investigations reveal that weak π-adsorption of ethylene on individual Ni sites in SA-Ni-NC facilitates its desorption and thus avoids its overhydrogenation. Eventually, under pure acetylene flow, SA-Ni-NC exhibits a high ethylene faradaic efficiency (FEethylene) of 91.3% and a large current density of −92.2 mA cm−2 at −0.6 V vs. the reversible hydrogen electrode (RHE). Even in crude ethylene stream containing 1% acetylene impurities (1 × 104 ppm), SA-Ni-NC still manifests a high acetylene conversion of 97.4% with a large space velocity (SV) of 2.4 × 104 mL gcat−1 h−1 and a high turnover frequency (TOF) of 22.9 h−1 for each Ni atom.
1. Introduction
During the ethylene polymerization process, residual acetylene impurities (∼5 × 103 to 3 × 104 parts per million (ppm)) in an ethylene feedstock will adversely degrade the quality of target polymers by deactivating ethylene polymerization catalysts (e.g., Ziegler–Natta catalysts).1,2 Thus, selectively removing acetylene from ethylene stream dominantly produced by petroleum hydrocarbon cracking is indispensable for achieving a polymer-grade ethylene feedstock.3–5 Currently, the industrial implementation and laboratory research mainly focus on thermocatalytic hydrogenation of acetylene.6–10 Unfortunately, for such thermocatalytic processes, there are several serious drawbacks: (1) the applied thermocatalysts are precious metal Pd-based materials; (2) high reaction temperatures (∼100–250 °C) result in intensive energy consumption; (3) excess hydrogen gas as a hydrogen source causes safety issues; (4) competing side reactions include carbon–carbon coupling, overhydrogenation, etc. Therefore, in comparison with the traditional thermocatalytic process, electrocatalytic acetylene semihydrogenation under ambient conditions is highly promising due to its environmental friendliness, low cost, easy operation, as well as integration with electricity generated from sustainable energy sources such as solar energy.11,12 However, in the past few decades, the lack of high-performance electrocatalysts with superior activity, ethylene selectivity and stability essentially hampers the development of electrocatalytic acetylene semihydrogenation.13–16For enhancing ethylene selectivity, weakening ethylene adsorption to avoid its over-hydrogenation is crucial during acetylene semihydrogenation.17–19 Among different ethylene adsorption modes ethylidyne mode, di-σ-mode, and π-bonded mode, adsorption strength of the π-bonding configuration on one metal atom is the lowest.1,20,21 Meanwhile, isolated metal atoms can suppress the carbon–carbon coupling reaction between two neighboring acetylene molecules. Accordingly, electrocatalysts featuring single-atom active sites are promising for facilitating electrocatalytic acetylene semihydrogenation to ethylene but remain unexplored until now.
In this work, we synthesize single-atom Ni supported on N-doped carbon (SA-Ni-NC) by pyrolyzing nickel doped zeolitic imidazolate framework-8 (ZnNi-ZIF) precursors.22,23 When the SA-Ni-NC serves as an electrocatalyst for acetylene semihydrogenation, it shows an excellent electrocatalytic performance with an ethylene faradaic efficiency (FEethylene) of 91.3%, a partial ethylene current density of −92.2 mA cm−2, and a turnover frequency (TOF) of 3847.8 h−1 at −0.6 V vs. the reversible hydrogen electrode (RHE) under a pure acetylene flow. For crude ethylene flow containing 1 × 104 ppm acetylene, SA-Ni-NC presents a high conversion of 97.4% and a large space velocity of 2.4 × 104 mL gcat−1 h−1. Density functional theory calculations combined with in situ electrochemical Raman analyses demonstrate that the superior electrocatalytic performance of SA-Ni-NC is attributed to weak π-adsorption of ethylene on single Ni atoms, which favors the rapid ethylene desorption and eventually promotes selective semihydrogenation of acetylene to ethylene.
2. Results and discussion
The precursor bimetallic ZnNi-ZIF was fabricated in methanol through a typical synthesis method following the preparation procedure in the ESI.† X-ray diffraction (XRD), scanning electron microscopy (SEM), and thermogravimetric analysis (TGA) reveal that the ZnNi-ZIF has a similar morphology to that of ZIF-8 (Fig. S1–S3, ESI†). Subsequently, after the carbonization of the ZnNi-ZIF at 950 °C in a N2 atmosphere, the derived SA-Ni-NC was confirmed to be a carbon material by the Raman and XRD analyses (Fig. 1a, S4 and S5, ESI†). Obviously, no XRD diffraction peaks of Ni nanoparticles were observed. As references, the samples at different carbonization temperatures were also prepared. By increasing the Ni/Zn ratio of the ZnNi-ZIF to 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Ni nanoparticle immobilized N-doped carbon (Ni NP/N-C) was fabricated by increasing the Ni/Zn ratio of the ZnNi-ZIF to 9:1 (Fig. S6–S9, ESI†). The specific surface area of SA-Ni-NC was measured to be ∼727.9 m2 g−1 based on Brunauer–Emmett–Teller (BET) adsorption/desorption isotherms (Fig. S10, ESI†).
:1, Ni nanoparticle immobilized N-doped carbon (Ni NP/N-C) was fabricated by increasing the Ni/Zn ratio of the ZnNi-ZIF to 9:1 (Fig. S6–S9, ESI†). The specific surface area of SA-Ni-NC was measured to be ∼727.9 m2 g−1 based on Brunauer–Emmett–Teller (BET) adsorption/desorption isotherms (Fig. S10, ESI†).
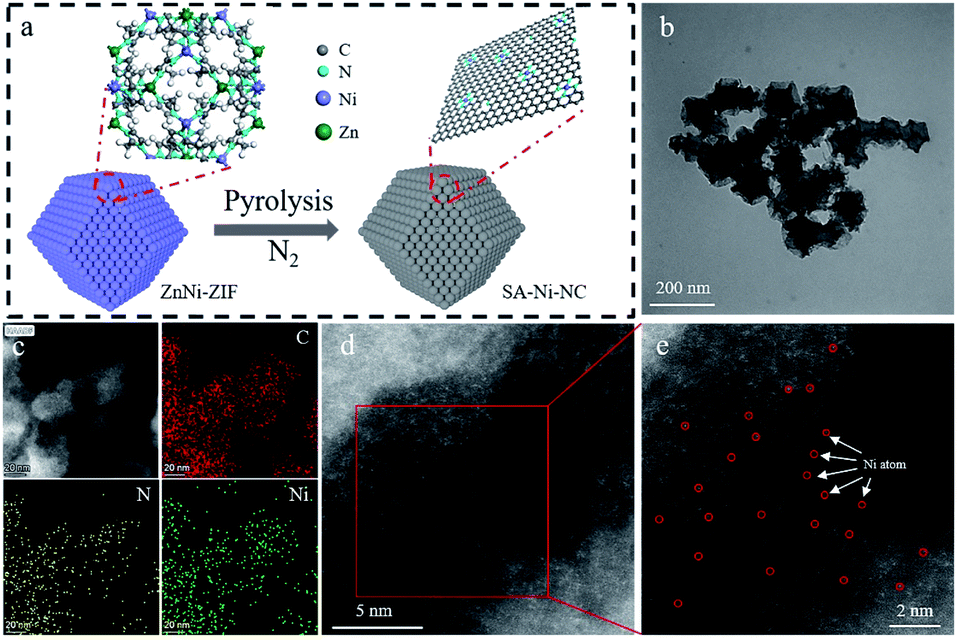 | ||
| Fig. 1 Schematic illustrations of the preparation and structural characterization of SA-Ni-NC. (a) The synthesis process of SA-Ni-NC. (b) TEM, (c) HAADF-STEM and corresponding C, N, Ni elemental mapping, and (d, e) magnified HAADF-STEM images of SA-Ni-NC. | ||
X-ray photoelectron spectroscopy (XPS) was employed to study the chemical structure of SA-Ni-NC (Fig. S11, ESI†). In the XPS spectra of Ni in SA-Ni-NC, two characteristic peaks at 855.3 and 872.5 eV correspond to Ni 2p1/2 and Ni 2p3/2, respectively.24 The high-resolution XPS spectrum of N 1s revealed the coexistence of pyridinic (398.6 eV), pyrrolic (400.8 eV) and graphitic (403.8 eV) nitrogen species.25 The Ni content in SA-Ni-NC was determined to be ∼2.39 wt% by utilizing inductively coupled plasma analysis (ICP, Table S1, ESI†). Transmission electron microscopy (TEM) was conducted to scrutinize the morphology of SA-Ni-NC and no Ni particles existed (Fig. 1b and S12, ESI†). Fig. 1c and S13d (ESI†) show the corresponding elemental mapping images of C, N, and Ni, evidencing their homogeneous distributions over SA-Ni-NC. The high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) showed the atomic dispersion of Ni in the carbon matrix of SA-Ni-NC (Fig. 1d, e and S13a–c, ESI†). The electron energy loss spectroscopy (EELS) analysis was further conducted. As shown in Fig. S14,† the bright atoms are single-atom Ni in SA-Ni-NC.
To further unveil the bonding environment of Ni atoms in SA-Ni-NC, the X-ray absorption fine structure (XAFS) analysis was employed. With the Ni foil and Ni porphyrin complex (NiPc) as references, the Ni K-edge X-ray absorption near-edge structure (XANES) of SA-Ni-NC is depicted in Fig. 2a. Clearly, the XANES spectra of the Ni K-edge indicated that the energy absorption edge of SA-Ni-NC was located between those of Ni foil and NiPc, and close to NiPc, indicating that the valence state of Ni species in SA-Ni-NC was about +2. Furthermore, the Fourier transform (FT) k3-weighted extended X-ray absorption fine structure (EXAFS) curve of SA-Ni-NC is shown in Fig. 2b. The main peak of SA-Ni-NC at 1.41 Å corresponded to the first shell scattering of the Ni–N bonds rather than the Ni–Ni bonds at 2.18 Å. The EXAFS fitting analysis revealed that the isolated Ni atoms were in a Ni–N4 configuration with a Ni–N bond length of 1.83 Å (Fig. 2c and Table S2, ESI†).26–28 The wavelet transform (WT) contour plot of SA-Ni-NC had only one intensity maximum at 5.6 Å−1, which was assigned to the Ni–N bonds rather than Ni–Ni scattering (8.2 Å−1). These results clearly prove the dispersion of Ni–N4 species in SA-Ni-NC (Fig. 2d–f).29,30
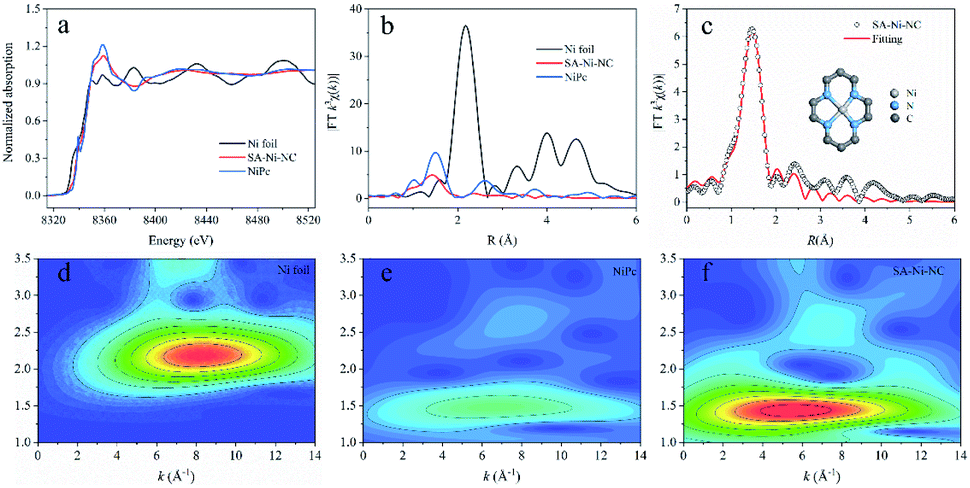 | ||
| Fig. 2 Chemical state and coordination information of SA-Ni-NC. (a) Normalized XANES spectra at the Ni K-edge of Ni foil, NiPc, and SA-Ni-NC. (b) Fourier transform (FT) magnitude of the experimental EXAFS spectra of Ni foil, NiPc, and SA-Ni-NC. (c) EXAFS fitting curve of SA-Ni-NC. The inset in (c) shows the proposed Ni–N4 coordination environment. (d–f) The WT of the k3-weighted EXAFS signals of Ni foil, NiPc, and SA-Ni-NC. | ||
For electrocatalytic acetylene semihydrogenation, SA-Ni-NC and Ni NP/N-C were tested using a three-electrode flow cell in 1 M KOH aqueous solution under pure acetylene flow with a flow rate of 20 mL min−1 (Fig. S15–S17, ESI†). As shown in the linear sweep voltammetry (LSV) curve in Fig. 3a, the cathodic current density of Ni NP/N-C at −0.5 V considerably decreases to −31.9 mA cm−2 from −47.8 mA cm−2 when the gas flow switched from Ar to pure acetylene. By contrast, SA-Ni-NC exhibited an increased current density of −55.3 mA cm−2 at −0.5 V in acetylene flow, which was 3.1 times higher than that under Ar. These results undoubtedly reflected the outstanding electrocatalytic activity of SA-Ni-NC toward acetylene. Based on gas chromatography (GC) analyses, the total faradaic efficiencies of gaseous products (H2 and ethylene) were close to 100% and no liquid products were detected (Fig. S18b, ESI†). Noticeably, as shown in Fig. 3b, SA-Ni-NC achieves a FEethylene of 91.3% at −0.6 V, which is dramatically higher than 71.7% for Ni NP/N-C. Meanwhile, SA-Ni-NC also exhibited an enhanced ethylene partial current density (jethylene) over all applied potentials (Fig. 3c). The jethylene of SA-Ni-NC reached −84.2 mA cm−2 at −0.6 V, which was approximately 1.8 times higher than that for Ni NP/N-C. Impressively, according to the inset in Fig. 3c, the calculated TOF of SA-Ni-NC was up to 3847.8 h−1 at a potential of −0.6 V. Then, long-term electrocatalytic acetylene semihydrogenation at −30 mA cm−2 was carried out (Fig. S18c, ESI†). Apparently, over 12 h, SA-Ni-NC presented a stable potential of about −0.5 V and an average FEethylene of ∼95%. For deeply probing the underlying mechanism of superior electrocatalytic acetylene semihydrogenation on SA-Ni-NC, electrochemical active surface area (ECSA) and electrochemical impedance spectroscopy (EIS) measurements were evaluated (Fig. S19, ESI†). The ECSA of SA-Ni-NC and Ni NP/N-C was determined by measuring the electrochemical double-layer capacitance (Cdl), which was derived from the cyclic voltammograms at different scan rates from 20 to 100 mV s−1. As indicated in Fig. S19c,† the Cdl of SA-Ni-NC was about 34.1 mF cm−2, which was much higher than 20.0 mF cm−2 for Ni NP/N-C, suggesting that SA-Ni-NC exposed more active sites. The EIS plot of SA-Ni-NC showed a smaller charge transfer resistance of ∼29 Ω than >120 Ω for Ni NP/N-C, indicating its fast electron transfer kinetics.
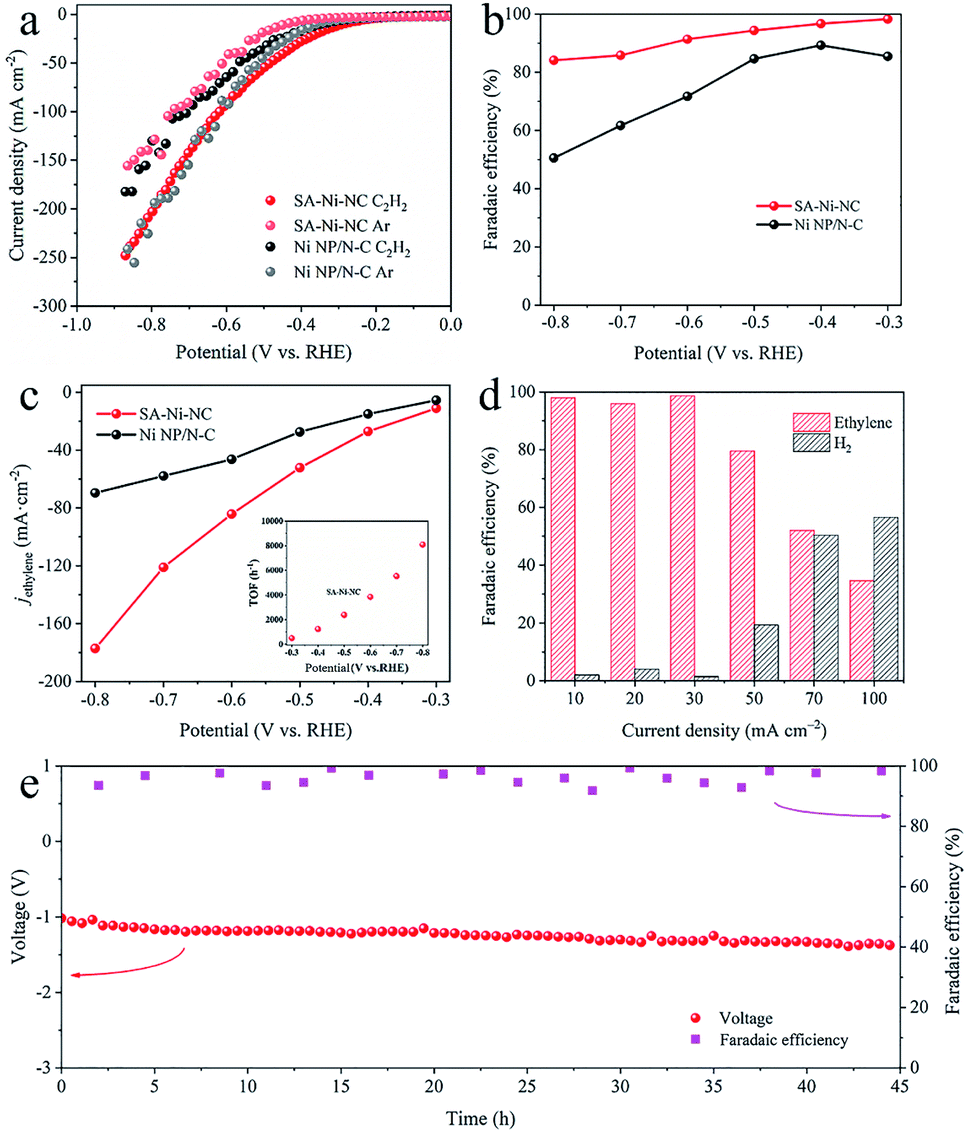 | ||
| Fig. 3 Electrocatalytic acetylene semihydrogenation measurements under pure acetylene flow. (a) The LSV curves of SA-Ni-NC and Ni NP/N-C in an acetylene and Ar atmosphere at a scan rate of 1 mV s−1. (b) Faradaic efficiencies of ethylene for SA-Ni-NC and Ni NP/N-C. (c) Ethylene partial current density of SA-Ni-NC and Ni NP/N-C. The inset in (c) shows the TOF of SA-Ni-NC. (d) Faradaic efficiencies of ethylene and H2 for SA-Ni-NC in a two-electrode flow cell. (e) Long-term stability test results of SA-Ni-NC at a current density of −30 mA cm−2 in a two-electrode flow cell. | ||
To assess the practical application feasibility of SA-Ni-NC, a two-electrode full cell was constructed by integrating NiFe-LDH on Ni foam as the anode for the oxygen evolution reaction. As shown in Fig. S20a,† in 1 M KOH aqueous solution, the current density of the full cell was as high as −100 mA cm−2 at a voltage of 3.25 V under pure acetylene flow. The FEethylene of SA-Ni-NC was ∼99% at a current density of −30 mA cm−2 (Fig. 3d). In particular, during a 45 h long-term durability at −30 mA cm−2, the FEethylene of SA-Ni-NC was steadily >90% (Fig. 3e).
Encouraged by such outstanding semihydrogenation performance of SA-Ni-NC under a pure acetylene atmosphere, its performance in the crude ethylene feedstock containing 1% acetylene impurities (1 × 104 ppm) was further assessed in a flow cell. Limited by the large input flow rate and short gas residence time, more than 70% acetylene impurities still existed in output ethylene flow. Therefore, we customized a flow cell with a 5 × 5 cm2 electrode area as a preliminary demonstration for further evaluating the application possibility of SA-Ni-NC (Fig. S21–S23, ESI†). Fig. 4a shows the contents of acetylene impurities at different current densities. The acetylene concentration in output ethylene flow was considerably decreased to 263.6 ppm at −40 mA (−1.6 mA cm−2), corresponding to an acetylene conversion of ∼97.4%. Accordingly, the space velocity (SV) of SA-Ni-NC was up to 2.4 × 104 mL gcat−1 h−1 with a high TOF of 22.9 h−1, which was much higher than those for the state-of-the-art thermocatalysts, as shown in Table S3,† for example, 3.26 × 102 mL gcat−1 h−1 for Pd/PPS,31 1.2 × 103 mL gcat−1 h−1 for Pd SAs,32 and 3 × 103 mL gcat−1 h−1 for Cu1/ND@G.5 Next, during a 500 min stability test in crude ethylene flow, acetylene conversion was stably above 97.0% (Fig. 4b). These results unambiguously demonstrate the promising applications of SA-Ni-NC for electrocatalytic acetylene semihydrogenation.
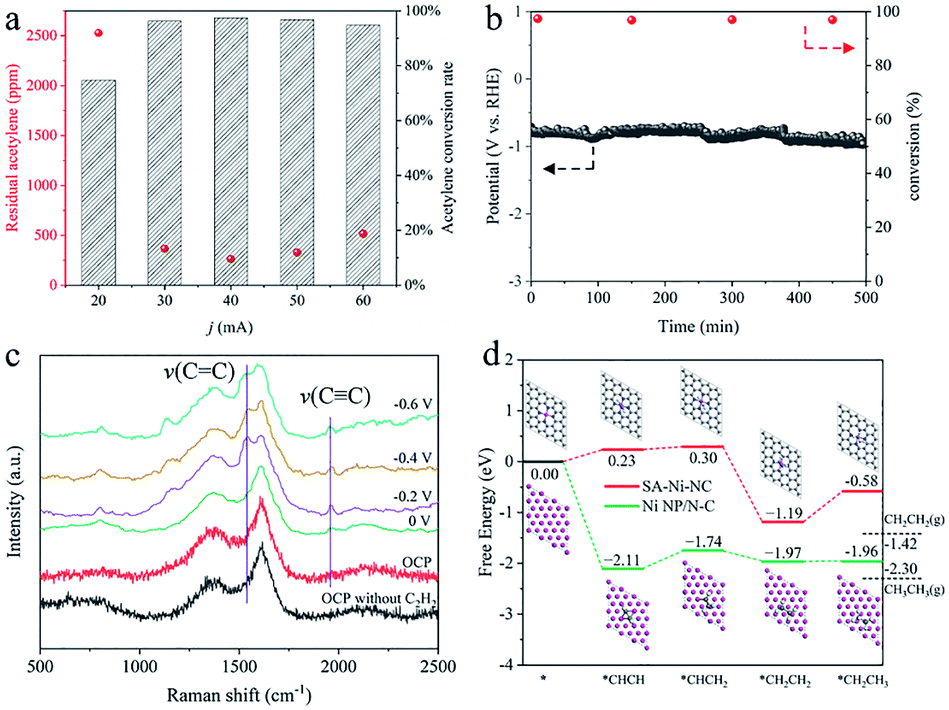 | ||
| Fig. 4 Electrochemical performances and DFT calculations of SA-Ni-NC. (a) The residual acetylene concentrations versus the cathodic current in 1 M KOH aqueous solution at a flow rate of 10 mL min−1. (b) Long-term stability test results for continuous semihydrogenation of 1 × 104 ppm acetylene at a cathodic current of 40 mA. (c) In situ electrochemical Raman spectra of SA-Ni-NC in 1 M KOH aqueous solution. (d) Free energy profile of acetylene hydrogenation on the SA-Ni-NC catalyst and Ni NP/N-C (111) surface, with the corresponding atomic structures of important reaction intermediates. | ||
To reveal the nature of superior acetylene semihydrogenation performance of SA-Ni-NC, in situ electrochemical Raman (Fig. S24, ESI†) and density functional theory (DFT) investigations were employed. As illustrated in Fig. 4c, in comparison with that of SA-Ni-NC under open circuit potential (OCP), a new Raman peak representing the ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) stretching vibration appeared at 1961 cm−1 at 0 V under acetylene flow, verifying acetylene adsorption on SA-Ni-NC.33,34 When the potential increased from 0 to −0.6 V, the ν(C
C) stretching vibration appeared at 1961 cm−1 at 0 V under acetylene flow, verifying acetylene adsorption on SA-Ni-NC.33,34 When the potential increased from 0 to −0.6 V, the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) stretching vibration of π-bound ethylene gradually emerged at 1546 cm−1.34–37 Weak π-adsorption of ethylene is beneficial for ethylene desorption, eventually facilitating acetylene semi-hydrogenation kinetics. As illustrated by the DFT simulations in Fig. 4d, the adsorbed acetylene molecules were first hydrogenated into *CHCH2 with a lower free energy change of 0.07 eV on SA-Ni-NC than that on Ni NP/N-C (0.37 eV) (the main crystal planes of Ni NP/N-C are (111)), implying a lower activity of Ni NP/N-C than SA-Ni-NC. More importantly, the positive free energy change (0.61 eV) for the *CH2CH3 further hydrogenation step over SA-Ni-NC indicates that deep hydrogenation can be effectively suppressed. Meanwhile, the weak desorption of the produced ethylene is exothermic by 0.23 eV on SA-Ni-NC, implying a rapid ethylene desorption. In addition, energy barriers for water dissociation on both catalysts were calculated to further evaluate the competitive hydrogen evolution reaction (HER) under alkaline conditions. As shown in Fig. S25a,† there is a kinetic energy barrier of 1.02 eV for water dissociation on the Ni (111) surface indicating a sluggish kinetics for water splitting. For Ni SAC shown in Fig. S25b,† the H2O molecule only shows very weak physical adsorption (−0.07 eV), which clearly indicates an even harsher kinetics for water splitting. Therefore, under alkaline conditions, the SA-Ni-NC catalyst exhibits a higher catalytic selectivity (i.e., faradaic efficiency) for acetylene semihydrogenation by suppressing the HER process. These DFT results clearly confirm the efficient occurrence of acetylene semihydrogenation to ethylene on SA-Ni-NC.
C) stretching vibration of π-bound ethylene gradually emerged at 1546 cm−1.34–37 Weak π-adsorption of ethylene is beneficial for ethylene desorption, eventually facilitating acetylene semi-hydrogenation kinetics. As illustrated by the DFT simulations in Fig. 4d, the adsorbed acetylene molecules were first hydrogenated into *CHCH2 with a lower free energy change of 0.07 eV on SA-Ni-NC than that on Ni NP/N-C (0.37 eV) (the main crystal planes of Ni NP/N-C are (111)), implying a lower activity of Ni NP/N-C than SA-Ni-NC. More importantly, the positive free energy change (0.61 eV) for the *CH2CH3 further hydrogenation step over SA-Ni-NC indicates that deep hydrogenation can be effectively suppressed. Meanwhile, the weak desorption of the produced ethylene is exothermic by 0.23 eV on SA-Ni-NC, implying a rapid ethylene desorption. In addition, energy barriers for water dissociation on both catalysts were calculated to further evaluate the competitive hydrogen evolution reaction (HER) under alkaline conditions. As shown in Fig. S25a,† there is a kinetic energy barrier of 1.02 eV for water dissociation on the Ni (111) surface indicating a sluggish kinetics for water splitting. For Ni SAC shown in Fig. S25b,† the H2O molecule only shows very weak physical adsorption (−0.07 eV), which clearly indicates an even harsher kinetics for water splitting. Therefore, under alkaline conditions, the SA-Ni-NC catalyst exhibits a higher catalytic selectivity (i.e., faradaic efficiency) for acetylene semihydrogenation by suppressing the HER process. These DFT results clearly confirm the efficient occurrence of acetylene semihydrogenation to ethylene on SA-Ni-NC.
3. Experimental
3.1 Materials and characterization
Nickel nitrate (Ni(NO3)2·6H2O, 98%), methanol, and ethanol (99.7%) were purchased from Guangdong Guanghua Sci-Tech Co., Ltd. Zinc nitrate (Zn(NO3)2·6H2O, 98%) was purchased from Sinopharm Chemical Reagent Co., Ltd. 2-Methylimidazole (90%) was purchased from Shanghai Macklin Biochemical Co., Ltd. Potassium hydroxide (KOH, 99.99%) and potassium bicarbonate (KHCO3, 99.99%) were purchased from Shanghai Aladdin Bio-Chem Technology Co., Ltd. Nafion solution (5 wt%) was purchased from Sigma-Aldrich LLC. All chemical reagents were used as received without further purification. Ultrapure water (>18.25 MΩ cm) was used for the experiments. The XRD patterns were recorded by using a X-ray diffractometer with Cu Kα radiation (XRD, PANalytical B.V., Netherlands). A scanning electron microscope (SEM, FEI-Verios G4) operated at 15 kV was used to analyze the morphologies of the as-prepared electrocatalysts. The transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) images were acquired on a FEI Talos F200X at an acceleration voltage of 200 kV. X-ray photoelectron spectroscopy (XPS, Kratos-Axis Supra) was performed to determine the chemical composition of the electrocatalysts. Raman spectra were recorded with an Alpha300R or Bruker RFS 100/S spectrometer (laser wavelength 532 nm).3.2 Synthesis of SA-Ni-NC
In a typical procedure, 0.01 mol of Ni(NO3)2·6H2O and 0.01 mol of Zn(NO3)2·6H2O, as well as 1 g 2-methylimidazole were separately dissolved in 15 mL methanol to form a clear solution. After ultrasonication for 5 min, the above two solutions were mixed to obtain a stable green solution. Then the solution was ultrasonicated for 20 min at room temperature. The formed precipitates were then washed with methanol three times and dried in a vacuum at 60 °C for 8 h. The powder samples were eventually placed in a furnace and heated to the desired temperature (950 °C) at a rate of 5 °C min−1 for 3 h under N2 gas. After cooling to room temperature, SA-Ni-NC was achieved.3.3 Synthesis of Ni NP/N-C
In a typical procedure, 0.018 mol of Ni(NO3)2·6H2O and 0.002 mol of Zn(NO3)2·6H2O, as well as 1 g 2-methylimidazole were separately dissolved in 15 mL methanol to form a clear solution. After ultrasonication for 5 min, these two solutions were mixed to obtain a stable green solution. Then, the solution was ultrasonicated for 20 min at room temperature. The formed precipitates were washed with methanol three times and dried in a vacuum at 60 °C for 8 h. The powders were placed in a furnace and then heated to the desired temperature (950 °C) at a rate of 5 °C min−1 for 3 h under N2 gas. After cooling to room temperature, Ni NP/N-C was achieved.3.4 Electrochemical tests
Electrochemical measurements were performed using an electrochemical workstation (CHI760E). The gas-diffusion carbon paper loaded with electrocatalysts (1 mg cm−2) served as the working electrode. The Hg/HgO electrode and Ni foam were used as the reference and counter electrodes, respectively. Alkaline solutions with various KOH concentrations at a flow rate of 10 mL min−1 were utilized as electrolytes at both cathode and anode chambers. An anion exchange membrane was sandwiched between the catholyte and anolyte compartments to avoid product crossover. The C2H2 (99.99%) flow was kept constant at 20 mL min−1 using a mass flow controller and fed to the cathode. For electrochemical tests, all the potentials were referred to a reversible hydrogen electrode (RHE) based on the following equation:| E(vs. RHE) = E(vs. Hg/HgO) + 0.099 V + 0.059 V × pH |
Two-electrode flow cells were also constructed SA-Ni-NC loaded on a gas diffusion electrode as the cathode and NiFe-LDH loaded on nickel foam as the anode for electrocatalytic acetylene semihydrogenation in a 1 M KOH aqueous solution.
3.5 Product analyses
Gas phase products were detected by using a gas chromatograph (GC) with a thermal conductivity detector (TCD) and a flame ionization detector (FID). Ultra-pure Ar was used as a carrier gas with a flow rate of 20 mL min−1. The faradaic efficiency (FE) of gas products was calculated through the concentration (C) detected by GC using the following equation:where n is the number of transferred electrons; F is the Faraday constant; Vm is the molar volume of gas when the temperature is 298 K; jtotal is the total current density.
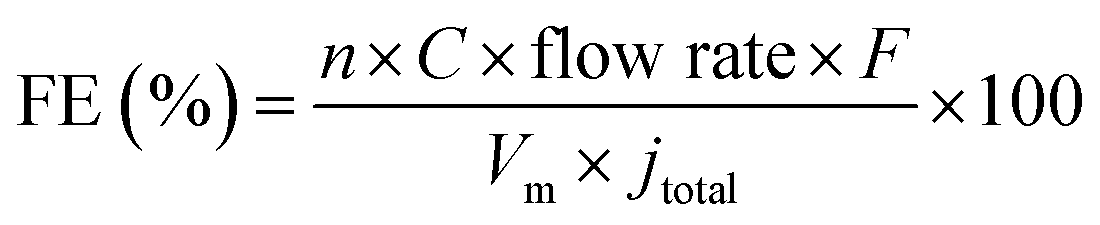
3.6 Turnover frequencies and space velocity calculation
Turnover frequencies (TOFs) have been calculated using the following formulas for each experiment:TOF: turnover frequency, h−1. Iproduct: partial current for a certain product, A. n: the number of transferred electrons for product formation, which is 2 for C2H4 production. F: Faraday constant, 96
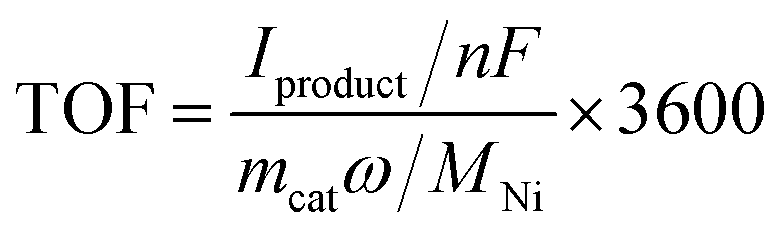 485 C mol−1. mcat: catalyst mass in the electrode, g. ω: Ni loading in the catalyst. MNi: atomic mass of Ni, 58.69 g mol−1.
485 C mol−1. mcat: catalyst mass in the electrode, g. ω: Ni loading in the catalyst. MNi: atomic mass of Ni, 58.69 g mol−1.
The space velocity (SV) of the catalysts was calculated based on the amount of feed gas according to the following equation:
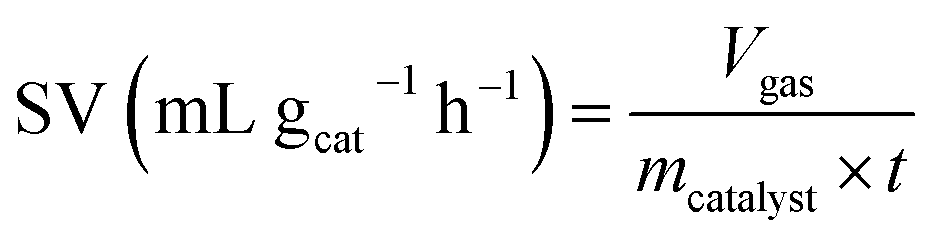
3.7 Computational details
All spin-polarized DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP) code.38 The revised Perdew–Burke–Ernzerhof (RPBE) functional was employed to describe the exchange correlation interactions within the generalized gradient approximation (GGA).39,40 The electron–ion interactions were represented by the projector augmented wave (PAW) method.41 The kinetic energy cutoff of the plane wave was set to be 500 eV and the convergence criterion for the residual forces and total energies were set to be 0.03 eV Å−1 and 10−5 eV, respectively. A 5 × 5 × 1 Monkhorst–Pack k-point grid is used to sample the Brillouin zone. The empirical correction in Grimme's method (DFT+D3) was adopted to describe van der Waals interactions.42The SA-Ni-NC catalyst was constructed by embedding the NiN4 moiety in a (5 × 5) supercell of graphene. As for the Ni (111) surface, a (3 × 3) supercell composed of 4 layers of Ni atoms was used, in which the bottom two atomic layers were fixed, while atoms in the other layers were fully relaxed during geometry optimization. For both models, a vacuum layer of around 15 Å was added in the z direction to eliminate the interactions between periodic images.
The Gibbs free energy change (ΔG) of each elementary step during the acetylene hydrogenation process was calculated by using the computational hydrogen electrode (CHE) model proposed by Nørskov et al.43 The chemical potential of the proton–electron pair in aqueous solution is related to one-half of the chemical potential of an isolated hydrogen molecule. Based on this model, the ΔG value can be obtained using the formula ΔG = ΔE + ΔZPE − TΔS, where ΔE is the reaction energy of the reactant and product species adsorbed on the catalyst directly obtained from DFT calculations; ΔZPE and ΔS are the changes between the adsorbed species and the gas phase molecules in zero-point energies and entropy at 298.15 K, which can be calculated from the vibrational frequencies.
4. Conclusions
In summary, SA-Ni-NC has been fabricated via the pyrolysis of nickel doped zeolitic imidazolate framework-8 precursors. The high-angle annular dark field-scanning transmission electron microscopy and X-ray absorption fine structure analyses confirm the chemical structure of a Ni–N4 coordination in the SA-Ni-NC matrix. Serving as an electrocatalyst for acetylene semihydrogenation in alkaline aqueous solution, SA-Ni-NC exhibits superior electrochemical activity and selectivity towards ethylene. The experimental and theoretical studies prove that the outstanding acetylene semihydrogenation performance of SA-Ni-NC is attributed to its atomically dispersed Ni active sites, which favors weak π-adsorption of ethylene and thus facilitates rapid ethylene desorption. Therefore, this work will not only open a new window for electrocatalytic acetylene semihydrogenation, but also initiate the rapid exploration of high efficiency electrocatalysts for selective hydrogenation reactions.Author contributions
W. X. M. and Z. C. contributed equally to this work. J. Z. designed the project. W. X. M. conducted the catalyst syntheses and performance evaluations. Z. C. and T. W. performed the DFT calculations. J. C. Z. performed the X-ray absorption fine structure analyses. J. B., Z. P. L., J. J. L., C. Y., L. Z. and H. P. Z. performed the related materials characterization. L. C. performed the in situ electrochemical Raman analyses. W. X. M. and J. Z. co-wrote the manuscript. All the authors contributed to the discussions and analyses of the results in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Fundamental Research Funds for the Central Universities (Grant No. 310201911cx028), the Natural Science Foundation of Shaanxi Province (No. 2020JQ-141), the National Natural Science Foundation of China (No. 22005245), and Synergy Innovation Foundation of the University and Enterprise for Graduate Students in Northwestern Polytechnical University (No. CX2021037). We would like to thank the Analytical & Testing Center of Northwestern Polytechnical University for the morphological and spectroscopic characterization studies. We thank beamline BL14W1 and BL15U1 of the Shanghai Synchrotron Radiation Facility (SSRF, China) for providing the beamtime. T. W. acknowledges the start-up fund of Westlake University and Westlake University HPC Center for computation support.Notes and references
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS
.
- B. Bridier and J. Perez-Ramirez, J. Am. Chem. Soc., 2010, 132, 4321–4327 CrossRef CAS
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen and J. K. Nørskov, Science, 2008, 320, 1320–1322 CrossRef CAS PubMed
- C. W. A. Chan, A. H. Mahadi, M. M.-J. Li, E. C. Corbos, C. Tang, G. Jones, W. C. H. Kuo, J. Cookson, C. M. Brown and P. T. Bishop, Nat. Commun., 2014, 5, 1–10 Search PubMed
- F. Huang, Y. Deng, Y. Chen, X. Cai, M. Peng, Z. Jia, J. Xie, D. Xiao, X. Wen, N. Wang, Z. Jiang, H. Liu and D. Ma, Nat. Commun., 2019, 10, 1–7 CrossRef
- S. Wang, Z. J. Zhao, X. Chang, J. Zhao, H. Tian, C. Yang, M. Li, Q. Fu, R. Mu and J. Gong, Angew. Chem., Int. Ed., 2019, 58, 7668–7672 CrossRef CAS PubMed
- A. Primo, F. Neatu, M. Florea, V. Parvulescu and H. Garcia, Nat. Commun., 2014, 5, 1–9 Search PubMed
- K. Ding, D. A. Cullen, L. Zhang, Z. Cao, A. D. Roy, I. N. Ivanov and D. Cao, Science, 2018, 362, 560–564 CrossRef CAS PubMed
- M. Hu, S. Zhao, S. Liu, C. Chen, W. Chen, W. Zhu, C. Liang, W. C. Cheong, Y. Wang and Y. Yu, Adv. Mater., 2018, 30, 1801878 CrossRef
- F. Huang, Y. Deng, Y. Chen, X. Cai, M. Peng, Z. Jia, P. Ren, D. Xiao, X. Wen and N. Wang, J. Am. Chem. Soc., 2018, 140, 13142–13146 CrossRef CAS
- R. S. Sherbo, A. Kurimoto, C. M. Brown and C. P. Berlinguette, J. Am. Chem. Soc., 2019, 141, 7815–7821 CrossRef CAS
- R. P. Jansonius, A. Kurimoto, A. M. Marelli, A. Huang, R. S. Sherbo and C. P. Berlinguette, Cell Rep. Phys. Sci., 2020, 1, 100105 CrossRef
- B. Huang, C. Durante, A. A. Isse and A. Gennaro, Electrochem. Commun., 2013, 34, 90–93 CrossRef CAS
- G. Vilé, B. Bridier, J. Wichert and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2012, 124, 8748–8751 CrossRef
- K. Werner, X. Weng, F. Calaza, M. Sterrer, T. Kropp, J. Paier, J. Sauer, M. Wilde, K. Fukutani and S. Shaikhutdinov, J. Am. Chem. Soc., 2017, 139, 17608–17616 CrossRef CAS PubMed
- M. Tejeda-Serrano, J. R. Cabrero-Antonino, V. Mainar-Ruiz, M. López-Haro, J. C. Hernández-Garrido, J. J. Calvino, A. Leyva-Pérez and A. Corma, ACS Catal., 2017, 7, 3721–3729 CrossRef CAS
- I. Horiuti and M. Polanyi, Trans. Faraday Soc., 1934, 30, 1164–1172 RSC
- J. McCue, A. Guerrero-Ruiz, C. Ramirez-Barria, I. Rodríguez-Ramos and J. A. Anderson, J. Catal., 2017, 355, 40–52 CrossRef
- H. Zhou, X. Yang, L. Li, X. Liu, Y. Huang, X. Pan, A. Wang, J. Li and T. Zhang, ACS Catal., 2016, 6, 1054–1061 CrossRef CAS
- G. Hamm, T. Schmidt, J. Breitbach, D. Franke, C. Becker and K. Wandelt, Z. Phys. Chem., 2009, 223, 209–232 CrossRef CAS
- M. Li and J. Shen, Thermochim. Acta, 2001, 379, 45–50 CrossRef CAS
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS
- R. Li, X. Ren, X. Feng, X. Li, C. Hu and B. Wang, Chem. Commun., 2014, 50, 6894–6897 RSC
- L. Zhang, D. Liu, Z. Muhammad, F. Wan, W. Xie, Y. Wang, L. Song, Z. Niu and J. Chen, Adv. Mater., 2019, 31, 1903955 CrossRef CAS PubMed
- M. Zhou, Y. Jiang, G. Wang, W. Wu, W. Chen, P. Yu, Y. Lin, J. Mao and L. Mao, Nat. Commun., 2020, 11, 3188 CrossRef CAS PubMed
- J. Zhu, L. Xia, W. Yang, R. Yu, W. Zhang, W. Luo, Y. Dai, W. Wei, L. Zhou and Y. Zhao, Energy Environ. Mater., 2021 DOI:10.1002/eem2.12219
- S. Chen, B. Wang, J. Zhu, L. Wang, H. Ou, Z. Zhang, X. Liang, L. Zheng, L. Zhou and Y.-Q. Su, Nano Lett., 2021, 21, 7325–7331 CrossRef CAS
- Y. Quan, R. Yu, J. Zhu, A. Guan, X. Lv, C. Yang, S. Li, J. Wu and G. Zheng, J. Colloid Interface Sci., 2021, 601, 378–384 CrossRef CAS PubMed
- H. Funke, A. Scheinost and M. Chukalina, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 094110 CrossRef
- H. Funke, M. Chukalina and A. C. Scheinost, J. Synchrotron Radiat., 2007, 14, 426–432 CrossRef CAS
- S. Lee, S.-J. Shin, H. Baek, Y. Choi, K. Hyun, M. Seo, K. Kim, D.-Y. Koh, H. Kim and M. Choi, Sci. Adv., 2020, 6, eabb7369 CrossRef CAS
- S. Wei, A. Li, J.-C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W.-C. Cheong, Y. Wang and L. Zheng, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS PubMed
- J. Heaviside, P. J. Hendra, P. Tsai and R. P. Cooney, J. Chem. Soc., Faraday Trans. 1, 1978, 74, 2542–2549 RSC
- K. Manzel, W. Schulze and M. Moskvits, Chem. Phys. Lett., 1982, 85, 183–186 CrossRef CAS
- C. S. Wondergem, T. Hartman and B. M. Weckhuysen, ACS Catal., 2019, 9, 10794–10802 CrossRef CAS
- M. F. Mrozek and M. J. Weaver, J. Phys. Chem. B, 2001, 105, 8931–8937 CrossRef CAS
- C. S. Wondergem, J. J. G. Kromwijk, M. Slagter, W. L. Vrijburg, E. J. Hensen, M. Monai, C. Vogt and B. M. Weckhuysen, ChemPhysChem, 2020, 21, 625 CrossRef CAS
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413 CrossRef
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08002d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |