
Guest-boosted phosphorescence efficiency of a supramolecular cage†
Zhi-Yin
Zhang
a,
Dong-Qin
Ye
a,
Qi-Qi
Gao
a,
Zhi-Chun
Shi
a,
Mo
Xie
 a,
Shun-Ze
Zhan
b,
Yong-Liang
Huang
c,
Guo-Hong
Ning
*a and
Dan
Li
*a
a,
Shun-Ze
Zhan
b,
Yong-Liang
Huang
c,
Guo-Hong
Ning
*a and
Dan
Li
*a
aCollege of Chemistry and Materials Science, Guangdong Provincial Key Laboratory of Functional Supramolecular Coordination Materials and Applications, Jinan University, Guangzhou, Guangdong 510632, P. R. China. E-mail: guohongning@jnu.edu.cn; danli@jnu.edu.cn
bDepartment of Chemistry and Key Laboratory for Preparation and Application of Ordered Structural Materials of Guangdong Province, Shantou University, Shantou 515063, P. R. China
cDepartment of Chemistry, Shantou University Medical College, Shantou, Guangdong 515041, P. R. China
First published on 1st March 2021
Abstract
Phosphorescent organic light emitting diodes (PhOLEDs) are required to achieve sufficiently high performance of devices; however, the synthesis of a phosphorescent emitter with high quantum efficiency and a short lifetime remains a great challenge. In this work, we synthesized a series of inclusion complexes composed of a phosphorescent Cu6L3 trigonal prismatic cage host and halobenzene guests. Interestingly, the quantum yield (QY) and emission lifetime of the inclusion complexes can be altered with the variation of halogen atoms in the guests due to the increment of spin–orbit coupling (SOC) and the promotion of intersystem crossing (ISC) processes via external heavy-atom effects (HAEs). For instance, on increasing the atomic number of the halogen from F to I, the QY enhanced six times (from 12.6 to 74.3%), while the lifetime reduced nearly two times (33.11 to 18.39 μs) at room temperture. Time-dependent density functional theory (TDDFT) calculations revealed that the encapsulation of iodobenzene guests can greatly promote the ISC processes, which are in good agreement with experimental results. Our studies demonstrated that a simple and efficient approach for designing promising phosphorescent emitters might be useful for PhOLED applications.
Introduction
The discovery of novel luminescent materials and development of new strategies to tune the photoluminescence (PL) properties are crucial for fabricating organic light emitting diodes (OLEDs) with superior quantum efficiency, brightness, long-term stability and chromaticity.1–4 Compared with conventional organic fluorescent emitters with a maximum of 25% internal quantum efficiency (IQE), phosphorescent emitters with noble metals such as platinum and iridium could deliver 100% IQE and lead to outstanding performance of OLEDs.5 Therefore, phosphorescent OLEDs (PhOLEDs) are promising for commercial applications and are attracting much attention.6–8 To obtain efficient high-brightness PhOLEDs, a high quantum yield (QY) and a short emission lifetime (e.g., in the range of submicroseconds to microseconds) at room temperature (rt) are particularly desired.9 In principle, promoting the intersystem crossing (ISC) processes from singlet excited states to triplet states would be beneficial for enhancing the phosphorescence QY or shortening the lifetime.10–13Cyclic trinuclear complexes (CTCs) containing heavy metal atoms, such as Hg(I), Au(I), Ag(I) and Cu(I), are able to add organic aromatics via π-acid⋯base interactions and the phosphorescence of an organic luminophore can be triggered by CTCs due to the external heavy-atom effect (HAE) (Scheme 1a).14–16 In addition, Omary et al. have reported that the phosphorescence QY and emission lifetimes of CTC/organic arene adducts can be fine-tuned through the gradual increase of the size of halogen substituents (i.e., from Cl and Br to I atoms) on the aromatics because of simultaneous external and internal HAEs.17 By utilizing these strategies, the highest phosphorescence QY of 70% with a long lifetime of 1.25 ms17 or the shortest lifetime of 29 μs with a low QY of 44%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 at rt can be achieved for CTC/organic arene adducts; so far, however, they are still far away from the ideal requirement for PhOLED applications. Moreover, the preparation of CTC/organic arene adducts is quite difficult and unpredictable; therefore, few examples have been reported.14–17
18 at rt can be achieved for CTC/organic arene adducts; so far, however, they are still far away from the ideal requirement for PhOLED applications. Moreover, the preparation of CTC/organic arene adducts is quite difficult and unpredictable; therefore, few examples have been reported.14–17
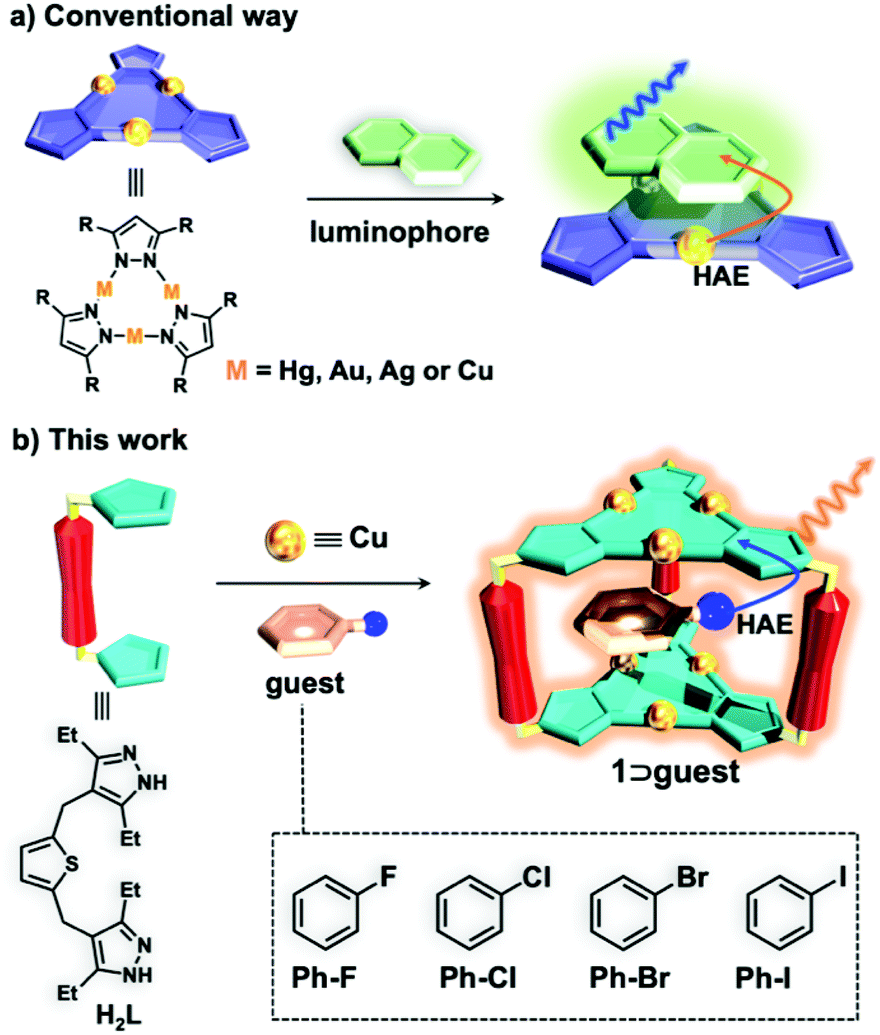 | ||
| Scheme 1 Conceptual representation of (a) triggering the phosphorescence of organic arenes utilizing the external HAEs of CTCs and (b) enhancing the phosphorescence of Cu6L3 cage 1 by the encapsulation of halogen aromatic guests. | ||
A supramolecular coordination cage with a confined cavity is an excellent platform for the investigation of host–guest interactions and the encapsulation of the guest can readily affect the photophysical and photochemical properties of inclusion complexes.19–24 Recently, our group synthesized a phosphorescent trigonal prismatic Cu6L3 cage 1 with two Cu(I) CTC units as the roof and floor (Scheme 1b).25 By tailoring the ionization potential and volume occupancy of aromatic guests, the PL properties of 1 including the maximum emission peak and QY can be fine-tuned. However, only a very low QY of 14% has been demonstrated.25 Inspired by the tunable phosphorescent cage host 1, we envision that the inclusion of halogen aromatic guests could promote the ISC process of cage 1via external HAEs (Scheme 1b), thereby enhancing the phosphorescence QY and reducing the emission lifetime.
Herein, we prepared four inclusion complexes composed of Cu6L3 cage host 1 and halogen aromatic guests and their structures were all confirmed by single X-ray crystallographic analyses. With the increment of the spin–orbit coupling constant (ζ) of halobenzene (i.e., 272, 587, 2460 and 5060 cm−1 for fluorobenzene (Ph-F), chlorobenzene (Ph-Cl), bromobenzene (Ph-Br), and iodobenzene (Ph-I), respectively),26 the phosphorescence QY of the inclusion complexes is enhanced 6 times from 12.6% to 74.3% and the lifetimes are reduced 2 times from 30.11 to 18.39 μs at rt, which provide potential for phosphorescent OLED applications. To the best of our knowledge, such a high phosphorescence QY with a microsecond lifetime has never been achieved in the field of metal–organic supramolecular cages (Table S1† and Fig. 2).25,27–30 More interestingly, the phosphorescence QY of the inclusion complexes exhibits a good linear relationship with the atomic number of the halogen atom. Furthermore, time-dependent density functional theory (TDDFT) calculations revealed that the encapsulation of iodobenzene guests can greatly promote the ISC processes, which are in good agreement with experimental results. Our work demonstrated a new approach for great enhancement of the phosphorescence QY and the reduction of lifetimes by a simple alteration of guest molecules, which might be explored for other luminescent materials for fabricating PhOLEDs with high performance.
Results and discussion
To verify our hypothesis, a series of halobenzene guests (i.e., Ph-F, Ph-Cl, Ph-Br, and Ph-I) have been chosen (Scheme 1b). According to our previous synthetic method,25 single crystals of the inclusion complexes 1⊃Ph-X (X = F, Cl, Br, and I) were obtained by the solvothermal reaction of H2L and Cu2O in acetonitrile/halobenzene for 72 h with high yields of up to 62% (see the ESI† for details). As expected, the 1H-NMR spectra of the inclusion complexes in CD2Cl2 confirmed the 1:1 stoichiometry between host 1 and guests, which consisted of the crystal structures (Table S5 and Fig. S2–5†). In addition, the phase purity and thermal stability of the inclusion complexes were confirmed by powder X-ray diffraction (PXRD) analysis, elemental analysis, infrared spectroscopy and thermogravimetric analysis (TGA) (Fig. S6–8†).
The inclusion complexes of 1⊃Ph-Br and 1⊃Ph-I were crystallized in the triclinic space group of P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , while 1⊃Ph-F and 1⊃Ph-Cl were crystallized in the monoclinic space group of P21/c and P21, respectively. The single crystal X-ray diffraction (SCXRD) analysis of these host–guest complexes revealed a similar hexanuclear prismatic cage 1 composed of two bending and distorted Cu(I) CTC units and three ligands (Fig. 1). The distance between the center of two Cu3N6 rings was changed in a range of 6.724 to 7.000 Å (Table S4†) with the alteration of guest molecules, showing no intertrimer Cu⋯Cu interaction within the cage.14 In addition, the cavity volume of cage 1 was changed in a range between 248.76 Å3 and 272.16 Å3 with the variation of guests due to the flexibility of host 1. The halobenzene guests were encapsulated within the cavity of cage 1 to give a CTC–halobenzene–CTC structure and the distances between the plane of aromatic rings and the center of Cu3N6 rings ranged from 2.939 to 3.911 Å, suggesting strong Cu3⋯π interactions (Fig. 1a–d).14 Moreover, cage 1 units in all inclusion crystals were stacked tightly to form an infinite column structure through supramolecular interactions including intermolecular Cu⋯Cu interactions, CH/π interactions, and van der Waals force (Fig. 1a–d). Specifically, the shortest intermolecular Cu⋯Cu distance (dCu⋯Cu) between two neighboring cage units in 1⊃Ph-F and 1⊃Ph-Cl is 2.962 and 2.974 Å, respectively, which is significantly shorter than those in 1⊃Ph-Br (3.155 Å) and 1⊃Ph-I (3.151 Å). Such structural features suggest that 1⊃Ph-Br and 1⊃Ph-I exhibit weaker intermolecular Cu⋯Cu interactions compared to 1⊃Ph-F and 1⊃Ph-Cl. More importantly, short intermolecular Cu⋯X distances (dCu⋯X) (X = F, Cl, Br, and I) in a range from 3.496 to 3.775 Å are observed (Fig. 1e and f), which is close enough to facilitate the ISC process of host 1via external HAEs.31 The DFT-calculated electrostatic potential (ESP) of the inclusion complexes based on their crystal structures shows that the charges on the isosurface of 1⊃Ph-F and 1⊃Ph-Cl are relatively well distributed and indicate weak π-basicity (Fig. S18†). It is beneficial to reduce the electrostatic repulsion, thereby forming a more compact intermolecular Cu⋯Cu interaction in 1⊃Ph-F and 1⊃Ph-Cl. The accommodation of Ph-Br and Ph-I guests significantly increases the electron density on cage surfaces, resulting in strong π-basicity and weaker intermolecular Cu⋯Cu interactions.
, while 1⊃Ph-F and 1⊃Ph-Cl were crystallized in the monoclinic space group of P21/c and P21, respectively. The single crystal X-ray diffraction (SCXRD) analysis of these host–guest complexes revealed a similar hexanuclear prismatic cage 1 composed of two bending and distorted Cu(I) CTC units and three ligands (Fig. 1). The distance between the center of two Cu3N6 rings was changed in a range of 6.724 to 7.000 Å (Table S4†) with the alteration of guest molecules, showing no intertrimer Cu⋯Cu interaction within the cage.14 In addition, the cavity volume of cage 1 was changed in a range between 248.76 Å3 and 272.16 Å3 with the variation of guests due to the flexibility of host 1. The halobenzene guests were encapsulated within the cavity of cage 1 to give a CTC–halobenzene–CTC structure and the distances between the plane of aromatic rings and the center of Cu3N6 rings ranged from 2.939 to 3.911 Å, suggesting strong Cu3⋯π interactions (Fig. 1a–d).14 Moreover, cage 1 units in all inclusion crystals were stacked tightly to form an infinite column structure through supramolecular interactions including intermolecular Cu⋯Cu interactions, CH/π interactions, and van der Waals force (Fig. 1a–d). Specifically, the shortest intermolecular Cu⋯Cu distance (dCu⋯Cu) between two neighboring cage units in 1⊃Ph-F and 1⊃Ph-Cl is 2.962 and 2.974 Å, respectively, which is significantly shorter than those in 1⊃Ph-Br (3.155 Å) and 1⊃Ph-I (3.151 Å). Such structural features suggest that 1⊃Ph-Br and 1⊃Ph-I exhibit weaker intermolecular Cu⋯Cu interactions compared to 1⊃Ph-F and 1⊃Ph-Cl. More importantly, short intermolecular Cu⋯X distances (dCu⋯X) (X = F, Cl, Br, and I) in a range from 3.496 to 3.775 Å are observed (Fig. 1e and f), which is close enough to facilitate the ISC process of host 1via external HAEs.31 The DFT-calculated electrostatic potential (ESP) of the inclusion complexes based on their crystal structures shows that the charges on the isosurface of 1⊃Ph-F and 1⊃Ph-Cl are relatively well distributed and indicate weak π-basicity (Fig. S18†). It is beneficial to reduce the electrostatic repulsion, thereby forming a more compact intermolecular Cu⋯Cu interaction in 1⊃Ph-F and 1⊃Ph-Cl. The accommodation of Ph-Br and Ph-I guests significantly increases the electron density on cage surfaces, resulting in strong π-basicity and weaker intermolecular Cu⋯Cu interactions.
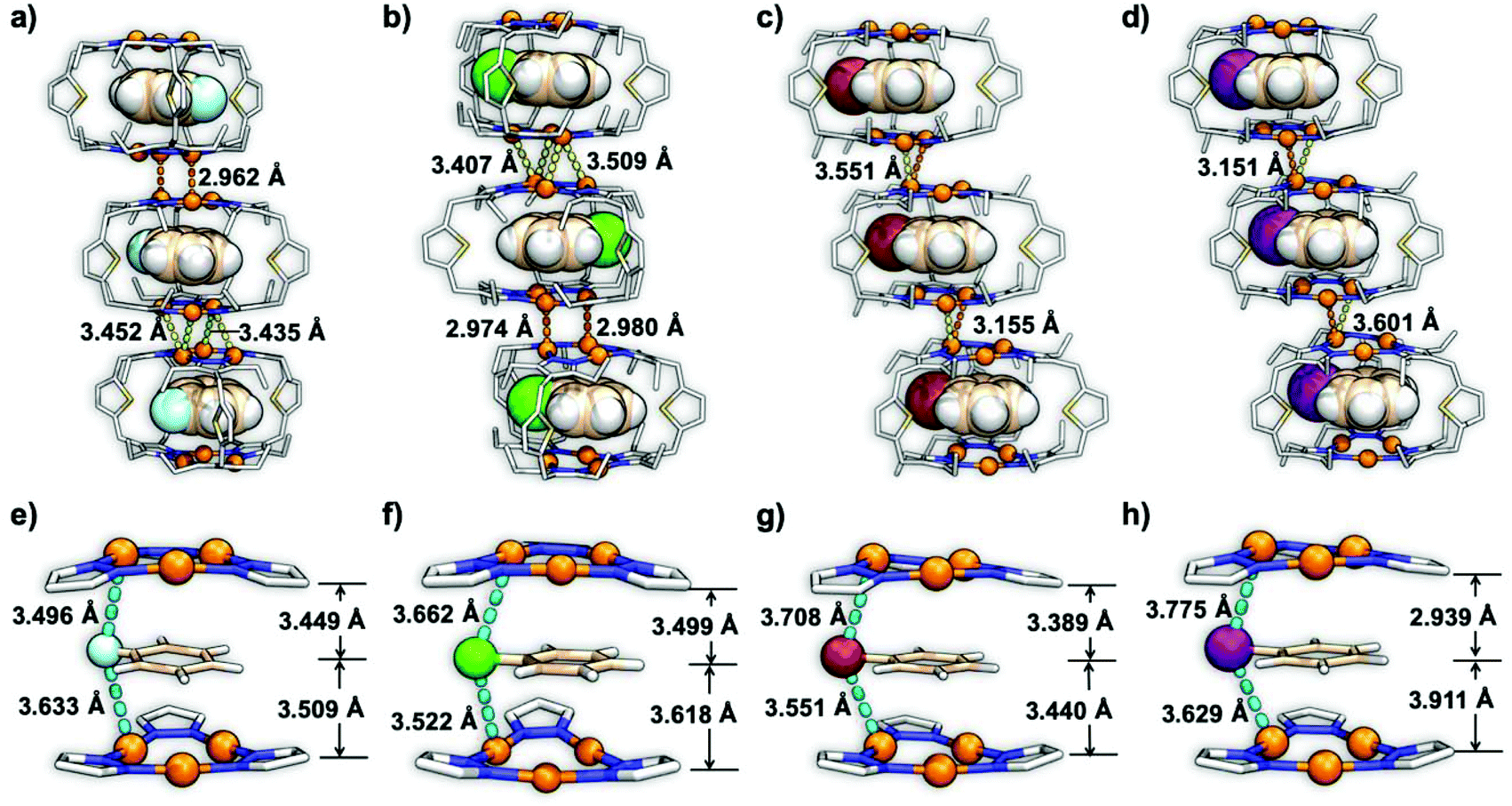 | ||
| Fig. 1 Crystal structures of (a) 1⊃Ph-F, (b) 1⊃Ph-Cl, (c) 1⊃Ph-Br, and (d) 1⊃Ph-I, showing the Cu3L6 cage units in one packed column with intermolecular Cu⋯Cu distances of 2.962, 3.452 and 3.435 Å in 1⊃Ph-F, 3.407, 3.509, 2.974 and 2.980 Å in 1⊃Ph-Cl, 3.551 and 3.155 Å in 1⊃Ph-Br, and 3.151 and 3.601 Å in 1⊃Ph-I, respectively (Cu(I) ions, cage hosts 1, and halobenzene guests are shown as the ball, line and space-filling model, respectively; hydrogen atoms in cage hosts 1 are omitted for clarity; C, N, S, and Cu atoms in the cage are shown in gray, blue, yellow, and orange colours, respectively; C, H, F, Cl, Br and I atoms in the guests are shown in wheat, white, light cyan, green, brown and purple colours, respectively); CTC–guest–CTC structures of (e) 1⊃Ph-F, (f) 1⊃Ph-Cl, (g) 1⊃Ph-Br, and (h) 1⊃Ph-I, showing intermolecular Cu⋯X distances of 3.496 and 3.633 Å in 1⊃Ph-F, 3.662 and 3.522 Å in 1⊃Ph-Cl, 3.778 and 3.551 Å in 1⊃Ph-Br, and 3.775 and 3.629 Å in 1⊃Ph-I, respectively (Cu(I) ions, halogen atoms, CTC units, and guests are shown as the ball and line model, respectively). | ||
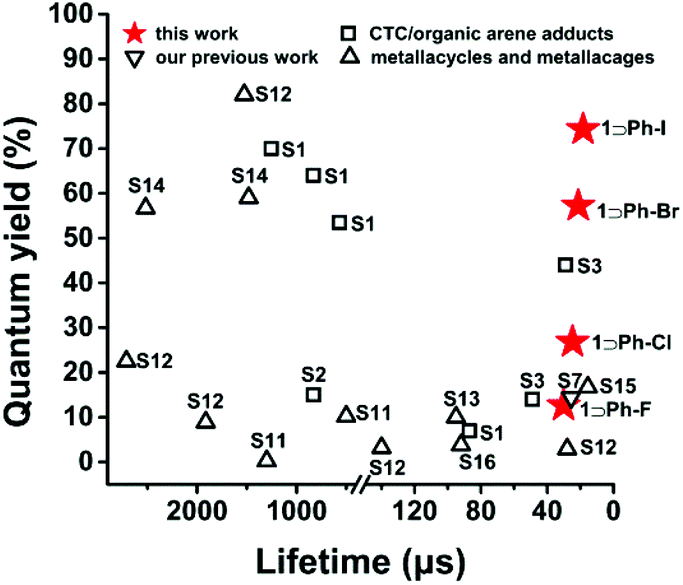 | ||
| Fig. 2 A comparison of the phosphorescence quantum yield and emission lifetime for reported CTC complexes and coordination macrocylces and cages (S1 to S16 represent the reference S1–S16 in ESI†). | ||
The UV-vis absorption spectra (Fig. S9†) of all inclusion complexes in the solid state show strong absorption bands at 250 to 280 nm corresponding to π–π* transitions of the ligand. The broad absorption bands appearing at 325 to 350 nm can be ascribed to metal-to-ligand charge-transfer (MLCT) transitions. All these inclusion complexes presented phosphorescence behavior at rt with lifetimes (τ) at the microsecond level. The solid-state photoluminescence (PL) spectrum of 1⊃Ph-I at rt exhibits low-energy (LE) dominated broad emission bands at 640 nm attributed to metal–metal interaction based phosphorescence and weak high-energy (HE) structural emission bands in a range of 450–550 nm assigned to the ligand-centred (LC) emission, which are similar to those of the proligand (Fig. 3b). On increasing the excitation energy, LE bands gradually quenched concomitant with a slight enhancement of HE bands, as shown in the varied-excitation wavelength PL spectra (Fig. 3b). 1⊃Ph-F, 1⊃Ph-Cl, and 1⊃Ph-Br show similar PL behaviors to 1⊃Ph-I, and display an LE dominated broad emission peak located at 672, 673, and 645 nm, respectively (Fig. 3a). It is worth mentioning that the red-shift emission of 1⊃Ph-F and 1⊃Ph-Cl compared with that of 1⊃Ph-Br and 1⊃Ph-I can be explained by cooperative effects between intermolecular Cu⋯Cu interactions and the ionization potential of guests, which have been demonstrated in our previous studies.25 Furthermore, the emission intensity of 1⊃Ph-F, 1⊃Ph-Cl, 1⊃Ph-Br, and 1⊃Ph-I slightly reduced with the increase of the temperature from 77 to 375 K.
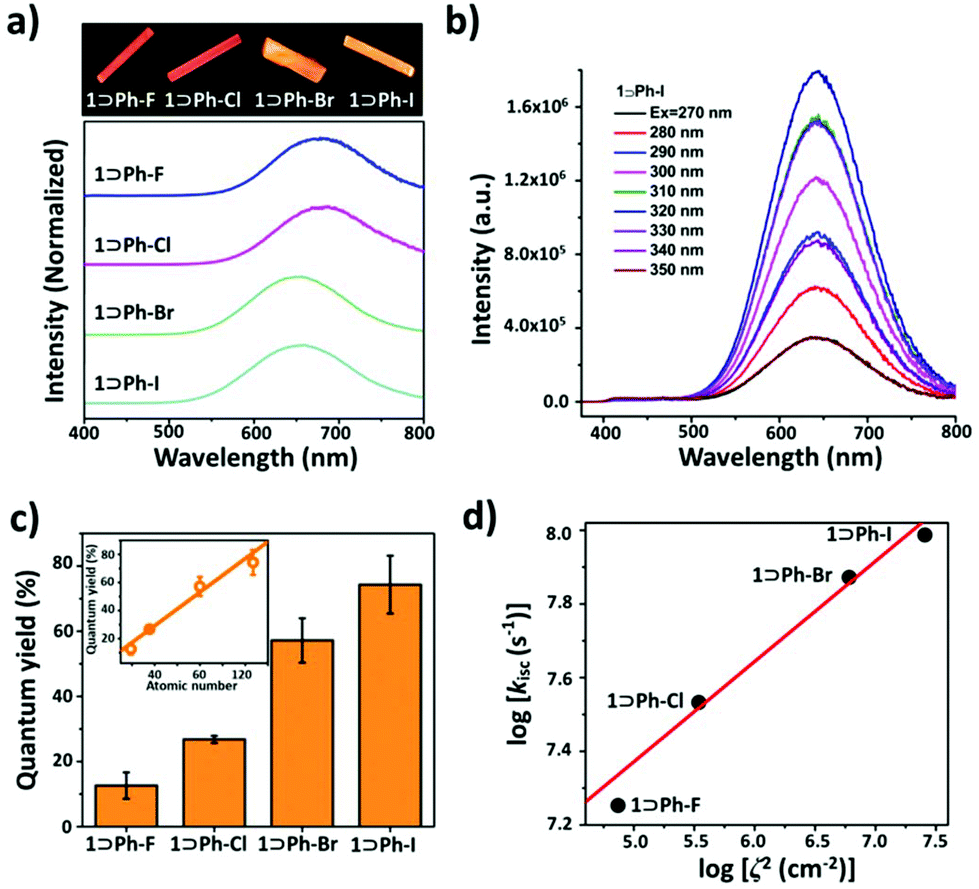 | ||
| Fig. 3 (a) Luminescence photographs of inclusion crystals under 365 nm UV irradiation and emission spectra of the inclusion complexes; (b) excitation-energy-varied emission spectra of 1⊃Ph-I; (c) quantum yield of the inclusion complexes (inset: linear relationship between the quantum yield and atomic number of halogen in the guest); and (d) linear relationship between the logarithm of intersystem crossing rate constants (log(kisc)) in the inclusion complexes and logζ2. | ||
The introduction of the halogen atom greatly affects the phosphorescence properties of cage 1. By replacing one hydrogen atom with fluorine on benzene, the phosphorescence QY remains almost unchanged (12.6% for 1⊃Ph-F, and 14.4% for the inclusion complex accommodating benzene25). However, the phosphorescence QY remarkably enhanced and followed the order of 1⊃Ph-Cl (26.8%) < 1⊃Ph-Br (57.2%) < 1⊃Ph-I (74.3%) by utilizing heavier halogen atoms (Table 1). Interestingly, the phosphorescence QY of the inclusion complexes exhibits a linear relationship with the atomic number of halogen atoms (Fig. 3c). In addition, the QY can further increase with the decrease of temperature, especially, the QY of 1⊃Ph-I can reach a near-unit QY of 97% (Table 1) at 77 K. More importantly, their lifetimes are also significantly reduced from 30.11 to 18.39 μs at 298 K upon the change of halogen substituents on the benzene ring from F to I, which further confirms their phosphorescence nature. These results demonstrated that the phosphorescence QY and lifetime of the inclusion complexes can be fine-tuned with the alteration of halogen atoms on guests.
| Complexes | Shortest Cu⋯X distance (Å) | ζ /cm | λ ex (nm) | λ em (nm) | τ P (μs) | τ F (ns) | Φ (%) | k r × 104 (s−1) | k nr × 104 (s−1) | k isc × 107 (s−1) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 298 K | 77 K | 298 K | 77 K | |||||||||
| a Spin–orbit coupling constant; see ref. 26. b τ P is the average phosphorescence decay lifetime, and τF is the average fluorescence decay lifetime. c Φ is the average quantum yield measured at 298 K. d Radiative decay rate constant: kr = Φ/τP. e Non-radiative decay rate constant: knr = (1 − Φ)/τP. f Intersystem crossing rate constant: kisc = Φ/τF the temperature effects did not consider here. | ||||||||||||
| 1⊃Ph-F | 3.496 | 272 | 316 | 672 | 30.11 | 31.01 | 7.04 | 12.6 | 19.6 | 0.42 | 2.90 | 1.79 |
| 1⊃Ph-Cl | 3.522 | 587 | 310 | 673 | 24.75 | 24.49 | 7.86 | 26.8 | 46.9 | 1.08 | 2.96 | 3.41 |
| 1⊃Ph-Br | 3.551 | 2460 | 310 | 645 | 21.29 | 20.73 | 7.68 | 57.2 | 87.0 | 2.69 | 2.01 | 7.45 |
| 1⊃Ph-I | 3.629 | 5060 | 320 | 640 | 18.39 | 18.36 | 7.65 | 74.3 | 96.9 | 4.04 | 1.40 | 9.71 |
It has been proved that, in the case of external HAEs, the atomic number or SOC constant of the heavy atom is a crucial factor for strengthening ISC in the luminophore reported by Kasha.32 Thus, to better understand the mechanism, the ISC rate constants (kisc) were calculated on the basis of the measured phosphorescence QY and lifetime.33 The kisc value of 1⊃Ph-I (9.71 × 107 s−1) increased 5.4 times than that of 1⊃Ph-F (1.79 × 107 s−1) (Table 1). As shown in Fig. 3d, the logarithm of kisc of the inclusion complexes increases with the increment of logζ2, showing good linear correlation. Such results indicated that the SOC in the inclusion complexes was enhanced with the increment of the ζ values of guests; thus, ISC processes between singlet and triplet states were accelerated via external HAEs. In addition, the logarithm of the triplet radiative decay rate constants (kr) and logζ2 also shows a linear relationship, suggesting that the transition from triplet excited states to ground states is remarkably promoted by the SOC in halobenzene guests (Fig. S16†). For instance, kr increased almost 10 times from 4.18 × 103 s−1 to 4.04 × 104 s−1 and followed the order of 1⊃Ph-F < 1⊃Ph-Cl < 1⊃Ph-Br < 1⊃Ph-I (Table 1), implying that radiative decay processes from triplet excited states to ground states in the inclusion complexes are enhanced by external HAEs. In particular, 1⊃Ph-I features the fastest ISC rate and radiative decay rate, resulting in the highest quantum efficiency of 74.3% and the shortest lifetime of 18.39 μs.
To further understand the mechanism of external HAEs, time-dependent density functional theory (TDDFT) calculations were performed to reveal the nature of singlet and triplet states of the inclusion complex 1⊃Ph-X. Generally, the small energy difference and matching transition components of singlet and triplet states are conducive to the ISC process.34 As shown in Fig. 4, the inclusion complex 1⊃Ph-I exhibited more than five triplet states (e.g., T32, T33, T34, T35, and T36) close to the energy of its S1, suggesting the presence of many potentially effective ISC channels in the excited state. In contrast, the potential ISC channels for 1⊃Ph-F and 1⊃Ph-Cl are much less than those of 1⊃Ph-I (Fig. S19–22†), further confirming that the promotion of the ISC processes followed the order of 1⊃Ph-F < 1⊃Ph-Cl < 1⊃Ph-Br < 1⊃Ph-I. More importantly, the electron density difference (EDD) diagram between the excited state and the ground state (Tables S7–S14†) revealed that the guests Ph-F and Ph-Cl did not participate in the charge transfer transition of the triplet state (3XLCT), and the guest Ph-Br slightly participated in 3XLCT, while the guest Ph-I greatly contributed to 3XLCT, suggesting a great increment of the SOC and promotion of the ISC processes with the increase of the halogen atom size. Theoretical results further supported that the introduction of the halogen atom on the guests can accelerate the ISC processes in the excited state and enhance the efficiency of phosphorescence.
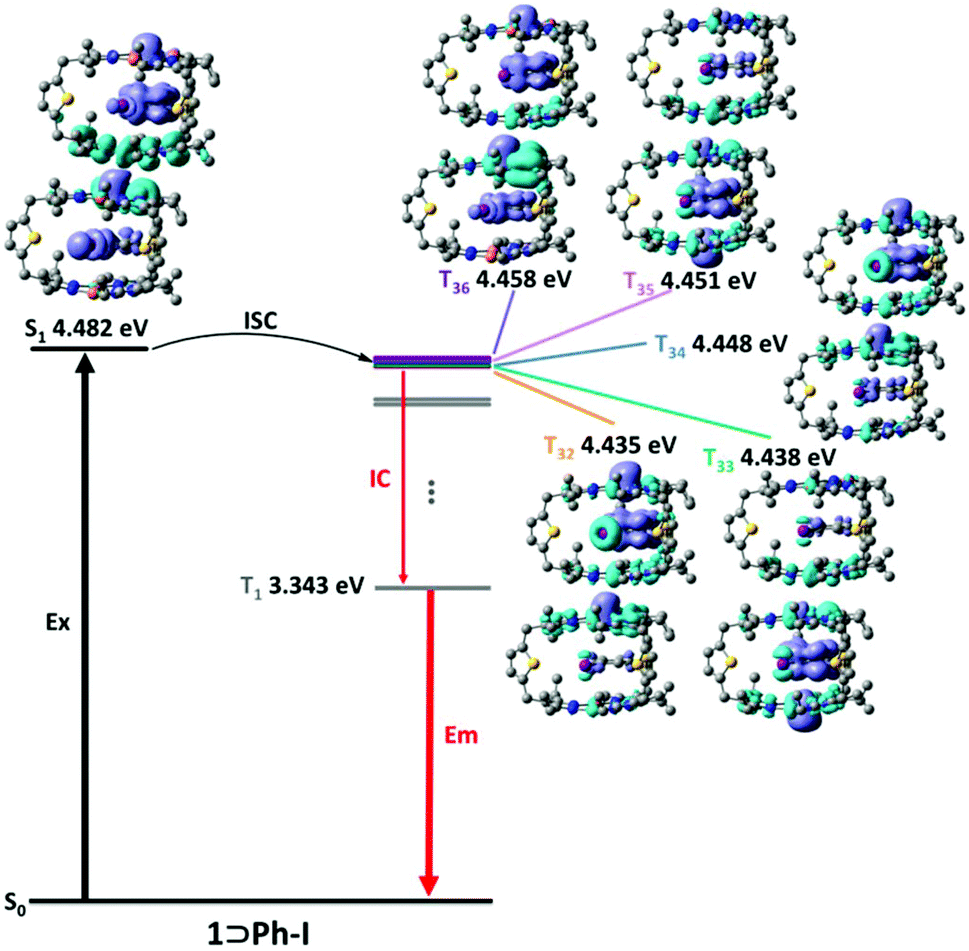 | ||
| Fig. 4 Proposed intersystem crossing approaches for 1⊃Ph-I. The energy level and electron density difference (EDD) of the S1 state and selected triplet states are illustrated. | ||
Conclusions
In summary, we designed four inclusion complexes by encapsulating halobenzene guests into a phosphorescent Cu6L3 prismatic cage. By mediating the halogen atom, the phosphorescence QY and lifetime of the inclusion complexes can be fine-tuned through external HAEs. More importantly, the linear relationship between the logarithm of ISC rate constants and logζ2 revealed that heavy atoms played an important role in the enhancement of QY and shortening of lifetime. For instance, the phosphorescence QY increased 6 times from 12.6% to 74.3%, while the lifetimes reduced 1.6 times from 30.11 to 18.39 μs for 1⊃Ph-F and 1⊃Ph-I, respectively. To the best of our knowledge, 1⊃Ph-I with such high phosphorescence QY and short lifetime has rarely been achieved in the supramolecular cage. Furthermore, the TDDFT simulation suggested that the inclusion of iodobenzene guests can narrow the energy difference between the excited singlet and triplet states and allow more effective ISC channels, thus greatly promoting the ISC processes. By combining the experimental and theoretical approaches, we demonstrated a novel and simple way to tuning the PL properties of the phosphorescent emitter that might be suitable for PhOLED applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21731002, 21975104 and 21801095), the Guangdong Major Project of Basic and Applied Research (2019B030302009), and Jinan University. The computation in this work was supported by the high-performance public computing service platform of Jinan University. G.-H. Ning is thankful for the financial support from the Guangdong Basic and Applied Basic Research Foundation (2019B151502024), the Guangdong Province Pearl River Scholar Funded Scheme (2019), and the Fundamental Research Funds for the Central Universities (21619315).Notes and references
- H. Yersin, Triplet Emitters for OLED Applications. Mechanisms of Exciton Trapping and Control of Emission Properties, Top. Curr. Chem., 2004, 241, 1–26 CrossRef CAS.
- Y. Chi and P.-T. Chou, Transition-metal phosphors with cyclometalating ligands: fundamentals and applications, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- E. Holder, B. M. W. Langeveld and U. S. Schubert, New Trends in the Use of Transition Metal−Ligand Complexes for Applications in Electroluminescent Devices, Adv. Mater., 2005, 17, 1109–1121 CrossRef CAS.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, High-efficiency fluorescent organic light-emitting devices using a phosphorescent sensitizer, Nature, 2000, 403, 750–753 CrossRef CAS PubMed.
- H. Noda, H. Nakanotani and C. Adachi, Excited state engineering for efficient reverse intersystem crossing, Sci. Adv., 2018, 4, 1–7 Search PubMed.
- Y.-J. Su, H.-L. Huang, C.-L. Li, C.-H. Chien, Y.-T. Tao, P.-T. Chou, S. Datta and R.-S. Liu, Highly Efficient Red Electrophosphorescent Devices Based on Iridium Isoquinoline Complexes: Remarkable External Quantum Efficiency Over a Wide Range of Current, Adv. Mater., 2003, 15, 884–888 CrossRef CAS.
- Y. Wang, Dramatic effects of hole transport layer on the efficiency of iridium-based organic light-emitting diodes, Appl. Phys. Lett., 2004, 85, 4848–4850 CrossRef CAS.
- W. Song and J. Y. Lee, Degradation Mechanism and Lifetime Improvement Strategy for Blue Phosphorescent Organic Light–Emitting Diodes, Adv. Opt. Mater., 2017, 5, 1600901–16009012 CrossRef.
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. A. Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, High–performance light–emitting diodes based on carbene-metal-amides, Science, 2017, 356, 159–163 CrossRef CAS PubMed.
- D. Lee, X. Ma, J. Jung, E. J. Jeong, H. Hashemi, A. Bregman, J. Kieffer and J. Kim, The effects of extended conjugation length of purely organic phosphors on their phosphorescence emission properties, Phys. Chem. Chem. Phys., 2015, 17, 19096–19103 RSC.
- X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi, J. Jiang, X. Zhang, J. N. Demas, C. O. Trindle, Y. Luo and G. Zhang, Versatile Room-Temperature-Phosphorescent Materials Prepared from N-Substituted Naphthalimides: Emission Enhancement and Chemical Conjugation, Angew. Chem., Int. Ed., 2016, 55, 9872–9876 CrossRef CAS PubMed.
- X. Sun, B. Zhang, X. Li, C. O. Trindle and G. Zhang, External Heavy-Atom Effect via Orbital Interactions Revealed by Single-Crystal X–ray Diffraction, J. Phys. Chem. A, 2016, 120, 5791–5797 CrossRef CAS PubMed.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, Crystallization-Induced Phosphorescence of Pure Organic Luminogens at Room Temperature, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS.
- J. Zheng, Z. Lu, K. Wu, G.-H. Ning and D. Li, Coinage-Metal-Based Cyclic Trinuclear Complexes with Metal-Metal Interactions: Theories to Experiments and Structures to Functions, Chem. Rev., 2020, 120, 9675–9742 CrossRef CAS PubMed.
- O. Elbjeirami, M. A. Rawashdeh-Omary and M. A. Omary, Phosphorescence sensitization via heavy-atom effects in d10 complexes, Res. Chem. Intermed., 2011, 37, 691–703 CrossRef CAS.
- S.-Z. Zhan, F. Ding, X.-W. Liu, G.-H. Zhang, J. Zheng and D. Li, White Light from Blue Fluorescence and Sensitized Yellow Long–Afterglow Phosphorescence of o-Terphenyl in Its π–Acid⋯Base Adduct with Ag3Pz3, Inorg. Chem., 2019, 58, 12516–12520 CrossRef CAS PubMed.
- O. Elbjeirami, C. N. Burress, F. P. Gabbaï and M. A. Omary, Simultaneous External and Internal Heavy-Atom Effects in Binary Adducts of 1-Halonaphthalenes with Trinuclear Perfluoro-ortho-phenylene Mercury(II): A Structural and Photophysical Study, J. Phys. Chem. C, 2007, 111, 9522–9529 CrossRef CAS.
- C. Burress, O. Elbjeirami, M. A. Omary and F. P. Gabbaï, Five-Order-of-Magnitude Reduction of the Triplet Lifetimes of N-Heterocycles by Complexation to a Trinuclear Mercury Complex, J. Am. Chem. Soc., 2005, 127, 12166–12167 CrossRef CAS PubMed.
- T. R. Cook and P. J. Stang, Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- M. L. Saha, X. Yan and P. J. Stang, Photophysical Properties of Organoplatinum(II) Compounds and Derived Self-Assembled Metallacycles and Metallacages: Fluorescence and its Applications, Acc. Chem. Res., 2016, 49, 2527–2539 CrossRef CAS PubMed.
- K. Ono, J. K. Klosterman, M. Yoshizawa, K. Sekiguchi, T. Tahara and M. Fujita, ON/OFF Red Emission from Azaporphine in a Coordination Cage in Water, J. Am. Chem. Soc., 2009, 131, 12526–12527 CrossRef CAS PubMed.
- P. R. Neelakandan, A. Jiménez and J. R. Nitschke, Fluorophore incorporation allows nanomolar guest sensing and white-light emission in M4L6 cage complexes, Chem. Sci., 2014, 5, 908–915 RSC.
- G.-F. Gao, M. Li, S.-Z. Zhan, Z. Lv, G.-H. Chen and D. Li, Confined Metallophilicity within a Coordination Prism, Chem. – Eur. J., 2011, 17, 4113–4117 CrossRef CAS PubMed.
- Z.-C. Shi, D.-X. Zhang, S.-Z. Zhan, M. Li, J. Zheng, H. Yang, X.-P. Zhou and D. Li, Trigonal Prismatic Cu6L3 Coordination Cage: Encapsulation of Aromatic Molecules and Tuned Photoluminescence, Isr. J. Chem., 2019, 59, 317–322 CrossRef CAS.
- Z.-C. Shi, W. Chen, S.-Z. Zhan, M. Li, M. Xie, Y. Y. Li, S. W. Ng, Y.-L. Huang, Z. Zhang, G.-H. Ning and D. Li, Guest effects on crystal structure and phosphorescence of a Cu6L3 prismatic cage, Inorg. Chem. Front., 2020, 7, 1437–1444 RSC.
- D. S. McClure, Triplet-Singlet Transitions in Organic Molecules. Lifetime Measurements of the Triplet State, J. Phys. Chem., 1949, 17, 905–913 CrossRef CAS.
- C.-L. Liu, R.-L. Zhang, C.-S. Lin, L.-P. Zhou, L.-X. Cai, J.-T. Kong, S.-Q. Yang, K.-L. Han and Q.-F. Sun, Intraligand Charge Transfer Sensitization on Self-Assembled Europium Tetrahedral Cage Leads to Dual-Selective Luminescent Sensing toward Anion and Cation, J. Am. Chem. Soc., 2017, 139, 12474–12479 CrossRef CAS PubMed.
- S.-Y. Wu, X.-Q. Guo, L.-P. Zhou and Q.-F. Sun, Fine-Tuned Visible and Near-Infrared Luminescence on Self-Assembled Lanthanide-Organic Tetrahedral Cages with Triazole-Based Chelates, Inorg. Chem., 2019, 58, 7091–7098 CrossRef CAS PubMed.
- Y. Zhang, M. R. Crawley, C. E. Hauke, A. E. Friedman and T. R. Cook, Phosphorescent Decanuclear Bimetallic Pt6M4 (M = Zn, Fe) Tetrahedral Cages, Inorg. Chem., 2017, 56, 4258–4262 CrossRef CAS PubMed.
- Z. Wang, L. He, B. Liu, L.-P. Zhou, L.-X. Cai, S.-J. Hu, X.-Z. Li, Z. Li, T. Chen, X. Li and Q.-F. Sun, Coordination-Assembled Water-Soluble Anionic Lanthanide Organic Polyhedra for Luminescent Labeling and Magnetic Resonance Imaging, J. Am. Chem. Soc., 2020, 142, 16409–16419 CrossRef CAS PubMed.
- A. Bondi, Van der Waals Volumes and Radii, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- M. Kasha, Collisional Perturbation of Spin-Orbital Coupling and the Mechanism of Fluorescence Quenching. A Visual Demonstration of the Perturbation, J. Chem. Phys., 1952, 20, 71–74 CrossRef CAS.
- Z.-Y. Zhang and Y. Liu, Ultralong room-temperature phosphorescence of a solid-state supramolecule between phenylmethylpyridinium and cucurbit[6]uril, Chem. Sci., 2019, 10, 7773–7778 RSC.
- J. Zhao, W. Wu, J. Sun and S. Guo, Triplet photosensitizers: from molecular design to applications, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2047942–2047945. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qi00033k |
| This journal is © the Partner Organisations 2021 |