DOI:
10.1039/D1TA04911A
(Paper)
J. Mater. Chem. A, 2022,
10, 6031-6037
Catalytic conversion of NO and CO into N2 and CO2 by rhodium–aluminum oxides in the gas phase†
Received
10th June 2021
, Accepted 13th September 2021
First published on 14th September 2021
Abstract
The increasingly stringent emission regulations make it urgent to get a fundamental understanding on the catalytic behaviors of rhodium, the most efficient component in three-way catalysts (TWCs) to convert NOx into N2. Herein, the catalytic conversion of NO and CO into N2 and CO2 by gas-phase rhodium-aluminum oxide species (RhAlO0–3 and RhAl2O1–4) was identified by mass spectrometry and quantum chemical calculations. This finding represents a significant step in the field of gas-phase catalysis, given that only elementary reactions for this conversion are commonly observed because it is extremely challenging to rupture the strong N–O bond and couple the N–N bond as well as drive the resulting product to oxidize CO. It emphasized that a polarized Rh–Al bond in gas-phase species is indispensable and the RhAlO0,1 and RhAl2O1,2 species with direct Rh–Al bonding can be involved in catalytic NO reduction by CO.
1. Introduction
The catalytic conversion of toxic NO and CO gases, produced during the processes of fossil-fuel combustion in vehicles, power plants, and chemical industries, into harmless N2 and CO2 (reaction (1)) has long been an important subject due to their negative impacts on human health and the environment.1,2 Automobile three-way catalysts (TWCs) represent a breakthrough in this field since their recognizable application in the 1970s for the clean-out of CO, NOx, and hydrocarbons from exhaust gases.3 Commercial TWCs typically consist of noble metals (Rh, Pt, and Pd) highly dispersed on porous alumina.4,5 Among these noble metals, Rh is especially regarded as the most efficient and irreplaceable component to convert NOx into N2 (principal reaction described in eqn (1)).6,7 However, Rh is significantly scarce and expensive. Due to the increasingly high demand for Rh in automobile catalysis over the past few decades,8 maximizing its use becomes one of the most urgent global issues to satisfy the economic and environmental requirements. In this regard, fundamental understanding of Rh catalysis is paramount to guide the rational design of desired catalysts.
Catalytic bond activation and formation behaviors take place microscopically on active sites typically composed of a limited number of atoms.9 Confining individual active species on isolated gas-phase atomic clusters that can be studied under well-controlled and reproducible conditions is an important way to reveal the nature of active sites on related catalysts.10–14 In the field of gas phase studies, the elementary reactions of Rhx0,±,15–17 RhxM+ (M = Ta, Al, V, and Co),18,19 RhxOy,20 and RhAl2O6+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 species with NO or CO have been extensively investigated, while the catalysis of NO reduction by CO mediated by Rh-containing species has not been established. Herein, benefiting from a homemade time-of-flight mass spectrometer (TOF-MS) coupled with a vacuum ultraviolet (VUV) laser system,22 the catalytic conversion of NO and CO into N2 and CO2 by neutral rhodium-aluminum oxides RhAlO0–3 and RhAl2O1–4 has been identified for the first time in the gas phase. Note that it is experimentally challenging to study the reactivity of neutral heteronuclear metal oxides due to the difficulty of cluster ionization and detection without fragmentation.23 The outstanding roles of Rh atoms in driving the catalysis are emphasized and the molecular-level mechanisms have been captured. The identification of reaction (1) makes big progress in the field of cluster science because it is substantially challenging to capture two NO molecules tightly and then induce the rupture of the strong N–O bond (bond enthalpy: N–O = 6.55 eV)24 as well as the subsequent coupling of the N–N bond. In contrast, the closely related catalytic reactions (2) and (3) involving N2O and O2 as oxidants (bond enthalpies: N2–O = 1.73 eV; O–O = 5.16 eV) have been extensively explored.25–27
21 species with NO or CO have been extensively investigated, while the catalysis of NO reduction by CO mediated by Rh-containing species has not been established. Herein, benefiting from a homemade time-of-flight mass spectrometer (TOF-MS) coupled with a vacuum ultraviolet (VUV) laser system,22 the catalytic conversion of NO and CO into N2 and CO2 by neutral rhodium-aluminum oxides RhAlO0–3 and RhAl2O1–4 has been identified for the first time in the gas phase. Note that it is experimentally challenging to study the reactivity of neutral heteronuclear metal oxides due to the difficulty of cluster ionization and detection without fragmentation.23 The outstanding roles of Rh atoms in driving the catalysis are emphasized and the molecular-level mechanisms have been captured. The identification of reaction (1) makes big progress in the field of cluster science because it is substantially challenging to capture two NO molecules tightly and then induce the rupture of the strong N–O bond (bond enthalpy: N–O = 6.55 eV)24 as well as the subsequent coupling of the N–N bond. In contrast, the closely related catalytic reactions (2) and (3) involving N2O and O2 as oxidants (bond enthalpies: N2–O = 1.73 eV; O–O = 5.16 eV) have been extensively explored.25–27
2. Methods
2.1 Experimental methods
The gas-phase neutral RhxAly16Oz or RhxAly18Oz species were generated by pulsed laser ablation of a mixed-metal disk compressed with Rh and Al powders (molar radio: Rh/Al = 1/1) in the presence of pure He, 16O2 (1%) or 18O2 (0.05%) seeded in a He carrier gas (99.999%) with a backing pressure of about 4.0 atm. The species generated in a gas channel were expanded to the fast flow reactor, where they reacted separately with each kind of reactant gas (Ar, NO, N2O, CO, 16O2, or 18O2 diluted in the buffer gas He) that was controlled by the pulsed valve. An inert gas (He) was used to clean the gas handling system without any residues left before one kind of reactant gas was changed to another. The partial pressures of the reactants ranged from about 0.2 Pa to 3.0 Pa, depending on the relative reactivity of gas-phase species. After the reactions, the charged species were removed from the molecular beams with two deflection plates. The neutral reactants and products were skimmed into the vacuum system of the TOF-MS and ionized with four VUV laser beams (118 nm, 10.5 eV per photon) generated with an intense λ = 355 nm laser beam in a gas cell containing an Ar/Xe mixture. After single photon ionization, the charged species passed through the reflector and then were detected by a dual microchannel plate detector. The signals from the detector were recorded with a digital oscilloscope. Note that most of the metal oxides have ionization energies below 10.5 eV and the VUV photoionization is widely considered as a soft ionization process,28 and thus, the neutral metal oxide species can be gently ionized and generally detected without fragmentation or isomerization.29 The details of the experimental setup can be found in our previous work.22
2.2 Theoretical methods
Density functional theory (DFT) calculations using the Gaussian 09 program30 were carried out to investigate the structures of RhAlO0–3 and RhAl2O1–4 as well as the mechanisms of the reactions with NO, N2O, CO, O2, and H2O. TZVP basis sets31 for H, C, N, O, and Al atoms were used and D95V basis sets combined with Stuttgart/Dresden relativistic effective core potentials (denoted as SDD in Gaussian software)32 for Rh atoms were adopted. The M06L functional33 had been tested to perform well for the Rh–Al–O system;21 thus, the results from M06L were given throughout this work. Note that the isomers of smaller species RhAlO0–2 and RhAl2O1,2 were obtained by intuitive guess of initial structures, while the isomers of larger clusters RhAlO3 and RhAl2O3,4 were obtained from a Fortran code based on a genetic algorithm.34 Small basis sets (LANL2DZ for all atoms)35 and Coarse convergence thresholds were used to reduce the computational costs. More than 200 initial structures were generated by the genetic algorithm, among which more than 20 low-lying isomers were re-optimized with larger basis sets (SDD for Rh and TZVP for O and Al). Mechanism calculations involved the geometry optimization of reaction intermediates (Is) and transition states (TSs) through which the Is transfer to each other. The TSs were optimized by using the Berny algorithm method.36 The initial guess structures of TSs were obtained through relaxed potential energy surface scans using single or multiple internal coordinates. Intrinsic reaction coordinate calculations37 were employed to check that each TS connects two appropriate local minima. Vibrational frequency calculations were performed to check that the Is and TSs have zero and only one imaginary frequency, respectively. The zero-point vibration corrected energies (ΔH0 in eV) were reported in this work. Natural bond orbital analysis was performed with NBO 5.9.38
3. Results
3.1 Experimental results
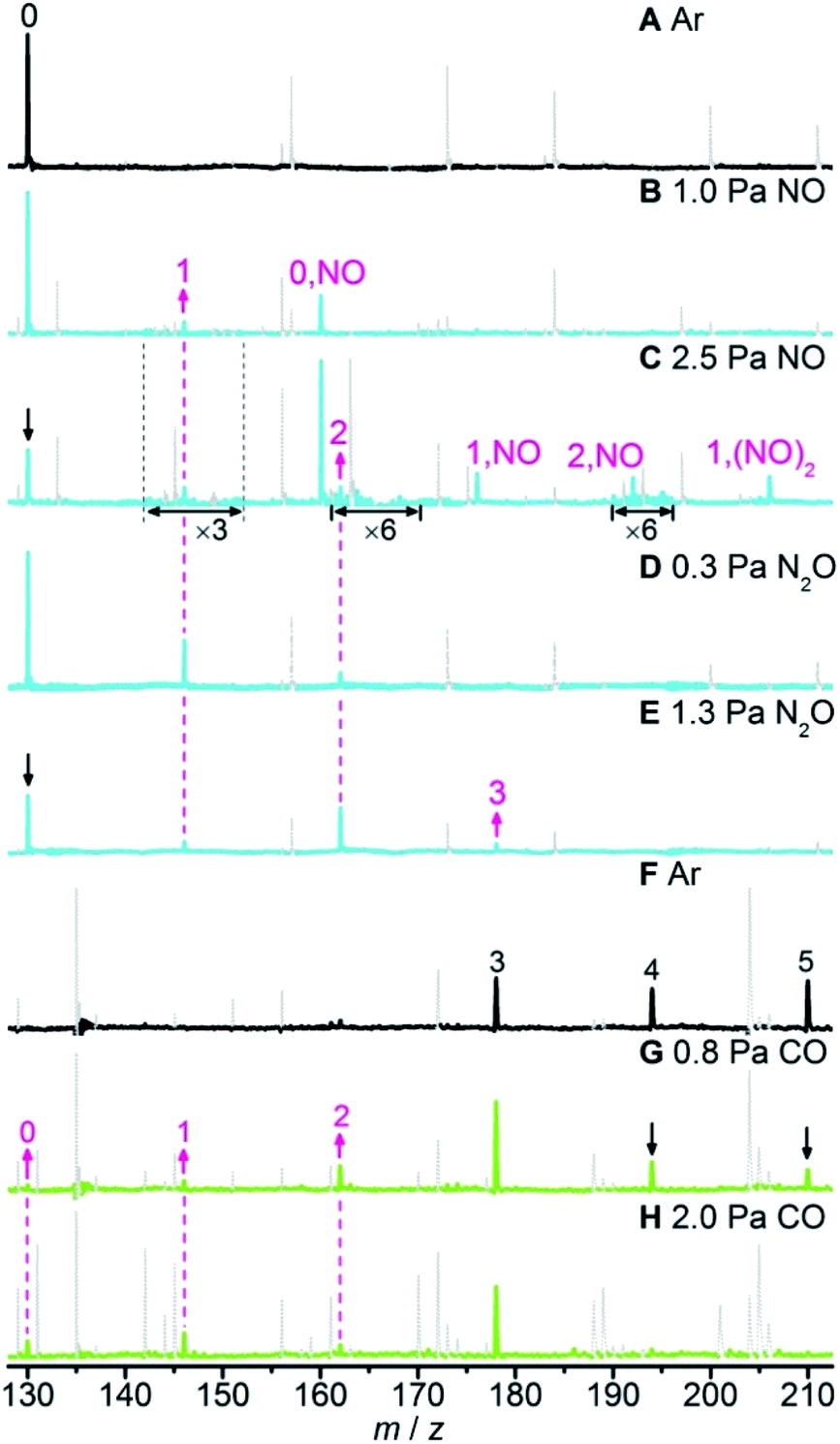
The selected mass spectra for the interactions of laser-ablation generated neutral RhAlOz species with NO, N2O, and CO in the fast flow reactor are presented in Fig. 1 and S1 in the ESI.†Fig. 1A and F show the reference spectra when the reactor was only filled with an inert bath gas (Ar). When the reactant gas (NO, N2O, or CO) was injected separately into the reactor, the reactivity of the RhAlOz species toward each kind of reactant gas can be identified by the intensity variation of mass peaks. Upon the interaction of RhAl with NO (Fig. 1B), new products RhAlNO (0,NO) and RhAlO (1) can be clearly observed, indicating that RhAl may pick up a NO molecule readily to generate RhAlNO (reaction (4a)), which then captures and reacts with a second NO molecule to produce RhAlO and N2O (reaction (4b)). With the increase of NO partial pressure (Fig. 1C), the intensities of products RhAlO(NO) (1,NO) and RhAlO(NO)2 (1,(NO)2) increase significantly while that of RhAlO does not change obviously, demonstrating that the product RhAlO from the reaction of RhAl with NO (reactions (4a) and (4b)) reacts easily with NO to generate adsorption products. It is relatively difficult for the adsorption product RhAlO(NO)2 to further transform because the signal intensities of products RhAlO2 (2) and RhAlO2NO (2,NO) that are amplified by a factor of 6 are still rather weak. When N2O was injected into the reactor (Fig. 1D and E), new signals RhAlOz (z = 1–3) appeared, indicating that N2O can be reduced by RhAlO0–2 to produce N2 (reaction (5)). Upon the interactions of RhAlOz with CO (Fig. 1G and H), the signal intensities of RhAlO5 and RhAlO4 decreased significantly, while those of RhAlO3 and RhAlO2 increased (Fig. 1G) and then decreased (Fig. 1H) with the increase of CO pressure. Moreover, new signals of RhAlO and RhAl appeared simultaneously. These experiments evidenced that RhAlO5 can oxidize five CO molecules consecutively to regenerate the product RhAl (reaction (6)). Note that the neutral product molecules N2O, N2, and CO2 that have ionization energies of 12.89, 15.58, and 13.78 eV,39 respectively, cannot be detected by single photon ionization (118 nm, 10.5 eV) in our current experimental apparatus. Moreover, the number of generated product molecules is very small (<105 s−1) and they can diffuse out of the cluster beam, so it is difficult to detect these product molecules by employing other experimental techniques.| | | RhAlOz + NO → RhAlOzNO (z = 0, 1) | (4a) |
| | | RhAlOzNO + NO → RhAlOz+1 + N2O (z = 0, 1) | (4b) |
| | | RhAlOz + N2O → RhAlOz+1 + N2 (z = 0–2) | (5) |
| | | RhAlOz + CO → RhAlOz–1 + CO2 (z = 5–1) | (6) |
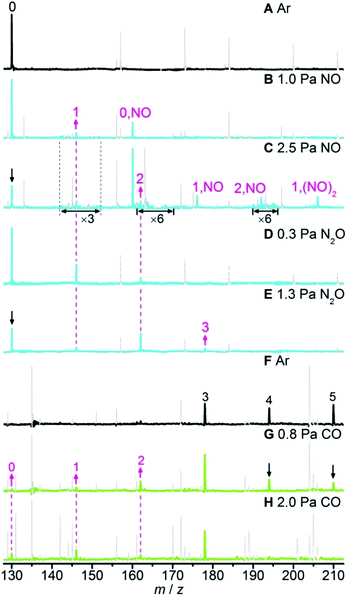 |
| | Fig. 1 Time-of-flight mass spectra for the interactions of neutral RhAlOz species with Ar (A) and (F), NO (B) and (C), N2O (D) and (E), and CO (G) and (H). The reactant gas pressures are shown and the reaction time is about 60 μs. The RhAlOz and RhAlOz,X species are denoted as z and z,X (X = NO or (NO)2), respectively. In panels B and C, the signal magnitudes for m/z in the range of 142–152 are amplified by a factor of 3 and those of 161–170 and 190–196 are amplified by a factor of 6. The upward and downward arrows indicate the increase and decrease of key mass peaks, respectively. To have a clear view on the reactivity of the RhAlOz species, unrelated peaks are shown in grey and dotted lines. See details in Fig. S1 in the ESI.† | |
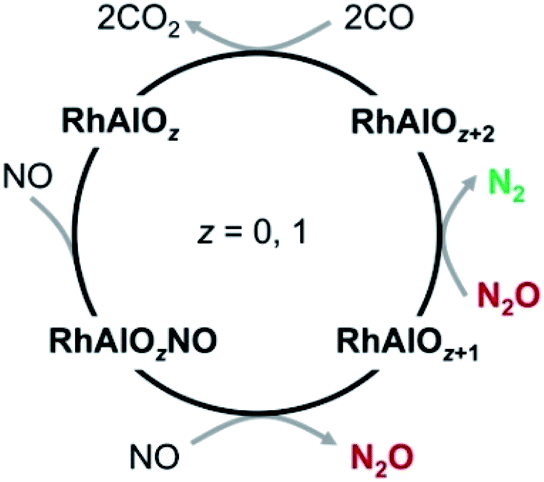
The rate constants for the above pseudo-first-order reactions have been roughly estimated and the order of magnitude is about 10−11 cm3 per molecule per s (Fig. S2†). For the reaction of RhAl with NO, the model RhAl → RhAlNO → RhAlO → RhAlONO + RhAlO(NO)2 + RhAlO2 + RhAlO2NO can fit the experimental results well (Fig. S2a†), providing solid evidence that RhAl can indeed reduce two NO molecules to generate RhAlO and N2O (reactions (4a) and (4b)). It is interesting to find that the RhAl2Oz (z = 1–6) clusters behave similarly to RhAlO0–5 in the reactions with NO, N2O, and CO (Fig. S3†). Furthermore, the RhAlO0–3 and RhAl2O1–4 species are also reactive toward O2 to produce RhAlO2–5 and RhAl2O3–6 (Fig. S4†), respectively. Thus, in principle, all the species mentioned above can be involved in the catalytic reactions. Moreover, these experiments indicated that most of the generated RhxAly16Oz or RhxAly18Oz species are reactive toward at least one kind of reactant gas (NO, N2O, O2, or CO), demonstrating that these neutral species are unstable under oxidative or reductive conditions in the gas phase. Note that the topic with O2 as the oxidant is not the focus of this study. The catalysis for the conversion of NO and CO into N2 and CO2 promoted by RhAlO0–3 is emphasized and shown in Scheme 1, and it is a similar case when this catalysis is driven by RhAl2O1–4.
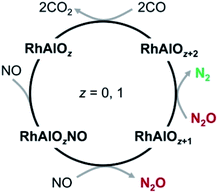 |
| | Scheme 1 Proposed catalytic cycle for the conversion of NO and CO into N2 and CO2 mediated by the neutral RhAlO0–3 species. This catalysis can also be driven by the RhAl2O1–4 clusters. | |
3.2 Theoretical results
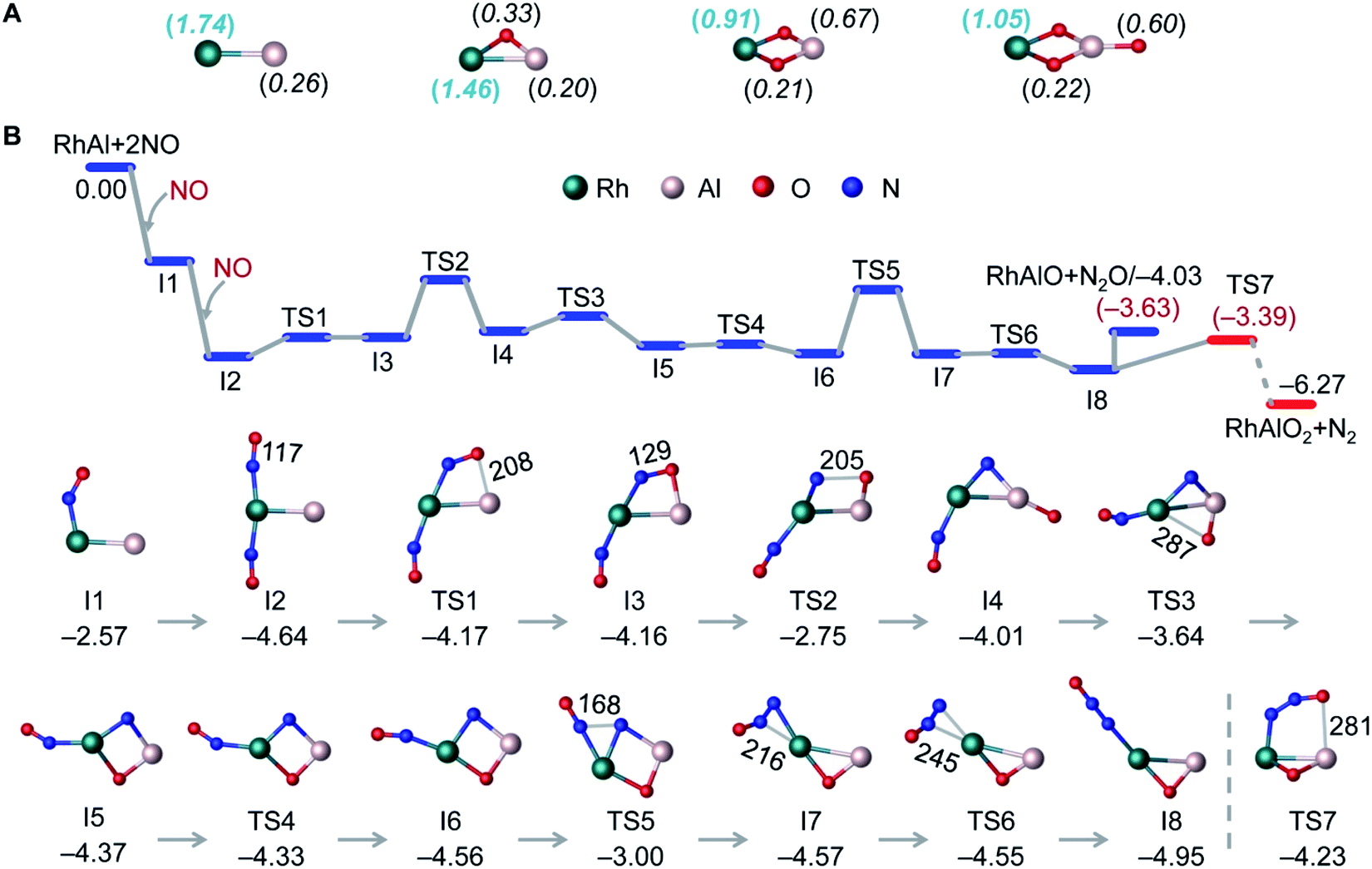
DFT calculations have been performed to explore the structures of RhAlO0–3 and RhAl2O1–4 (Fig. 2A and S5†) and the mechanisms of catalytic NO reduction by CO mediated by RhAlO0–3 and RhAl2O1–4 (Fig. 2B and S6–S15†). The lowest-lying isomers of RhAlO0–3 are in the triplet state (Fig. 2A). The direct bonding of Rh and Al drives the distribution of spin density dominantly on the Rh atom in RhAl and RhAlO, while the two unpaired electrons in RhAlO2,3 are delocalized extensively to more atoms. This special electronic structure of RhAlO0,1 may be the key factor to bring about the unique reactivity in NO reduction (Fig. 1B and C).
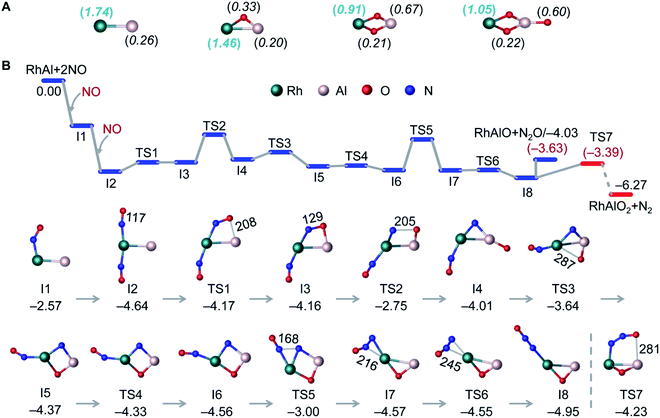 |
| | Fig. 2 DFT calculated lowest-lying isomers of RhAlO0–3 (A) and the potential energy profile for reaction RhAl + 2NO (B). In panel A, the unpaired electron spin density distributions of RhAlO0–3 are shown in the parentheses. In panel B, bond lengths (in pm) and relative energies (in eV) for intermediates (Ix) and transition states (TSx) are shown. The values in parentheses are calculated Gibbs free energies at T = 298 K. See details in Fig. S8b† for the pathway shown in the dotted line. | |
For reaction RhAl + 2NO (Fig. 2B), the bare Rh site can trap the first NO molecule tightly with a striking binding energy of 2.57 eV (I1). The direct dissociation of NO on RhAl (Fig. S6†) generates a product that is thermodynamically less favorable than that of I1, which has the chance to be stabilized under thermal collision conditions and then captures a second NO molecule to form I2. The large energy released (ΔH0 = −4.64 eV) in the formation of I2 is crucial to guarantee the N–O dissociation and the N–N coupling processes to take place. In the next step, the adjacent Al site in I2 is able to accept the O atom of NO to form I3, on which NO is apparently activated with the elongation of the N–O bond from 117 pm in I2 (115 pm in isolated NO) to 129 pm in I3. The complete cleavage of the N–O bond is the bottleneck of the whole reaction with an overall barrier of −2.75 eV (I3 → TS2 → I4). N–N recombination is another energetically demanding process (I6 → TS5 → I7) that needs to overcome an absolute barrier of 1.56 eV. The key step to determine the final products lies in the two competitive pathways after the formation of I8: the generation of N2O and RhAlO or N2 and RhAlO2. Note that the direct evaporation of gas-phase N2O from I8 is an entropically more favorable process (ΔG298 = −3.63 eV) with respect to N2 release (TS7, ΔG298 = −3.39 eV), which is in good agreement with the experimental result that only product RhAlO can be clearly observed at a lower pressure of NO (Fig. 1B). This is a similar case to NO reduction by RhAlO (Fig. 1C and S7†). Then N2O can be easily trapped and reduced to N2 by RhAlO0–2 (Fig. 1D, E and S8†). During the process of NO reduction by RhAlO0,1 (Fig. 2B and S7†), a deliberate analysis discovers that the Rh site functions as an antenna to anchor NO while the adjacent Al site is vital to assist the dissociation of NO and store the accumulated oxygen atom, underlining the importance of direct Rh–Al bonding for effective NO reduction. The half-naked Rh in RhAlO3–1 can capture and deliver CO for oxidation by nearby oxygen atoms favorably to regenerate RhAl (Fig. S9†), the reaction behavior of which has been extensively reported in both of the condensed-phase40,41 and gas-phase studies,25,26 and then a catalytic cycle can be established (Scheme 1). It also works when such catalysis is driven by the RhAl2O1–4 clusters (Fig. S10–S15†). Theoretical calculations well support and interpret the experimental observations shown in Fig. 1 and S1–S3.† Moreover, though the competing binding of environmental molecules (e.g., O2 and H2O) with the Rh–Al–O species will not occur under our current conditions (see details in the Experimental methods), the possible presence of this competing binding in real-life catalysis has also been considered by calculating the reactions of RhAl with O2 and H2O as examples (Fig. S16†). The potential reactivity of RhAl toward O2 and H2O demonstrates that the influence coming from these environmental molecules should not be neglected under real-life conditions.
4. Discussion
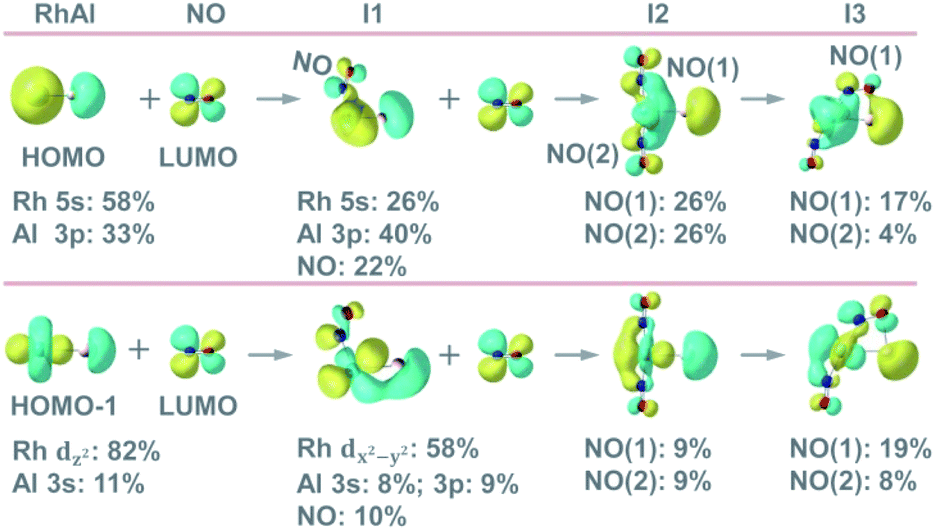
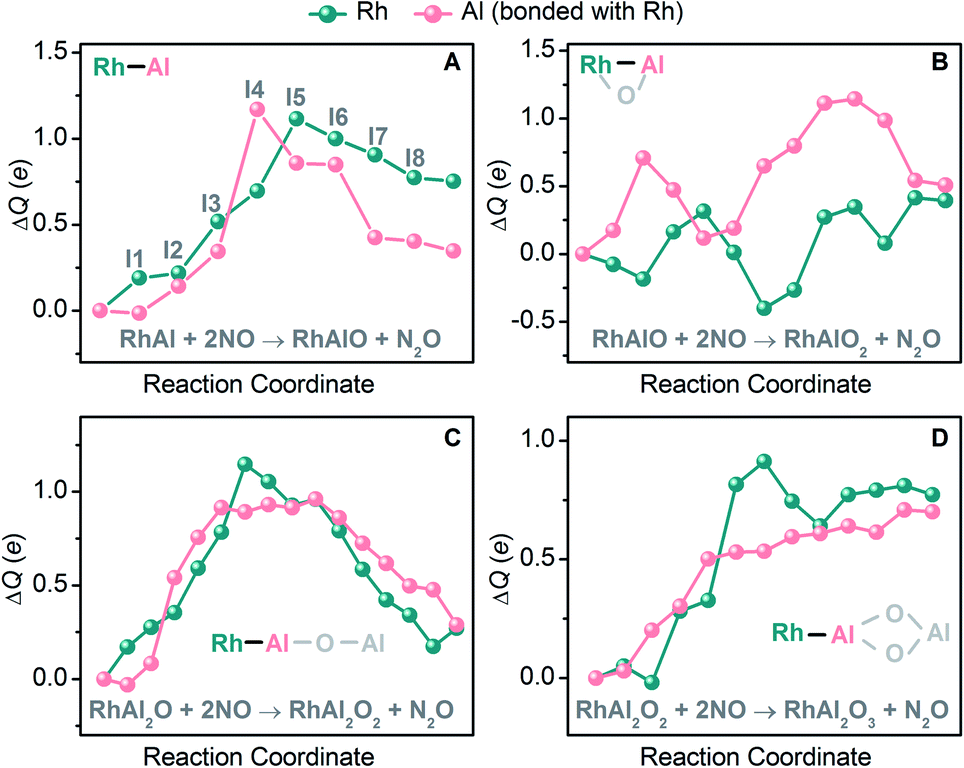
The reduction of NO is sensitive to the chemical environment of an active site and it has been evidenced that metallic Rh is more attractive to the coming NO molecules than the oxidized one.42 The remarkable difference of electronegativity between Rh (2.28) and Al (1.61)43 drives the accumulation of electrons on the Rh site of RhAl (Rh: −0.38 e, according to natural bond orbital analysis) and generates a polarized Rh–Al bond. Electron configuration analysis on RhAl further confirms the negatively charged Rh (4d8.575s0.80 in RhAl versus 4d85s1 in Rh), which functions as the preferred trapping site to capture two NO molecules tightly. Frontier orbital analysis on the interaction of RhAl with NO (Fig. 3 and S17†) evidences that the strong binding between Rh and NO can be traced back to the effective back-donation of 5s- and 4d-electrons of Rh in the occupied orbitals of RhAl to the π*-orbital of NO. In the next step, effective overlap between the Al-3p and O-2p orbitals in I3 leads to the pronounced activation of NO, as reflected by the fact that a large amount of negative charge is accumulated on the bridged NO (−0.76 e) and the N–O bond order has been decreased to about 1.5. Then the N–O bond ruptures favorably, accompanied by the disruption of the Rh–Al bond (I3 → I5, Fig. 2B). Note that two electrons (2NO + 2e− → N2O + O2−) are required to reduce two NO to one N2O and the Rh atom in RhAl alone is not sufficient to complete this task. In this case, the Al atom in proximity to Rh is vital to relieve the burden of Rh for electron control, not only acting as a reservoir to store oxygen after NO dissociation. A deep exploration discovers that the Al site in RhAl has already taken part in the reaction during the capture of the second NO through electron transfer interaction (I1 → I2, Fig. 4A). During the pathway of N–N coupling, the Rh–Al bond recovers to accumulate electrons and then drives this step to proceed (I6 → I8, Fig. 4A). A global view shows that both of the Rh and Al atoms experience vigorous changes in charge states during NO reduction (Fig. 4A), emphasizing the synergy of direct Rh–Al bonding that Rh triggers the reaction in the first step and then the nearby Al site facilitates the adsorption of NO, dissociation of the N–O bond, and the coupling of the N–N bond. The importance of this polarized Rh–Al bond can be further confirmed by the results that both the RhAlO0,1 and RhAl2O1,2 species (Fig. 2A and S5†) with direct Rh–Al bonding can be involved in NO reduction (Fig. 1B, C and S1, S3†). Moreover, severe fluctuation of charge states for both atoms can also be monitored obviously during NO reduction to dynamically adapt the structural changes of the reactive systems (Fig. 4B–D). It is previously believed that alumina appears to play an indirect role in influencing the reactivity of supported noble metal (NM) catalysts.44,45 Gas-phase studies on catalytic CO oxidation by O2 mediated with NM-doped aluminum oxide clusters, such as AuAl3O3–5+ (ref. 46) and PtAl3O5–7−,47 also confirmed that only NM atoms are highlighted to buffer electrons. This study provides new insights into NO reduction that an exposed and polarized Rh–Al bond is required to provide a characteristic binding site and distinct electronic structure for NO adsorption, dissociation, and N–N coupling.
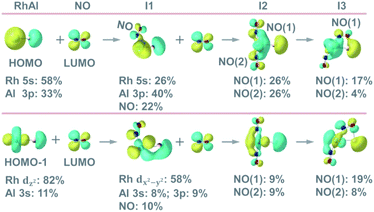 |
| | Fig. 3 Molecular orbital interactions between the occupied orbitals (HOMO and HOMO−1) of RhAl with the lowest unoccupied molecular orbital (LUMO) of NO as well as the occupied orbitals of RhAlNO (I1) with the LUMO of NO. The corresponding occupied orbitals for I2 and I3 (Fig. 2B) are also presented. See details in Fig. S17.† | |
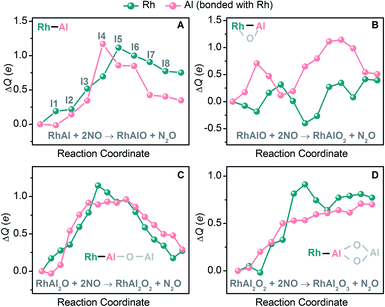 |
| | Fig. 4 DFT-calculated natural charge difference (ΔQ (e)) on the Rh and Al atoms with respect to those in RhAl (A), RhAlO (B), RhAl2O (C), and RhAl2O2 (D) along the reaction coordinates of NO reduction mediated by the four species (Fig. 2B and S7, S10, S11†). | |
Common knowledge on catalytic reactions is that each elementary step in the catalysis should be exothermic, and thus, a catalyst can be regenerated. The higher oxygen affinity of a catalyst favors NO reduction,48,49 while the strongly bonded oxygen may conversely result in the low activity of the resulting product for CO oxidation.50,51 Thus, the subsequent supply of the accumulated oxygen atom in RhAlO to oxidize CO is a vital criterion to validate the ability of RhAl as a catalyst. As shown in Fig. 2A, the presence of a polarized Rh–Al bond gives rise to a triangular structure of RhAlO, on which the oxygen atom is relatively weakly bonded (5.03 eV) and can be released to oxidize CO. In contrast, the oxygen atom is sandwiched between two Al atoms in Al2O (O-binding energy: 8.94 eV) with a linear structure, which is completely inert toward CO due to the thermodynamic impediment (Al2O + CO → Al2 + CO2, ΔH0 = 3.16 eV), though Al2 is more energetic in reducing NO (Fig. S18,† Al2 + 2NO → Al2O + N2O, ΔH0 = −7.93 eV). Thus, this heterobimetallic Rh–Al bond is crucial to regulate the reaction enthalpy of catalysis (2NO + CO → N2O + CO2, ΔH0 = −4.77 eV) to be distributed relatively evenly in each elementary step (NO reduction and CO oxidation) to guarantee that each step is exothermic, and then a catalytic reaction can be built. Complexes with heterometallic bonds are especially reactive toward the cleavage of conventionally inert bonds, such as O–H and N–H bonds, in organometallic chemistry.52,53 The importance of the polarized Rh–Al bond to rupture the thermodynamically strong C–F bond under mild conditions has been highlighted in homogeneous catalysis.54 Herein, in the field of gas-phase catalysis, the importance of a polarized bimetallic bond (Rh–Al) is emphasized to regulate a catalytic reaction.
The preciousness and indispensability of Rh in NO removal make it urgent to maximize the usage of Rh without the loss of catalytic activity.55,56 Using single atom catalysts (SACs) with isolated metal sites highly dispersed on oxide supports is the ultimate strategy to utilize NMs effectively.57–59 There have been several attempts to catalyze the conversion of NO and CO using oxide-supported single Rh atom catalysts,60–62 while these catalytic processes are subject to lower activity or selectivity at lower temperatures with respect to related clusters or nanoparticle counterparts. The key to activate Rh SACs lies in the fundamental understanding of related systems at a strictly molecular level. Though a gas-phase study can never account for the catalytic details that prevail in practical catalysis, the fundamental insights obtained at a strictly molecular level can be useful to guide the design of advanced catalysts. Benefiting from this gas-phase study, we expect that active sites with exposed and polarized Rh–M bonds (M = Al, Ce, Fe, and so on) on Rh-based catalysts can be essential in real-life catalysis. Thus, scientists should focus on the construction of energetic and flexible Rh–M bonds on the surface of catalysts to have effective conversion of NO under various conditions.
5. Conclusions
In conclusion, the catalytic conversion of NO and CO into N2 and CO2 mediated by gas-phase rhodium-aluminum oxide species (RhAlO0–3 and RhAl2O1–4) was identified by mass spectrometry and quantum chemical calculations. This finding is a sharp improvement in the field of gas-phase catalysis, given that only elementary steps for NO reduction or CO oxidation by Rh-doped species are commonly observed because it is dramatically challenging to rupture the strong N–O bond and couple the N–N bond. We demonstrated that N2O can be generated prior to N2 during NO reduction and then N2O can be recaptured and reduced. A mechanistic study highlights that the existence of a polarized Rh–Al bond on these gas-phase species is required to catalyze NO reduction by CO. This study uncovers the molecular-level origins of the puzzling reactivity of Rh-based three-way catalysts in NO reduction and it can be an important step to understand Rh catalysis in many chemical processes.
Author contributions
X. N. L. and S. G. H. conceived this work. J. J. C. conducted the experiments and theoretical calculations. J. J. C. wrote the original manuscript and provided the ESI.† X. N. L. contributed to manuscript revision. L. H. M. and Q. Y. L. provided helpful discussions on experimental and theoretical data. All authors discussed the results and commented on the manuscript.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Nos. 22022308 and 21773254), the China Postdoctoral Science Foundation (No. 2021M690153), and the K. C. Wong Education Foundation. X. N. L. thanks the Youth Innovation Promotion Association CAS (No. Y202007).
References
- Z. Gholami, G. Luo, F. Gholami and F. Yang, Catal. Rev.: Sci. Eng., 2020, 63, 68–119 CrossRef.
- S. Roy, M. S. Hegde and G. Madras, Appl. Energy, 2009, 86, 2283–2297 CrossRef CAS.
-
E. E. Weaver, J. W. Shiller, R. M. Campau and J. F. Macura, Ford 450 car catalyst field test, SAE Technical Paper 742061, 1974.
- P. Granger and V. I. Parvulescu, Chem. Rev., 2011, 111, 3155–3207 CrossRef CAS PubMed.
- J. Kašpar, P. Fornasiero and N. Hickey, Catal. Today, 2003, 77, 419–449 CrossRef.
- H. S. Gandhi, G. W. Graham and R. W. McCabe, J. Catal., 2003, 216, 433–442 CrossRef CAS.
- M. Shelef and G. W. Graham, Catal. Rev.: Sci. Eng., 1994, 36, 433–457 CrossRef CAS.
- H. Asakura, S. Hosokawa, T. Ina, K. Kato, K. Nitta, K. Uera, T. Uruga, H. Miura, T. Shishido, J. Ohyama, A. Satsuma, K. Sato, A. Yamamoto, S. Hinokuma, H. Yoshida, M. Machida, S. Yamazoe, T. Tsukuda, K. Teramura and T. Tanaka, J. Am. Chem. Soc., 2018, 140, 176–184 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS.
- G. Liu, P. Poths, X. Zhang, Z. Zhu, M. Marshall, M. Blankenhorn, A. N. Alexandrova and K. H. Bowen, J. Am. Chem. Soc., 2020, 142, 7930–7936 CrossRef CAS PubMed.
- H. Schwarz and K. R. Asmis, Chem.–Eur. J., 2019, 25, 2112–2126 CrossRef CAS PubMed.
- S. M. Lang and T. M. Bernhardt, Phys. Chem. Chem. Phys., 2012, 14, 9255–9269 RSC.
- R. A. J. O'Hair, Int. J. Mass Spectrom., 2015, 377, 121–129 CrossRef.
- C. Chi, H. Qu, L. Meng, F. Kong, M. Luo and M. Zhou, Angew. Chem., Int. Ed., 2017, 56, 14096–14101 CrossRef CAS.
- T. Nagata, K. Koyama, S. Kudoh, K. Miyajima, J. M. Bakker and F. Mafuné, J. Phys. Chem. C, 2017, 121, 27417–27426 CrossRef CAS.
- A. Fielicke, G. von Helden, G. Meijer, D. B. Pedersen, B. Simard and D. M. Rayner, J. Chem. Phys., 2006, 124, 194305 CrossRef PubMed.
- M. L. Anderson, M. S. Ford, P. J. Derrick, T. Drewello, D. P. Woodruff and S. R. Mackenzie, J. Phys. Chem. A, 2006, 110, 10992–11000 CrossRef CAS PubMed.
- M. Yamaguchi, S. Kudoh, K. Miyajima, O. V. Lushchikova, J. M. Bakker and F. Mafuné, J. Phys. Chem. C, 2019, 123, 3476–3481 CrossRef CAS.
- S. Hirabayashi and M. Ichihashi, J. Phys. Chem. A, 2017, 121, 2545–2551 CrossRef CAS.
- A. Yamada, K. Miyajima and F. Mafuné, Phys. Chem. Chem. Phys., 2012, 14, 4188–4195 RSC.
- X.-N. Li, H.-M. Zhang, Z. Yuan and S.-G. He, Nat. Commun., 2016, 7, 11404 CrossRef CAS.
- J.-J. Chen, Z. Yuan, X.-N. Li and S.-G. He, Int. J. Mass Spectrom., 2017, 422, 98–104 CrossRef CAS.
- J.-J. Chen, X.-N. Li, Q. Chen, Q.-Y. Liu, L.-X. Jiang and S.-G. He, J. Am. Chem. Soc., 2019, 141, 2027–2034 CrossRef CAS.
-
Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- L.-N. Wang, X.-N. Li and S.-G. He, Sci. China Mater., 2020, 63, 892–902 CrossRef CAS.
- X.-N. Li, L.-N. Wang, L.-H. Mou and S.-G. He, J. Phys. Chem. A, 2019, 123, 9257–9267 CrossRef CAS.
- H. Schwarz, Catal. Sci. Technol., 2017, 7, 4302–4314 RSC.
- E. R. Bernstein, Int. J. Mass Spectrom., 2015, 377, 248–262 CrossRef CAS.
- Y. Hu, J. Guan and E. R. Bernstein, Mass Spectrom. Rev., 2013, 32, 484–501 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1989, 90, 1730–1734 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- X.-L. Ding, Z.-Y. Li, J.-H. Meng, Y.-X. Zhao and S.-G. He, J. Chem. Phys., 2012, 137, 214311 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- H. B. Schlegel, J. Comput. Chem., 1982, 3, 214–218 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
-
E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2012, http://www.chem.wisc.edu/ Search PubMed.
-
NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg, MD, p. 20899, September 29, 2021, DOI:10.18434/T4D303.
- D. Widmann and R. J. Behm, Acc. Chem. Res., 2014, 47, 740–749 CrossRef CAS PubMed.
- D. Widmann and R. J. Behm, J. Catal., 2018, 357, 263–273 CrossRef.
- A. Srinivasan and C. Depcik, Catal. Rev.: Sci. Eng., 2010, 52, 462–493 CrossRef CAS.
- A. L. Allred, J. Inorg. Nucl. Chem., 1961, 17, 215–221 CrossRef CAS.
- M. Moses-DeBusk, M. Yoon, L. F. Allard, D. R. Mullins, Z. Wu, X. Yang, G. Veith, G. M. Stocks and C. K. Narula, J. Am. Chem. Soc., 2013, 135, 12634–12645 CrossRef CAS.
- J. H. Kwak, J. Hu, D. Mei, C.-W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed.
- Z.-Y. Li, Z. Yuan, X.-N. Li, Y.-X. Zhao and S.-G. He, J. Am. Chem. Soc., 2014, 136, 14307–14313 CrossRef CAS.
- X.-N. Li, Z. Yuan, J.-H. Meng, Z.-Y. Li and S.-G. He, J. Phys. Chem. C, 2015, 119, 15414–15420 CrossRef CAS.
- M. Yamaguchi, Y. Zhang, S. Kudoh, K. Koyama, O. V. Lushchikova, J. M. Bakker and F. Mafuné, J. Phys. Chem. Lett., 2020, 11, 4408–4412 CrossRef CAS.
- T. Nagata, K. Miyajima, R. A. Hardy, G. F. Metha and F. Mafuné, J. Phys. Chem. A, 2015, 119, 5545–5552 CrossRef CAS.
- Y. Zheng, S. Thampy, N. Ashburn, S. Dillon, L. Wang, Y. Jangjou, K. Tan, F. Kong, Y. Nie, M. J. Kim, W. S. Epling, Y. J. Chabal, J. W. P. Hsu and K. Cho, J. Am. Chem. Soc., 2019, 141, 10722–10728 CrossRef CAS.
- R. K. Grasselli, Top. Catal., 2002, 21, 79–88 CrossRef CAS.
- S. Rej, H. Tsurugi and K. Mashima, Coord. Chem. Rev., 2018, 355, 223–239 CrossRef CAS.
- P. Buchwalter, J. Rose and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- I. Fujii, K. Semba, Q.-Z. Li, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2020, 142, 11647–11652 CrossRef CAS.
- N. Zhang, C. Ye, H. Yan, L. Li, H. He, D. Wang and Y. Li, Nano Res., 2020, 13, 3165–3182 CrossRef CAS.
- A. Beniya and S. Higashi, Nat. Catal., 2019, 2, 590–602 CrossRef.
- Y. Lu, Z. Zhang, F. Lin, H. Wang and Y. Wang, Chemnanomat, 2020, 6, 1659–1682 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- H. Jeong, O. Kwon, B.-S. Kim, J. Bae, S. Shin, H.-E. Kim, J. Kim and H. Lee, Nat. Catal., 2020, 3, 368–375 CrossRef CAS.
- C. Asokan, Y. Yang, A. Dang, A. B. Getsoian and P. Christopher, ACS Catal., 2020, 10, 5217–5222 CrossRef CAS.
- S. Zhang, Y. Tang, L. Nguyen, Y.-F. Zhao, Z. Wu, T.-W. Goh, J. J. Liu, Y. Li, T. Zhu, W. Huang, A. I. Frenkel, J. Li and F. F. Tao, ACS Catal., 2018, 8, 110–121 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta04911a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ac,
Li-Hui
Mou
abc,
Qing-Yu
Liu
ac and
Sheng-Gui
He
*ac,
Li-Hui
Mou
abc,
Qing-Yu
Liu
ac and
Sheng-Gui
He