
Versatile, stable, and air-tolerant triplet–triplet annihilation upconversion block copolymer micelles†
Huanzhi
Yang
a,
Shaowei
Guo
a,
Bixin
Jin
a,
Yunjun
Luo
 ab and
Xiaoyu
Li
*abc
ab and
Xiaoyu
Li
*abc
aSchool of Materials Science and Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: xiaoyuli@bit.edu.cn
bKey Laboratory of High Energy Density Materials, MOE, Beijing Institute of Technology, Beijing 100081, China
cExperimental Center of Advanced Materials, Beijing Institute of Technology, Beijing 100081, China
First published on 27th July 2022
Abstract
Triplet–triplet annihilation photon upconversion (TTA-UC) systems based on organic molecules have emerged as a highly topical research area in the last decade. Despite their numerous advantages, however, the practical application of TTA-UC systems is often impeded by their limited dispersity in different solvents, low stability upon dilution, and poor air tolerance. Herein, we report the fabrication of a versatile, stable, and highly air-tolerant TTA-UC system based on block copolymer micelles. By polymerizing the 9,10-diphenylanthracene (DPA)-containing monomer for the core-forming block, and incorporating porphyrin platinum(II) (PtOEP) into the micellar core, spherical UC micelles were produced. The aggregated DPA moieties are in close contact with PtOEP in the micellar core, facilitating energy transfer and ensuring decent UC efficiency. Meanwhile, the choice of a poly(tert-butyl acrylate) coronal block endowed the micelles with high solubility and full functionality in various common organic solvents, and the low critical micellization concentration of the diblock copolymer ensured high stability of the micelles toward dilution. Moreover, this core–shell micellar morphology could remarkably shelter the chromophores from dissolved oxygen, which resulted in high air-tolerance for these micelles.
Introduction
Triplet–triplet annihilation photon upconversion (TTA-UC) has attracted tremendous research interest during the last decade, not only for fundamental interest, but also for its possible applications in photovoltaic,1–3 photocatalytic,4–7 and bioimaging technologies.8–10 TTA-UC systems are usually composed of multiple chromophores,11–13 in which the excited triplet energy of sensitizers can be transferred via the Dexter energy transfer process from donors to acceptors, where two low-energy state triplets can collide and annihilate to produce a high-energy singlet state and radiate delayed anti-Stokes fluorescence.14–16 Compared with the rare earth-based UC process, TTA-UC usually requires low excitation light energy and shows high upconversion quantum efficiency.17–19 Moreover, the excitation wavelength and emission wavelength can be adjusted independently by selecting different combinations of energy donors and acceptors, allowing for the conversion of light with different wavelengths into high-energy light.20 However, the wide applications of TTA-UC systems in the solution state have not been realized, and are mainly restricted by three factors: (1) the distinct solubilities of chromophore pairs in solvents, significantly impeding their wider applications; (2) limited stability of the chromophore pairs, especially under dilution, impairing the short-distance energy transfer between the donor and the acceptor; and (3) low tolerance toward air, since the triplet state is readily quenched by dissolved oxygen.12,21–25Among all combinations of acceptors and donors explored for TTA, the combination of 9,10-diphenylanthracene (DPA) as the acceptor and porphyrin platinum(II) (PtOEP) as the donor has been particularly intriguing.18,20,26 The two benzene rings of DPA, with a twisted configuration relative to the anthracene aromatic plane, effectively prevents the so-called concentration quenching and photochemical side reactions.23 In recent years, several approaches have been proposed to address the problems mentioned above by designing novel DPA-containing TTA-UC polymer-supramolecular systems.12,25,27–30 For example, mercapto-functionalized DPA luminophores, multifunctional thiols and a porphyrin platinum(II) sensitizer were combined into a homogeneous polymer network via photo-induced thiolene click chemistry to fabricate UC thin films.29 In another example, via the assembly of a nonionic surfactant, an upconversion sensitizer and an emitter, hydrogels could be obtained with a high UC efficiency of 13.5%, which were even stable under air.25 Despite these achievements toward decent air tolerance, the concentration of triplet sensitizers and emitters required by these studies is still relatively high, and the choice of solvents is very limited.25,29,31,32
Meanwhile, the combinations of luminophores and polymeric materials has offered an excellent solution for such problems.33–36 The chemical environment, density, and freedom of luminophores can be finely adjusted via the synthesis approach so that not only light intensity, but also emission colors can be arbitrarily tuned.37–39 In particular, block copolymer micelles with nicely adjusted dimensions and morphologies may provide an additional control over photoluminescence behaviours.40,41
Herein, we report a block copolymer micellization approach toward a versatile, stable, and highly air-tolerant TTA-UC system. A diblock copolymer containing a corona-forming block poly(tert-butyl acrylate) (PtBA), which is soluble in various organic solvents, and a core-forming block from DPA-containing methacrylate monomers (PDPAMA) was synthesized via atom transfer radical polymerization (ATRP). This diblock copolymer can self-assemble into a spherical micellar morphology in various solvents, including alcohol (n-butanol), ketone (acetone), ether (1,4-dioxane), and ester (n-butyl acetate), which will be desired for different application purposes. Consequently, through a co-assembly with a porphyrin platinum(II) sensitizer (PtOEP) into the micellar core, a TTA-UC from green to blue light was achieved from these micelles in various solvents. The donors and acceptors were closely packed within solvophobic micellar cores to ensure sufficient local concentration and overlapping of molecular orbitals as required by the UC process, facilitating an efficient energy transfer process. Furthermore, due to the relatively low critical micellization concentration, these micelles were colloidally stable even at very low concentrations in the solvent. More importantly, the densely packed polymer chains in the micellar core significantly improved its tolerance toward air and could maintain 70% of its original UC efficiency after exposure to air for over a month. Different studies have proposed various solutions to these three problems from different perspectives. However, to the best of our knowledge, no system has been developed to solve all three problems simultaneously. We proposed this block copolymer micelle approach, hoping to provide an easy and useful idea for the design of practical TTA-UC systems in the future.
Results and discussion
The DPA-containing methacrylate monomer (DPAMA) was synthesized according to the literature via a two-step method,42,43 which is depicted in Fig. S1,† and the molecular characteristics are included in Fig. S2–S4.† For the synthesis of the diblock copolymer, two-step ATRP was adopted, as shown in Fig. 1a and b. The monomer tert-butyl acrylate (tBA) was firstly polymerized to produce a PtBA macro-initiator, from which the DPAMA monomer was polymerized for the second block. The degree of polymerization (DP) was determined to be 81 for the PtBA block, and 31 for the PDPAMA block by combining the results from gel permeation chromatography (GPC) and proton nuclear magnetic resonance (1H NMR) spectra (Fig. S5†). The GPC test shows that the molecular weight is 26.5k Dalton and the polydispersity index (PDI) is 1.24 (Fig. 1c), agreeing with the results obtained from 1H NMR experiments. Thermal analysis of the block copolymer showed that its thermal decomposition consisted of two stages, with the first stage at 240 °C, corresponding to the decomposition of the PtBA block, and the second stage at 350–400 °C, from the PDPAMA block (Fig. S6†). The glass transition temperature (Tg) was determined to be 128 °C through the DSC test (Fig. S7†), which could be attributed to the glass transition of PDPAMA.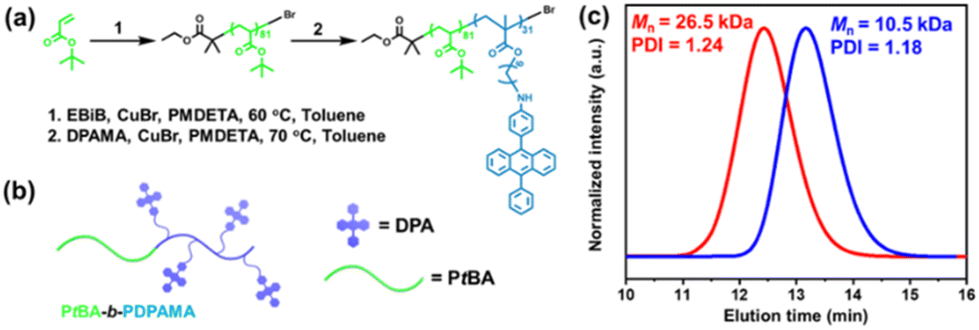 | ||
| Fig. 1 (a) Polymerization route of the PtBA-b-PDPAMA diblock copolymer. (b) Schematic illustrations of the PtBA-b-PDPAMA diblock copolymer. (c) GPC traces of the PtBA homopolymer (blue) and PtBA-b-PDPAMA diblock copolymer (red). | ||
In a typical self-assembly experiment (Fig. 2a), a mixture of PtBA-b-PDPAMA and PtOEP was dispersed in n-butanol at 105 °C for 1 h, and then leaving the solution to cool to room temperature (r.t., 21 °C). These micelles were spherical and uniform in size with a diameter of around 55 nm, as observed under a transmission electron microscope (EM, Fig. 2b). The core–shell structure of these micelles could be demonstrated by hydrolyzing the PtBA coronal chains on the micelles into poly(acrylic acid) and staining the micelles with uranyl acetate. As shown in Fig. S8,† these micelles appeared to be hollow spheres, clearly demonstrating their core–shell structure. Dynamic light scattering (DLS) data also suggested that these spherical micelles were very uniform, with hydrodynamic diameter (Dh) values around 61 nm (Fig. 2c) and decently low polydispersity values. The addition of a tiny amount (as discussed later) of PtOEP would not influence the micellar morphology. Since the PtBA block is highly soluble in most common organic solvents, self-assembly experiments were conducted in several typical solvents, including n-butanol, acetone, 1,4-dioxane, and n-butyl acetate. As seen from their EM images (Fig. S9†) and DLS data (Fig. 2c), the morphologies and sizes of these micelles were highly similar to each other (Table S1†).
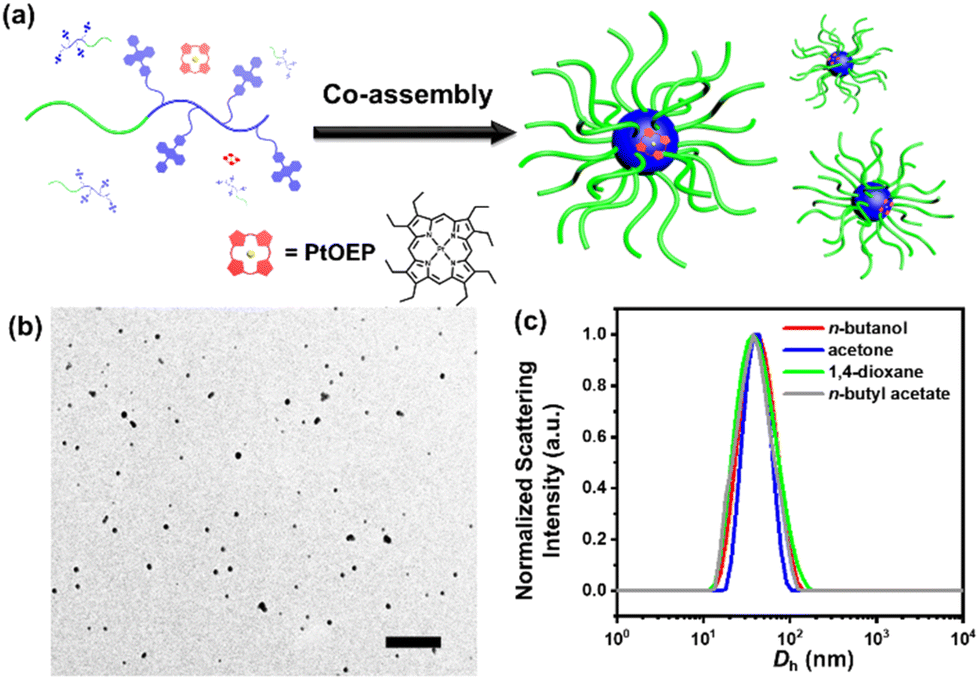 | ||
| Fig. 2 (a) Schematic illustration of the co-assembly process of the UC micelles. (b) EM image of the UC micelles co-assembled in n-butanol. The scale bar is 500 nm. (c) DLS profiles of the UC micelles in various solvents. | ||
The TTA-UC emission of the PtOEP/PDPAMA complex inside these UC micelles was proposed (Fig. 3a) and confirmed by simply irradiating the solutions with a 532 nm green laser, from which a blue beam was observed (Fig. 3b). In sharp contrast, for the micelles self-assembled from PtBA-b-PDPAMA without PtOEP, only a green beam was observed, simply from the Tyndall scattering from the micelles. In previously reported TTA-UC supramolecular systems,18,25,44 due to the limited solubility of DPA moieties, the UC process could only be realized in very limited solvents, with the assistance of surfactants or special molecular designs. However, in the current study, owing to the good solubility of the PtBA coronal chains, stable micelles could be produced in various solvents (Fig. 3b and c). Therefore, TTA-UC luminescence from the same micellar system can be realized in all these common solvents, greatly facilitating the application of these micelles.
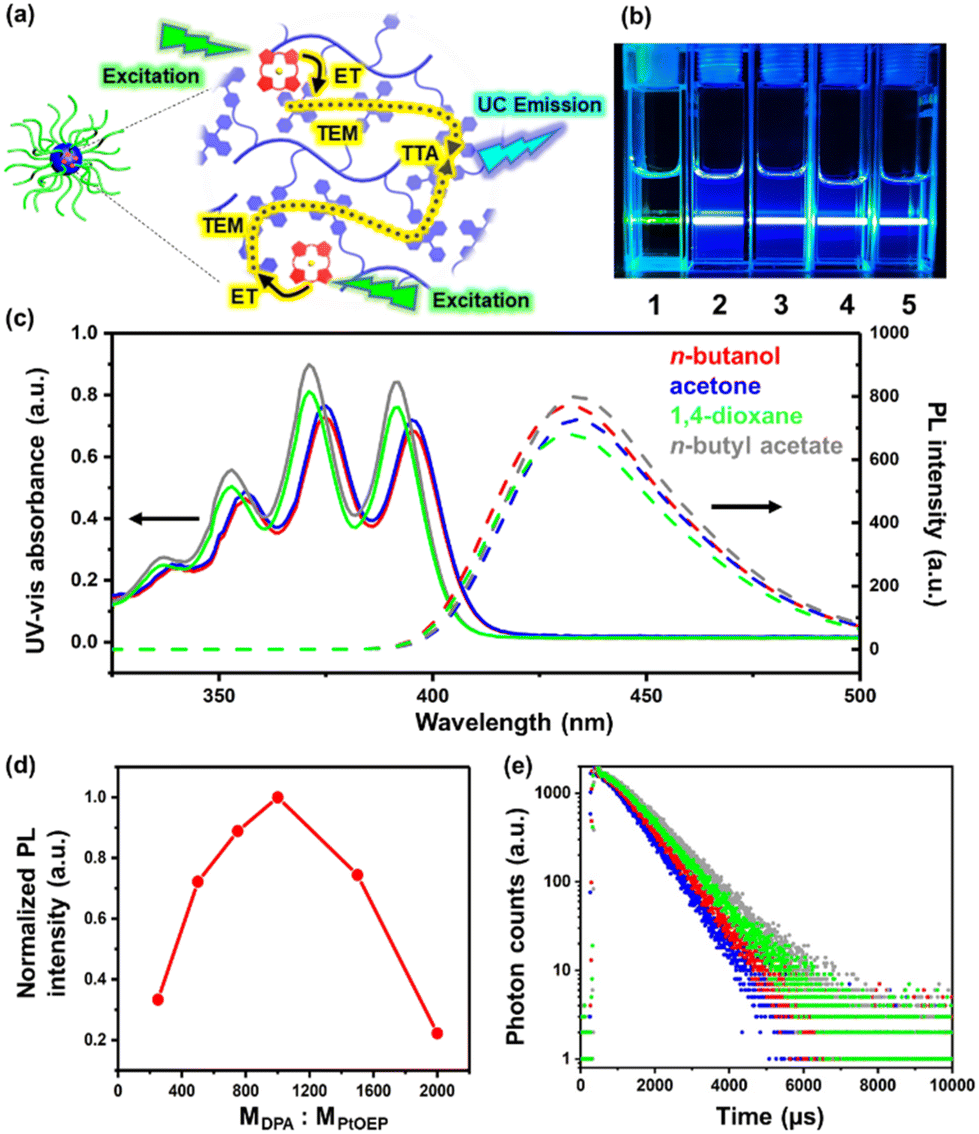 | ||
Fig. 3 (a) Schematic illustration of the TTA-UC process inside the UC micellar core. (b) Photographs of: (1) the PtBA-b-PDPAMA micelles in n-butanol, and (2)–(5) UC micelles in n-butanol (2), acetone (3), 1,4-dioxane (4), and n-butyl acetate (5) under exposure to a 532 nm laser beam at a fixed polymer concentration of 0.5 mg mL−1 and a DPA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PtOEP molar ratio of 1000. (c) Comparison of the absorption (solid line) and emission (dashed line) spectra of the UC micelles in various solvents. (d) Variation of the UC PL intensity versus the molar ratio of DPA:PtOEP in the n-butanol solution of UC micelles. (e) Phosphorescence decays of UC micelles in n-butanol (red), acetone (blue), 1,4-dioxane (green) and n-butyl acetate (grey) when irradiated at 532 nm. :PtOEP molar ratio of 1000. (c) Comparison of the absorption (solid line) and emission (dashed line) spectra of the UC micelles in various solvents. (d) Variation of the UC PL intensity versus the molar ratio of DPA:PtOEP in the n-butanol solution of UC micelles. (e) Phosphorescence decays of UC micelles in n-butanol (red), acetone (blue), 1,4-dioxane (green) and n-butyl acetate (grey) when irradiated at 532 nm. | ||
Considering the high Tg value of the PDPAMA block, the mobility of donors and acceptors inside the micellar core was significantly limited, and thus the TTA process occured more likely through the triplet energy migration (TEM) mechanism.45 The quantum efficiency of TTA-UC is an important characteristic and depends on many factors according to the mechanism of TTA-UC.23 For example, the molar ratio of the triplet donor to the acceptor is crucial for the luminous intensity of TTA-UC.46 While the sensitizers (donor) require a large Dexter distance for an effective diffusion, and thus sufficient spacing between sensitizers to ensure a sufficient triplet-to-triplet state energy transfer efficiency, the concentration of the sensitizers has to be high enough to allow for the full contact between sensitizers and acceptors.47–50 By varying the molar ratio between donors and acceptors, a maximum value for luminescence intensity was observed when MDPA:MPtOEP = 1000:1 with a fixed polymer concentration (Fig. 3d), agreeing with the values reported in the literature.43
Subsequently, by fixing the MDPA:MPtOEP ratio to 1000:1, the UV-vis absorbance and photoluminescence (PL) spectra (excitation wavelength λex = 532 nm) of UC micelles in the four solvents (n-butanol, acetone, 1,4-dioxane and n-butyl acetate) were obtained and are shown in Fig. 3c. All four solutions showed very similar UV-vis adsorption spectra, with obvious absorption peaks at 360, 370 and 385 nm, basically consistent with those reported in the literature.27,43 Moreover, upon excitation at 532 nm, all four solutions showed a clear PL emission at around 430 nm with a comparable emission intensity, strongly suggesting that the environment around the DPA and PtOEP moieties were very similar, despite the different solvents used. Moreover, the UC emission in n-butanol could still be observed even when the solution was frozen at 77 K, due to the confinement from the core–shell micellar structure and the resultant close contact between DPA and PtOEP units inside the micellar core (Fig. S10†). Meanwhile, the PL from the THF solution of the diblock copolymer and PtOEP was diminished to almost zero at 77 K.
The triplet lifetimes of the acceptors in the four solutions were measured via time-resolved photoluminescence experiments to be 0.935 ms, 0.855 ms, 1.032 ms and 1.125 ms for n-butanol, acetone, 1,4-dioxane and n-butyl acetate solutions, respectively (Fig. 3e). These values are close to each other, and are comparable to those from the literature,51 confirming the occurrence of the TTA-UC process. Meanwhile, for the n-butanol solution of pure PtBA-b-PDPAMA micelles, and UC micelles, when solely the DPA moieties were excited with 360 nm irradiation, the lifetime was determined to be 8.72 ns and 8.73 ns (Fig. S11†), respectively. This finding suggested that the back energy transfer from the acceptor to the donor was negligible.15
The UC quantum yield ΦUC of the system was determined using the THF solution of rhodamine B (RhB) as the standard sample, so as to minimize the different wavelength errors of the instrument caused by its optical response (Table 1). The quantum yield is defined as the ratio of the number of emitted photons to the number of absorbed photons, and so the theoretical maximum value for ΦUC is defined as 50%.52 The ΦUC values of our system were determined to be 1.43%, 1.41%, 1.46% and 1.25% for n-butanol, acetone, 1,4-dioxane and n-butyl acetate solutions, respectively. Again, these quantum yield values were close enough, due to the similar environment in the micellar core, and are similar to those obtained from other DPA/PtOEP supramolecular TTA-UC systems.27
| Sample | Maximum absorbance values | Integral value of fluorescence intensity | Relative quantum yields |
|---|---|---|---|
| THF solution (RhB) | 0.211 | 50992 (background subtraction) |
89% |
| n-Butanol solution (micelle) | 0.151 | 589 | 1.43% |
| Acetone solution (micelle) | 0.155 | 594 | 1.41% |
| 1,4-Dioxane solution (micelle) | 0.153 | 606 | 1.46% |
| n-Butyl acetate solution (micelle) | 0.156 | 530 | 1.25% |
By virtue of the macromolecular nature of the diblock copolymer, the UC micellar aggregates showed remarkable stability even in diluted solutions. In previously reported DPA-PtOEP systems, the concentration of the acceptor was set to around 10 mM at the lowest.53 In the current system, however, the acceptors and donors were confined inside the micellar core, ensuring a sufficient local concentration and a fixed ratio between the donor and the acceptor, as long as the polymer concentration was above the critical micellization concentration (CMC). Therefore, the CMC of the UC micelles was determined by gradually diluting the solution and monitoring the transmittance of the solution at 700 nm.54–56 As shown by the black squares and lines in Fig. 4, the CMC of UC micelles was determined to be 0.041 mg mL−1. Meanwhile, the PL intensity at 430 nm with an excitation wavelength of 532 nm was also monitored during the dilution process. A sharp turn-off of the PL intensity was also observed when the concentration was lowered to 0.045 mg mL−1, which is very close to the CMC value. This result suggested that the micellization indeed provided a nicely confined space to facilitate the TTA-UC process. Moreover, this low value of CMC corresponds to a DPA concentration of 0.0432 mM, which is two magnitudes lower than those values reported from similar TTA-UC systems.27 Similarly low CMC values were obtained for the UC micelles in the other three solvents: 0.037, 0.041 and 0.036 mg mL−1 for acetone, 1,4-dioxane, and n-butyl acetate, respectively. These findings definitely suggested a high colloidal stability of these TTA-UC micelles in a wide range of solvents.
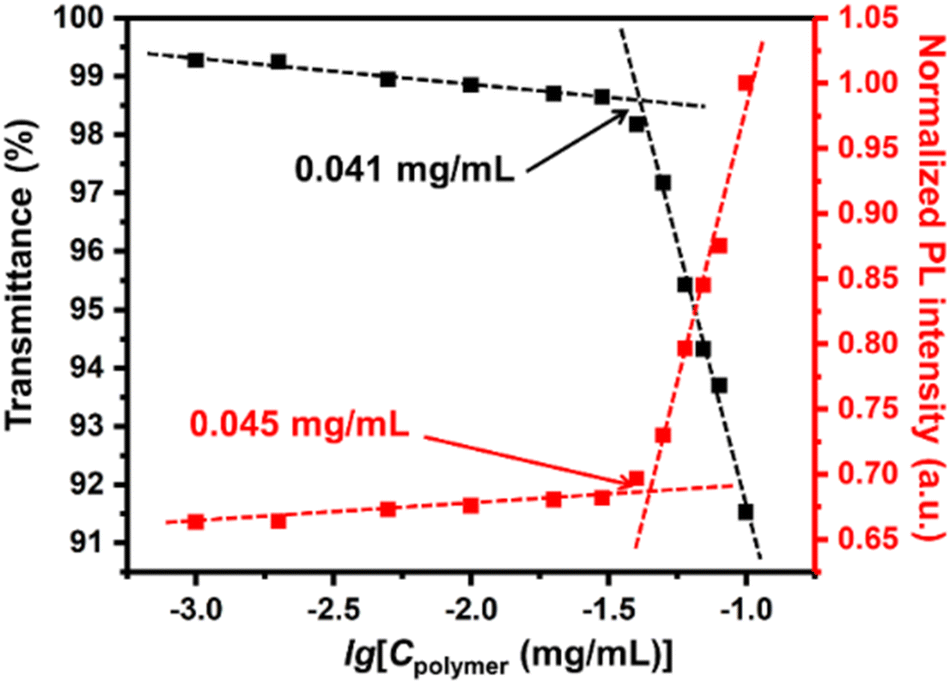 | ||
| Fig. 4 Plots of the transmittance at 700 nm and PL intensity of the UC micellar solution at 430 nm under excitation of a 532 nm laser versus the concentration of UC micelles. | ||
Another problem with TTA-UC emission systems is that they are usually extremely sensitive to air, since the triplet excited state can be easily quenched by oxygen molecules.21–24,57 As for our UC micelles, however, the aggregated core-forming block chains inside the micellar core could provide a quasi-solid state and thus efficiently shield the DPA/PtOEP complex from dissolved oxygen molecules in the solution. Although the dissolved oxygen molecules could quench some triplets near the micellar surface and a drop in the UC intensity to about 70% of their initial values was observed after the micellar solution was fully exposed to air for 12 h (Fig. 5a), the UC intensity remained unchanged with further exposure to air. It was found that a decent UC emission intensity (above 70% of the initial value) was retained even after exposing the solution to air for over a month, regardless of the solvent used (Fig. 5b). Meanwhile, when the PtBA-b-PDPAMA diblock copolymer and PtOEP were dissolved in THF instead of forming micelles, the UC emission would quickly be quenched by the dissolved oxygen molecules within 12 h (Fig. 5a), and only a green beam was observed from Tyndall scattering when irradiated with a 532 nm laser (inset in Fig. 5b). The air-tolerance performance of the current system is among the best that have been reported based on the same donor/acceptor pair.25,31,43,58 In addition, most of the better ones reported are based on bulk materials, which make it harder for oxygen molecules to diffuse into and quench the excited state. However, in our case, the micelles assembled were of a size of around 50–60 nm, and the micelles were dispersed in the solvent. Therefore, in an open vial, the oxygen can easily diffuse into the solution and quench the TTA-UC process, as shown in the THF solution case in Fig. 5. But due to the tightly packed polymer chains in the micellar core, the oxygen could not easily diffuse into the micellar core and a decent TTA-UC emission remained even after a month.
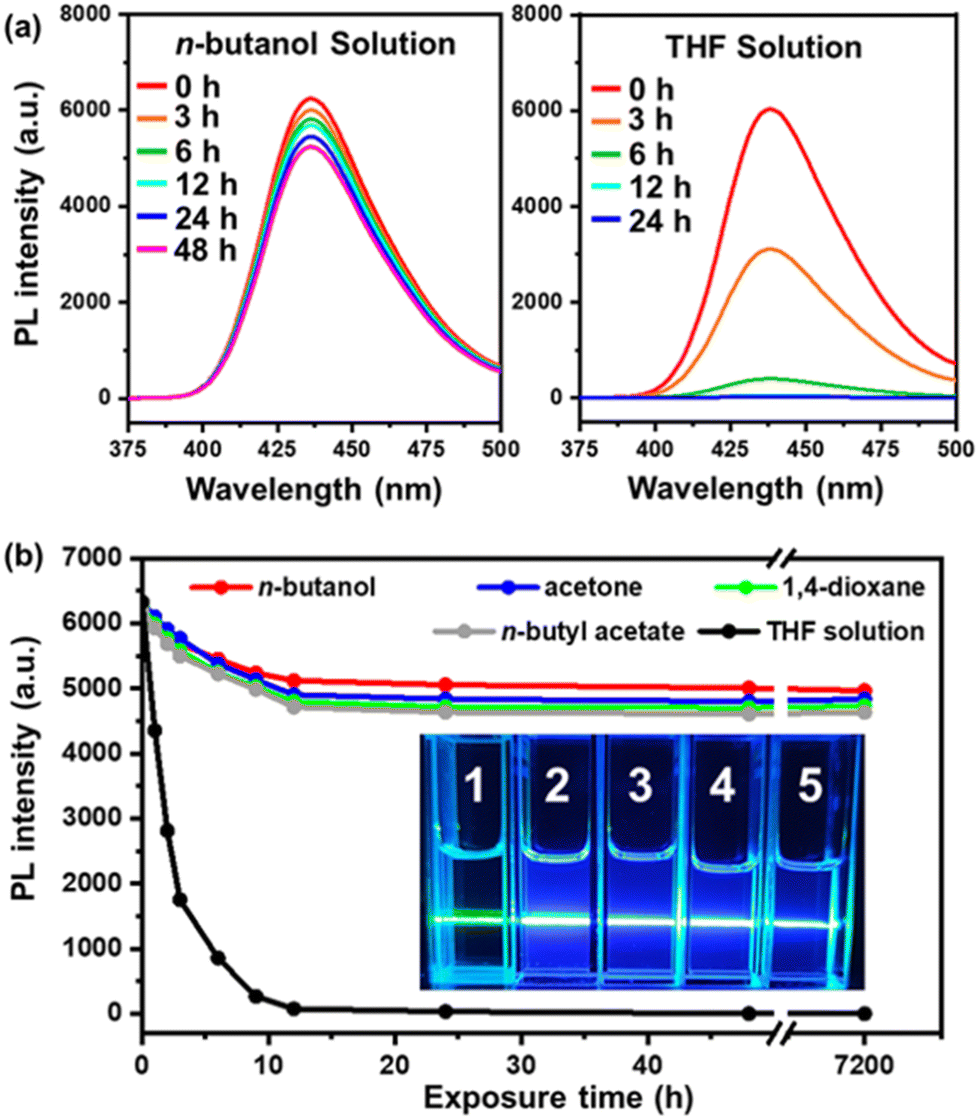 | ||
| Fig. 5 (a) PL spectra (λex = 532 nm) of the UC micelles in n-butanol, and the THF solution of the diblock copolymer and PtOEP after exposure to air for different periods of time. (b) Variation of the UC PL intensity versus the exposure period to air. Inset: A photograph of various solutions (1: THF solution of the mixture of the PtOEP and PtBA-b-PDPAMA; 2–5: UC micelles in n-butanol, acetone, 1,4-dioxane and n-butyl acetate) under exposure to a 532 nm laser beam and exposure to air for 12 h. | ||
Conclusions
In conclusion, in this paper we reported an interesting example of a block copolymer micellar system for triplet–triplet annihilation upconversion. By synthesizing and polymerizing the 9,10-diphenylanthracene-containing monomer as the core-forming block, poly(tert-butyl acrylate) as the corona-forming block and acceptor, and incorporating porphyrin platinum(II) into the micellar core as the donor, a stable upconversion micellar system was constructed. The diblock copolymer formed similar spherical micelles in various solvents, and the high solubility of PtBA coronal chains ensured the full functionality of this upconversion system in all these solvents, which will greatly enhance the feasibility of such TTA-UC systems in different practical scenarios. Meanwhile, this core–shell micellar structure was demonstrated to be crucial for achieving an efficient UC and could keep the donor/acceptor pairs tightly packed even under high dilution to ensure a decent UC efficiency. Moreover, the solvophobicity of the micellar core composed of acceptor and donor moieties could remarkably shelter them from molecular oxygen, allowing the micelles to maintain a high level of UC emission even after exposure to air for over a month. We believe these results suggest a novel and intriguing approach for the design and fabrication of a triplet–triplet annihilation upconversion system toward practical application purposes, and surely endow these TTA-UC micelles with potential advantages when applied in optogenetic manipulation, medical treatment, and in vivo imaging.18Experimental
Materials
Sodium tert-butoxide (99%), 9-(4-bromophenyl)-10-phenylanthracene (99%), 6-amino-1-hexanol (99%), tris(dibenzylideneacetone)dipalladium(0) (Pd2(DBA)3, 99%), (R)-(+)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl ((R)-BINAP, 99%), triethylamine (TEA, 99%), methacryoyl chloride (99%), the initiator ethyl 2-bromoisobutyrate (99%, EBiB), and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (99%, PMDETA) were purchased from Aldrich and were used as received unless otherwise stated. tert-Butyl acrylate (tBA), dichloromethane (DCM), tetrahydrofuran (THF) and toluene were distilled over CaH2 before use. All other solvents were used as received without further purification. For the atom transfer radical polymerization (ATRP), cuprous bromide (99%, CuBr) was purified with acetic acid before use. All of the self-assembly experiments were performed in HPLC grade solvents that were acquired from Fisher.Nuclear magnetic resonance (NMR)
NMR spectra were recorded using an Avance 500 (Bruker A.G.) instrument (operating at 400 MHz) at ambient temperatures.Gel permeation chromatography (GPC)
Molecular weights and polydispersity indexes of polymers were obtained with a Viscotek GPC max chromatograph equipped with styrene/divinylbenzene columns with pore sizes of 500 Å and 100000 Å, and a VE 3580 refractometer. THF (Fisher) was used as the eluent, with a flow rate of 1.0 mL min−1. Samples were dissolved in the eluent (2 mg mL−1) and filtered (Acrodisc, PTFE membrane, 0.45 mm) before analysis. The calibration of the refractive index detector was carried out using polystyrene standards (Viscotek).
Dynamic light scattering (DLS)
DLS experiments were performed using a nano series Malvern zetasizer instrument equipped with a 633 nm laser. Samples were analyzed in 1 cm glass cuvettes at 25 °C with a scattering angle of 173°. The refractive index of the block copolymers involved was assumed to be 1.60. The results of DLS studies were reported as the apparent hydrodynamic diameter (Dh), acknowledging that the particles have been modelled as hard spheres in the experiments.Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA)
DSC and TGA were performed on Q100 and Q500 equipment from TA Instruments, respectively, under nitrogen at a heating/cooling rate of 10 °C min−1.Transmission electron microscopy (EM)
The samples for EM analysis were prepared by drop-casting one drop (ca. 10 μL) of the micellar solution onto carbon coated copper grids, which were placed on a piece of filter paper to remove the excess solvent. Bright field EM images were obtained on a JEOL1200EX II microscope operating at 120 kV, which was equipped with an SIS MegaViewIII digital camera. No staining was applied to the EM samples. Images were analyzed using the ImageJ software package developed at the US National Institute of Health. For the statistical dimension analysis, over 300 micelles were carefully traced manually to determine their diameter.UV-visible and fluorescence spectrometry
UV-vis data were acquired with a Lambda 35 spectrometer using standard quartz cells from a wavelength of 200 nm to 700 nm. Fluorescence data were obtained with a PerkinElmer LS 45 fluorescence spectrometer at a wavelength of 365 nm. Solution state quantum yields were measured according to literature methods.30Transient steady-state fluorescence spectrometry
Transient steady-state fluorescence spectrometer experiments were performed on an Edinburgh FLS980 instrument using standard quartz cells at a wavelength of 532 nm. Solution state quantum yields were measured according to literature methods.30Synthesis of DPA(CH2)6OH
In a three-neck flask, 9-(4-bromophenyl)-10-phenylanthracene (2.00 g, 4.89 mmol), 6-amino-1-hexanol (3.44 g, 29.35 mmol), sodium tert-butoxide (3.29 g, 34.24 mmol), Pd2(DBA)3 (0.45 g, 0.49 mmol) and (R)-BINAP (0.91 g, 1.46 mmol) were stirred in toluene (20 mL) under a nitrogen atmosphere and heated at 80 °C for 6 h. The solvent was removed under vacuum and the crude product was purified by column chromatography (silica gel, DCM:ethyl acetate = 20:1). A light yellowish solid was obtained with a yield of 65% (1.42 g). 1H NMR δH (ppm) (400 MHz, CD2Cl2): 7.83–6.82 (m, 17H), 3.92 (s, 1H), 3.66–3.61 (t, 2H), 3.26–3.23 (t, 2H), 1.78–1.70 (m, 2H), 1.64–1.57 (m, 2H), 1.54–1.24 (m, 4H).
Synthesis of DPAMA
In a three-neck flask under a nitrogen atmosphere, DPA(CH2)6OH (1 g, 2.24 mmol) and TEA (0.69 mL, 4.93 mmol) were dissolved in dry DCM (50 mL) and cooled to 0 °C. Methacryoyl chloride (0.43 mL, 4.48 mmol) was added dropwise into the reaction mixture. The solution was allowed to warm up to r.t. and stirred for 10 h. After evaporation of the solvent, the crude product was purified by column chromatography (silica gel, light petroleum:DCM = 2:1). A light yellowish solid was obtained with a yield of 45% (0.52 g). 1H NMR δH (ppm) (400 MHz, CD2Cl2): 7.84–6.83 (m, 17H), 6.10(s, 1H), 5.57 (s, 1H), 4.19–4.16 (t, 2H), 3.98 (S, 1H), 3.27–3.24 (t, 2H), 1.95 (s, 3H), 1.77–1.73(m, 2H), 1.60–1.53 (m, 4H), 1.27 (s, 2H); 13C NMR δH (ppm) (101 MHz, CD2Cl2): 167.73, 148.47, 139.57, 138.26, 137.15, 136.85, 132.44, 131.70, 130.72, 130.33, 128.78, 127.80, 127.63, 127.16, 125.31, 125.18, 125.06, 65.02, 44.33, 29.92, 29.01, 27.25, 26.28, 18.50.
Polymerization of PtBA-Br
CuBr (11.6 mg, 0.08 mmol), PMDETA (14 mg, 0.08 mmol), tBA (1640 mg, 12.8 mmol), EBiB (15.6 mg, 0.08 mmol) and 1 mL of toluene were introduced into a Schlenk tube and degassed with three freeze–pump–thaw cycles. Subsequently, the polymerization solution was heated at 60 °C for 5 h under a nitrogen atmosphere with vigorous stirring. The cuprous salt was removed by filtering the reaction solution through Al2O3 columns, and the polymer was further purified via repeated precipitations from THF solution into a mixture of water and methanol (7/3, volume ratio), and dried under reduced pressure. A white solid was obtained as the final product (740 mg, yield 45%).Polymerization of PtBA-b-PDPAMA
Macroinitiator PtBA (230 mg, 0.02 mmol), CuBr (3 mg, 0.02 mmol), PMDETA (3.4 mg, 0.02 mmol), DPAMA (770 mg, 1.5 mmol) and 2 mL of toluene were introduced into a Schlenk tube and degassed with three freeze–pump–thaw cycles. Subsequently, the polymerization solution was heated at 70 °C for 12 h under a nitrogen atmosphere with vigorous stirring. The reaction mixture was purified by silica-gel column chromatography with DCM/ethyl acetate (4:1, volume ratio) as the eluent first to remove the residual trace monomer and then the polymer was eluted with ethyl acetate, with the concentrated crude product being further purified via repeated precipitations from THF solution into a mixture of water and methanol (7/3, volume ratio), and dried under reduced pressure. A light yellowish solid was obtained as the final product (356 mg, yield 36%).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. Y. L. is grateful for the financial support from the National Natural Science Foundation of China (Grant numbers 51973019 and 22175024).References
- D. Di, L. Yang, J. M. Richter, L. Meraldi, R. M. Altamimi, A. Y. Alyamani, D. Credgington, K. P. Musselman, J. L. MacManus-Driscoll and R. H. Friend, Adv. Mater., 2017, 29, 1605987 CrossRef PubMed.
- Y. Deng, L. Jiang, L. Huang and T. Zhu, ACS Energy Lett., 2022, 7, 847–861 CrossRef CAS.
- J. Pedrini and A. Monguzzi, J. Photonics Energy, 2018, 8, 022005 Search PubMed.
- Z. Jiang, M. Xu, F. Li and Y. Yu, J. Am. Chem. Soc., 2013, 135, 16446–16453 CrossRef CAS PubMed.
- B. S. Richards, D. Hudry, D. Busko, A. Turshatov and I. A. Howard, Chem. Rev., 2021, 121, 9165–9195 CrossRef CAS PubMed.
- F. Kishimoto, T. Wakihara and T. Okubo, ACS Appl. Mater. Interfaces, 2020, 12, 7021–7029 CrossRef CAS PubMed.
- S. N. Sanders, T. H. Schloemer, M. K. Gangishetty, D. Anderson, M. Seitz, A. O. Gallegos, R. C. Stokes and D. N. Congreve, Nature, 2022, 604, 474–478 CrossRef CAS PubMed.
- Q. Liu, B. Yin, T. Yang, Y. Yang, Z. Shen, P. Yao and F. Li, J. Am. Chem. Soc., 2013, 135, 5029–5037 CrossRef CAS PubMed.
- F. Xiao, X. Fang, H. Li, H. Xue, Z. Wei, W. Zhang, Y. Zhu, L. Lin, Y. Zhao, C. Wu and L. Tian, Angew. Chem., 2022, 61, e202115812 CAS.
- S. Chen, A. Z. Weitemier, X. Zeng, L. He, X. Wang, Y. Tao, A. J. Y. Huang, Y. Hashimotodani, M. Kano, H. Iwasaki, L. K. Parajuli, S. Okabe, D. B. L. Teh, A. H. All, I. Tsutsui-Kimura, K. F. Tanaka, X. Liu and T. J. McHugh, Science, 2018, 359, 679–683 CrossRef CAS PubMed.
- S. Amemori, N. Yanai and N. Kimizuka, Phys. Chem. Chem. Phys., 2015, 17, 22557–22560 RSC.
- C. Fan, W. Wu, J. J. Chruma, J. Zhao and C. Yang, J. Am. Chem. Soc., 2016, 138, 15405–15412 CrossRef CAS PubMed.
- A. B. Pun, L. M. Campos and D. N. Congreve, J. Am. Chem. Soc., 2019, 141, 3777–3781 CrossRef CAS PubMed.
- C. A. Parker and C. G. Hatchard, Trans. Faraday Soc., 1963, 59, 284–295 RSC.
- Y. Sasaki, S. Amemori, H. Kouno, N. Yanai and N. Kimizuka, J. Mater. Chem. C, 2017, 5, 5063–5067 RSC.
- D. Yildiz, C. Baumann, A. Mikosch, A. J. C. Kuehne, A. Herrmann and R. Goestl, Angew. Chem., Int. Ed., 2019, 58, 12919–12923 CrossRef CAS PubMed.
- J. Silver, M. I. Martinez-Rubio, T. G. Ireland, G. R. Fern and R. Withnall, J. Phys. Chem. B, 2001, 105, 948–953 CrossRef CAS.
- N. Yanai and N. Kimizuka, Chem. Commun., 2016, 52, 5354–5370 RSC.
- Y. Wei, K. Pan, X. Cao, Y. Li, X. Zhou and C. Yang, CCS Chem., 2022, 4, 1–12 CrossRef.
- N. Yanai and N. Kimizuka, Acc. Chem. Res., 2017, 50, 2487–2495 CrossRef CAS PubMed.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- W. Wu, H. Guo, W. Wu, S. Ji and J. Zhao, J. Org. Chem., 2011, 76, 7056–7064 CrossRef CAS PubMed.
- A. Monguzzi, R. Tubino, S. Hoseinkhani, M. Campione and F. Meinardi, Phys. Chem. Chem. Phys., 2012, 14, 4322–4332 RSC.
- V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345–10352 RSC.
- P. Bharmoria, S. Hisamitsu, H. Nagatomi, T. Ogawa, M.-a. Morikawa, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2018, 140, 10848–10855 CrossRef CAS PubMed.
- N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2020, 59, 10252–10264 CrossRef CAS PubMed.
- T. Ogawa, N. Yanai, A. Monguzzi and N. Kimizuka, Sci. Rep., 2015, 5, 10882 CrossRef CAS PubMed.
- C. Wohnhaas, A. Turshatov, V. Mailaender, S. Lorenz, S. Baluschev, T. Miteva and K. Landfester, Macromol. Biosci., 2011, 11, 772–778 CrossRef CAS PubMed.
- A. K. Williams, B. J. Davis, E. R. Crater, J. R. Lott, Y. C. Simon and J. D. Azoulay, J. Mater. Chem. C, 2018, 6, 3876–3881 RSC.
- S. Hisamitsu, N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2015, 54, 11550–11554 CrossRef CAS PubMed.
- P. Duan, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2013, 135, 19056–19059 CrossRef CAS PubMed.
- H. Kurz, C. Hils, J. Timm, G. Horner, A. Greiner, R. Marschall, H. Schmalz and B. Weber, Angew. Chem., 2022, 61, e202117570 CAS.
- N. R. Paisley, S. V. Halldorson, M. V. Tran, R. Gupta, S. Kamal, W. R. Algar and Z. M. Hudson, Angew. Chem., Int. Ed., 2021, 60, 18630–18638 CrossRef CAS PubMed.
- C. M. Tonge and Z. M. Hudson, J. Am. Chem. Soc., 2019, 141, 13970–13976 CrossRef CAS PubMed.
- A. Lorbach, M. Bolte, H. Li, H.-W. Lerner, M. C. Holthausen, F. Jaekle and M. Wagner, Angew. Chem., Int. Ed., 2009, 48, 4584–4588 CrossRef CAS PubMed.
- H. Li and F. Jaekle, Angew. Chem., Int. Ed., 2009, 48, 2313–2316 CrossRef CAS PubMed.
- J. Wang, B. Jin, N. Wang, T. Peng, X. Li, Y. Luo and S. Wang, Macromolecules, 2017, 50, 4629–4638 CrossRef CAS.
- J. Wang, N. Wang, G. Wu, S. Wang and X. Li, Angew. Chem., Int. Ed., 2019, 58, 3082–3086 CrossRef CAS PubMed.
- J. Wang, R. Yan, Y. Hu, G. Du, G. Liao, H. Yang, Y. Luo, X. Zheng, Y. Chen, S. Wang and X. Li, Angew. Chem., Int. Ed., 2022, 134, e202112290 Search PubMed.
- N. Zhang, H. Chen, Y. Fan, L. Zhou, S. Trepout, J. Guo and M.-H. Li, ACS Nano, 2018, 12, 4025–4035 CrossRef CAS PubMed.
- S. Wang, B. Jin, G. Chen, Y. Luo and X. Li, Polym. Chem., 2020, 11, 4706–4713 RSC.
- J. P. Wolfe, S. Wagaw and S. L. Buchwald, J. Am. Chem. Soc., 1996, 118, 7215–7216 CrossRef CAS.
- S. H. Lee, M. A. Ayer, R. Vadrucci, C. Weder and Y. C. Simon, Polym. Chem., 2014, 5, 6898–6904 RSC.
- K. Fujimoto, K. Kawai, S. Masuda, T. Mori, T. Aizawa, T. Inuzuka, T. Karatsu, M. Sakamoto, S. Yagai, T. Sengoku, M. Takahashi and H. Yoda, Langmuir, 2019, 35, 9740–9746 CrossRef CAS PubMed.
- M. J. Bennison, A. R. Collins, B. Zhang and R. C. Evans, Macromolecules, 2021, 54, 5287–5303 CrossRef CAS PubMed.
- M. Hosoyamada, N. Yanai, T. Ogawa and N. Kimizuka, Chem. – Eur. J., 2016, 22, 2060–2067 CrossRef CAS PubMed.
- A. Monguzzi, R. Tubino and F. Meinardi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 155122 CrossRef.
- C. Ye, V. Gray, J. Martensson and K. Borjesson, J. Am. Chem. Soc., 2019, 141, 9578–9584 CrossRef CAS PubMed.
- X. Yu, X. Cao, X. Chen, N. Ayres and P. Zhang, Chem. Commun., 2015, 51, 588–591 RSC.
- L. M. Yablon, S. N. Sanders, H. Li, K. R. Parenti, E. Kumarasamy, K. J. Fallon, M. J. A. Hore, A. Cacciuto, M. Y. Sfeir and L. M. Campos, J. Am. Chem. Soc., 2019, 141, 9564–9569 CrossRef CAS PubMed.
- N. Yanai, K. Suzuki, T. Ogawa, Y. Sasaki, N. Harada and N. Kimizuka, J. Phys. Chem. A, 2019, 123, 10197–10203 CrossRef CAS PubMed.
- S. Hisamitsu, N. Yanai, H. Kouno, E. Magome, M. Matsuki, T. Yamada, A. Monguzzi and N. Kimizuka, Phys. Chem. Chem. Phys., 2018, 20, 3233–3240 RSC.
- S. H. C. Askes, V. C. Leeuwenburgh, W. Pomp, H. Arjmandi-Tash, S. Tanase, T. Schmidt and S. Bonnet, ACS Biomater. Sci. Eng., 2017, 3, 322–334 CrossRef CAS PubMed.
- Y. Lu, Z. Yue, J. Xie, W. Wang, H. Zhu, E. Zhang and Z. Cao, Nat. Biomed. Eng., 2018, 2, 318–325 CrossRef CAS PubMed.
- R. Mandavi, S. K. Sar and N. Rathore, Orient. J. Chem., 2008, 24, 559–564 CAS.
- Y. Bakkour, V. Darcos, S. Li and J. Coudane, Polym. Chem., 2012, 3, 2006–2010 RSC.
- R. Vadrucci, A. Monguzzi, F. Saenz, B. D. Wilts, Y. C. Simon and C. Weder, Adv. Mater., 2017, 29, 1702992 CrossRef PubMed.
- H. Kouno, T. Ogawa, S. Amemori, P. Mahato, N. Yanai and N. Kimizuka, Chem. Sci., 2016, 7, 5224–5229 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data in Fig. S1–S11 and Table S1. See DOI: https://doi.org/10.1039/d2py00596d |
| This journal is © The Royal Society of Chemistry 2022 |