
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved emission of Yb(III) ions in triazacyclononane-based macrocyclic ligands compared to cyclen-based ones†
Salauat R.
Kiraev
 a,
Emilie
Mathieu‡
a,
Daniel
Kovacs
a,
Jordann A. L.
Wells
a,
Monika
Tomar
a,
Julien
Andres
*b and
K. Eszter
Borbas
*a
a,
Emilie
Mathieu‡
a,
Daniel
Kovacs
a,
Jordann A. L.
Wells
a,
Monika
Tomar
a,
Julien
Andres
*b and
K. Eszter
Borbas
*a
aDepartment of Chemistry, Ångström Laboratory, Uppsala University, Box 523, 75120, Uppsala, Sweden. E-mail: eszter.borbas@kemi.uu.se
bChemistry and Chemical Engineering Section, Ecole Polytechnique Fédérale de Lausanne (EPFL), BCH 3311, CH-1015, Lausanne, Switzerland. E-mail: julien.andres@epfl.ch
First published on 20th October 2022
Abstract
Yb(III) complexes based on ligands with a 1,4,7-triazacyclononane (tacn) macrocyclic core were synthesised. The complexes carry a 4-methoxymethyl-substituted carbostyril chromophore that serves as a light-harvesting antenna. The ligands supply 5 nitrogen and 3 oxygen donors via 1 methylenecarboxamide and 2 picolinate donors, creating +1 charged complexes with an octadentate binding environment. The electronic properties of the picolinates are modulated by varying the substitution at the 4 position with OMe, H, Cl, or CF3. Cyclic voltammetry indicated that the tacn-based Yb(III) complexes were easier to reduce than the analogous cyclen complexes. The first reductive event is likely picolinate-centred, followed by the formation of further reduced species. Antenna excitation yielded Yb(III) luminescence in the near-infrared (NIR) region in all cases. The antenna photophysical properties were consistent with intraligand photoinduced electron transfer from the excited carbostyril to the picolinate groups. The relative quantum yields of Yb(III) luminescence were determined. The lowest value was obtained for the complex with the most efficient antenna-to-picolinate photoinduced electron transfer. Despite intraligand electron transfer quenching of the antenna, the tacn-based Yb complexes were more emissive than their cyclen analogues, highlighting the influence of the ligand structure on the luminescence properties of NIR emissive lanthanide(III) ions.
Introduction
Compounds emitting in the near infrared (NIR) are of great interest for a wide variety of applications from telecommunications1 to thermometry2 and bioimaging.3,4 NIR emitting organic fluorophores and transition metal-based phosphors have several attractive properties. These include their tuneable excitation and emission wavelengths,5–8 and that they can be rendered aqueous,9 or fluorous soluble,10,11 environment sensitive or analyte responsive,4 and can incorporate reactive groups for labelling.12 A common drawback, however, is their oxygen sensitivity, which results in decreased brightness, rapid fluorophore degradation, and the generation of cytotoxic reactive oxygen species.13Several trivalent lanthanides (Ln) emit in the NIR.14 Of these, Nd and Yb have emissions that are sufficiently robust for applications in aqueous media. Numerous Yb and Nd-based coordination compounds,15–24 including some with inner-sphere solvent molecules,25 have useful luminescence outputs in water. Ln(III) luminescence originates from 4f–4f transitions,26,27 and direct Ln excitation is inefficient due to their low absorption coefficients. Ln(III) sensitisation is possible via a light-harvesting antenna.26 In the case of Nd and Yb, visible-absorbing antennae can be used,28–31 which is advantageous for minimising tissue damage in cellular applications. Even 2-photon excitation of antennae with only UV or blue absorption has been demonstrated using red light.18,19,32,33 Ln(III) luminescence spectra consist of one or more sharp peaks with well-defined positions that are minimally influenced by the coordination environment. Ln excited states are long-lived. Eu and Tb emit with ms lifetimes, but even the shorter lifetimes (hundreds of ns) of Yb and Nd emission are longer than what is typical for cellular autofluorescence. Therefore, time-resolved detection of Ln signals is typically straightforward and is used in e.g. confocal microscopy.34 Crucially, unlike organic-based fluorophores and transition metal-based phosphors, Ln emitters are usually not sensitive to oxygen quenching.35 The brightness of Ln luminescence depends on the absorption coefficient of the antenna at the excitation wavelength, the Ln(III) sensitisation, and the efficiency of radiative decay of the Ln excited state compared to the other deactivation.27 Modulation of these parameters enables the creation of analyte-sensitive probes.16,33,36,37
The Ln(III) sensitization mechanism depends on the Ln, the antenna, and their relative arrangement (Fig. 1). Resonance energy transfer is possible to Ln(III) receiving states that are not more than ∼5000 cm−1 lower in energy than the antenna (S1) or triplet (T1) excited states. For Yb(III) with its excited state at ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1, this means an antenna with S1 or T1 ∼15000 cm−1.27 A second mechanism, originally proposed by Horrocks, begins with photoinduced electron transfer (PeT) from the excited antenna to Yb(III); back electron transfer (BeT) yields the ground state antenna and the excited state Yb(III).38 A third mechanism was proposed by Crosby and Kasha in some cases when there is a large energy gap between a UV-absorbing antenna and the Yb(III) excited state:39 the dissipation of the excess energy to the solvent or the lattice vibrational modes.40 Such phonon-assisted energy transfer (PAEnT) could explain the observation of sensitised Yb emission in complexes wherein the first electron transfer of the Horrocks pathway is not thermodynamically favoured (Fig. 1).41
000 cm−1, this means an antenna with S1 or T1 ∼15000 cm−1.27 A second mechanism, originally proposed by Horrocks, begins with photoinduced electron transfer (PeT) from the excited antenna to Yb(III); back electron transfer (BeT) yields the ground state antenna and the excited state Yb(III).38 A third mechanism was proposed by Crosby and Kasha in some cases when there is a large energy gap between a UV-absorbing antenna and the Yb(III) excited state:39 the dissipation of the excess energy to the solvent or the lattice vibrational modes.40 Such phonon-assisted energy transfer (PAEnT) could explain the observation of sensitised Yb emission in complexes wherein the first electron transfer of the Horrocks pathway is not thermodynamically favoured (Fig. 1).41
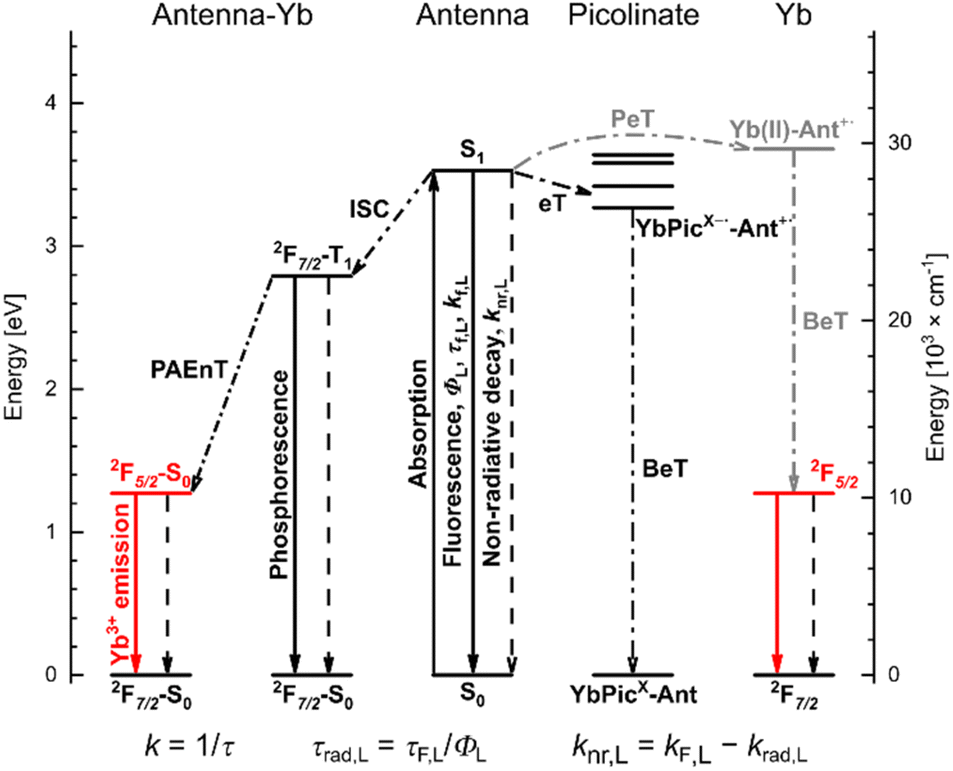 | ||
| Fig. 1 Competing sensitising and quenching processes in photoexcited YbLX complexes; solid and dashed lines indicate radiative and non-radiative processes, respectively. | ||
The efficiency of sensitised Ln(III) emission quantum yield (ΦLn) is dependent on the combined efficiencies of two consecutive stages. The first is the population of the Ln(III) excited state (Ln(III)*) via either the antenna's first singlet (S1) or first triplet (T1) excited states (ηsens in eqn (1)). The second stage is the emission of photons from the excited Ln ion. The latter is in competition with quenching pathways such as internal conversion, quenching by X–H oscillators (X = O, N, C), and, if the Ln(III)*–T1 energy gap is small (<2000 cm−1), thermal back energy transfer (BET).42–44 The vibrational overtones of C–H, N–H and O–H do not influence the energy transfer from the ligand to emitting level of lanthanide. The efficiency of photon emission by excited Ln ions is defined by the intrinsic quantum yield (ΦLnLn), which is characteristic of the ion in a particular environment. All else being equal, shorter radiative lifetimes (τrad) yield higher ΦLnLn, and lead to higher ΦLn. The τrad of an Ln ion is determined by the refractive index of the medium and by the dipole moments of the transitions, which are affected by the coordination geometry. Lns have high coordination numbers (CN), and Ln–ligand interactions are mostly Coulombic. Therefore, the Ln coordination environment is governed by steric factors, and is a function of the ligand and of the solvent. The shortening of τrad has been previously demonstrated for Eu(III) emitters as a viable strategy for improving Ln luminescence.45–47 The calculation of τrad for Eu(III) is possible from the corrected luminescence spectrum,48 however, this straightforward method is not available for the other Lns.27,49
| ΦLn = ΦLnLn·ηsens | (1) |
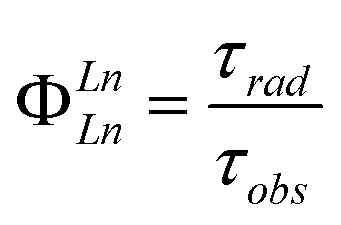 | (2) |
Octadentate ligands based on two different macrocycles [cyclen (1,4,7,10-tetraazacyclododecane), and tacn (1,4,7-triazacyclononane)] and completed with methylenecarboxylate or picolinate donors and 1 inner-sphere water molecule yield Eu(III) emitters with τrad ∼5.40 and ∼2.90 ms, respectively.46,47 The beneficial effect of the shorter τrad on ΦLn in the tacn complexes was masked by two quenching processes: PeT from the excited antenna (Ant*) to the reducible Eu(III), and intraligand electron transfer from Ant* to the picolinates.46 For Yb(III), antenna-to-Ln(III) PeT and the subsequent back electron transfer (BeT) can yield Yb(III)*, so unlike in the case of most Eu complexes,50 PeT can be sensitising (Fig. 1).38
In this study, we have prepared the Yb(III) complexes of 4 carbostyril-appended tacn-based octadentate ligands bearing 2 picolinate coordinating moieties (YbLX, Fig. 2a). We characterised them by 1H NMR spectroscopy and X-ray crystallography, measured their redox properties by cyclic voltammetry and evaluated their luminescence efficiencies relative to each other as well as to cyclen-based complexes carrying the same antenna (YbLc, Fig. 2b). We constructed model compounds (PyX in Fig. 2a) that enabled the study of the redox properties of the picolinate units in isolation. Based on these data, the possible contributions of the Horrocks sensitisation route (i.e. the combined PeT and BeT processes in Fig. 1), of the quenching intraligand PeT, and sensitisation by phonon-assisted energy transfer (PAEnT)39 were assessed.
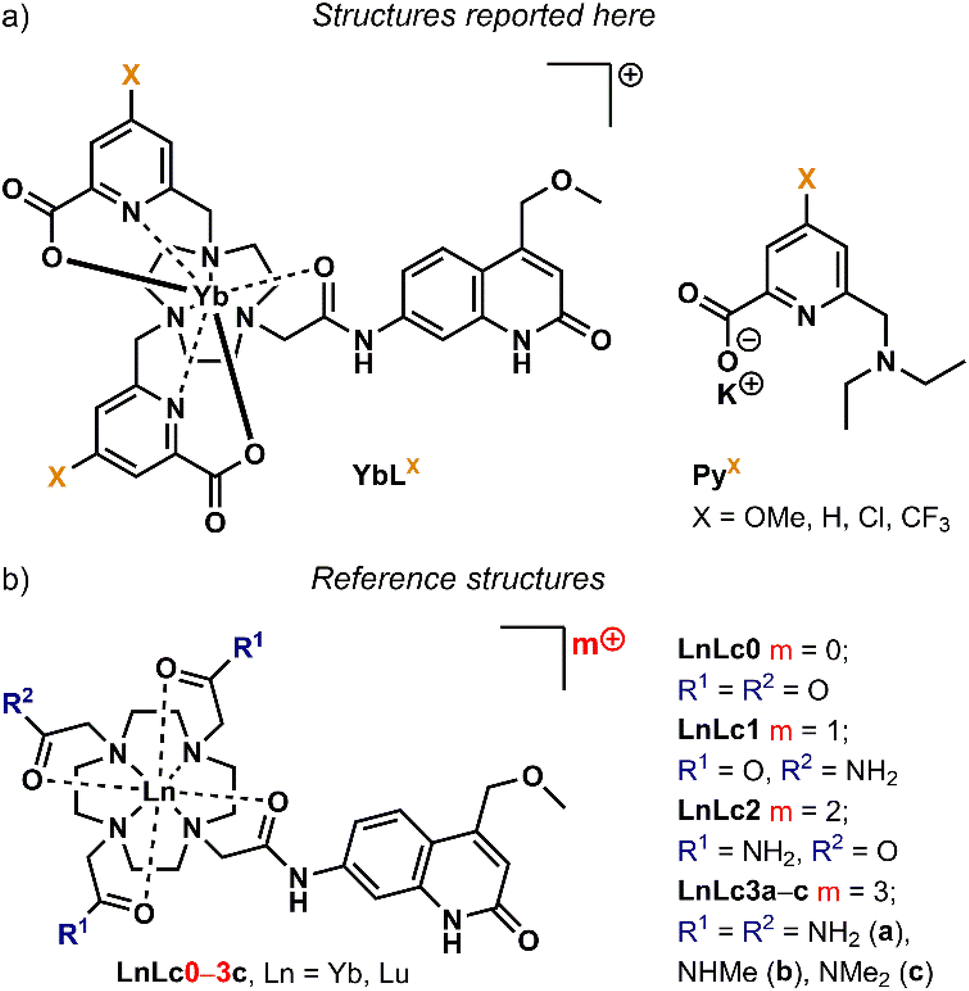 | ||
| Fig. 2 (a) Complexes studied here (YbLX) and pyridine model compounds (PyX), X is the pyridine p-substituent. (b) Cyclen-based Yb complexes used as the reference structures (YbLc), LnLc0–3c are cyclen-based complexes with 0 to +3 overall charge. | ||
Results and discussion
Solution and solid-state structures
Previously reported ligands LX (X = OMe, H, Cl, CF3)46 were reacted with YbCl3 in H2O:EtOH 1:1 mixture for 16 h at 45 °C to yield YbLX. The products were purified by column chromatography on neutral alumina.46,51 Potassium pyridine-2-carboxylate model compounds PyX were synthesised from known 4-substituted methyl 6-(bromomethyl)picolinates in two steps in excellent yield (Schemes S1 and S2†). PyX were fully characterised by 1H, 13C, and 19F NMR spectroscopies and high-resolution mass spectrometry (HR-MS), while the complexes were characterised using HPLC-MS, HR-MS, paramagnetic 1H NMR spectroscopy, and UV-Vis absorption and emission spectroscopies, as well as single crystal X-ray crystallography in the case of YbLH and YbLCF3. Analytical data confirmed the formulae and general structures of PyX and YbLX.
Paramagnetic 1H NMR spectroscopy of Yb(III) coordination compounds can be very informative, and several tacn-based Yb(III) chelates have recently been analysed.52 The 1H NMR spectra of YbLX consisted of 31–37 individual signals, which is consistent with the presence of both enantiomers (vide infra) of a single diastereomer for all four complexes, for which 34–36 individual signals are expected, or with several species rapidly interconverting on the NMR timescale (Fig. S5–S9†). Our attempts to fully assign the spectra were unsuccessful. Based on literature precedent, the broadened peaks >30 ppm are ascribed to protons on methylene spacers between tacn and picolinate groups, while the sharp signals at <−25 ppm are assigned to axial CH2 protons of the 9-membered ring.53 These signals are common to all four complexes. The pyridine para-substituents are far removed from the Yb(III), and their electronic effects are not expected to have a dramatic influence on picolinate coordination to the metal.46 Parker has demonstrated the high sensitivity of the 1H NMR shifts to solvation and the ligand field in tacn-based tris-picolinate Yb(III) complexes.54 Thus, the ∼10 ppm differences in the most and least deshielded signals in YbLH and YbLCF3 are unsurprising and consistent with the metals occupying similar coordination geometries. The presence of at least two species in solution is also confirmed by two resonances in 19F NMR spectrum of YbLCF3 both attributable to CF3-groups from different complex isomers (Fig. S10†).
FT-IR spectra were recorded on solid samples (Fig. S11–S14†) and displayed a broad absorption band between 3600–2800 cm−1 corresponding to O–H stretching vibration bands, with some sharp features that can be attributed to N–H and C–H stretching vibration bands. Strong absorption bands due to stretches of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, likely of the carboxylate groups, were also observed at 1637, 1610, 1654, 1651 cm−1 for YbLH, YbLOMe, YbLCl, and YbLCF3, respectively.
O bonds, likely of the carboxylate groups, were also observed at 1637, 1610, 1654, 1651 cm−1 for YbLH, YbLOMe, YbLCl, and YbLCF3, respectively.
Single crystals suitable for X-ray diffraction analysis were obtained by vapor diffusion of glyme and dioxane into concentrated aqueous solutions of YbLCF3–F, and YbLH, respectively. Crystals of YbLCF3–F were grown in the presence of 1 equiv. of potassium fluoride. Compounds YbLH (Fig. 3) and YbLCF3–F (Fig. S15†) possess a central Yb atom with a nine-coordinate geometry in a (heavily) distorted tricapped trigonal prismatic arrangement, as seen in related complexes.46 The trigonal prism is capped by two tethered carboxylate groups and a fluoride (for YbLCF3–F) or a water molecule (for YbLH). The two planes of the trigonal prism are comprised of the N-donors of the tacn ligand (N1, N2, N3 for YbLCF3–F/N4 YbLH; N3PL), or of the antenna amide O- and the pyridine N-donors (N3 for YbLH/N4 for YbLCF3–F, N5, O1; NNOPL). As previously observed, the two planes are not parallel with N3PL–Ln–NNOPL angles ranging 123–125°. These values are slightly wider than those of related Eu-, Gd- and Tb-complexes previously reported.46 In addition, the Yb centre sits further from the NNOPL (Ln–NNOPL ∼0.65 Å (Yb) vs. ∼0.3 Å (Eu, Gd, Tb)) and closer to the N3PL (Ln–N3PL 1.967(2) to 1.9860(15) Å (Yb) vs. 2.017(4) to 2.065(4) Å (Eu, Gd, Tb)). These differences are likely to arise from the shorter ionic radius of Yb3+ (for CN = 9, 1.04 Å) than that of lighter Ln3+ ions (CN = 9, Eu3+ 1.12 Å, Gd3+ 1.11 Å, Tb3+ 1.10 Å).55 Both YbLCF3–F and YbLH are racemic in the solid state, containing Λ(δδδ) and Λ(λλλ) isomers in the unit cell (Table S1†).52
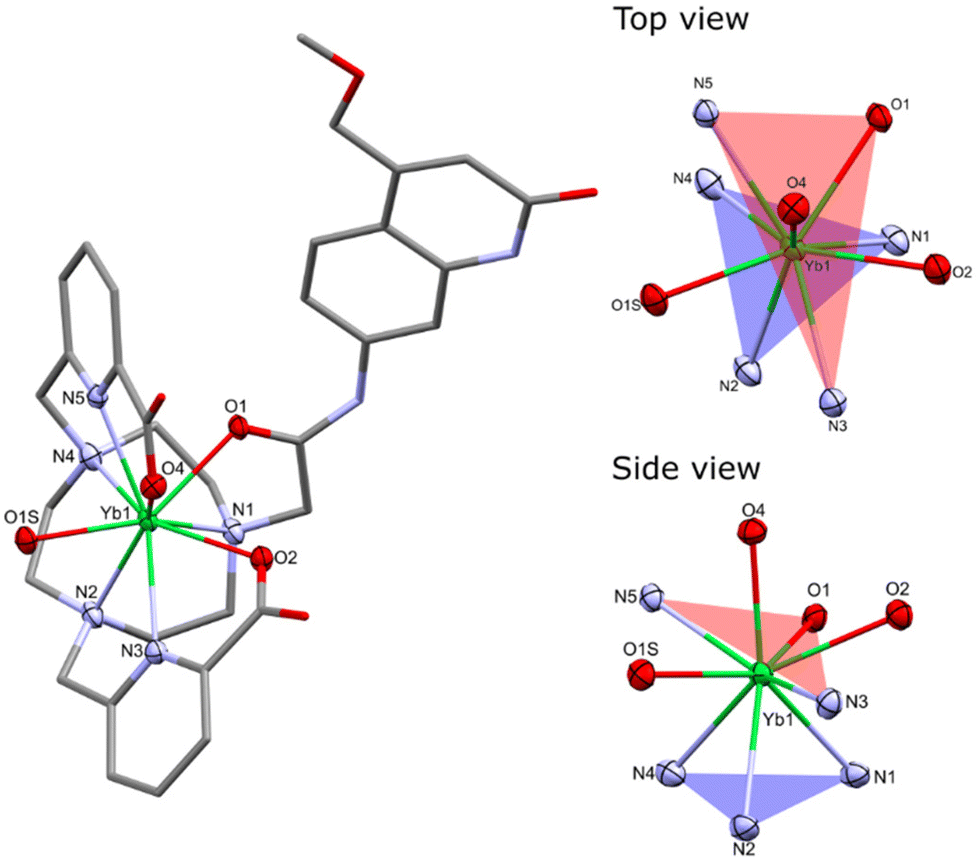 | ||
| Fig. 3 Solid-state structure (left) and the coordination environment of Yb from the top and side view (right) in YbLH. H atoms, non-coordinating Cl− counterions and water molecules were omitted for clarity. Ellipsoids displayed at 50% probability for atoms of the coordination environment, the rest of the molecule is depicted as capped sticks. | ||
The bond metrics are shorter than those reported for related Ln3+ carbostyril-substituted tacn and cyclen complexes,46,51 which is consistent with the smaller ionic radius of Yb3+. The Yb–O and Yb–N bond distances in YbLCF3–F are slightly elongated when compared to those in YbLH, which can be ascribed to the presence of the fluoride co-ligand shielding the Yb charge in the former, although electronic differences between the macrocyclic ligands may also play a role. The Yb–Ntacn distances range 2.513(3)–2.649(4) Å, while the Yb–NPY distances range 2.456(2)–2.512(2) Å. The Yb–O distances of the carboxylates range 2.306(2)–2.389(2) Å, and the Yb–O(amide) distances vary 2.355(2)–2.400(2) Å. The Yb–F and Yb–OH2 distances of 2.110(2) and 2.313(2)–2.326(2) Å, respectively, are shorter than those of related Ln3+ tacn- and cyclen-based complexes (averages: Ln3+–F, 2.203(3) Å; Ln3+–OH2, 2.404(4) Å), but the differences are consistent with the smaller ionic radius of Yb3+ (Table S2†).46,51
The CN and geometry of the complexes depend on the structure of the ligand. The X-ray structures of YbLCF3–F and YbLH show that these two species have CN = 9 and adopt a distorted tricapped trigonal prismatic arrangement, whereas the cyclen-based complexes were twisted square antiprismatic (TSAP) with CN = 8.56 The 1H NMR spectra of YbLX are consistent with all the complexes adopting the same geometry in solution. For both sets of ligands, we can assume that changes in photophysical properties within a series will be due to changes in redox properties rather than structural differences.
Electrochemistry
The metal- and ligand-based redox properties of the complexes were studied by cyclic voltammetry. Experiments were carried out on the complete library of Yb(III) complexes, as well as with a series of model compounds with only a picolinate moiety (Fig. 2a). The latter were designed to enable the identification of Yb and pyridine-based redox events. Analyses were performed at 0.1 V s−1 scan rate in DMF containing 0.1 M (n-Bu)4NClO4 as the electrolyte. DMF has a more suitable solvent window than water to study Yb(III) reduction. Voltammograms were recorded by scanning first towards more negative potential values (reduction). A glassy carbon electrode and a Ag/Ag+ reference electrode (0.01 M AgNO3 in MeCN) were used. Ferrocene was used as a pseudo-reference and was added at the end of the experiment. The peak anodic and cathodic potentials (Epa, Epc) values vs. Fc/Fc+ and vs. NHE are reported in Table S4.†The electrochemical properties of YbLX were explored along with those of p-substituted picolinate models PyX (Fig. S17 and S18†). Compounds PyX display an irreversible reduction wave with Epc values from −1.66 to −1.46 V vs. NHE that follow the order PyOMe < PyH < PyCl < PyCF3. This wave was attributed to the substituted pyridine reduction, which is easier for the picolinate with a stronger electron withdrawing group (Table 1).57,58 An oxidation wave at ∼0.94 V vs. NHE is common to all PyX, and may correspond to the oxidation of the tertiary 6-amino group (Table S4†).59YbLX display several reductive events and no oxidation wave (Fig. S18†). The first reduction wave in these complexes range from −1.88 to −1.51 V vs. NHE and similarly follows the order YbLOMe < YbLH < YbLCl < YbLCF3. As a comparison, picolinate reduction in GdLX in 100 mM aqueous NH4Cl solution under Ar occurred between Ered = −1.43 V vs. NHE (for X = OMe) and −1.13 V vs. NHE (for X = CF3),46 and shows a similar trend in Epc value of the first reduction wave (OMe < H < Cl < CF3, Table 1). The acetylated 4-methoxymethyl carbostyril antenna has a reduction potential of −2.22 V vs. NHE measured in identical conditions as YbLX.56 This entity is probably not involved in the reductive event observed in YbLX complexes. The first reduction wave may be either due to the reduction of a picolinate moiety or of Yb(III).
| Complex | E red [V vs. NHE]a | Model compound | E red [V vs. NHE]a | Complex | E red [V vs. NHE]b |
|---|---|---|---|---|---|
| a Measured in DMF in glovebox with 0.1 M NBu4ClO4 as a supporting electrolyte and at 0.1 V s−1 scan rate. b Measured in H2O with 0.1 M NH4Cl as a supporting electrolyte under Ar at 0.1 V s−1 scan rate, from ref. 46. | |||||
| YbLOMe | −1.88 | PyOMe | −1.66 | GdLOMe | −1.43 |
| YbLH | −1.82 | PyH | −1.61 | GdLH | −1.36 |
| YbLCl | −1.66 | PyCl | −1.52 | GdLCl | −1.21 |
| YbLCF3 | –1.51 | PyCF3 | −1.46 | GdLCF3 | −1.13 |
A linear variation of Epcversus Hammett substituent constants σp is observed for YbLX and PyX, similarly to what has previously been observed for GdLX (Fig. S19†).46 This indicates that the p-substituents have a strong influence on the electron-accepting ability of YbLX, and the greater electron accepting ability of the picolinate moieties shifts Epc to more positive values. The impact of the substituent is larger in YbLX and GdLX complexes compared to the model compounds and seems to depend on the Lewis acidity of the metal ion as ΔEpc(CF3vs. OMe) = 0.37 V for YbLX, 0.30 V for GdLX, and 0.20 V for PyX.
All tacn-based YbLX cyclic voltammograms contain several irreversible reductive events that happen at close potentials (Fig. S18†), and could correspond to picolinate or Yb(III) reductions. The irreversibility of the first reduction wave in YbLX suggest that the reduced YbLXred is not stable and that a chemical step takes place after the electron transfer. Due to this, it is impossible to determine the reduction potential of the Yb(III)/Yb(II) couple for YbLX. Thus, the value of −1.92 V vs. NHE (Epc for YbLc1) was used for the calculations of ΔGeT for antenna to Yb(III) PeT, while Ered values of YbLX (Table 1) were utilised to calculate intraligand PeT driving force in YbLX complexes (vide infra).
Photophysical properties
The photophysical properties of the complexes were determined in aqueous 10 mM PIPES buffer at pH = 6.5 ([LnL] = 10 μM) to enable comparison with LnLc.56 The results are summarised in Tables 2, 3, S5–S7, and Fig. 4, 5 and S20–S35.†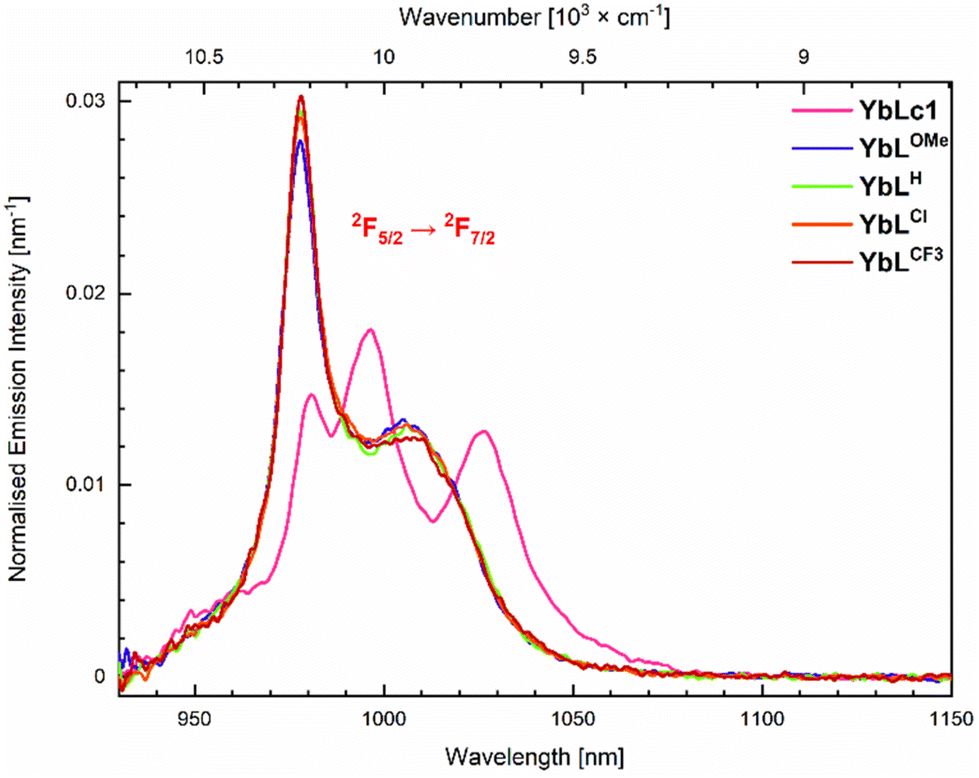 | ||
| Fig. 4 Emission spectra of the Yb(III) complexes (A = 0.10) in 10 mM PIPES in H2O, pH 6.5. λex = 323 nm. | ||
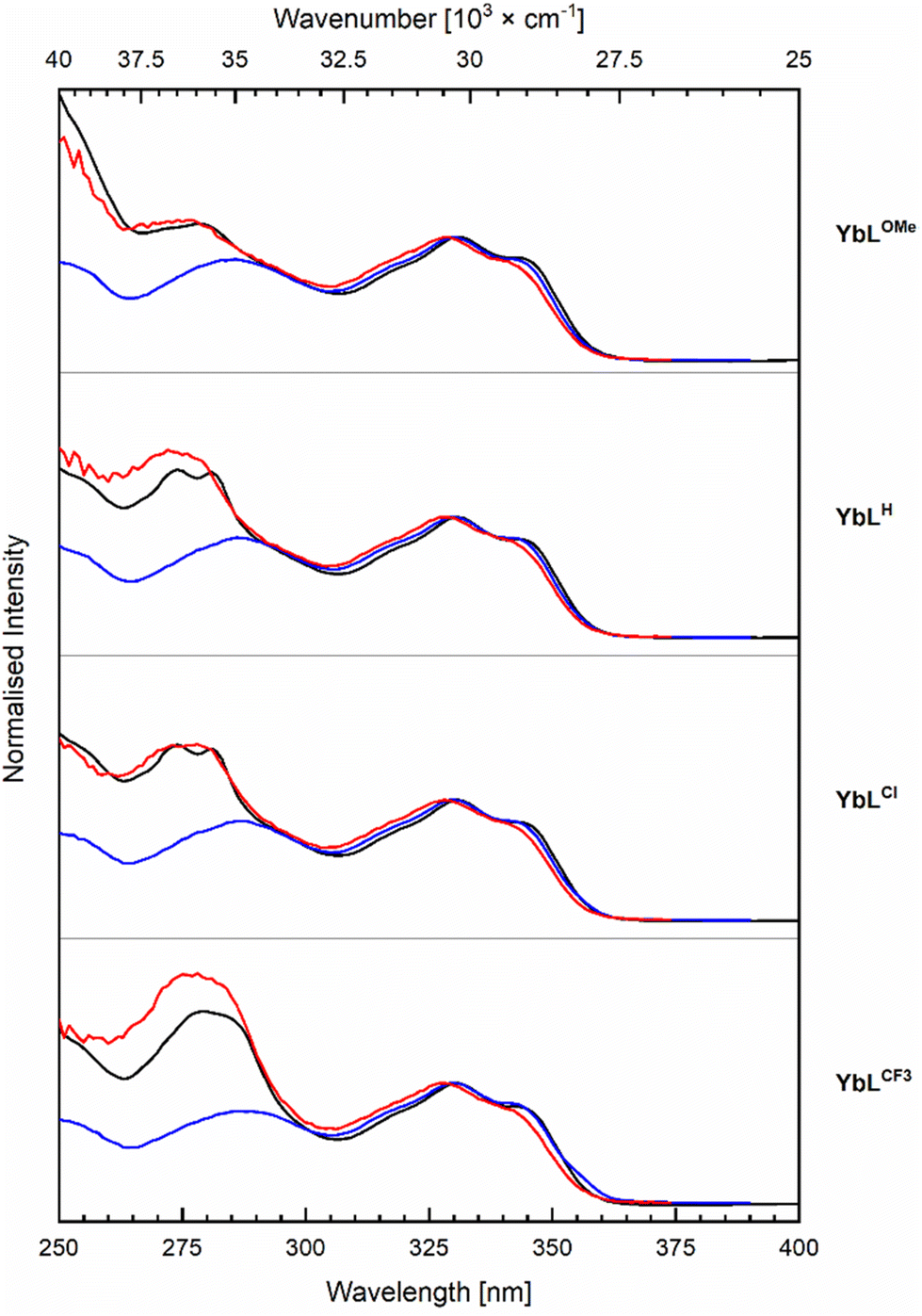 | ||
| Fig. 5 Normalised excitation spectra for the Yb (red, λem = 978 nm) and antenna (blue, λem = 405 nm) emission with corresponding absorption (black) spectra of YbLX complexes. Recorded in PIPES buffered (10 mM, pH 6.5, A = 0.10) aqueous solutions at r.t. | ||
| Complex |
Φ
La,b [%] (Rel. ΦL [%]) |
Complex |
Φ
La,c [%] (Rel. ΦL [%]) |
τ f,L [ns] (YbLX) |
|---|---|---|---|---|
| a Determined relative to quinine sulfate (Φ = 0.59) in H2SO4 (0.05 M) in H2O.61 b Mean ± standard deviation for three independent measurements. c From ref. 51 (GdLc1) and ref. 46 (GdLX). d χ 2 = 1.5600. e χ 2 = 1.5215. f Too short to measure. | ||||
| YbLOMe | 4.69 ± 0.18 (100) | GdLOMe | 6.85 (100) | 0.26d |
| YbLH | 3.68 ± 0.04 (79) | GdLH | 6.42 (94) | 0.20e |
| YbLCl | 2.01 ± 0.02 (43) | GdLCl | 4.64 (68) | <0.15f |
| YbLCF3 | 0.99 ± 0.04 (21) | GdLCF3 | 2.11 (31) | <0.15f |
| YbLc1 | 5.43 ± 0.05 | GdLc1 | 7.01 | 0.34 |
| Complex | Relative φLna [%] |
|---|---|
| a Measured in PIPES 10 mM, pH 6.5, upon excitation at 323 nm. b The luminescence intensity of YbLX is related to the cyclen-based series by a factor 0.89(2), i.e. φ(YbLc)/φ(YbLCl) = 0.89(2) × φ(YbLc)/φ(YbLc3c). | |
| YbLOMe | 96 ± 2 |
| YbLH | 94 ± 2 |
| YbLCl | 100 |
| YbLCF3 | 68 ± 1 |
| YbLc1 | 81 (rel. to YbLc3c) |
| 72 (rel. to YbLCl)b | |
Antenna excitation at λex = 329 nm yielded antenna fluorescence with λem = 376 nm (Fig. S29†), along with Yb(III) luminescence in the NIR (vide infra). The shapes of the YbLX carbostyril fluorescence spectra differed from those of cyclen-based complexes with the same antenna. Specifically, in the case of YbLX an additional band is observed between 400–600 nm compared to YbLc (Fig. S22†). This band becomes stronger in the order YbLH < YbLOMe < YbLCl < YbLCF3, and could indicate aggregation, or could be a twisted intramolecular charge transfer state of the carbostyril induced by steric, electrostatic, and/or photodynamic effects. A similar extra band has been observed in Ln complexes bearing multiple coumarin antennae in the coordination sphere,60 and was shown to impact the photophysical properties of the Ln ions by facilitating either EnT or BET.
The antenna fluorescence residual quantum yields (ΦL) were determined relative to quinine sulfate (Table 2). YbLOMe with the most electron-rich OMe-substituted pyridines had the largest ΦL, 4.69%, that of YbLCF3 was only 0.99%. These trends follow the ones seen in the Eu(III), Tb(III), and Gd(III) complexes of the same ligands.46 The differences in ΦL can be ascribed to PeT from the excited antenna to the pyridines, in addition to any PeT that may take place to Yb(III) (vide infra).46 The larger ΦL of YbLc1 than that of YbLX is consistent with intraligand PeT in the latter, which is also supported by the antenna fluorescence lifetimes (τf,L). The τf,L are longer in YbLc1 than in YbLX, 0.34 ns and <0.26 ns, respectively (Tables 2, S7, Fig. S31 and S32†). For YbLX with the most electron-deficient pyridines (X = Cl, CF3) τf,L were too short to measure with the experimental setup available to us (the lowest measurable lifetime value was 0.15 ns).
The steady-state emission spectra at 77 K of YbLX revealed structured fluorescence bands, and no phosphorescence was observed, unlike in the case of the Gd analogues (Fig. S33†). Hence, T1 is completely quenched in YbLX as it was also demonstrated for YbLc series. Notably, S1/T1 were ∼0.36 for GdLX which is indicative of similar intersystem crossing rates across the set of complexes with Lx ligands (Table S7†).
The photostabilities of YbLX were determined by irradiation of their samples in the presence of atmospheric oxygen. The extent of complex degradation was estimated from the antenna fluorescence emission spectrum. Within the YbLX series, YbLOMe had the lowest emission intensity (78% from initial) after continuous light irradiation for 2 h (Fig. S34 and S35†). The other compounds retained 86–91% of their original emission intensity. These values are comparable to what has been observed for EuLX for which both PeT and intraligand PeT are possible.47YbLX are more photostable than TbLX, the degradation of the latter likely proceeds through antenna T1 repopulation via BET, which is not possible for YbLX.47
The spectra of YbLX are similar and only a small variation of the most energetic and intense transition is observed. Upon increasing the electron-withdrawing character of the picolinate para-substituent, the intensity of 979 nm transition increases, whereas that of 1006 nm is unchanged within the experimental error. The spectra of YbLH and YbLCl are very close to each other.
The Yb(III) excitation spectra contain features ascribed to the antenna as well as the picolinates (Fig. 5). The high-energy component is picolinate-dependent, and the intensity of this excitation band increases from the YbLOMe to YbLCl, YbLH and YbLCF3, which is 1.7-fold higher than the excitation of YbLOMe. YbLCl and YbLH have approximately the same excitation intensity but with YbLH slightly higher and blue-shifted compared to YbLCl. The absorption spectra display a similar trend, so that the higher intensity may come only from the increased amount of absorbed light. If the picolinates are better sensitisers than the carbostyril antenna, the effect should be in the order of 10–30% according to the peak intensity difference observed between the absorption and the excitation.
The relative Ln(III) emission quantum yields (φLn) were calculated compared to the strongest emitter YbLCl (Table 3). With one exception, YbLX are overall more luminescent than YbLc, despite intraligand PeT quenching the antenna excited state, and an inner-sphere water molecule. YbLOMe and YbLH have similar quantum yields within experimental error, and YbLCl was found to have the largest quantum yield, albeit only 2–3 standard deviations higher than YbLOMe and YbLH. The electron-withdrawing picolinate p-CF3 group is unfavourable to Yb(III) luminescence, and YbLCF3 has the lowest quantum yield, even lower than YbLc1. While electron-withdrawing groups in the ligand should induce a stabilisation of the more electron rich Yb(II), such effect appears to be overpowered by the negative consequences of the intraligand eT which is the most thermodynamically downhill process for YbLCF3 in the YbLX series (Table S8†). The calculation of intraligand ΔGeT was done with Ered(Pic/Pic˙−) ranging from −1.88 V (YbLOMe) to −1.51 V vs. NHE (YbLCF3). The results were consistent with intraligand eT being least and most favourable in YbLOMe (−0.04 eV) and YbLCF3 (−0.41 eV), respectively, which is consistent with the lowest φLn and ΦL for the latter complex.
This result suggests that having electron-withdrawing groups on the picolinates is indeed beneficial to the PeT antenna–Yb(III) pathway, but that the balance between the two PeT mechanisms, one quenching and one sensitising, is subtle. There is therefore probably not a lot of room for improvement of the PeT sensitised Yb(III) quantum yield by incorporating extra chromophores that are susceptible to photoredox processes.
Ln(III) are often assumed to have the antenna T1 as the major feeding level,26,62 although EnT from S1 is also possible.25,63–65 Neither T1 (∼22500 cm−1)25 nor S1 (>27500 cm−1) in YbLX appreciably overlap with the Yb(III) receiving level (10260 cm−1),66 which excludes resonance EnT sensitisation. Therefore, PeT38,41 or PAEnT39 (Fig. 1) must be operating.
We have recently shown that in YbLc1 and all its 0 to +3 charged analogues sensitisation can happen by PAEnT and by PeT. The contribution from PeT was probably small even in those +2 and +3 charged complexes where the initial PeT step of the mechanism was feasible. The thermodynamic feasibility of PeT as the first stage of this pathway was calculated (Table S9†). Due to the uncertainty of assigning Ered(Yb(III)/Yb(II)) in the cyclic voltammograms of YbLX, calculations were performed with Ered(Yb(III)/Yb(II)) equal to −1.92 V vs. NHE. The latter value is Ered(Yb(III)/Yb(II)) for YbLc1. The reduction potential of Ln(III) are strongly dependent on the overall charge of the complex,50,51,67 so the Yb(III) centre in YbLc1 is a reasonable model for a +1 charged species. With Ered(Yb(III)/Yb(II)) of −1.92 V vs. NHE a ΔG(PeT) = 0 eV was obtained. The other, less negative Ered for Yb(III)/Yb(II) reduction values allowed for increasingly thermodynamically downhill processes.
The major sensitisation pathway of Yb(III) emission was still a question despite the suggested mechanisms (Fig. 1). Thus, we calculated the ratios of the Franck–Condon (FC) factors for the processes: (2F7/2–T1) → (2F5/2–S0), which ultimately leads to Yb(III) luminescence, and (2F7/2–T1) → (2F7/2–S0) that is responsible for the non-radiative deactivation of the complexes. The former was found to be 8 (YbLH,Cl,CF3) to 9 (YbLOMe) orders of magnitude faster than the latter (Tables S10 and S11†). Moreover, the ratio of FC factors for the same processes in YbLc3c, the strongest emitter of the YbLc series, was found to be identical to that of YbLH,Cl,CF3. Hence, intraligand eT quenching in YbLCF3 (vide supra) offsets the improved φLn, which was observed for the rest of the YbLX series compared to the YbLc.
Another pathway to consider is the population of the 2F5/2 receiving level via the Pic˙−–Ant˙+ state. This process would be feasible if eT yielding Yb(II)–Ant˙+ from the initially formed Pic˙−–Ant˙+ is possible. The estimated energy difference E(Yb(II)–Ant˙+) = EMeox (AcCSMOM)46 − EYbred (YbLc1) = 3.68 eV is larger than the potential difference for the intraligand eT, ranging from 3.64 eV (YbLOMe) to 3.27 eV (YbLCF3) (Table S8†). The difference is small for YbLOMe,H,Cl (ΔE = 0.04–0.26 eV) and considerable for YbLCF3 (ΔE = 0.41 eV). Thus, the improved φLn may come from the intermediate Pic˙−–Ant˙+ state formed after intraligand eT, which could contribute with additional sensitisation.
In short, ΔrG for the direct Yb(III)–antenna PeT reaction is approximately 0, whereas for the intraligand PeT it is slightly to moderately negative and hence favourable. So intraligand PeT is more likely to happen, and is especially supposed to be prevalent for YbLCF3, the least luminescent complex. While intraligand PeT thus does appear to be unfavourable for Yb(III) luminescence, YbLCl is the second most favoured for intraligand PeT and the most luminescent YbLX. Without additional experiments the actual contribution of this process to the overall sensitisation mechanism in YbLX is difficult to establish with certainty.
Conclusions
A series of Yb(III) coordination compounds based on a tacn macrocycle functionalised by a carbostyril antenna and two identical para-substituted picolinate derivatives was prepared. Solution and solid-state studies of the Yb(III) complexes revealed the presence of similar species with a nonadentate distorted tricapped trigonal prism coordination environment of the metal centre. The picolinate-based reductions in Yb(III) compounds took place from −1.88 to −1.51 V vs. NHE for the least and most electron-deficient ligands, respectively, which is in accordance with the expected substitution effect. Yb(III) emission was differently shaped compared to the one of the cyclen series, which is expected as the tacn- and cyclen-based complexes have different symmetries. The fluorescence of the antenna was altered by the different substituents on the picolinate, and the antenna ΦL were consistent with PeT quenching of the carbostyril excited state by the electron-poor pyridines.The quantum yields of the Yb(III) emission were higher than those of cyclen-based emitters. This was surprising as in YbLX Yb(III) sensitisation was competing with antenna quenching by the picolinates. The higher Yb(III) luminescence could be the result of a an improvement in either of the two components contributing to the overall emission, namely (1) better sensitization, or (2) more efficient radiative decay of excited-state Yb(III), i.e. higher intrinsic quantum yield of LX-bound Ln(III). The possibility of the latter is suggested by the comparison with the analogous Eu species for which ΦLnLn is readily determined: ΦEuEu(EuLX) is larger than ΦEuEu(EuLc), ∼17.5 and 11%, respectively.7,51
Yb(III) sensitisation was unlikely to proceed via resonance EnT due to the lack of spectral overlap between the antenna and the Yb(III) excited states. The initial antenna-to-Yb(III) PeT step of Horrock's PeT mechanism seemed to be thermodynamically neutral. Other PeT pathways may be sensitising, and could contribute to the increased Yb luminescence of YbLX. According to our calculations the intraligand process is thermodynamically favourable, and should be upon recombination or further electron transfers sensitising for Yb. T1 decay to Yb(III)*–S0via PAEnT appears to be a viable sensitisation pathway in these emitters. Future experiments to determine the Yb(III) radiative lifetimes and intrinsic quantum yields could determine the contribution made by the coordination environment to the overall improvement in luminescence. The measurement of Yb(III), and antenna S1 and T1 lifetimes and rise times, and an analysis of triplet T–T absorption could show how the Yb(III) excited state is populated, and could directly show the contributions of the various sensitisation and quenching pathways.
Taken together, these results are relevant for emitter design in several ways. First, incorporating multiple antennae in the same complex is often employed to increase emitter brightness. Our results suggest that quenching by intraligand PeT should be considered, and if possible, avoided so as not to lose the benefit of the improved absorption. Second, the emission intensity from the Eu(III) and Tb(III) complexes of these tacn-based ligands were clearly inferior to the cyclen-based ones; the situation was reversed for Yb(III). This may be due to increased ΦLnLn resulting from a change in the complex coordination geometry, or an improvement in sensitisation, e.g. the fact that specifically for Yb, intraligand PeT could also be sensitising.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Research Council (project grant 2017-04077 to K. E. B.), Carl Tryggers Stiftelse för vetenskaplig forskning (post doc fellowship to E. M.), and the Knut och Alice Wallenbergs Stiftelse (Dnr: 2018.0066 and Dnr: KAW 2019.0071).References
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2009, 39, 189–227 RSC.
- A. V. Orlova, V. Y. Kozhevnikova, A. S. Goloveshkin, L. S. Lepnev and V. V. Utochnikova, Dalton Trans., 2022, 51, 5419–5425 RSC.
- L.-L. Chen, L. Zhao, Z.-G. Wang, S.-L. Liu and D.-W. Pang, Small, 2022, 18, 2104567 CrossRef CAS PubMed.
- M. Zhao, B. Li, H. Zhang and F. Zhang, Chem. Sci., 2021, 12, 3448–3459 RSC.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS PubMed.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2014, 9, 855–866 CrossRef CAS PubMed.
- D. Ra, K. A. Gauger, K. Muthukumaran, T. Balasubramanian, V. Chandrashaker, M. Taniguchi, Z. Yu, D. C. Talley, M. Ehudin, M. Ptaszek and J. S. Lindsey, J. Porphyrins Phthalocyanines, 2015, 19, 547–572 CrossRef CAS PubMed.
- K. M. Faries, J. R. Diers, J. W. Springer, E. Yang, M. Ptaszek, D. Lahaye, M. Krayer, M. Taniguchi, C. Kirmaier, J. S. Lindsey, D. F. Bocian and D. Holten, J. Phys. Chem. B, 2015, 119, 7503–7515 CrossRef CAS PubMed.
- K. E. Borbas, V. Chandrashaker, C. Muthiah, H. L. Kee, D. Holten and J. S. Lindsey, J. Org. Chem., 2008, 73, 3145–3158 CrossRef CAS.
- E. M. Sletten and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 13574–13577 CrossRef CAS.
- I. Lim, A. Vian, H. L. van de Wouw, R. A. Day, C. Gomez, Y. Liu, A. L. Rheingold, O. Campàs and E. M. Sletten, J. Am. Chem. Soc., 2020, 142, 16072–16081 CrossRef CAS.
- S. I. Reja, M. Minoshima, Y. Hori and K. Kikuchi, Chem. Sci., 2021, 12, 3437–3447 RSC.
- A. P. Demchenko, Methods Appl. Fluoresc., 2020, 8, 022001 CrossRef CAS.
- Y. Ning, M. Zhu and J.-L. Zhang, Coord. Chem. Rev., 2019, 399, 213028 CrossRef CAS.
- E. R. Trivedi, S. V. Eliseeva, J. Jankolovits, M. M. Olmstead, S. Petoud and V. L. Pecoraro, J. Am. Chem. Soc., 2014, 136, 1526–1534 CrossRef CAS PubMed.
- S. Shuvaev and D. Parker, Dalton Trans., 2019, 48, 4471–4473 RSC.
- A. Beeby, S. Faulkner, D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 2001, 1268–1273 RSC.
- A. T. Bui, M. Beyler, A. Grichine, A. Duperray, J.-C. Mulatier, Y. Guyot, C. Andraud, R. Tripier, S. Brasselet and O. Maury, Chem. Commun., 2017, 53, 6005–6008 RSC.
- A. D'Aleo, A. Bourdolle, S. Brustlein, T. Fauquier, A. Grichine, A. Duperray, P. L. Baldeck, C. Andraud, S. Brasselet and O. Maury, Angew. Chem., Int. Ed., 2012, 51, 6622–6625 CrossRef PubMed.
- A. D'Aleo, F. Pointillart, L. Ouahab, C. Andraud and O. Maury, Coord. Chem. Rev., 2012, 256, 1604–1620 CrossRef.
- A. D'Aléo, A. Bourdolle, S. Brustlein, T. Fauquier, A. Grichine, A. Duperray, P. L. Baldeck, C. Andraud, S. Brasselet and O. Maury, Angew. Chem., Int. Ed., 2012, 51, 6622–6625 CrossRef PubMed.
- M. F. K. Trautnitz, C. Doffek and M. Seitz, ChemPhysChem, 2019, 20, 2179–2186 CrossRef CAS PubMed.
- C. Kruck, P. Nazari, C. Dee, B. S. Richards, A. Turshatov and M. Seitz, Inorg. Chem., 2019, 58, 6959–6965 CrossRef CAS PubMed.
- C. Doffek and M. Seitz, Angew. Chem., Int. Ed., 2015, 54, 9719–9721 CrossRef CAS PubMed.
- D. Kovacs, X. Lu, L. S. Mészáros, M. Ott, J. Andres and K. E. Borbas, J. Am. Chem. Soc., 2017, 139, 5756–5767 CrossRef CAS PubMed.
- A. de Bettencourt-Dias, in Luminescence of Lanthanide Ions in Coordination Compounds and Nanomaterials, John Wiley & Sons Ltd, 2014, pp. 1–48 Search PubMed.
- J.-C. G. Bünzli and S. V. Eliseeva, in Lanthanide Luminescence: Photophysical, Analytical and Biological Aspects, ed. P. Hänninen and H. Härmä, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 1–45 Search PubMed.
- N. M. Shavaleev, L. P. Moorcraft, S. J. A. Pope, Z. R. Bell, S. Faulkner and M. D. Ward, Chem. Commun., 2003, 1134–1135 RSC.
- S. J. A. Pope and R. H. Laye, Dalton Trans., 2006, 3108–3113 RSC.
- J.-Y. Hu, Y. Ning, Y.-S. Meng, J. Zhang, Z.-Y. Wu, S. Gao and J.-L. Zhang, Chem. Sci., 2017, 8, 2702–2709 RSC.
- R. Xiong, D. Mara, J. Liu, R. Van Deun and K. E. Borbas, J. Am. Chem. Soc., 2018, 140, 10975–10979 CrossRef CAS PubMed.
- N. Hamon, A. Roux, M. Beyler, J.-C. Mulatier, C. Andraud, C. Nguyen, M. Maynadier, N. Bettache, A. Duperray, A. Grichine, S. Brasselet, M. Gary-Bobo, O. Maury and R. Tripier, J. Am. Chem. Soc., 2020, 142, 10184–10197 CrossRef CAS PubMed.
- J. H. S. K. Monteiro, N. R. Fetto, M. J. Tucker and A. de Bettencourt-Dias, Inorg. Chem., 2020, 59, 3193–3199 CrossRef CAS PubMed.
- S. Pandya, J. Yu and D. Parker, Dalton Trans., 2006, 2757–2766 RSC.
- A. Beeby, D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1996, 1565–1580 RSC.
- L. Norel, O. Galangau, H. Al Sabea and S. Rigaut, ChemPhotoChem, 2021, 5, 393–405 CrossRef CAS.
- Y. Ning, Y.-W. Liu, Y.-S. Meng and J.-L. Zhang, Inorg. Chem., 2018, 57, 1332–1341 CrossRef CAS PubMed.
- W. D. Horrocks Jr., J. P. Bolender, W. D. Smith and R. M. Supkowski, J. Am. Chem. Soc., 1997, 119, 5972–5973 CrossRef.
- C. Reinhard and H. U. Güdel, Inorg. Chem., 2002, 41, 1048–1055 CrossRef CAS PubMed.
- G. A. Crosby and M. Kasha, Spectrochim. Acta, 1958, 10, 377–382 CrossRef CAS.
- A. Beeby, S. Faulkner and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 2002, 1918–1922 RSC.
- D. Mara, F. Artizzu, P. F. Smet, A. M. Kaczmarek, K. Van Hecke and R. Van Deun, Chem. – Eur. J., 2019, 25, 15944–15956 CrossRef CAS PubMed.
- I. Hernández and W. P. Gillin, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. Bünzli and V. K. Pecharsky, Elsevier, 2015, vol. 47, pp. 1–100 Search PubMed.
- E. Kreidt, C. Kruck and M. Seitz, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, 2018, vol. 53, pp. 35–79 Search PubMed.
- S. V. Eliseeva, D. N. Pleshkov, K. A. Lyssenko, L. S. Lepnev, J.-C. G. Bunzli and N. P. Kuzmina, Inorg. Chem., 2011, 50, 5137–5144 CrossRef CAS PubMed.
- D. Kovacs, D. Kocsi, J. A. L. Wells, S. R. Kiraev and K. E. Borbas, Dalton Trans., 2021, 50, 4244–4254 RSC.
- D. Kocsi, D. Kovacs, J. A. L. Wells and K. E. Borbas, Dalton Trans., 2021, 50, 16670–16677 RSC.
- M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542–1548 RSC.
- A. Aebischer, F. Gumy and J.-C. G. Bünzli, Phys. Chem. Chem. Phys., 2009, 11, 1346–1353 RSC.
- D. Kovacs and K. E. Borbas, Coord. Chem. Rev., 2018, 364, 1–9 CrossRef CAS.
- D. Kovacs, E. Mathieu, S. R. Kiraev, J. A. L. Wells, E. Demeyere, A. Sipos and K. E. Borbas, J. Am. Chem. Soc., 2020, 142, 13190–13200 CrossRef CAS PubMed.
- J. W. Walton, R. Carr, N. H. Evans, A. M. Funk, A. M. Kenwright, D. Parker, D. S. Yufit, M. Botta, S. De Pinto and K.-L. Wong, Inorg. Chem., 2012, 51, 8042–8056 CrossRef CAS PubMed.
- E. R. Neil, A. M. Funk, D. S. Yufit and D. Parker, Dalton Trans., 2014, 43, 5490–5504 RSC.
- K. Mason, A. C. Harnden, C. W. Patrick, A. W. J. Poh, A. S. Batsanov, E. A. Suturina, M. Vonci, E. J. L. McInnes, N. F. Chilton and D. Parker, Chem. Commun., 2018, 54, 8486–8489 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- E. Mathieu, S. R. Kiraev, D. Kovacs, J. A. L. Wells, M. Tomar, J. Andres and K. E. Borbas, submitted.
- M. D. C. Teixeira, F. S. Felix, S. S. Thomasi, Z. M. Magriotis, J. M. da Silva, L. L. Okumura and A. A. Saczk, Microchem. J., 2019, 148, 66–72 CrossRef CAS.
- J. A. Therrien and M. O. Wolf, Inorg. Chem., 2017, 56, 1161–1172 CrossRef CAS PubMed.
- J. Andrez, G. Bozoklu, G. Nocton, J. Pécaut, R. Scopelliti, L. Dubois and M. Mazzanti, Chem. – Eur. J., 2015, 21, 15188–15200 CrossRef CAS PubMed.
- J. Andres and K. E. Borbas, Inorg. Chem., 2015, 54, 8174–8176 CrossRef CAS PubMed.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodriguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- J. Andres and A.-S. Chauvin, Phys. Chem. Chem. Phys., 2013, 15, 15981–15994 RSC.
- G. A. Hebbink, S. I. Klink, L. Grave, P. G. B. Oude Alink and F. C. J. M. Van Veggel, ChemPhysChem, 2002, 3, 1014–1018 CrossRef CAS PubMed.
- C. Yang, L.-M. Fu, Y. Wang, J.-P. Zhang, W.-T. Wong, X.-C. Ai, Y.-F. Qiao, B.-S. Zou and L.-L. Gui, Angew. Chem., Int. Ed., 2004, 43, 5010–5013 CrossRef CAS PubMed.
- W. T. Carnall, G. L. Goodman, K. Rajnak and R. S. Rana, J. Chem. Phys., 1989, 90, 3443–3457 CrossRef CAS.
- S. R. Kiraev, E. Mathieu, F. Siemens, D. Kovacs, E. Demeyere and K. E. Borbas, Molecules, 2020, 25, 5282 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2095001 and 2095002. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02266d |
| ‡ Current address: Laboratoire de Chimie de Coordination du CNRS, UPR 8241, 31077 Toulouse, France. |
| This journal is © The Royal Society of Chemistry 2022 |