
Efficient hydrogen evolution by reconstruction of NiMoO4–CoO via Mo recombination†
Juejin
Teng
,
Zhixin
Dong
,
Daokun
Liu
,
Xiao
Zhang
 * and
Jinxue
Guo
*
* and
Jinxue
Guo
*
Key Laboratory of Optic-Electric Sensing and Analytical Chemistry for Life Science, MOE, Key Laboratory of Biochemical Analysis, Shandong Province, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, PR China. E-mail: xiaozhang@qust.edu.cn; gjx1213@126.com; Fax: +86 532 84023927; Tel: +86 532 84022681
First published on 25th April 2022
Abstract
Surface reconstruction of oxygen evolution reaction electrocatalysts with enhanced catalysis activity has been intensively reported recently. However, the surface reconstruction has seldom been observed from hydrogen evolution reaction (HER) catalysts. Here, we report the surface reconstruction with enhancement of catalysis activity in the HER catalyst of NiMoO4–CoO nanosheets on carbon cloth. Mo species dissolve from NiMoO4–CoO by activating the pristine catalyst at certain potential, which enables the formation of K2Mo3O10 on the catalyst surface. The surface reconstruction not only brings large numbers of active sites and enhanced charge transfer, but also boosts the intrinsic activity per catalysis site. Together with the above advantages and the benefits of nanosheet structure, the activated catalyst exhibits high HER activity with the ultralow overpotentials of 130 and 192 mV at high current densities of 100 and 400 mA cm−2. This finding provides new insight for the exploration on surface reconstruction of transition metal-based HER catalysts and may enable further understanding on the origin of catalysis activity improvement.
Introduction
To address the environmental crisis caused by fossil fuel overuse, one of the promising solutions is the utilization of high-energy-density hydrogen fuels supplied via water electrolysis.1 Aiming at affordable hydrogen production, a large number of research studies have focused on developing earth-abundant transition-metal-based materials for highly efficient hydrogen evolution reaction (HER) and oxygen evolution reaction (OER).2–6 A recent study shows that when a certain voltage is reached, transition-metal-based catalysts may be reduced or oxidized in a harsh electrochemical environment.7 For OER electrocatalysts, it is found that the structure and component of catalysts are not stable after a long-time oxygen evolution at certain voltage, suggesting that such kinds of materials actually act as precatalyst.8 Therefore, unveiling the in situ reconstruction of catalysts and real situation of the active surface during catalysis is crucial for designing catalysts with high efficiency, and it has recently attracted increasing research effort.9,10 For instance, Duan et al. reported that Cr3+ leaches from the surface of CoCr2O4 catalyst at 1.7 V during OER process, inducing the transformation of real active centres of Co (oxy)hydroxides for much improved OER performance.11 Wang and co-workers reported the structural self-optimization process of LiCoO1.8Cl0.2 into amorphous (oxy)hydroxide phase, which delivered increased OER catalytic activity in alkaline condition.12Recently, some findings on the reconstruction of HER catalysts were reported. Laursen and co-workers revealed the surface reconstruction of Ni3P into Ni4P4, which served as the active surface site for enhanced HER activity.13 Chen et al. reported that chronopotentiometry treatment could drive the in situ transformation of Cu2O@MnO2 into Cu@Mn(OH)2 for much boosted HER catalysis activity.14 Unfortunately, compared with OER materials, less research effort has been paid to the reconstruction of HER catalysts. Therefore, it is significant to focus on the reconstruction of HER catalyst and unveil the corresponding origin of the catalytic performance.
Among non-precious metal catalysts, Ni–Co–Mo based materials demonstrate good HER performance and excellent stability.15–18 Most of the literature shows that Mo serves as dopant to regulate the catalytic activities and kinetics of the Ni and/or Co centres. Very recently, Zhang and co-workers found out that Mo in the Ni4Mo alloy could be oxidized and dissolved in the form of MoO42−, which is adsorbed and polymerized into Mo2O72− to boost the HER activity of the Ni species.19
Inspired by the above analysis, herein, we employ carbon-cloth-supported NiMoO4–CoO nanosheets (NiMoO4–CoO/CC) as a model sample to demonstrate the surface reconstruction with catalytic activity improvement of NiMoO4–CoO, towards promising HER performance. When NiMoO4–CoO is electrochemically activated at potential of −0.25 V for 1.5 hours, Mo in the NiMoO4–CoO dissolves and then transforms into a new phase of K2Mo3O10 crystals, which anchor on the surface of the catalyst. The reconstructed catalyst (act-NiMoO4–CoO/CC) exhibits remarkably improved HER performance compared to the precatalyst NiMoO4–CoO/CC. This study demonstrates an effective method for the controllable electrochemical activation-induced surface reconstruction of transition metal oxides for HER application, broadening the understanding on the origin of reconstruction and activity improvement of HER catalysts.
Results and discussion
The SEM image in Fig. S1† depicts the smooth nanosheet array structure of the NiCoMo-LDH precursor, which grows vertically and forms various micropores through crosslinking. As shown in Fig. 1a, NiMoO4–CoO still inherits the nanosheet structure of NiCoMo-LDH after calcination under H2/Ar, but the nanosheet surface turns rough. The XRD pattern (Fig. 1b) of NiMoO4–CoO reveals the coexistence of monoclinic NiMoO4 (JCPDS No. 45-0142) and cubic CoO (JCPDS No. 70-2855). The TEM image (Fig. 1c) of NiMoO4–CoO also shows the nanosheet structure, which possesses rough texture that is advantageous for the exposure of active sites. The high-resolution TEM (HRTEM) of NiMoO4–CoO nanosheet is shown in Fig. 1d. The clear lattice fringes with spacing distances of 0.21 and 0.24 nm are assigned to the (200) and (111) planes of CoO. The lattice fringes with interplanar spacing of 0.33 nm correspond to NiMoO4. This result shows that these nanosheets are hybrid NiMoO4–CoO. Notably, a large number of hetero interfaces and defects are detected on the NiMoO4–CoO nanosheets, which may supply additional active sites for electrocatalysis. The corresponding EDX elemental mapping images (Fig. 1e) show that Ni, Mo, O, and Co distribute uniformly on the NiMoO4–CoO nanosheets, suggesting the homogeneously formed hetero nanocrystals that are derived from the NiCoMo-LDH precursor.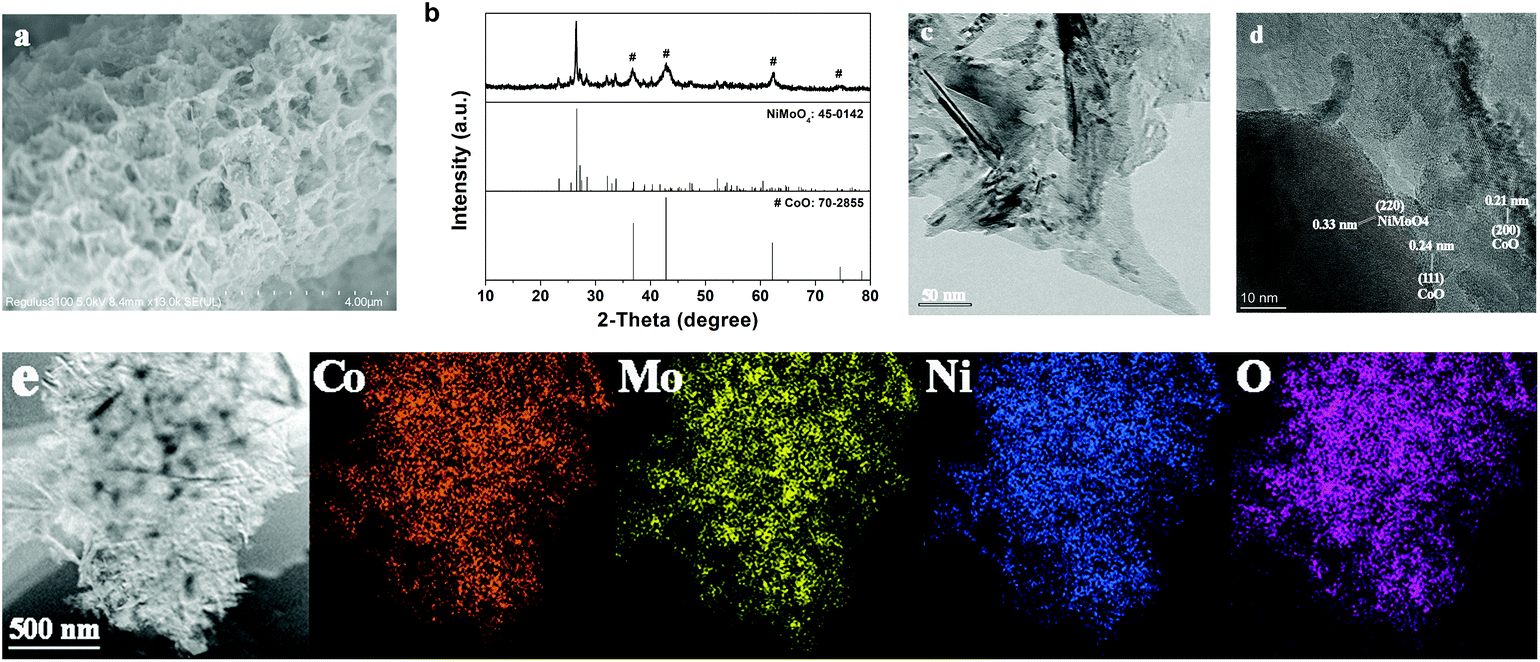 | ||
| Fig. 1 (a) SEM image, (b) XRD, (c) TEM, (d) HRTEM, and (e) EDX elemental mapping of Co, Mo, Ni, and O elements of NiMoO4–CoO/CC. | ||
The HER polarization curve of NiMoO4–CoO/CC is displayed in Fig. 2a, and the curves of NiMoO4/CC, CoMoO4/CC, and NiCoO/CC were also collected for comparison. NiMoO4–CoO/CC exhibits a current density of 100 mA cm−2 at the low overpotential of 222 mV, which is much better than those of NiMoO4/CC (320 mV), CoMoO4/CC (278 mV), and NiCoO/CC (292 mV). The extremely low overpotential of 279 mV is achieved to afford a high current density of 400 mA cm−2, showing promising potential for industrial applications. The remarkable HER catalysis efficiency of NiMoO4–CoO/CC should be due to the numerous active sites and synergisms between hetero materials, as well as the nanosheet structure benefits. The chronoamperometric test (Fig. 2b) obtained at a fixed overpotential of 222 mV shows that NiMoO4–CoO/CC performs with stable durability for 30 hours. Notably, there is an interesting activation process at the initial ∼1 h, during which the current density increases linearly from 100 to ∼120 mA cm−2, suggesting the possible reconstruction of NiMoO4–CoO catalyst during the HER.
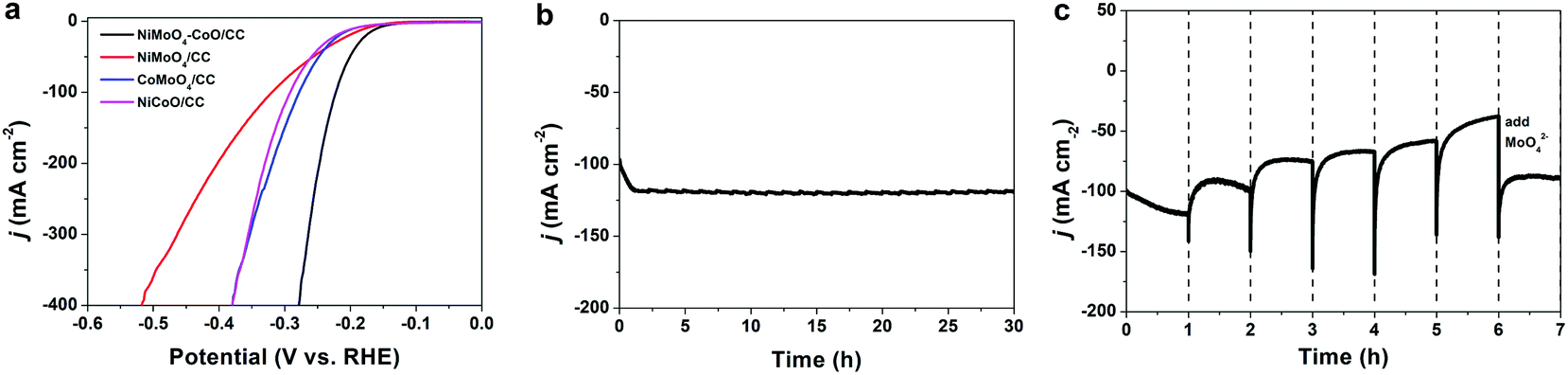 | ||
| Fig. 2 (a) LSV curves of NiMoO4–CoO/CC, NiMoO4/CC, CoMoO4/CC, and NiCoO/CC. (b) J–t curve of NiMoO4–CoO/CC in 1.0 M KOH. (c) J–t curve of NiMoO4–CoO/CC obtained by renewing the electrolyte every 1 hour. | ||
To further unveil the activation process, a controllable current–time (j–t) test of NiMoO4–CoO/CC was performed by renewing the electrolyte every 1 hour during the chronoamperometric test. As shown in Fig. 2c, a clear HER activation process is observed at the initial 1 h, and the current density increases from 100 to about 120 mA cm−2. After the electrolyte is renewed with KOH solution, the current density shows a slight fade to about 100 mA cm−2 after 1 hour. When the electrolyte is renewed every 1 hour, a similar current fading is observed. After six hours, the electrolyte is renewed with KOH solution that contains 0.1 M MoO42−. Interestingly, the catalysis current recovers to 90 mA cm−2. Such results indicate that Mo species could dissolve into the electrolyte during the chronoamperometric test, which greatly contributes to the improved HER efficiency of NiMoO4–CoO/CC.19 To reveal the possible leaching, the contents of Mo, Ni, and Co in the electrolyte were measured by inductively coupled plasma mass spectrometry (ICP-MS). Fig. S2† shows the time-dependent concentration of dissolved Mo, Ni, and Co in the electrolyte. The concentrations of Ni and Co remain at low levels, showing that Ni and Co do not leach throughout the process. Notably, Mo concentration increases with time and reaches the maximum value (0.125 mmol L−1) after 1 hour, confirming the Mo dissolution.
Based on the aforementioned analysis, the freshly obtained NiMoO4–CoO/CC was subjected to activation at the various potentials of −0.2, −0.25, and −0.3 V for 1.5 h, respectively. Fig. 3a depicts the HER polarization curves of pristine NiMoO4–CoO/CC and act-NiMoO4–CoO/CC samples at different activation potentials. The act-NiMoO4–CoO/CC at −0.2 V shows no obvious activation, delivering similar HER efficiency compared with pristine NiMoO4–CoO/CC. The act-NiMoO4–CoO/CC at −0.25 V and act-NiMoO4–CoO/CC at −0.3 V exhibit improved HER activities over pristine NiMoO4–CoO/CC, highlighting the remarkable effect of activation. The corresponding overpotentials of these catalysts at current densities of 100 and 400 mA cm−2 are summarized in Fig. 3b. Clearly, the optimized activation potential of −0.25 V is obtained. For instance, the act-NiMoO4–CoO/CC at −0.25 V only needs an ultralow overpotential of 130 mV at 100 mA cm−2, showing impressively high HER efficiency that is better than most of the reported HER catalysts. Notably, the small value of 192 mV at the high current density of 400 mA cm−2 is obtained, which is promising to fulfil the demands for practical water splitting application. The corresponding Tafel plots (Fig. 3c) show that the catalysts activated at −0.25 and −0.3 V acquire enhanced catalysis kinetics. In comparison with pristine NiMoO4–CoO/CC, the Tafel slope of act-NiMoO4–CoO/CC at −0.25 V decreases from 65 to 41 mV dec−1, indicating the accelerated reaction kinetics. In the Nyquist plots (Fig. S3†), the act-NiMoO4–CoO/CC at −0.25 V exhibits decreased charge transfer compared to NiMoO4–CoO/CC, showing that the activation could facilitate charge transfer during electrocatalysis. In addition, the act-NiMoO4–CoO/CC at −0.25 V shows improved double-layer capacitance (Cdl) compared with NiMoO4–CoO/CC (Fig. S4†), suggesting more active sites after activation. Moreover, the HER current densities of various catalysts are normalized by ECSA, in which (Fig. S5†) the act-NiMoO4–CoO/CC at −0.25 V still affords better HER efficiency than pristine NiMoO4–CoO/CC, showing that the activation not only generates more active sites but also boosts the intrinsic activity per site.
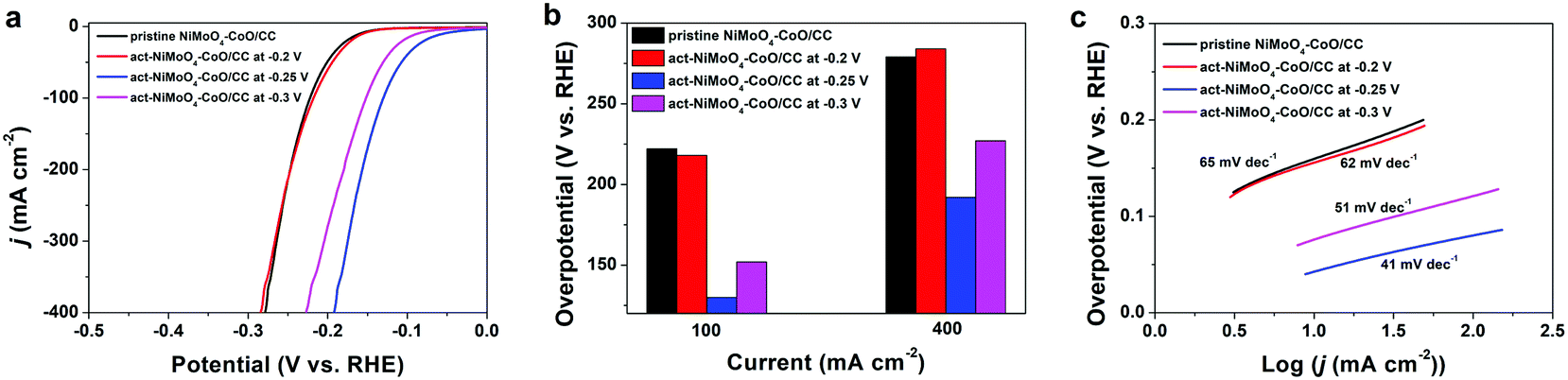 | ||
| Fig. 3 (a) LSV curves, (b) overpotentials at current densities of 100 and 400 mA cm−2, and (c) Tafel plots of pristine NiMoO4–CoO/CC, act-NiMoO4–CoO/CC at −0.2 V, act-NiMoO4–CoO/CC at −0.25 V, and act-NiMoO4–CoO/CC at −0.3 V. | ||
To investigate catalyst transformation during the activation process, a series of characterizations were employed. The HRTEM image (Fig. 4) reveals that the nanosheet structure of the catalyst is maintained after the activation process. Notably, new lattice fringes with spacing distances of 0.30 nm are detected, attributed to the (303) plane of K2Mo3O10·3H2O (JCPDS No. 32-0815). The analysis indicates the formation of K2Mo3O10 during the activation process, which is further revealed by the XRD pattern (Fig. S6†). Under the potential of −0.25 V (vs. RHE), Mo species exist as MoO42− in 1.0 M KOH.19,20 When the concentration increases, MoO42− transforms into K2Mo3O10via the reaction 3MoO42− + 2K+ + 2H2O = K2Mo3O10 + 4OH−. In addition, the possible element content changes on the surface of the catalyst were detected by EDX and are summarized in Table S1.† After the activation process, the Mo content decreases obviously from 7.83% to 3.31% due to the dissolution of Mo into the electrolyte. The detection of K in the sample of act-NiMoO4–CoO/CC at −0.25 V should be assigned to the formation of K2Mo3O10 on the catalyst surface, which is also shown in the EDX images (Fig. S7†). To probe the role of K2Mo3O10, the HER activity of K2Mo3O10/CC was collected. In addition, the mixture of K2Mo3O10 with NiMoO4–CoO on CC (K2Mo3O10 + NiMoO4–CoO/CC) was also tested as HER catalyst. In Fig. S8,† K2Mo3O10/CC delivered negligibly improved HER activity in comparison with pristine CC, suggesting that pristine K2Mo3O10 is inactive for HER. The HER performance of K2Mo3O10 + NiMoO4–CoO/CC is lower than NiMoO4–CoO/CC due to the addition of inactive K2Mo3O10. A comparative experiment testing the HER performance of NiMoO4–CoO/CC in a series of electrolytes, adding 0.01, 0.05, 0.10, and 0.15 M MoO42− without activation, was conducted to determine the contribution of MoO42− to the improved HER performance. As shown in Fig. S9,† the addition of MoO42− to the electrolyte indeed improves the HER performance of the pristine NiMoO4–CoO/CC electrode, and 0.10 M is the optimized concentration of MoO42−. However, the HER performance of NiMoO4–CoO/CC in the optimized concentration of 0.10 M is much lower than that of act-NiMoO4–CoO/CC at −0.25 V. It can be concluded that the in situ-formed K2Mo3O10 on the activated composite catalyst supplies synergistic effects to make a main contribution on the improved HER activity of NiMoO4–CoO.
 | ||
| Fig. 4 HRTEM of act-NiMoO4–CoO/CC at −0.25 V. | ||
To unveil the influences of activation potential on HER activity, the concentrations of Mo, Ni, and Co were determined by ICP-MS. It was found that Ni and Co do not dissolve at all activation potentials, serving as the stable active sites for HER. Under −0.2 V, there is no Mo detected in the electrolyte, showing that Mo dissolution and catalyst transformation cannot occur under such potential. At −0.3 V, Mo is detected in the electrolyte, and the maximum value of 0.149 mmol L−1 is obtained, which is higher than 0.125 mmol L−1. The higher concentration of MoO42− induces the formation of excessive inactive K2Mo3O10 in the activated composite, which should be responsible for the reduced HER activity at −0.3 V.
XPS analysis was employed to investigate the possible changes in the element chemical states of the catalyst after activation. Fig. 5a depicts the Mo 3d XPS spectra of the initial NiMoO4–CoO/CC and act-NiMoO4–CoO/CC at −0.25 V, both of which consist of two deconvoluted peaks. The peaks at 231.9 and 235.0 eV for the initial NiMoO4–CoO/CC are assigned to the Mo 3d5/2 and Mo 3d3/2 of Mo6+ species.21 After activation, the Mo 3d spectrum shows a negative shift (0.25 eV) in comparison with the initial catalyst, suggesting the reduction of Mo species during the formation of K2Mo3O10. In the Ni 2p spectrum of the initial NiMoO4–CoO/CC (Fig. 5b), the peaks at 854.0 and 871.4 eV are due to Ni2+ 2p3/2 and Ni2+ 2p1/2. The peaks at 855.7 and 872.8 eV are assigned to Ni3+ 2p3/2 and Ni3+ 2p1/2.22,23 The activated catalyst shows a negatively shifted Ni 2p spectrum (0.2 eV) compared with the initial catalyst. The Co 2p spectrum of the initial NiMoO4–CoO/CC (Fig. 5c) shows peaks at 778.9 and 794.2 eV, corresponding to Co3+ 2p3/2 and Co3+ 2p1/2.24,25 The peaks at Co2+ 2p3/2 and Co2+ 2p1/2 are related to 780.4 and 796.2 eV. The Co 2p spectrum of act-NiMoO4–CoO/CC shifts to the positive region (0.15 eV) in comparison with the initial sample. The above analysis indicates the electron donation from Co to Ni in the activated composite catalyst, which is accompanied by Mo dissolution. The O 1s spectrum of the initial NiMoO4–CoO/CC (Fig. 5d) consists of two peaks of 529.7 and 531.1 eV, which are indexed to lattice oxygen (O2−) and hydroxide (M-OH).26 After activation, a new peak related to the adsorbed water molecules (H2O) is observed at 532.7 eV.12 Interestingly, the peak of lattice oxygen in the activated catalyst shows significant decrease and negative shift (0.7 eV) compared with the initial NiMoO4–CoO/CC. The loss of lattice oxygen should be induced by the leaching of Mo species, which generates abundant oxygen vacancies on the surface of act-NiMoO4–CoO/CC.11 In addition, the leaching of Mo in NiMoO4 reduces the electron donation from O2− to metal, thus resulting in the negative shift of lattice oxygen peak. The XPS K 2p spectrum (Fig. S10†) was detected in the activated catalyst and confirms the existence of K after activation. Two peaks of K 2p3/2 (293.1 eV) and K 2p1/2 (295.8 eV) correspond to the K+ in K2Mo3O10. The XPS analysis unveils the Mo dissolution and lattice oxygen loss during the electrochemical activation process.
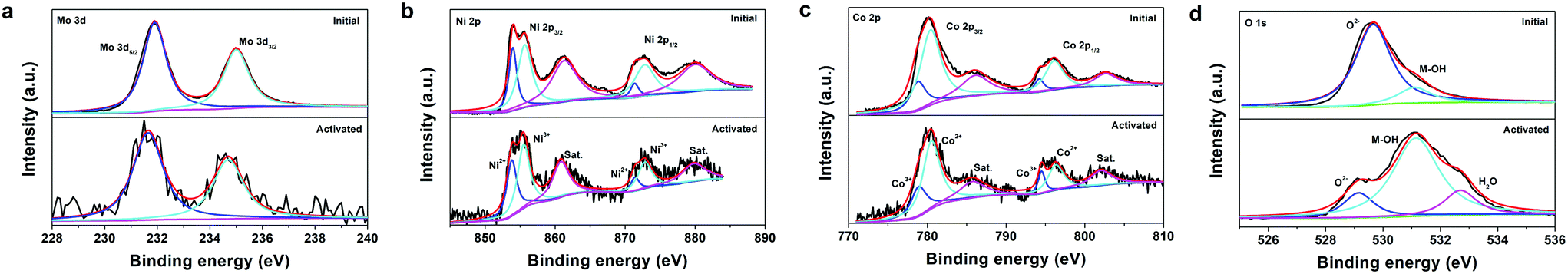 | ||
| Fig. 5 High-resolution XPS spectra of (a) Mo 3d, (b) Ni 2p, (c) Co 2p, and (d) O 1s regions for the initial NiMoO4–CoO/CC and act-NiMoO4–CoO/CC at −0.25 V. | ||
Additionally, the boosting effect of such activation process was also observed from the other Mo-based catalysts, NiMoO4/CC (Fig. S11†) and CoMoO4/CC (Fig. S12†). In contrast, the activation process shows no positive influence on the HER activity of NiCoO/CC (Fig. S13†). The above results further suggest that the dissolution and recombination of Mo species play a key role in the improved HER activity of such Mo-based catalysts.
Experimental
All chemicals were of analytical grade and used as purchased without further purification. The carbon cloth (CC)-supported NiCoMo layered double hydroxide (NiCoMo-LDH/CC) precursor was obtained via a hydrothermal method. In a specific synthesis route, the CC is firstly cleaned with concentrated H2SO4 and HNO3 in turn to remove impurities on the surface, then rinsed with deionized water and ethanol several times. Ni(NO3)2·6H2O (1 mmol), Co(NO3)2·6H2O (1 mmol), Na2MoO4·2H2O (1 mmol), urea (10 mmol), and NH4F (4 mmol) are dissolved in 40 mL water under ultrasonication. The obtained mixture is transferred into a 50 mL Teflon-steel autoclave and then heated at 100 °C for 10 h. After cooling to room temperature, the collected NiCoMo-LDH/CC sample is washed with water and ethanol, which is then dried under vacuum at 60 °C for 12 h. To prepare NiMoO4–CoO/CC, the dried NiCoMo-LDH are loaded in a porcelain boat and heated in a tube furnace at 550 °C for 2 h at a ramp rate of 2 °C min−1 under H2/Ar (5%, v/v) atmosphere. A similar synthesis procedure was employed for the preparation of NiMoO4/CC, CoMoO4/CC, and NiCoO/CC, with ratios of the Ni/Mo (1.5/1.5 mmol), Co/Mo (1.5/1.5 mmol), and Ni/Co (1.5/1.5 mmol).The NiMoO4–CoO/CC samples were characterized by powder X-ray diffraction (Bruker, D8 advance, diffractometer with Cu Kα radiation, λ = 1.5408 Å). The morphology and microstructure of NiMoO4–CoO/CC were recorded with a scanning electron microscope (SEM, Regulus SU8100) and transmission electron microscope (TEM, FEI TalosF200x) equipped with energy dispersive X-ray spectroscopy (EDS). X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha) was used to detect the surface element states of NiMoO4–CoO/CC, and all binding energies were modulated to the C 1s peak at 284.80 eV. Inductively coupled plasma mass spectrometry (ICP-MS) was performed on an Agilent 7800.
All the electrochemical data were collected on a CHI 660E instrument (CH Instruments, Shanghai, China), using a three-electrode cell with 1.0 M KOH as electrolyte. The as-obtained NiMoO4–CoO/CC was directly used as working electrode; the Hg/HgO electrode and platinum foil served as reference electrode and counter electrode, respectively. Linear sweep voltammetry (LSV) was performed with a scan rate of 5 mV s−1 to evaluate the HER performance with 90% iR compensation. The voltage potentials were calibrated with a reversible hydrogen electrode (vs. RHE); the equation is: E(RHE) = E(Hg/HgO) + pH × 0.0591 V + 0.098 V. The Tafel plots are derived from the LSV curves with the Tafel equation: η = a + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(j), where η is the overpotential, b is the Tafel slope, and j is the current density. The cyclic voltammetry (CV) curves were collected at different scan rates in the potential range without faradaic processes, and used to derive the electrochemical double-layer capacitance (Cdl). The electrochemical active surface area (ECSA) of the electrocatalyst was determined with the equation ECSA = Cdl/Cs (Cs is the specific capacitance, which is 0.040 mF cm−2 in alkaline media). The electrochemical impedance spectroscopy (EIS) was performed in the frequency range of 105 Hz to 10−1 Hz.
log(j), where η is the overpotential, b is the Tafel slope, and j is the current density. The cyclic voltammetry (CV) curves were collected at different scan rates in the potential range without faradaic processes, and used to derive the electrochemical double-layer capacitance (Cdl). The electrochemical active surface area (ECSA) of the electrocatalyst was determined with the equation ECSA = Cdl/Cs (Cs is the specific capacitance, which is 0.040 mF cm−2 in alkaline media). The electrochemical impedance spectroscopy (EIS) was performed in the frequency range of 105 Hz to 10−1 Hz.
The electrochemical activation was conducted in a three-electrode cell. NiMoO4–CoO/CC served as working electrode. A Hg/HgO electrode and platinum foil were used as reference electrode and counter electrode, respectively. NiMoO4–CoO/CC was activated with chronoamperometry method at −0.2, −0.25, and −0.3 V (vs. RHE) for 1.5 hours in 1 M KOH solution.
Conclusions
In summary, for the first time, this study demonstrates the electrochemical activation-promoted surface reconstruction of NiMoO4–CoO/CC for improved HER application. TEM, XRD, and XPS reveal that the activation induces the dissolution of Mo species, which transform into K2Mo3O10 on the surface of NiMoO4–CoO/CC. The electrochemical characterizations show that the surface reconstruction endows the catalyst with the accelerated charge transfer, more active sites, and higher intrinsic catalytic activity. With the help of such advantages and the nanosheet structure benefits, the developed catalyst, via activation, shows remarkable HER activity in terms of the extremely low overpotentials of 130 and 192 mV at the current densities of 100 and 400 mA cm−2. Moreover, such activation-induced improvement on HER activity was obtained from other Mo-based catalysts, namely, NiMoO4/CC and CoMoO4/CC. This work brings forward the electrochemical activation-induced surface reconstruction of Mo-based transition metal oxides, providing specific insight into the surface reconstruction and the catalysis activity enhancement of HER electrocatalysts. This finding may offer a paradigm to enlighten the design of HER electrocatalysts with transition-metal-activated surface reconstruction ability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from Shandong Provincial Natural Science Foundation (ZR2019MEM035).Notes and references
- D. G. Li, E. J. Park, W. L. Zhu, Q. R. Shi, Y. Zhou, H. Y. Tian, Y. H. Lin, A. Serov, B. Zulevi, E. D. Baca, C. Fujimoto, H. T. Chung and Y. S. Kim, Highly quaternized polystyrene ionomers for high performance anion exchange membrane water electrolysers, Nat. Energy, 2020, 5, 378–385 CrossRef CAS.
- C. Q. Dong, T. Y. Kou, H. Gao, Z. Q. Peng and Z. H. Zhang, Eutectic-derived mesoporous Ni-Fe-O nanowire network catalyzing oxygen evolution and overall water splitting, Adv. Energy Mater., 2018, 8, 9 Search PubMed.
- X. X. Wang, G. W. She, L. X. Mu and W. S. Shi, Amorphous Co-Mo-P-O bifunctional electrocatalyst via facile electrodeposition for overall water splitting, ACS Sustainable Chem. Eng., 2020, 8, 2835–2842 CrossRef CAS.
- S. W. Wen, T. Yang, N. Q. Zhao, L. Y. Ma and E. Z. Liu, Ni-Co-Mo-O nanosheets decorated with NiCo nanoparticles as advanced electrocatalysts for highly efficient hydrogen evolution, Appl. Catal., B, 2019, 258, 7 CrossRef.
- J. Yao, W. Huang, W. Fang, M. Kuang, N. Jia, H. Ren, D. Liu, C. Lv, C. Liu, J. Xu and Q. Yan, Promoting electrocatalytic hydrogen evolution reaction and oxygen evolution reaction by fields: effects of electric field, magnetic field, strain, and light, Small Methods, 2020, 4, 2000494 CrossRef CAS.
- M. Kuang, W. Huang, C. Hedge, W. Fang, X. Tan, C. Liu, J. Ma and Q. Yan, Interface engineering in transition metal carbides for electrocatalytic hydrogen generation and nitrogen fixation, Mater. Horiz., 2020, 7, 32–53 RSC.
- N. Heidary, M. Morency, D. Chartrand, K. H. Ly, R. Iftimie and N. Kornienko, Electrochemically triggered dynamics within a hybrid metal-organic electrocatalyst, J. Am. Chem. Soc., 2020, 142, 12382–12393 CrossRef CAS PubMed.
- W. Zheng and L. Y. S. Lee, Metal-organic frameworks for electrocatalysis: catalyst or precatalyst?, ACS Energy Lett., 2021, 6, 2838–2843 CrossRef CAS.
- Q. Chen, Y. L. Fu, J. L. Jin, W. J. Zang, X. Liu, X. Y. Zhang, W. Z. Huang, Z. K. Kou, J. Wang, L. Zhou and L. Q. Mai, In situ surface self-reconstruction in ternary transition metal dichalcogenide nanorod arrays enables efficient electrocatalytic oxygen evolution, J. Energy Chem., 2021, 55, 10–16 CrossRef.
- Y. Y. Wu, Z. J. Xie, Y. Li, Z. Lv, L. L. Xu and B. Wei, In situ self-reconstruction of Ni–Fe–Al hybrid phosphides nanosheet arrays enables efficient oxygen evolution in alkaline, Int. J. Hydrogen Energy, 2021, 46, 25070–25080 CrossRef CAS.
- Y. Duan, J. Y. Lee, S. B. Xi, Y. M. Sun, J. J. Ge, S. J. H. Ong, Y. B. Chen, S. Dou, F. X. Meng, C. Z. Diao, A. C. Fisher, X. Wang, G. G. Scherer, A. Grimaud and Z. C. J. Xu, Anodic oxidation enabled cation leaching for promoting surface reconstruction in water oxidation, Angew. Chem., Int. Ed., 2021, 60, 7418–7425 CrossRef CAS PubMed.
- J. Wang, S. J. Kim, J. P. Liu, Y. Gao, S. Choi, J. Han, H. Shin, S. Jo, J. Kim, F. Ciucci, H. Kim, Q. T. Li, W. L. Yang, X. Long, S. H. Yang, S. P. Cho, K. H. Chae, M. G. Kim, H. Kim and J. Lim, Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation, Nat. Catal., 2021, 4, 212–222 CrossRef CAS.
- A. B. Laursen, R. B. Wexler, M. J. Whitaker, E. J. Izett, K. U. D. Calvinho, S. Hwang, R. Rucker, H. Wang, J. Ji, E. Garfunkel, M. Greenblatt, A. M. Rappe and G. C. Dismukes, Climbing the volcano of electrocatalytic activity while avoiding catalyst corrosion: Ni3P, a hydrogen evolution electrocatalyst stable in both acid and alkali, ACS Catal., 2018, 8, 4408–4419 CrossRef CAS.
- L. Chen, X. Zhang, W. J. Jiang, Y. Zhang, L. B. Huang, Y. Y. Chen, Y. G. Yang, L. Li and J. S. Hu, In situ transformation of Cu2O@MnO2 to Cu@Mn(OH)2 nanosheet-on-nanowire arrays for efficient hydrogen evolution, Nano Res., 2018, 11, 1798–1809 CrossRef CAS.
- S. M. N. Jeghan, N. Kim and G. Lee, Mo-incorporated three-dimensional hierarchical ternary nickel-cobalt-molybdenum layer double hydroxide for high-efficiency water splitting, Int. J. Hydrogen Energy, 2021, 46, 22463–22477 CrossRef CAS.
- X. Q. Du, G. Y. Ma and X. S. Zhang, Mo-doped Co9S8 nanorod array as a high performance electrochemical water splitting catalyst in alkaline solution, Int. J. Hydrogen Energy, 2019, 44, 27765–27771 CrossRef CAS.
- K. Y. Tao, H. M. Dan, Y. Hai, L. Liu and Y. Gong, Controllable synthesis of Mo-O linkage enhanced CoP ultrathin nanosheet arrays for efficient overall water splitting, Appl. Surf. Sci., 2019, 493, 852–861 CrossRef CAS.
- G. Solomon, A. Landstrom, R. Mazzaro, M. Jugovac, P. Moras, E. Cattaruzza, V. Morandi, I. Concina and A. Vomiero, NiMoO4@Co3O4 core-shell nanorods: in situ catalyst reconstruction toward high efficiency oxygen evolution reaction, Adv. Energy Mater., 2021, 11, 2101324 CrossRef CAS.
- W. Du, Y. M. Shi, W. Zhou, Y. F. Yu and B. Zhang, Unveiling the in situ dissolution and polymerization of Mo in Ni4Mo alloy for promoting the hydrogen evolution reaction, Angew. Chem., Int. Ed., 2021, 60, 7051–7055 CrossRef CAS PubMed.
- M. Nishimoto, I. Muto, Y. Sugawara and N. Hara, Morphological characteristics of trenching around MnS inclusions in type 316 stainless steel: The role of molybdenum in pitting corrosion resistance, J. Electrochem. Soc., 2019, 166, C3081–C3089 CrossRef CAS.
- D. Cui, R. D. Zhao, J. Q. Dai, J. Xiang and F. F. Wu, A hybrid NiCo2O4@NiMoO4 structure for overall water splitting and excellent hybrid energy storage, Dalton Trans., 2020, 49, 9668–9679 RSC.
- J. Yan, Z. Fan, W. Sun, G. Ning, T. Wei, Q. Zhang, R. Zhang, L. Zhi and F. Wei, Advanced asymmetric supercapacitors based on Ni(OH)2/graphene and porous graphene electrodes with high energy density, Adv. Funct. Mater., 2012, 22, 2632–2641 CrossRef CAS.
- A. L. Wang, H. Xu and G. R. Li, NiCoFe layered triple hydroxides with porous structures as high-performance electrocatalysts for overall water splitting, ACS Energy Lett., 2016, 1, 445–453 CrossRef CAS.
- B. D. Chen, C. X. Peng and Z. Cui, Ultrasonic synthesis of CoO/graphene nanohybrids as high performance anode materials for lithium-ion batteries, Trans. Nonferrous Met. Soc. China, 2012, 22, 2517–2522 CrossRef CAS.
- J. Yang, H. W. Liu, W. N. Martens and R. L. Frost, Synthesis and characterization of cobalt hydroxide, cobalt oxyhydroxide, and cobalt oxide nanodiscs, J. Phys. Chem. C, 2010, 114, 111–119 CrossRef CAS.
- Y. M. Hu, Z. L. Wang, W. J. Liu, L. Xu, M. L. Guan, Y. P. Huang, Y. Zhao, J. Bao and H. M. Li, Novel cobalt-iron-vanadium layered double hydroxide nanosheet arrays for superior water oxidation performance, ACS Sustainable Chem. Eng., 2019, 7, 16828–16834 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qi00597b |
| This journal is © the Partner Organisations 2022 |