
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient CRISPR-Cas9-based knockdown of RUNX2 to induce chondrogenic differentiation of stem cells†
Hye Jin
Kim‡
a,
Jong Min
Park‡
a,
Sujin
Lee
a,
Hui Bang
Cho
a,
Ji-In
Park
a,
Jae-Hwan
Kim
*b,
Ji Sun
Park
*a and
Keun-Hong
Park
 *a
*a
aLaboratory of Nano-regenerative Medicine, Department of Biomedical Science, College of Life Science, CHA University, CHA Biocomplex, Sampyeong-Dong, Bundang-gu, Seongnam-si, 13488, Republic of Korea. E-mail: pjs09@cha.ac.kr; pkh0410@cha.ac.kr
bMolecular Genetics, Department of Biomedical Science, College of Life Science, CHA University, 605, CHA Biocomplex, Sampyeong-Dong, Bundang-gu, Seongnam-si, 13488, Republic of Korea. E-mail: jaehwan_k@cha.ac.kr
First published on 14th December 2021
Abstract
The clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system recognizes and deletes specific nucleotide sequences in cells for gene editing. This study aimed to edit and knockdown the RUNX2 gene, a key transcription factor that is directly involved in all stages of stem cell differentiation into osteoblasts. The RUNX2 gene was depleted using the CRISPR-Cas9 system to inhibit osteoblast differentiation of stem cells. shRNA vectors targeting RUNX2 were used as a control. The surface of nanoparticles (NPs) was coated with the cationic polymer linear polyethyleneimine. Thereafter, negatively charged CRISPR-Cas9 and shRNA vectors were complexed with positively charged NPs via ionic interactions. Several analytical methods were used to determine the size, surface charge, and morphology of NPs and to characterize the complexed genes. NPs complexed with CRISPR-Cas9 and shRNA vectors were delivered into human mesenchymal stem cells (hMSCs) via endocytosis. The mRNA and protein expression patterns of various genes in hMSCs were measured over time following internalization of NPs complexed with CRISPR-Cas9 and shRNA vectors in two- and three-dimensional culture systems. Knockdown of the RUNX2 gene decreased osteogenic differentiation and increased chondrogenic differentiation of hMSCs. As a result of investigating the efficiency of NPs complexed with CRISPR-Cas9 (CASP-NPs), Runx2 effectively knocked down in mesenchymal stem cells to enhance differentiation into chondrocytes, therefore CASP-NPs proved to be an effective gene carrier in hMSCs.
Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR) are sequences specifically found in prokaryotes such as bacteria.1–3 They are a type of memory element created to protect organisms against external invading elements such as viruses and bacteriophages.4–6 CRISPR sequences prevent reinfection of external factors by specifically binding to DNA during reinvasion.7 CRISPR-related protein 9 (Cas9) uses the CRISPR sequence as a guide to recognize and cut a specific strand of DNA that is complementary to the CRISPR sequence.8 Due to their ability to remove specific genes, CRISPR-Cas9 can be used in a variety of biological applications for research and therapeutic purposes.9–14 Stem cells are undifferentiated cells found in embryonic and adult tissues.15,16 Among them, mesenchymal stem cells (MSCs) are pluripotent cells that can differentiate into various types of mesenchymal cells such as osteoblasts, chondrocytes, muscle cells, and adipocytes.17–20 Due to their self-renewal ability and pluripotency, MSCs are used as a cell source in various cell therapy fields including regenerative medicine. However, the use of MSCs is limited by their differentiation into unwanted cell types. This problem can be solved by deleting specific genes.21–24 CRISPR-Cas9 can reduce expression of and delete certain genes in stem cells. Silencing of certain genes can interfere with upstream signaling pathways and thereby regulate or prevent differentiation of stem cells into specific lineages.25 Consequently, decreased expression of specific genes can induce differentiation of stem cells into a desired lineage.26RUNX2 is a major transcription factor involved in all stages of stem cell differentiation into osteoblasts.27 Expression of RUNX2 is essential for differentiation of stem cells into bone cells. Thus, silencing of RUNX2 perturbs osteogenic differentiation of stem cells.28 Consequently, RUNX2 silencing facilitates differentiation of human MSCs (hMSCs) into other lineages such as cartilage and fat.
Our group previously tried to deliver a single shRNA vector targeting RUNX2 into hMSCs to reduce their osteogenic differentiation.29–31 In addition, shRNA vectors targeting RUNX2 and miRNA vectors have been simultaneously delivered.32 The single shRNA vector and both the shRNA and miRNA vectors were well delivered into hMSCs and yielded knockdown efficiencies of 30% and 50%, respectively. Both approaches reduce bone formation and promote cartilage formation by hMSCs.
In this study, we constructed a CRISPR-Cas9 vector to silence RUNX2. In addition, a shRNA vector targeting RUNX2 was constructed and delivered into hMSCs. The abilities of the CRISPR-Cas9 and shRNA vectors to knockdown RUNX2 and alter differentiation of hMSCs were compared. Dexamethasone-loaded nanoparticles (NPs, DNPs) with the ability to alleviate cytotoxicity and enhance differentiation were produced. The surface of DNPs was modified with linear polyethyleneimine (LPEI) and then the CRISPR-Cas9 and shRNA vectors were complexed with the DNPs. The sizes and shapes of CRISPR-Cas9 plasmid vector-complexed NPs (CASP-NPs) and shRNA plasmid vector-complexed NPs (shP-NPs) were analyzed by dynamic light scattering (DLS) and scanning electron microscopy (SEM). The delivery efficiencies of CASP-NPs and shP-NPs internalized by endocytosis were analyzed by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy according to the expression level of green fluorescent protein (GFP), which was encoding in each vector. The mRNA and protein levels of various markers were measured in two-dimensional (2D) and three-dimensional (3D) culture systems over time by reverse transcription-polymerase chain reaction (RT-PCR) and western blotting, respectively. Delivery of CASP-NPs into hMSCs rapidly increased the expression levels of chondrogenic markers such as collagen type II (COL II), SOX9, and aggrecan compared with the control group. The experimental methods and results are summarized in Scheme 1.
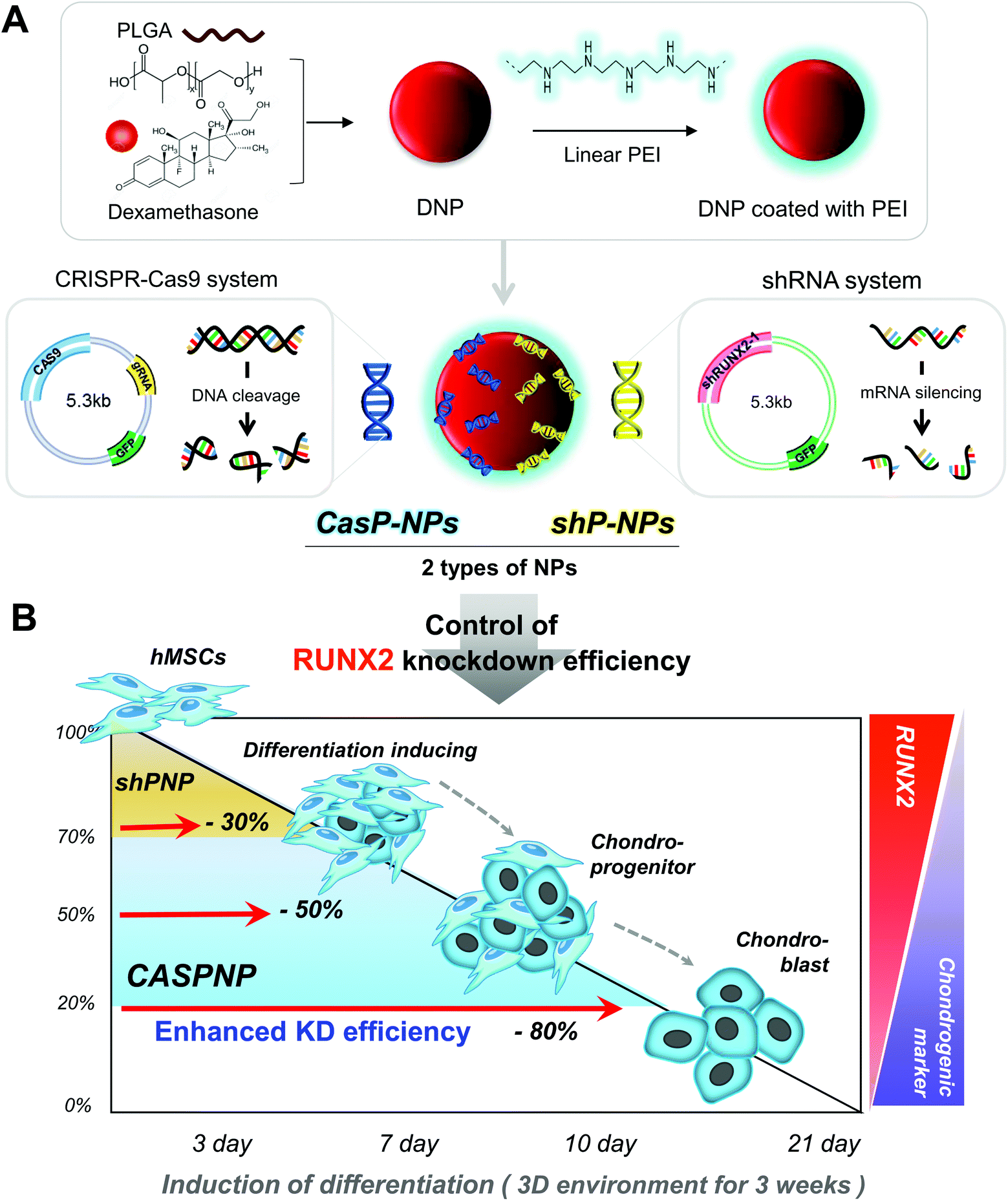 | ||
| Scheme 1 Preparation of vectors to knockdown RUNX2, their complexation with DNPs, and induction of chondrogenic differentiation of stem cells. A: Preparation of shRNA and CRISPR-Cas9 vectors to knockdown RUNX2 and their complexation with DNPs. B: Induction of chondrogenic differentiation by complexing various vectors with DNPs and introducing these DNPs into stem cells to knockdown RUNX2. | ||
Experimental
Materials
The biodegradable polymer PLGA (molecular weight of 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Evonik (Essen, Germany). Dexamethasone (molecular weight of 492.53) was purchased from Sigma-Aldrich (St Louis, MO, USA). The cationic polymer LPEI (25 kDa) was purchased from Polysciences (Warrington, PA, USA). Dulbecco's modified Eagle's medium, high glucose (DMEM-high), fetal bovine serum (FBS), and Dulbecco's phosphate-buffered saline (DPBS) were purchased from HyClone (Logan, Canada). Antibiotic–antimycotic solution and trypsin–EDTA were purchased from Gibco (Waltham, MA, USA). An EnGen mutation detection kit (NEB #E3321) was purchased from New England BioLabs (Ipswich, MA, USA).
000) was purchased from Evonik (Essen, Germany). Dexamethasone (molecular weight of 492.53) was purchased from Sigma-Aldrich (St Louis, MO, USA). The cationic polymer LPEI (25 kDa) was purchased from Polysciences (Warrington, PA, USA). Dulbecco's modified Eagle's medium, high glucose (DMEM-high), fetal bovine serum (FBS), and Dulbecco's phosphate-buffered saline (DPBS) were purchased from HyClone (Logan, Canada). Antibiotic–antimycotic solution and trypsin–EDTA were purchased from Gibco (Waltham, MA, USA). An EnGen mutation detection kit (NEB #E3321) was purchased from New England BioLabs (Ipswich, MA, USA).
Preparation of shRNA and CRISPR-Cas9 vectors
The vectors used in this study were synthesized by recombinant PCR methods and confirmed by nucleotide sequencing. The GFP-encoding shRNA vector was obtained by annealing a human RUNX2 oligonucleotide into the multiple cloning sites of pGSH1-GFP-Red2. The GFP-encoding CRISPR-Cas9 vector was obtained by annealing a human RUNX2 gRNA oligonucleotide into the multiple cloning sites of pSpCas9 (BB)-2A-GFP (RX458).Fabrication of shP-NPs and CASP-NPs
NPs were fabricated via a water-in-oil-in-water solvent evaporation technique using 100 mg of PLGA and 1 mg of dexamethasone. The surface of NPs was modified with LPEI to complex plasmid DNA via ionic interactions. DNPs, LPEI (6.5 μg), and plasmid DNA (shRNA and CRISPR-Cas9 vectors, 2 μg of each) were mixed to generate shP-NPs and CASP-NPs.Characterization of shP-NPs and CASP-NPs
The sizes and surface charges of shP-NPs and CASP-NPs were measured using Zetasizer Nano ZS apparatus (Malvern Panalytical, Malvern, UK). NPs were dispersed in distilled water and prepared in disposable capillary cells. Measurements were conducted three times per sample. The mean diameter, size distribution, and zeta-potential of NPs were determined.Cell culture
Bone marrow-derived hMSCs were purchased from Lonza (Walkersville, MD, USA) and cultured in DMEM-high (SH30022, HyClone) containing 10% FBS and 1% antibiotic–antimycotic solution in 5% CO2 at 37 °C. The culture medium was replaced every 2 days. Most experiments were conducted with cells at passage 7–8. Human MG63 cell line was purchased from ATCC (Virginia, USA) and cultured in DMEM-high containing 10% FBS and 1% antibiotic–antimycotic solution in 5% CO2 at 37 °C. The culture medium was replaced every 2 days.Assessment of cellular uptake of NPs
hMSCs were seeded in 6-well plates (1.5 × 105 per well) the day before FACS. Cells were pre-incubated in serum- and antibiotic–antimycotic-free medium for 30 min and then incubated with shP-NPs and CASP-NPs for 4 h at 37 °C. Thereafter, hMSCs were detached using trypsin–EDTA and suspended in DPBS containing 0.1% bovine serum albumin (BSA). Suspended cells were analyzed by FACS (Beckman Coulter, California, USA). Green fluorescence was detected in 10000 cells per sample.
Transfection
hMSCs were pre-incubated in DMEM-high lacking serum and antibiotics for 30 min prior to transfection of NPs. The optimal amount of NPs was complexed with shRNA and CRISPR-Cas9 vectors, added to hMSCs, and incubated for 4 h in 5% CO2 at 37 °C. The transfection efficiency was analyzed by detecting GFP signals using a confocal microscope (FV3000; Olympus, Japan) 1 day after transfection.RT-PCR
Total RNA was extracted from hMSCs using TRIzol according to a standard protocol. The concentration of RNA was measured using a spectrophotometer (Agilent Technologies, Amstelveen, Netherlands). Total RNA was reverse-transcribed into complementary DNA. PCR was performed under conditions suitable for each primer. The PCR products were electrophoresed and detected using a gel documentation imaging system (BR170-8265; Bio-Rad Laboratories, Korea).Western blotting
Total proteins were extracted with radioimmunoprecipitation cell lysis buffer. The concentration of extracted proteins was determined using a BCA protein assay kit. Proteins (35–40 μg per well) were loaded into a 10% SDS-polyacrylamide gel, electrophoresed at 80–100 V, and then transferred to a polyvinylidene fluoride membrane. After transfer of proteins, the membrane was blocked with 3% skim milk, incubated with a primary antibody diluted to a suitable concentration with 3% skim milk, and then incubated with a secondary antibody diluted 1:5000. Signals were detected on film using enhanced chemiluminescence.
3D cell culture
A total of 1 × 106 hMSCs were seeded into a 100 mm dish 1 day before transfection, transfected for 4 h, detached with trypsin–EDTA, transferred to a 15 ml conical tube, and centrifuged at 1300 rpm for 3 min. Centrifuged hMSCs were pelleted on the wall of the tube. Cell pellets were cultured for 3 weeks in DMEM-high containing 2% FBS and 1% antibiotic–antimycotic solution. The culture medium was changed every 3 days.Histology and immunofluorescence
Pellets of hMSCs cultured in a 3D system for 3 weeks were harvested, fixed with 4% paraformaldehyde for 1 day, embedded in optimal cutting temperature compound (TISSUE-TEK 4583; Sakura Finetek Inc., Torrance, CA, USA), and frozen at −20 °C for 1 h. Samples were sliced into sections (10 μm thick) using a cryotome (Leica CM3050S; Leica Biosystems). Sections were stained with hematoxylin and eosin to observe the morphology. Alcian blue, Safranin O, and Trichrome blue staining was performed according to standard protocols. Alternatively, sections were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 for 5 min, blocked with 1% BSA for 30 min, incubated with a primary antibody at room temperature for 2 h, and then stained with a fluorescent secondary antibody for 30 min. DAPI staining was performed for 5 min to label nuclei. Samples were visualized using a confocal microscope (FV3000, Olympus).Statistical analysis
Statistical analysis was performed using the Student's t-test. Statistical significance was defined as *P < 0.05, **P < 0.01, and ***P < 0.001.Results and discussion
Preparation of shRNA and CRISPR-Cas9 vectors targeting RUNX2 and characterization of CASP-NPs and shP-NPs
CRISPR-Cas9 and shRNA vectors were constructed to knockdown RUNX2 and thereby induce chondrogenic differentiation of hMSCs. Two different types of sgRNA and shRNA expression vectors were used for solid verification of experiments. The corresponding sequences are presented in Fig. 1. Briefly, hRUNX2 sgRNA-1 (nucleotide 434 to 453) and sgRNA-2 (nucleotide 534 to 553 from ATG start codon) were inserted into pSpCas9(BB)-2A-GFP (PX458) vector (Fig. S1A†). hRUNX2 shRNA-1 (nucleotide 1439 to 1457) and shRNA-2 (nucleotide 534 to 553) were inserted into pGSH1-GFP shRNA expression vector (Fig. S1B†). All sequences were confirmed by DNA sequencing.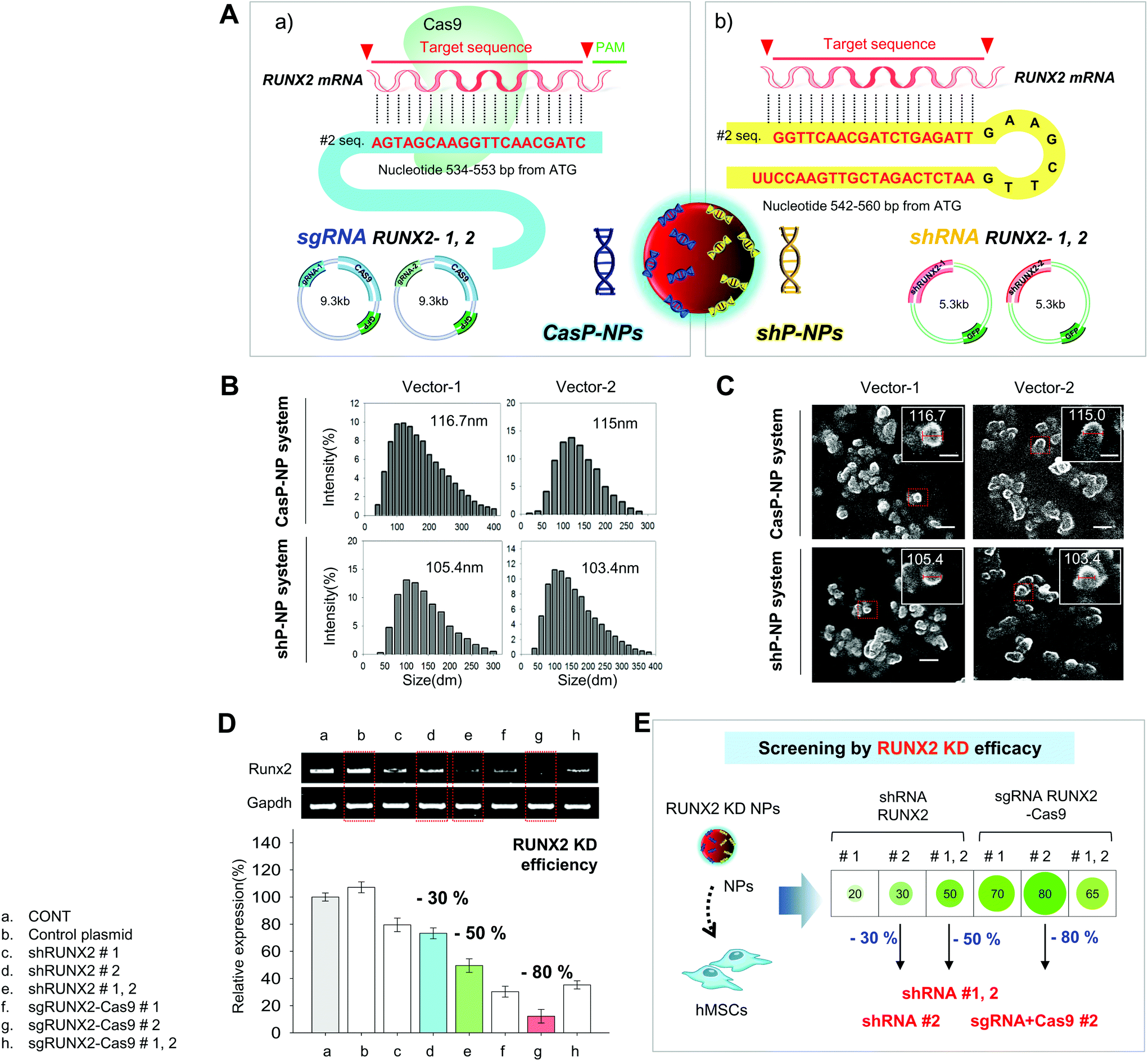 | ||
| Fig. 1 Characteristics and knockdown efficiencies of CASP-NPs and shP-NPs. A: Schematic diagram of the complexation of CRISPR-Cas9 (a) and shRNA (b) vectors with NPs. B: Analysis of the sizes of NPs complexed with various vectors by DLS. C: Analysis of the shapes of NPs complexed with various vectors by SEM. D: Analysis of the knockdown efficiencies of NPs complexed with various vectors by RT-PCR. E: Schematic diagram of RUNX2 knockdown using various shRNA and CRISPR-Cas9 vectors and combinations thereof. | ||
The constructed vectors were complexed with DNPs for delivery into cells (Fig. 1Aa and b). Before complexation with the constructed vectors, the surface of DNPs was coated with LPEI, a cationic polymer, and thus became positively charged. The modified DNPs were easily complexed with the vectors via ionic interactions. The constructed vectors were confirmed by electrophoresis (Fig. S2†).
DLS and SEM were performed to confirm that the resulting NPs were of a suitable size for delivery into cells. This revealed that the NPs were evenly dispersed and suitably sized. DLS demonstrated that the average diameters of NPs complexed with CRISPR-Cas9 vector #1, CRISPR-Cas9 vector #2, shRNA vector #1, and shRNA vector #2 were approximately 116, 115, 105, and 103 nm, respectively (Fig. 1B). As shown in the SEM images, it showed a change in the size of the complex consistent with the above DLS results (Fig. 1C).
A gel retardation assay was performed to analyze formation of complexes between NPs and each vector (Fig. S3†). An absent or faint band indicates that the NPs and vector readily complexed with each other. Since NP-complexed vectors can induce cytotoxicity, a concentration that does not induce cytotoxicity while easily complex to vector was used.
We measured the sizes and surface charges of NPs complexed with various vectors (Fig. S4†). NPs formed complexes with each of the six vector combinations tested, namely, each of the two shRNA vectors and a combination thereof and each of the two CRISPR-Cas9 vectors and a combination thereof. We further determined the transfection efficiency of these NPs (Fig. S5†). The transfection efficiency was higher than 70% in all groups except for the control group including an empty vector.
Knockdown of the Runx2 gene using the various NPs was determined by RT-PCR analysis (Fig. 1D). The knockdown efficiency was better using two shRNA vectors than using one shRNA vector and was much better using a CRISPR-Cas9 vector than using one or two shRNA vectors.
Fig. 1E shows a simplified representation of the knockdown efficiency achieved with each of the vectors and combinations thereof. The knockdown efficiency was highest (80%) using CRISPR-Cas9 vector #2 and was must better using CRISPR-Cas9 vectors than using shRNA vectors.
Cellular internalization of NPs and knockdown efficiencies in MG63 cells
A single shRNA vector and a combination of two shRNA vectors were selected as controls (Fig. 2A). NPs complexed with one and two shRNA vectors were called shPNP and shPNPs, respectively. NPs complexed with the CRISPR-Cas9 vector with the best knockdown efficiency were called CasPNP. MG63 cells cultured in a 2D system were transfected with these NPs. Each vector contained the sequence encoding GFP to track its intracellular delivery. GFP expression in the cytoplasm was indicative of cellular internalization of NPs. GFP expression was analyzed by confocal microscopy and FACS the day after transfection.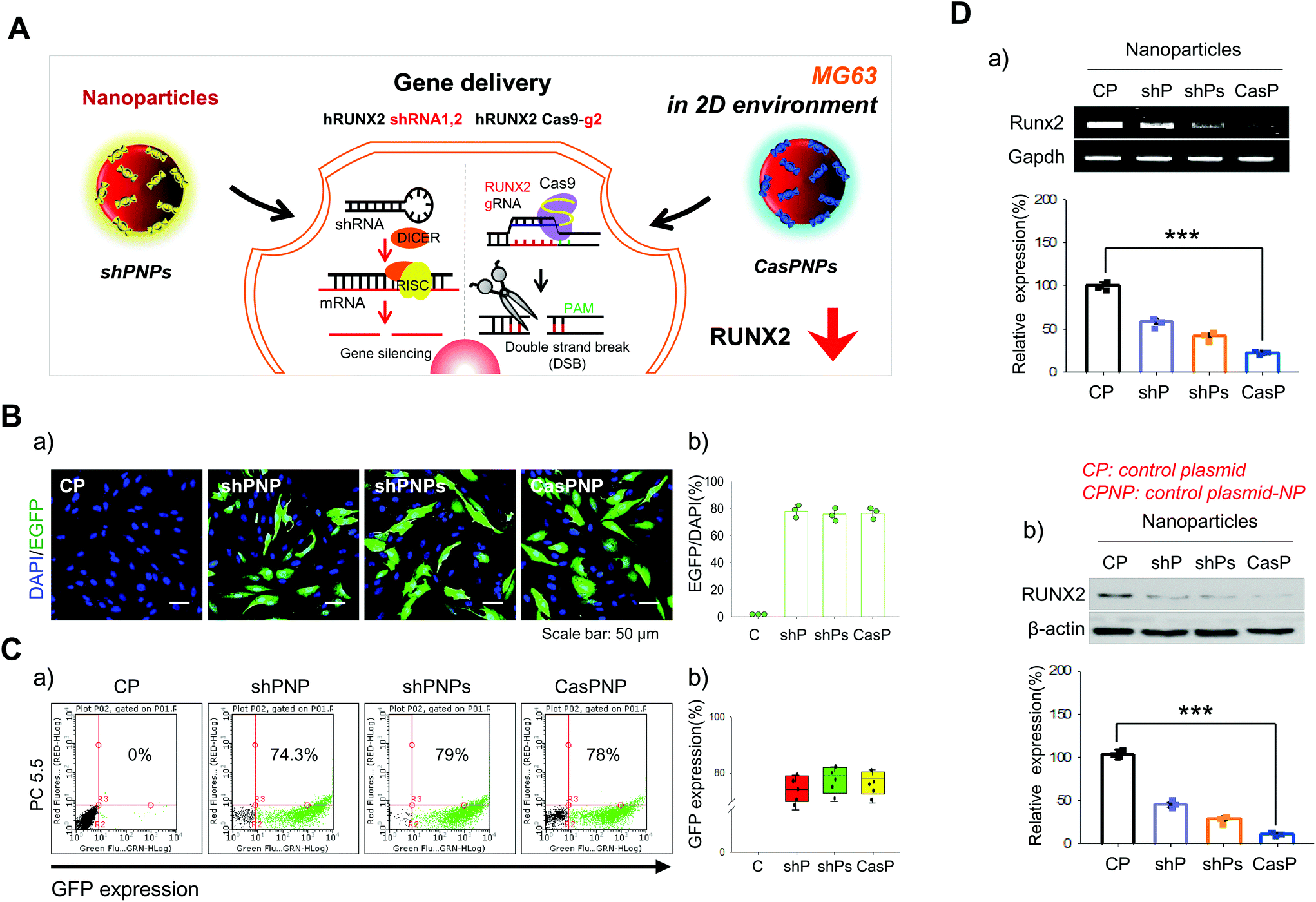 | ||
| Fig. 2 Internalization of shPNP, shPNPs, and CasPNP by MG63 cells and quantification of the knockdown efficiencies by RT-PCR and western blot analysis. A: Schematic diagram of internalization of CASP-NPs and shP-NPs and subsequent knockdown of RUNX2 in MG63 cells. B and C: Visualization (a) and quantification (b) of expression of EGFP-encoding shRNA and CRISPR-Cas9 vectors by confocal microscopy (B) and FACS (C). D: Analysis of mRNA and protein expression of RUNX2 in MG63 cells treated with control NPs (CP), shPNP, shPNPs, and CasPNP by RT-PCR (a) and western blotting (b). | ||
Green fluorescence was clearly observed in cells incubated with shPNP, shPNPs, and CasPNP (Fig. 2Ba) and was quantitated (Fig. 2Bb). Green fluorescence was observed in almost 80% of cells incubated with these NPs. FACS confirmed that 74%, 79%, and 78% of cells incubated with shPNP, shPNPs, and CasPNP expressed GFP, respectively (Fig. 2Ca). Quantitative analysis showed that GFP was expressed in almost 75% or a higher percentage of cells in the three groups (Fig. 2Cb). The LIVE/DEAD assay demonstrated that shPNP, shPNPs, and CasPNP did not cause cell death (Fig. S6†).
The knockdown efficiencies of the various NPs were compared by RT-PCR and western blot analysis (Fig. 2D). Treatment with shPNP, shPNPs, and CasPNP reduced expression of the RUNX2 gene by about 30%, 50%, and 70%, respectively, compared with the control. This confirmed that intracellular delivery of these NPs depleted expression of RUNX2.
We compared RUNX2 expression over time between cells that had internalized control NPs and CasPNP (Fig. S7†). Expression of RUNX2 was 70% lower in cells treated with CasPNP than in cells treated with control NPs at 72 h. Taken together, these results demonstrate that shPNP, shPNPs, and CasPNP were well delivered into cells and that the complexed vectors separated from NPs and suppressed expression of RUNX2.
Knockdown efficiencies in hMSCs
shPNP, shPNPs, and CasPNP were delivered to hMSCs in a 2D environment (Fig. 3A) and the knockdown efficiencies was investigated by several methods. To determine whether each vector was delivered into hMSCs, NPs were loaded with the fluorescent probe TRITC and tracked by detecting this fluorescent probe. Red fluorescence of TRITC was located throughout the cytoplasm, implying that NPs were delivered into hMSCs and localized to the cytoplasm (Fig. 3D).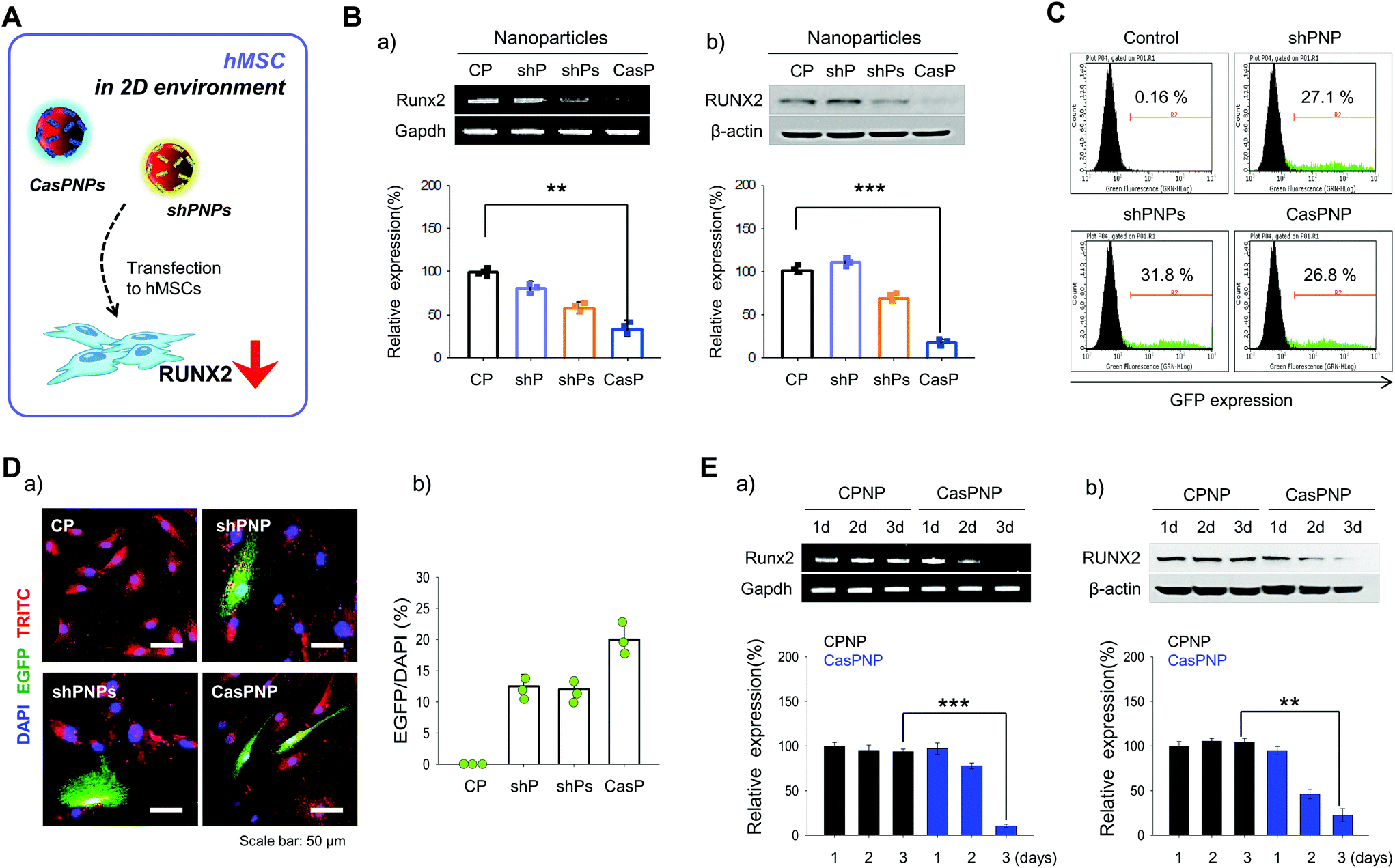 | ||
| Fig. 3 Internalization of shPNP, shPNPs, and CasPNP by hMSCs and quantification of the knockdown efficiencies by RT-PCR and western blot analysis. A: Schematic diagram of internalization of CASP-NPs and shP-NPs and subsequent knockdown of RUNX2 in hMSCs. B: Visualization (a) and quantification (b) of expression of EGFP-encoding shRNA and CRISPR-Cas9 vectors in hMSCs. C: Analysis of the transfection efficiencies of EGFP-encoding shRNA and CRISPR-Cas9 vectors in hMSCs by FACS. D: Analysis of mRNA and protein expression of RUNX2 in hMSCs treated with control NPs (CP), shPNP, shPNPs, and CasPNP by RT-PCR (a) and western blotting (b). E: Analysis of mRNA and protein expression of RUNX2 over time in hMSCs treated with control NPs (CPNP) and CasPNP by RT-PCR (a) and western blotting (b). | ||
Expression of the vectors complexed with NPs in hMSCs was investigated by FACS based on expression of GFP (Fig. 3C). In total, 27%, 31%, and 27% of cells expressed GFP upon treatment with shPNP, shPNPs, and CasPNP, respectively. This demonstrates that each vector could be complexed with NPs and delivered into hMSCs, and thereby silence RUNX2 expression.
The knockdown efficiencies of shPNP, shPNPs, and CasPNP were compared by RT-PCR and western blot analysis. Each vector complexed with NPs depleted RUNX2 expression in hMSCs (Fig. 3Ba and b). Delivery of shPNP, shPNPs, and CasPNP reduced RUNX2 expression by approximately 30%, 50%, and 70%, respectively, compared with the control. These results are similar to those obtained with MG63 cells. In summary, the vectors present in shPNP, shPNPs, and CasPNP suppressed expression of the RUNX2 gene in hMSCs.
Based on the previous finding that CasPNP were most efficiently delivered into cells, we compared RUNX2 expression between hMSCs treated with control NPs and CasPNP over time to determine the optimal incubation duration (Fig. 3Ea and b). RUNX2 expression in hMSCs decreased over time following delivery of CasPNP and was lowest after 3 days (Fig. S8†).
Internalization of NPs by hMSCs reduces bone formation by depleting RUNX2
Expression of the RUNX2, Dlx5, and ATF4 transcription factors, which are involved in differentiation of stem cells into bone cells, was investigated in hMSCs cultured in 2D and 3D systems to determine how knockdown of RUNX2 affects osteogenic differentiation (Fig. 4A). The effect of RUNX2 knockdown on osteoblast differentiation of hMSCs cultured in a 2D system was investigated by RT-PCR and western blot analysis. Delivery of shPNPs and CasPNP reduced mRNA and protein expression of RUNX2, Dlx5, and ATF4 in hMSCs (Fig. 4Ba and b). Quantitative analysis revealed that shPNPs and CasPNP reliably suppressed mRNA and protein expression of these transcription factors. Knockdown of RUNX2 was greatest using CasPNP.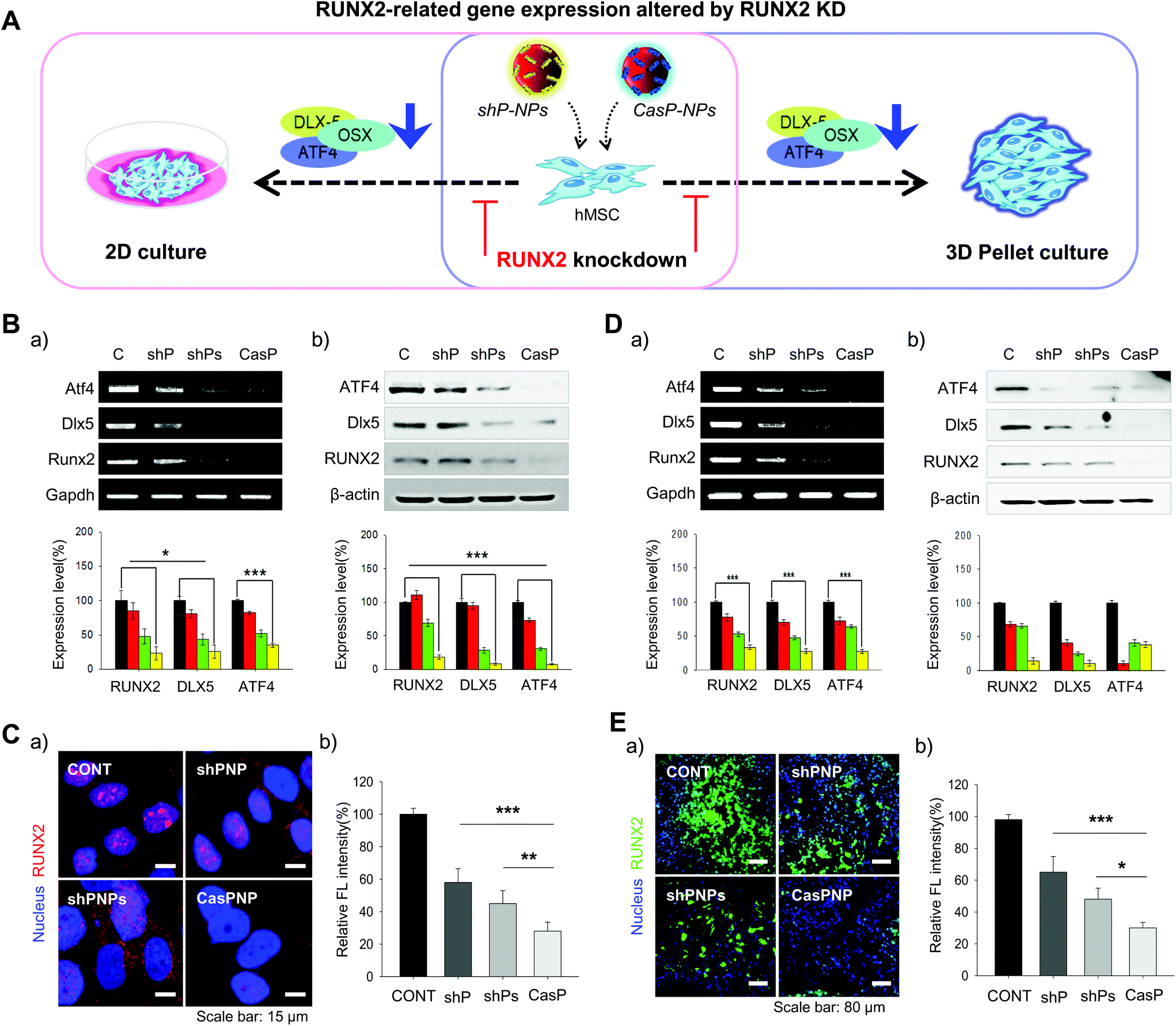 | ||
| Fig. 4 Osteogenic differentiation-related gene expression in hMSCs treated with shPNP, shPNPs, and CasPNP. A: Schematic diagram of inhibition of osteogenic differentiation-related gene expression in hMSCs cultured in 2D and 3D systems using shP-NPs and CASP-NPs. B and D: Analysis of mRNA and protein expression of ATF4, Dlx5, and RUNX2 in hMSCs treated with shPNP, shPNPs, and CasPNP by RT-PCR (a) and western blotting (b) under 2D (B) and 3D (D) culture conditions. C and E: Visualization (a) and quantification (b) of RUNX2 protein expression in hMSCs treated with shPNP, shPNPs, and CasPNP under 2D (C) and 3D (E) culture conditions. | ||
To investigate knockdown of RUNX2, we detected its nuclear expression via immunostaining (Fig. 4C). Red staining indicative of RUNX2 was detected in nuclei of control hMSCs, but was barely detected in nuclei of hMSCs treated with CasPNP. The CRISPR-Cas9 vector released from CasPNP depleted RUNX2 expression in hMSCs. The relative fluorescence intensity of RUNX2 immunostaining was nearly 80% lower in hMSCs treated with CasPNP than in control hMSCs.
The knockdown efficiency was also investigated in cells cultured in a 3D system. The effect of RUNX2 knockdown on osteoblast differentiation of hMSCs in the 3D system was investigated by RT-PCR and western blot analysis (Fig. 4Da and b). Delivery of shPNPs and CasPNP reduced mRNA and protein expression of RUNX2, Dlx5, and ATF4. Quantitative analysis revealed that shPNPs and CasPNP reliably suppressed mRNA and protein expression of these transcription factors. Knockdown of RUNX2 was greatest using CasPNP.
RUNX2 secreted by hMSCs in 3D pellets was stained and observed to confirm that it was reliably depleted (Fig. 4Ea). Green labeling indicative of RUNX2 was detected in all parts of pellets of control hMSCs, but was only faintly detected in pellets of hMSCs treated with CasPNP. These results demonstrate that delivery of CasPNP reduces expression of the RUNX2 gene and suppresses secretion of RUNX2 protein in hMSCs. The fluorescence intensity of RUNX2 immunolabeling was almost 80% lower in hMSCs treated with CasPNP than in control hMSCs (Fig. 4Eb).
Induction of chondrogenic differentiation of hMSCs by knockdown of RUNX2
Induction of chondrogenic differentiation of hMSCs by depletion of the RUNX2 gene was investigated. shP-NPs and CASP-NPs were used to knockdown RUNX2, which is the most important factor in osteoblast differentiation. This downregulated Dlx5, OSX, and ATF4, which are bone formation markers, and thus inhibited bone differentiation. Inhibition of osteogenic differentiation induced chondrogenic differentiation of hMSCs. This process is depicted in Fig. 5A.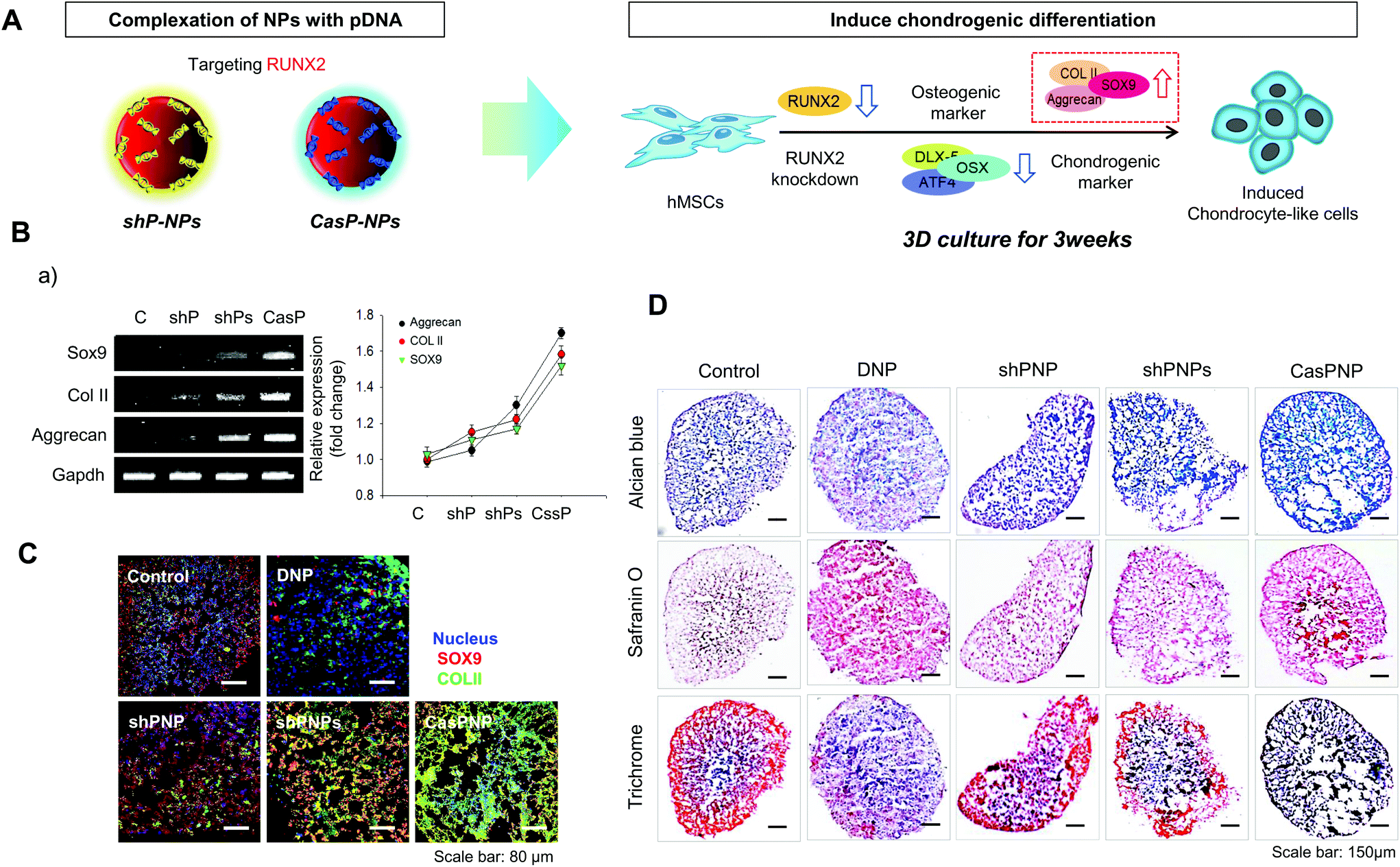 | ||
| Fig. 5 Chondrogenic differentiation-related gene expression in hMSCs treated with DNP, shPNP, shPNPs, and CasPNP. A: Schematic diagram of enhancement of chondrogenic differentiation-related gene expression upon knockdown of RUNX2 in hMSCs cultured in a 3D system. B: RT-PCR analysis (a) and quantification (b) of mRNA expression of cartilage-related genes in hMSCs treated with shPNP, shPNPs, and CasPNP in a 3D culture system. C: Immunofluorescence analysis (a) and quantitation (b) of SOX9 and COL II expression in hMSCs treated with DNP, shPNP, shPNPs, and CasPNP. D: Alcian blue, Safranin O, and Trichrome blue staining of hMSCs treated with DNP, shPNP, shPNPs, and CasPNP in a 3D culture system. | ||
Expression of SOX9, COL II, and aggrecan, which are markers of chondrogenic differentiation, was investigated by RT-PCR (Fig. 5B). Expression of these factors rapidly increased in hMSCs treated with shPNPs and CasPNP (Fig. 5Ba) and this was confirmed by quantitative analysis. The effects of CasPNP were greatest (Fig. 5Bb). SOX9 (red) and COL II (green) were barely detected by confocal laser microscopy in control hMSCs, but were readily detected in hMSCs treated with shPNPs and CasPNP (Fig. 5Ca). The expression level of each protein per cell was quantified and plotted. This revealed that the chondrogenic differentiation markers were highly expressed in hMSCs treated with CasPNP. In particular, expression of COL II, which is an important extracellular matrix component for construction of cartilage tissue, was 6-fold higher in hMSCs treated with CasPNP than in control hMSCs (Fig. 5Cb). These results demonstrate that knockdown of RUNX2 induces chondrogenic differentiation of hMSCs.
Histological analysis confirmed that hMSCs treated with CasPNP in a 3D system generated proteoglycans and polysaccharides (Fig. 5D). In addition, Trichrome blue staining revealed that hMSCs treated with CasPNP secreted a large amount of collagen, indicative of chondrogenic differentiation. Alizarin Red staining demonstrated that hMSCs treated with shPNP, shPNPs, and CasPNP in a 3D system did not differentiate into bone cells (Fig. S9†).
Conclusion
In this study, we constructed several vectors to knockdown the RUNX2 gene and thereby induce chondrogenic differentiation of stem cells and complexed these vectors with NPs to successfully deliver them into MG63 cells and hMSCs. Among them, CasPNP most effectively reduced RUNX2 expression and induced chondrogenic differentiation of hMSCs. These results suggest that knockdown of RUNX2 induces chondrogenic differentiation of hMSCs and is expected to have therapeutic applications such as cartilage regeneration.Author contributions
Keun-Hong Park, Ji Sun Park, and Jae-Hwan Kim devised the concept. Hye Jin Kim, Jong Min Park, and Ji Sun Park designed the experiments. Hye Jin Kim, and Jong Min Park carried out the experiments. Hye Jin Kim, Sujin Lee, and Hui Bang Cho drew the schematic figures. Ji-In Park cultured the cells. Hye Jin Kim, Jong Min Park, and Ji Sun Park, and Keun-Hong Park, wrote the paper.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean Government (NRF-2017M3A9C6061360, NRF-2019R1A6A1A03032888, and NRF-2020R1A2C3009783).Notes and references
- O. O. Abudayyeh, J. S. Gootenberg, S. Konermann, J. Joung, I. M. Slaymaker, D. B. Cox, S. Shmakov, K. S. Makarova, E. Semenova, L. Minakhin, K. Severinov, A. Regev, E. S. Lander, E. V. Koonin and F. Zhang, Science, 2016, 353, aaf5573 CrossRef PubMed.
- R. Barrangou, C. Fremaux, H. Deveau, M. Richards, P. Boyaval, S. Moineau, D. A. Romero and P. Horvath, Science, 2007, 315, 1709–1712 CrossRef CAS PubMed.
- L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini and F. Zhang, Science, 2013, 339, 819 CrossRef CAS PubMed.
- W. Jiang, I. Maniv, F. Arain, Y. Wang, B. R. Levin and L. A. Marraffini, PLoS Genet., 2013, 9, e1003844 CrossRef CAS PubMed.
- G. G. R. Sapranauskas, C. Fremaux, R. Barrangou, P. Horvath and V. Siksnys, Nucleic Acids Res., 2011, 39, 9275–9282 CrossRef PubMed.
- A. Szczepankowska, Adv. Virus Res., 2012, 82, 289–339 CAS.
- M. Terns, Mol. Cell, 2018, 72, 404–412 CrossRef CAS PubMed.
- P. S. Daisy, K. S. Shreyas and T. S. Anitha, Mol. Biotechnol., 2021, 63, 93–108 CrossRef CAS PubMed.
- A. Barman, B. Deb and S. Chakraborty, Curr. Genet., 2020, 66, 447–462 CrossRef CAS PubMed.
- C. Liu, L. Zhang, H. Liu and K. Cheng, J. Controlled Release, 2017, 266, 17–26 CrossRef CAS PubMed.
- F. Memi, A. Ntokou and I. Papangeli, Semin. Perinatol., 2018, 42, 487–500 CrossRef PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
- X. Xu, T. Wan, H. Xin, D. Li, H. Pan, J. Wu and Y. Ping, J. Gene Med., 2019, 21, e3107 CrossRef PubMed.
- X. Gao, Z. Jin, X. Tan, C. Zhang, C. Zou, W. Zhang, J. Ding, B. C. Das, K. Severinov, I. I. Hitzeroth, P. R. Debata, D. He, X. Ma, X. Tian, Q. Gao, J. Wu, R. Tian, Z. Cui, W. Fan, Z. Huang, C. Cao, Y. Bao, S. Tan and Z. Hu, J. Controlled Release, 2020, 321, 654–668 CrossRef CAS PubMed.
- M. F. Pittenger, A. M. Mackay, S. C. Beck, R. K. Jaiswal, R. Douglas, J. D. Mosca, M. A. Moorman, D. W. Simonetti, S. Craig and D. R. Marshak, Science, 1999, 284, 143–147 CrossRef CAS PubMed.
- C. C. Shih, S. J. Forman, P. Chu and M. Slovak, Stem Cells Dev., 2007, 16, 893–902 CrossRef CAS PubMed.
- G. Abagnale, M. Steger, V. H. Nguyen, N. Hersch, A. Sechi, S. Joussen, B. Denecke, R. Merkel, B. Hoffmann, A. Dreser, U. Schnakenberg, A. Gillner and W. Wagner, Biomaterials, 2015, 61, 316–326 CrossRef CAS PubMed.
- B. Hodgson, R. Mafi, P. Mafi and W. S. Khan, Curr. Stem Cell Res. Ther., 2018, 384–407 CrossRef CAS PubMed.
- S. N. Li and J. F. Wu, Stem Cell Res. Ther., 2020, 29, 41 CrossRef PubMed.
- X. Xu, Y. Liang, X. Li, K. Ouyang, M. Wang, T. Cao, W. Li, J. Liu, J. Xiong, B. Li, J. Xia, D. Wang and L. Duan, Biomaterials, 2021, 269, 120539 CrossRef CAS PubMed.
- V. Bunpetch, Z. Y. Zhang, X. Zhang, S. Han, P. Zongyou, H. Wu and O. Hong-Wei, Biomaterials, 2019, 203, 96–110 CrossRef PubMed.
- C. Brown, C. McKee, S. Bakshi, K. Walker, E. Hakman, S. Halassy, D. Svinarich, R. Dodds, C. K. Govind and G. R. Chaudhry, J. Tissue Eng. Regener. Med., 2019, 13, 1738–1755 CrossRef CAS PubMed.
- H. Lin, J. Sohn, H. Shen, M. T. Langhans and R. S. Tuan, Biomaterials, 2019, 203, 96–110 CrossRef CAS PubMed.
- B. Mathew, S. Ravindran, X. Liu, L. Torres, M. Chennakesavalu, C. C. Huang, L. Feng, R. Zelka, J. Lopez, M. Sharma and S. Roth, Biomaterials, 2019, 197, 146–160 CrossRef CAS PubMed.
- F. L. Cardenas-Diaz, J. A. Maguire, P. Gadue and D. L. French, J. Visualized Exp., 2019, 151, 60085 Search PubMed.
- J. Xiao, S. Qin, W. Li, L. Yao, P. Huang, J. Liao, J. Liu and S. Li, Life Sci., 2020, 253, 117660 CrossRef CAS PubMed.
- S. Wei, Q. Zou, S. Lai, Q. Zhang, L. Li, Q. Yan, X. Zhou, H. Zhong and L. Lai, Sci. Rep., 2016, 6, 19648 CrossRef CAS PubMed.
- T. K. T, Front. Biosci., 2008, 13, 898–903 CrossRef PubMed.
- S. Y. Jeon, J. S. Park, H. N. Yang, H. J. Lim, S. W. Yi, H. Park and K. H. Park, Biomaterials, 2014, 35, 8236–8248 CrossRef CAS PubMed.
- S. Y. Jeon, J. S. Park, H. N. Yang, D. G. Woo and K. H. Park, Biomaterials, 2012, 33, 4413–4423 CrossRef CAS PubMed.
- H. J. Kim, S. W. Yi, H. J. Oh, J. S. Lee, J. S. Park and K. H. Park, Biomaterials, 2018, 177, 1–13 CrossRef CAS PubMed.
- S. M. Kim, S. W. Yi, H. J. Kim, J. S. Park, J. H. Kim and K. H. Park, J. Biomed. Nanotechnol., 2019, 15, 113–126 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1bm01716k |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2022 |