
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptical sensing of anions by macrocyclic and interlocked hosts
Hui Min
Tay
and
Paul
Beer
 *
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: paul.beer@chem.ox.ac.uk
First published on 6th May 2021
Abstract
The ubiquity of anions in biological and environmental systems has motivated the development of many novel anion receptors and sensors over the past two decades. Optical anion sensors, which undergo a spectral change in response to anion binding, are particularly desirable due to the technical simplicity and fast response time of such systems. A myriad of macrocyclic host molecules have been shown to be effective anion receptors and present a promising platform for elaboration into optical anion sensors by incorporation of an appropriate fluorogenic or chromogenic group. The enhanced anion binding properties of the three-dimensional binding cavities in mechanically interlocked host molecules have also been exploited to design anion sensors that exhibit remarkable selectivity and sensitivity. This review summarises recent progress in the development of optical anion sensors based on macrocyclic and interlocked hosts. The major classes of macrocyclic receptors possessing neutral, cationic and metal-based binding motifs are examined, followed by a survey of optically-responsive interlocked anion hosts.
1. Introduction
Prompted by the pivotal role of anions in multiple biological1–3 and environmental4–6 processes, the field of anion recognition has grown significantly in recent years. A particularly exciting development is the design of increasingly sophisticated sensors capable of reporting the presence of specific anions in a rapid and sensitive manner.7Macrocyclic hosts are particularly well-suited to act as anion receptors due to the relative ease with which they can be functionalised with multiple anion binding motifs.8 This enables the use of a variety of synergistic non-covalent interactions, including hydrogen bonding, electrostatic interactions, metal coordination, halogen bonding and anion–π interactions, to achieve the recognition of a target guest species. By careful choice and placement of these binding groups, it is possible to design cavities that are complementary to specific anions – often a prerequisite for strong and selective recognition. Perhaps unsurprisingly, the earliest anion receptor, reported by Park and Simmons in 1968, was a cryptand-like macrobicyclic host that encapsulated halides between the ammonium groups located at the poles of its cavity.9 More recently, mechanically-interlocked molecules, such as catenanes and rotaxanes, have emerged as promising molecular hosts due to their highly pre-organised binding cavities, which have been shown to enhance the affinity and selectivity for target guests.10
The modular synthesis of macrocyclic and mechanically interlocked hosts facilitates the incorporation of various reporter groups, opening up possibilities for optical and/or electrochemical sensing of anions.11 The development of colorimetric and fluorescent probes for anions is especially desirable due to the sensitivity, fast response and technical simplicity of such systems.12,13 Many examples of optical anion sensors have been reported, including acyclic14 and metal-based systems15 which have been comprehensively discussed in other reviews. This review highlights recent developments in macrocyclic and interlocked optical anion sensors, with a particular focus on examples in which the macrocyclic/interlocked nature of the host significantly influences its sensing properties, either by conferring selectivity for specific anions, and/or by facilitating the spectral changes that occur upon anion binding. Macrocycles containing various neutral, cationic and metal-based binding motifs are discussed, followed by a survey of advances in the field of mechanically interlocked anion sensors.
2. Macrocycles with neutral anion-binding motifs
2.1. Calix[n]arene-based macrocycles
Calixarenes and their derivatives are versatile scaffolds for guest binding due to their highly pre-organised arene core which can be readily functionalised with a range of binding and reporter groups. As such, calixarenes have been widely employed as chemo-sensors,16 catalysts17 and biomimetic models.18 A slew of functionalised calixarene derivatives have also been employed as optical anion sensors, in which a common design feature involves attaching one or more fluorogenic and/or chromogenic groups to the lower rim of the calixarene core. The linkers that connect the reporter group to the calixarene core typically contain hydrogen bond donors like amide or urea, with the aim of encapsulating the target anion in the cavity formed between the lower rim and the reporter groups.Of the available fluorophores, pyrene has been most frequently used in calixarene-based sensors, possibly due to the propensity of pyrene units to form an excimer, which is typically accompanied by the appearance of a diagnostic emission band. This effect was exploited for anion sensing by Kim and co-workers, who synthesised a calix[4]arene based receptor 1 functionalised with two pyrenyl moieties (Fig. 1).19 Upon binding of fluoride to the amide linkers in acetonitrile, a new band at 470 nm was observed in the emission spectrum of 1. The authors attributed this to the formation of a static excimer when the pyrenyl groups, which previously enjoyed a high degree of conformational freedom, were forced to reside in close proximity to each other by their mutual interaction with fluoride. A similar principle was employed in a calix[6]arene sensor 2, which contained three pyrenyl units appended to the core via urea linkages (Fig. 1). The receptor selectively binds SO42− in DMSO via multiple hydrogen bonds involving the urea groups, resulting in slight quenching of the monomer emission while the excimer emission increased, suggesting that the pyrenyl moieties were brought closer together. Interestingly, 2-SO42− was shown to cooperatively bind an ammonium cation as a contact ion pair in chloroform, whereas the free receptor displayed no affinity for ammonium.20 In contrast, the pyrene-functionalised 1,3-alternate tetraoxacalix[2]arene[2]triazine azacrown receptor 3 (Fig. 1) exhibited a decrease in the excimer fluorescence at 477 nm and increase in the monomer bands at 378–396 nm upon addition of fluoride in 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 acetonitrile/chloroform. This was attributed to deprotonation of a triazine-appended amino group by the basic fluoride ion, resulting in an intramolecular hydrogen bond that pulled the triazine moieties closer to each other. A concomitant increase in the distance between the alternate benzene rings caused the pyrene excimer to break apart.21
:5 acetonitrile/chloroform. This was attributed to deprotonation of a triazine-appended amino group by the basic fluoride ion, resulting in an intramolecular hydrogen bond that pulled the triazine moieties closer to each other. A concomitant increase in the distance between the alternate benzene rings caused the pyrene excimer to break apart.21
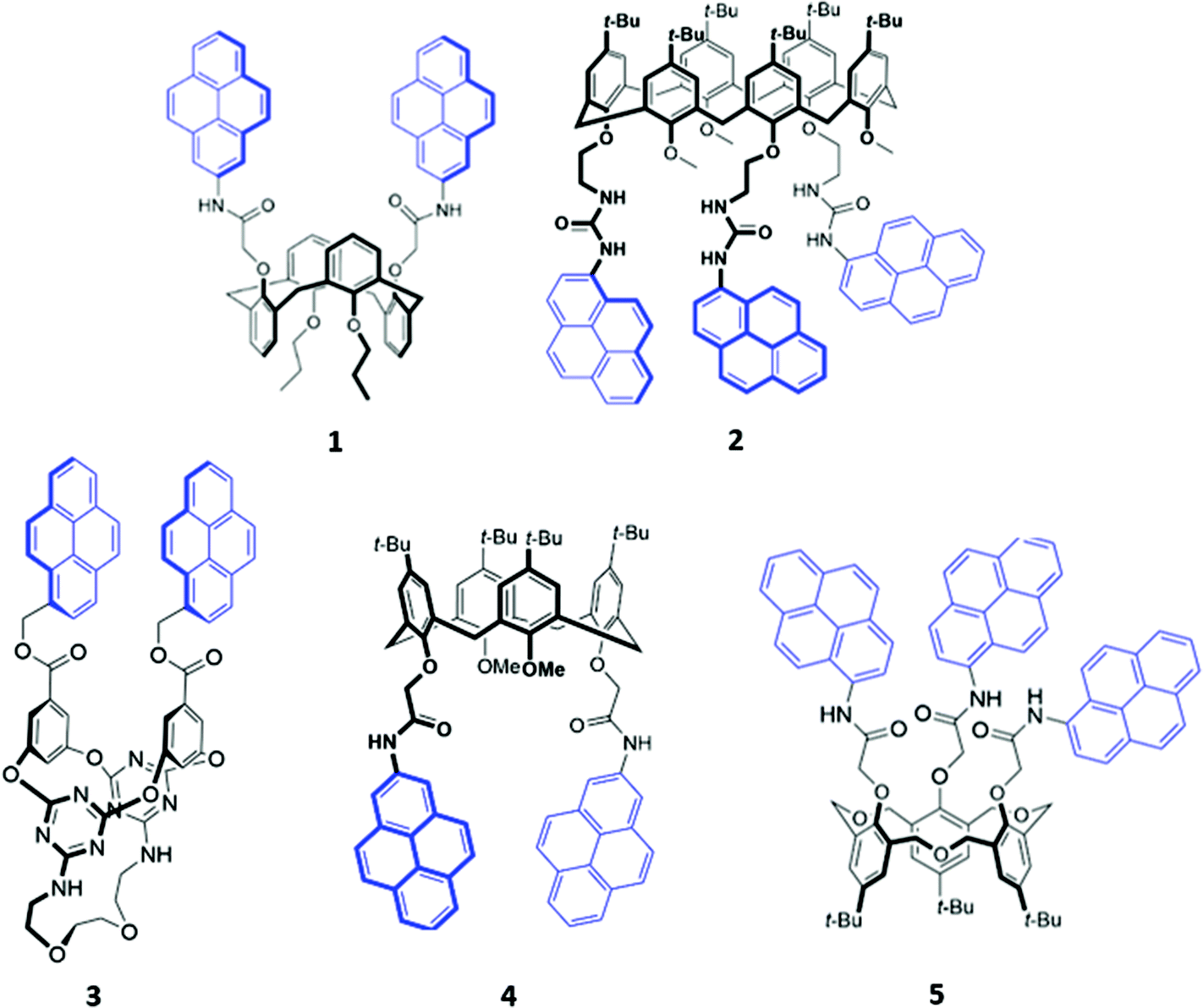 | ||
| Fig. 1 Structures of pyrene-appended calix[n]arenes 1–5 with the pyrenyl moieties highlighted in blue. | ||
Recently, two further examples of pyrenyl-appended derivatives of calixarenes were developed for optical sensing of halides. A calix[4]arene receptor 4 decorated with tert-butyl groups on the upper rim and amide-pyrene groups on the lower rim (Fig. 1) served as a dual cation and anion sensor specific for As(III), Nd(III) and Br−. Impressively, 4 exhibited a binding constant of 6.31 × 108 M−1 for Br− in a competitive solvent mixture of acetonitrile/aqueous phosphate buffer (8:2, v/v). The formation of hydrogen bonds between Br− and the amide linkers increased the partial negative charge on the pyrenyl-adjacent nitrogen atom, switching on photoinduced electron transfer (PET) and quenching the pyrenyl fluorescence.22 A hexahomotrioxacalix[3]arene receptor 5 similarly underwent fluorescence quenching upon addition of iodide in THF/H2O (7:3, v/v) (Fig. 1). The authors suggested that iodide was bound by synergistic hydrogen bonding interactions involving the amide groups. The heavy atom effect associated with iodine led to increased intersystem crossing to the non-emissive triplet state of pyrene.23
The fluorenone-appended calix[4]arene receptor 6 exhibits a high affinity for fluoride, with a binding constant of 1.18 × 106 M−1 in acetonitrile, attributed to the formation of multiple OH and NH hydrogen bonds (Fig. 2). Addition of small quantities of fluoride resulted in significant fluorescence quenching due to an increase in PET from the partially deprotonated amide nitrogen atoms to the fluorenone groups, as well as a ‘naked eye’ colour change from yellow to orange-red.24 A clear trend towards preferential fluoride binding was observed in a related series of calix[4]arene-based receptors functionalised on the lower rim with various fluorophores or chromophores (Fig. 2), including benzooxadiazole (7),25 naphthalene (8),26 anthraquinone (9),27 nitrophenyl (10)28 and naphthol (11).29 In these systems, anion binding occurs in the pocket formed by the lower rim and the hydrogen-bonding linker, suggesting that size complementarity exists between this binding site and the small fluoride anion. In a slightly unusual synthesis, the calix[4]arene receptor 12 was functionalised at the upper rim with thiourea linkers connected to coumarin groups.30 This receptor once again displayed a high affinity for fluoride, with NMR studies in DMSO-d6 suggesting the deprotonation of the thiourea groups and formation of HF2− at high fluoride concentration. This resulted in fluorescence quenching due to enhanced PET. In addition, a new absorption band appeared at 475 nm, causing a colour change from colourless to orange.
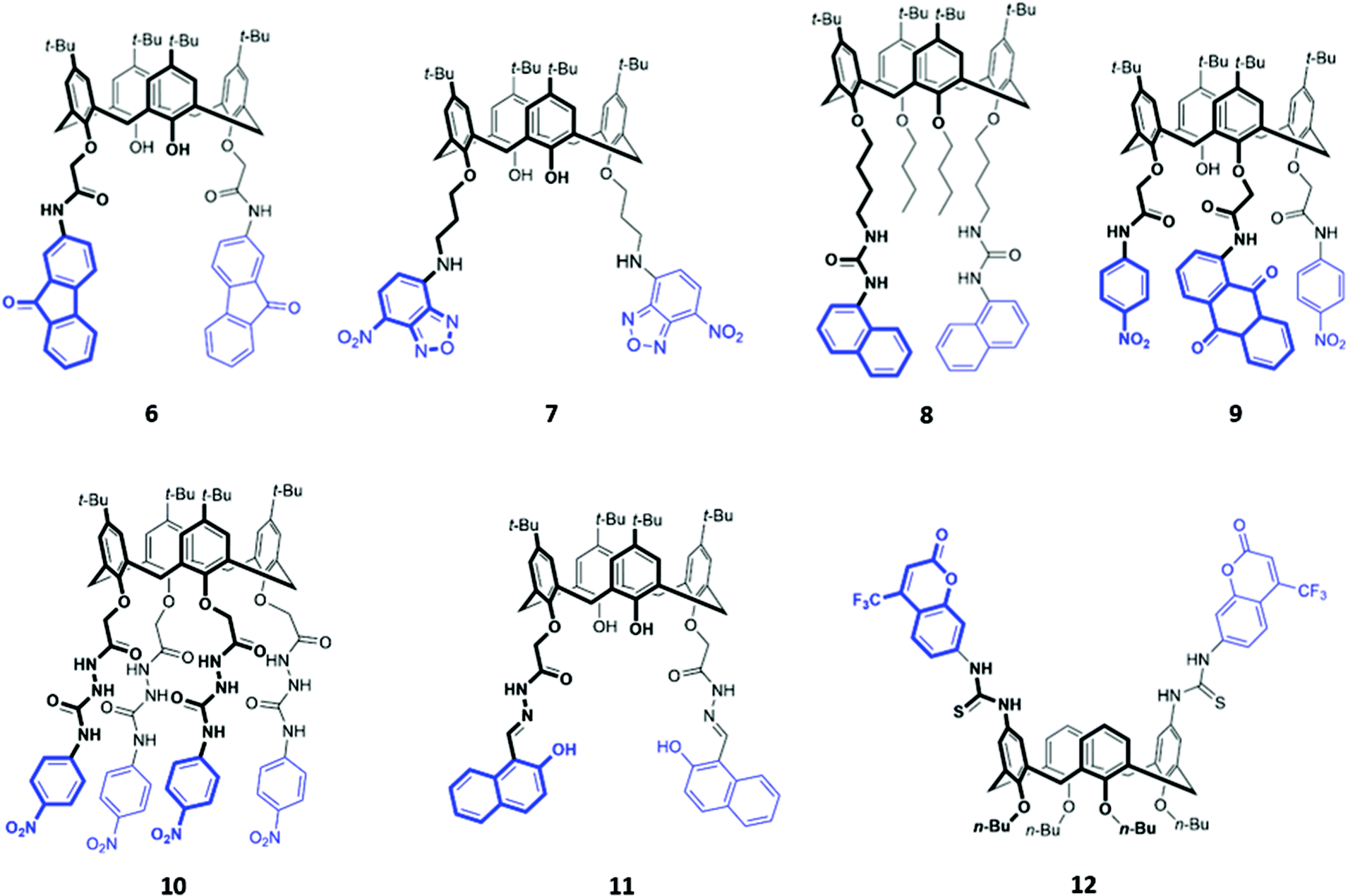 | ||
| Fig. 2 Structures of calix[n]arenes 7–12 functionalised with various chromophores/fluorophores highlighted in blue. | ||
A unique bis(bora)calix[4]arene 13 similarly displayed selectively for fluoride (Fig. 3).31 Unlike the previous examples, in which hydrogen bonding interactions were responsible for anion binding, 13 binds fluoride in chloroform via Lewis acid–base interactions with the boron centres. The authors suggested that the fluoride selectivity arose from endo binding of fluoride in the pocket between the two boron centres, which is too small to accommodate other anions. However, a subsequent computational study suggested that cooperative endo and exo binding modes were responsible.32 The fluorescence of the boron-adjacent phenyl groups was quenched upon fluoride binding, which may be due to increased PET from boron to the phenyl groups or internal conversion to a long-lived ‘dark state’ that suppressed the ‘prompt’ fluorescence from the higher excited states.
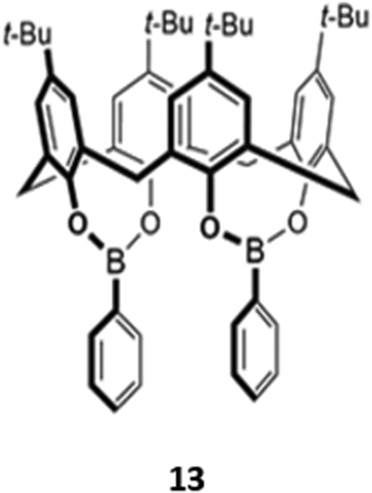 | ||
| Fig. 3 Structure of a bis(bora)calix[4]arene for fluoride sensing (Ka = 2.00 × 106 M−1 in CHCl3). | ||
While many calix[4]arene receptors recognised fluoride selectively, tweaking the nature and position of the binding groups can alter the selectivity for anions. A resin consisting of calix[4]amido-crown-5 receptor 14 attached to 4-benzyloxy benzyl alcohol polymer was found to preferentially bind iodide (Fig. 4). The addition of iodide to a suspension of the resin in CH2Cl2/CH3OH (v/v 1:1) resulted in an enhancement of fluorescence emission, which was attributed to increased configurational rigidity of the amido-crown macrocycle upon anion binding.33 A calix[4]arene host 15 was synthesised by Singh and co-workers, which bore long aliphatic linkers ending in urea groups for an enlarged binding pocket that was complementary for iodide (Fig. 4). Unlike the majority of calixarene-based optical sensors, 15 did not contain a fluorophore tag; instead, the receptor was coupled to the surface of gold nanoparticles. Binding of iodide to a suspension of 15 in water induced the aggregation of nanoparticles, as seen by TEM, resulting in a colour change from light pink to blue.34
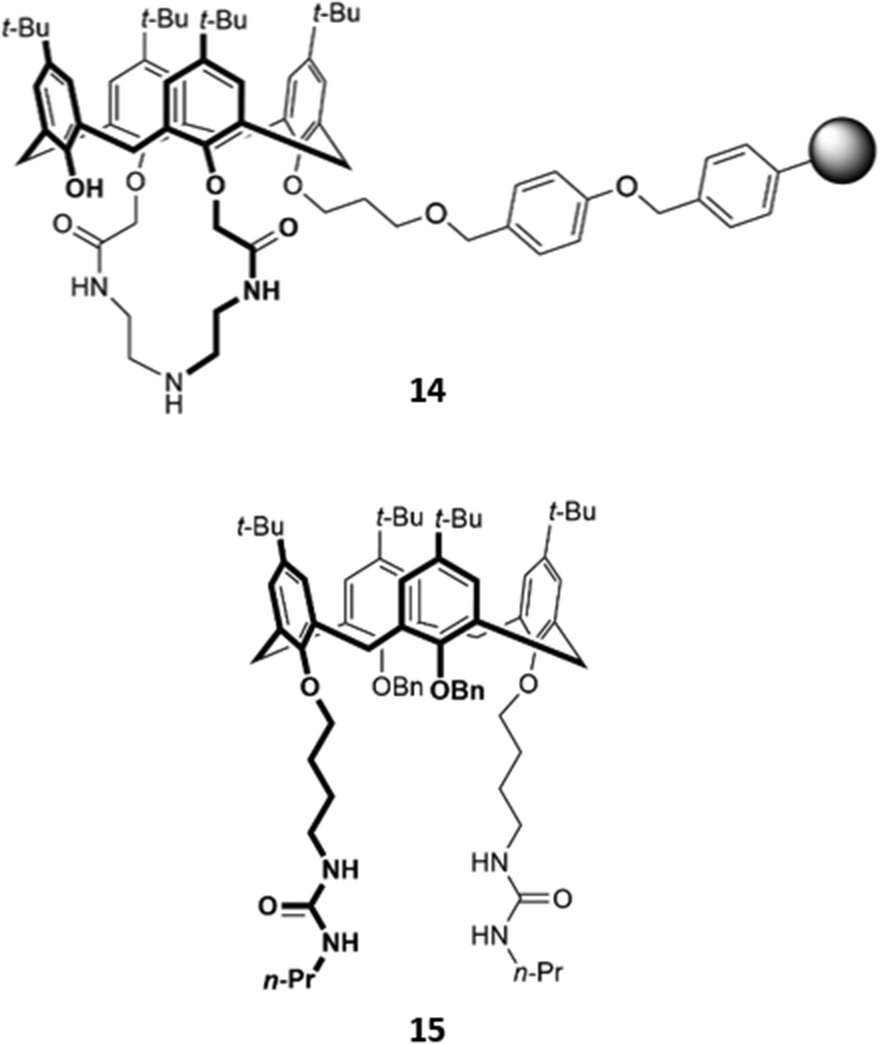 | ||
| Fig. 4 Structures of iodide-selective calix[4]arene receptors 14 (where the grey sphere represents a resin of 4-benzyloxybenzyl alcohol polymer) and 15. | ||
An unusual oxacalix[2]arene[2]triazine host 16, in which two of the benzene rings in the calix[4]arene core were replaced by triazines, was used as a sensor for H2PO4− in acetonitrile (Fig. 5). The binding of the anion was due to a combination of hydrogen bonds with the pendant amide groups as well as anion–π interactions with the electron-poor triazine rings. Binding of H2PO4− to 16 was accompanied by a quenching of the triazine fluorescence at 310 nm and the appearance of a new band at 412 nm.35
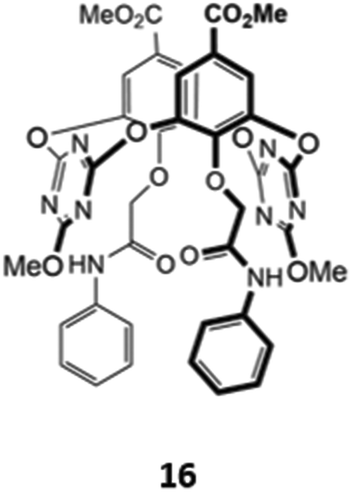 | ||
| Fig. 5 Structure of an oxacalix[2]arene[2]triazine capable of sensing H2PO4− (Ka = 7.98 × 103 M−1 in CH3CN). | ||
2.2. Calix[n]pyrrole-based macrocycles
Calix[n]pyrroles are a well-studied class of macrocycles first synthesised by Baeyer in 1886,36 and subsequently repurposed as an anion receptor by Sessler in 1996.37 To elaborate these anion receptors into functional optical sensors, a natural approach is to attach a fluorophore or chromophore to the macrocyclic ring. This was first demonstrated by Sessler and co-workers, who prepared a series of β-functionalized calix[4]pyrrole anthracene conjugates (17a–c), which contain amide linkers of varying length (Fig. 6). These systems formed 1:1 complexes with various anions, which were shown by 1H NMR studies to bind via multiple hydrogen bonds in the macrocyclic cavity. The addition of F−, Cl− and H2PO4− in dichloromethane resulted in significant fluorescence quenching, attributed to an increase in PET, with electrons being transferred from the pyrrole rings to the anthracene fluorophore. Consistent with this mechanism, the quenching effect was most pronounced in 17a, which contained the shortest linker.38 Anzenbacher and co-workers recently reported a series of five calix[n]pyrrole-based macrocycles of varying ring sizes and bearing different fluorophores (18a–e) (Fig. 6). The macrocycles showed surprisingly high cross-reactivity for different anions, with little size discrimination observed. For example, in acetonitrile, the expanded calix[2]benzo[4]pyrrole 18d exhibited similar or lower affinities for large carboxylates compared to the smaller calix[4]pyrrole analogue 18a. Nonetheless, subtle differences in the photophysical responses of the five sensors towards different anions enabled the establishment of a fluorescence-based microarray that successfully classified 18 anions by linear discriminant analysis.39
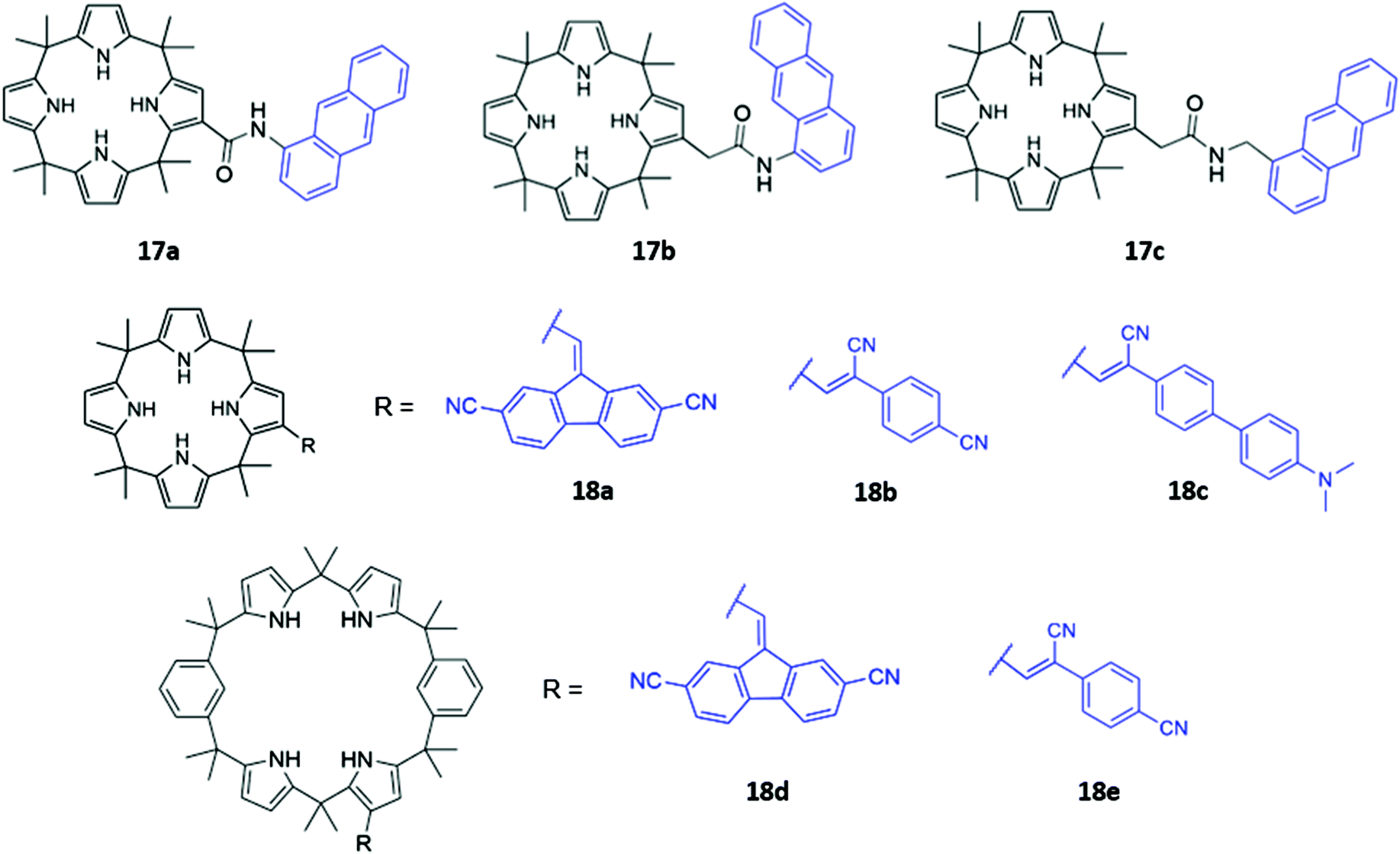 | ||
| Fig. 6 Structures of calixpyrrole-based anion receptors 17–18 with chromophores highlighted in blue. | ||
In a bid to improve the binding affinity of calix[4]pyrroles for anions, Lee and co-workers synthesised a bicyclic or ‘strapped’ calix[4]pyrrole 19, which bears a dipyrrolylquinoxaline strap that serves the dual roles of providing additional sites for receptor–anion interactions as well as reporting anion binding via colour changes in the quinoxaline chromophore (Fig. 7). 1H NMR anion binding studies in CD3CN/DMSO-d6 (9:1 v/v) showed that fluoride formed hydrogen bonds with the NH groups of the calix[4]pyrrole, but not the pyrrole groups of the strap, which did not undergo any appreciable downfield shift upon fluoride addition. Instead, the β-pyrrolic protons of the pyrrole groups in the strap shifted downfield by up to 0.3 ppm, which the authors attributed to an anion–π interaction between the electron-deficient pyrrole rings and the anion in the cavity. The binding of fluoride and dihydrogen phosphate to the receptor in CH3CN/DMSO (97:3 v/v) was accompanied by a ‘naked eye’ colour change from orange to dark blue.40
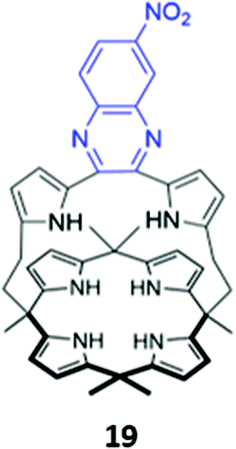 | ||
| Fig. 7 Structure of ‘strapped’ calix[4]pyrrole 19 with quinoxaline fluorophore shown in blue. 19 binds F− with Ka = 8.97 × 106 M−1 in 97:3 CH3CN/DMSO. | ||
A novel bis-calix[4]pyrrole-appended phenazine sensor 20, which uses vibration induced emission (VIE) to differentiate between anions, was designed by Sessler and co-workers (Fig. 8). VIE is a phenomenon in which the fluorophore in question undergoes reversible vibrations between bent and planar configurations, resulting in multiple and tunable emission wavelengths from a single system.41 The free receptor adopts a planar configuration which has a long π-conjugation pathway and therefore emits orange-red fluorescence. Dubbed a ‘molecular calliper’ by the authors, 20 binds dicarboxylate anions in a bridging fashion between the two calix[4]pyrrole units, imposing strain on the phenazine core and inducing a bending vibration that causes the emission to blueshift. In general, the shorter the dicarboxylate anion, the greater the degree of bending and therefore the observed blueshift, allowing structurally-similar anions to be distinguished in acetonitrile based on the colour of the fluorescence signal produced.42
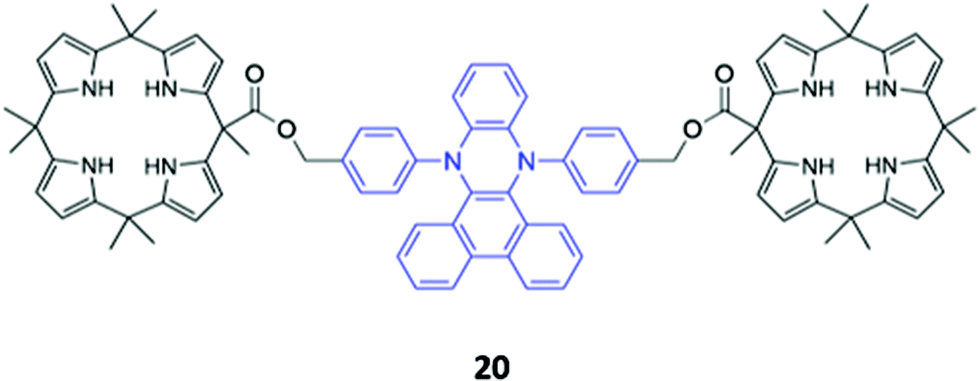 | ||
| Fig. 8 Structure of a bis-calix[4]pyrrole-appended phenazine for anion sensing using VIE, with the phenazine core shown in blue. 20 binds a range of dicarboxylates with Ka values ranging from 105–107 M−1 in CH3CN. | ||
2.3. Amide- and urea-based macrocycles
The incorporation of hydrogen bonding donor groups directly into the core of the macrocycle is a common strategy for designing potent anion receptors, with amides, ureas and thioureas being some of the most widely utilised motifs.12 An added advantage of the aforementioned groups is their ease of formation, allowing them to be readily constructed during the macrocyclization process, which greatly simplifies synthetic efforts. A diverse range of anion sensors based on amide or urea groups have been reported over the years,43–45 with some recent examples highlighted below.Yoon and co-workers reported a chiral urea-based macrocycle 21 which contains an (S)-binaphthol (BINOL) unit that serves both as a chiral recognition site and a chromophore (Fig. 9). 21 bound D-Boc-alanine via hydrogen bonding interactions between the deprotonated carboxylate of the amino acid and the urea moieties of the macrocycle, resulting in marked enhancements in the UV–Vis absorption at 258 and 370 nm as well as a decrease at 342 nm in DMSO. Addition of L-Boc-alanine, however, induced only small changes in the absorption at these wavelengths, allowing discrimination of the two enantiomers.46 In the following year, Anzenbacher and co-workers synthesised three chiral macrocycles (22a–c) decorated with multiple amide and/or sulfonamide groups (Fig. 9). BINOL was once again chosen as the chiral fluorophore, with both the (R)- and (S)-enantiomers of each macrocycle prepared. In addition, 22a also possesses a chromogenic nitrophenyl group that enabled colorimetric detection of anions. The three sensors displayed significant cross-reactivity for simple inorganic ions as well as various chiral nucleoside phosphates, exhibiting moderate enantioselectivities. Promisingly, a multi-sensor array of the six sensors was capable of discriminating seven biologically relevant anions (AMP, ADP, ATP, CMP, GMP, PO43− and P2O74−) in DMSO.47
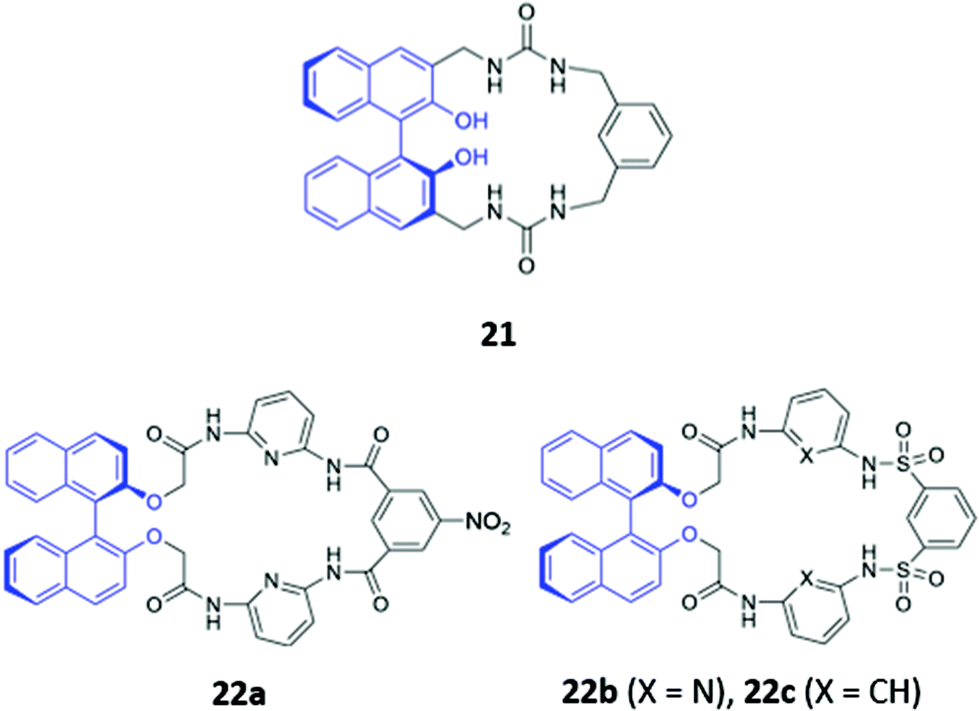 | ||
| Fig. 9 Structures of the chiral amide-based anion receptors 21–22, with BINOL fluorophores shown in blue. | ||
Two macrocyclic tetraamide receptors containing one (23a) or two (23b) azulene units were found to be selective for phosphate-containing anions (H2PO4− and HP2O73−) (Fig. 10). The addition of these anions to a DMSO solution of the asymmetrical receptor 23a produced a redshift of the UV–vis absorption, which led to observable colour changes. Crystallographic studies showed that both the free and complexed receptors adopt a bent-sheet conformation that allows all four amides to participate in hydrogen bonding with the anionic guest, displaying a high degree of pre-organisation that favours anion binding. Notably, acyclic analogues of both receptors showed reduced affinity for phosphate, with no detectable spectroscopic changes.48
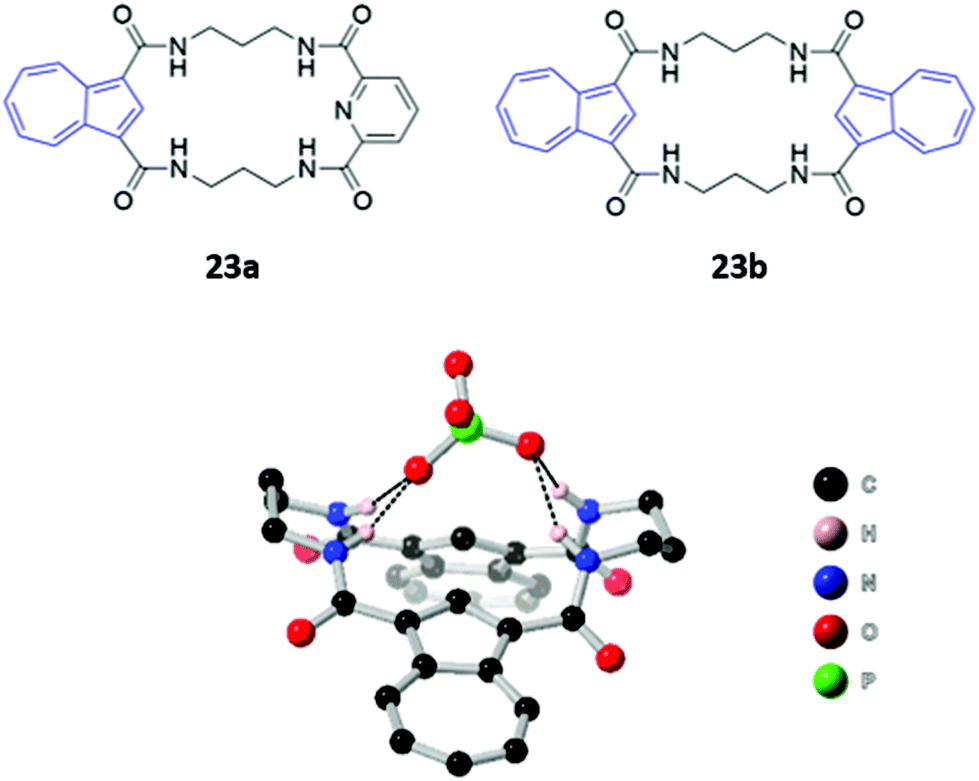 | ||
| Fig. 10 (Top) Structures of tetraamide macrocyclic anion receptors with azulene units shown in blue. (Bottom) Crystal structure of 23b·H2PO4−, with hydrogen bonds between the receptor amide groups and H2PO4− shown as dashed lines. All other hydrogen atoms have been omitted for clarity. Ka (H2PO4−) = 4800 M−1 for 23a and >104 M−1 for 23b in 9:1 DMSO/MeOH. | ||
Jolliffe and co-workers synthesised two semi-flexible tetrathiourea macrocycles 24a–b as receptors for ditopic anions, in order to study the effects of chelate cooperativity in multitopic anion receptors (Fig. 11). Although not the main focus of the study, fluorescence studies of 24a–b in H2O/DMSO (1:9 v/v) showed that the emission of the 1,8-diaminocarbazole groups was significantly quenched in the presence of dicarboxylates and sulfate, but not when mono-anions were added. This was attributed to increased PET from the thiourea moieties to the 1,8-diaminocarbazole groups and demonstrates the preference of the receptors for binding ditopic anions.49
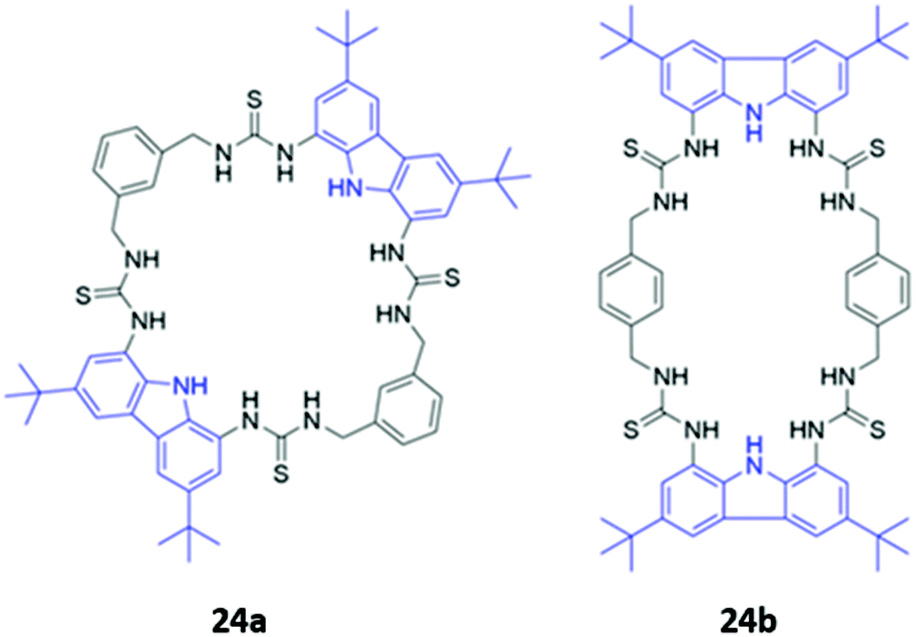 | ||
| Fig. 11 Structures of semi-flexible tetrathiourea macrocyclic anion receptors 24a–b with 1,8-diaminocarbazole fluorophores shown in blue. | ||
Amide or urea functionality can also be inserted into the pendant groups of the macrocycle, as demonstrated in a series of hybrid polyether/polyaza macrocycles 25a–d synthesised by Núñez and co-workers (Fig. 12). Anion recognition in 25a–d typically proceeds via the formation of hydrogen bonds involving the amide/urea/thiourea groups on the arms in DMSO or acetonitrile. The receptors displayed significant cross-reactivity for a range of anions and also showed affinity for some metal cations, which were coordinated by the lone pairs of the oxygen or nitrogen groups forming the macrocyclic cavity. Binding events were signalled by colour changes associated with changes in the absorption of the nitrophenyl chromophore.50–52
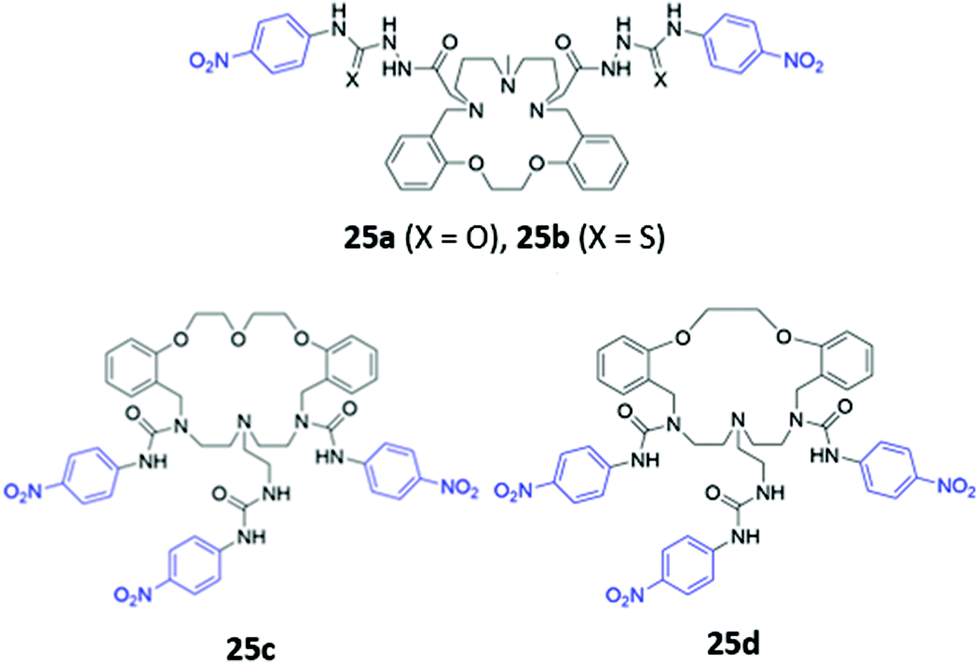 | ||
| Fig. 12 Structures of macrocyclic receptors with pendant amides with nitrophenyl chromophores shown in blue. | ||
2.4. Miscellaneous neutral macrocycles
Although the literature on optical anion sensors based on macrocyclic hosts with neutral binding groups has been dominated by the types discussed in the preceding sections, there is certainly scope to design new macrocycles that contain novel anion binding groups or employ unusual sensing mechanisms.Anion–π interactions, which occur between the HOMO of the anion and the LUMO of an electron-deficient aromatic ring, have been increasingly identified in the crystal structures of small molecules and proteins.53 Recently, several examples of anion receptors based on anion–π interactions have been reported,54 including two s-tetrazine-based macrocycle examples that possess optical sensing capabilities. Li and co-workers synthesised a tetrazine-containing diamide macrocycle 26, which bound fluoride selectively through a combination of hydrogen bonding involving the amide NH groups as well as anion–π interactions involving the electron-poor tetrazine (Fig. 13). The addition of fluoride to a solution of 26 dissolved in DMSO led to a rapid colour change from red to green, which the authors attributed to the formation of a tetrazine radical anion due to electron transfer from the fluoride guest. The formation of the radical anion was further confirmed by the appearance of a diagnostic signal in the EPR spectrum.55 Subsequently, Wang and co-workers synthesised four tetrahomocorona[4]arenes 27a–d, which contained rectangular box-like cavities complementary in size and shape to linear anions such as azide and thiocyanate (Fig. 13). The receptors exhibited low binding strengths even for these anions, as evidenced by stability constants in the range 23–166 M−1 for azide and 3–22 M−1 for thiocyanate in acetonitrile. This is likely due to the relatively weak nature of anion–π interactions compared to other non-covalent interactions like hydrogen bonding. Nonetheless, 27a–d showed a detectable fluorescence response to azide and thiocyanate, with gradual quenching of the tetrazine emission observed when these anions were introduced into acetonitrile solutions of the receptors.56
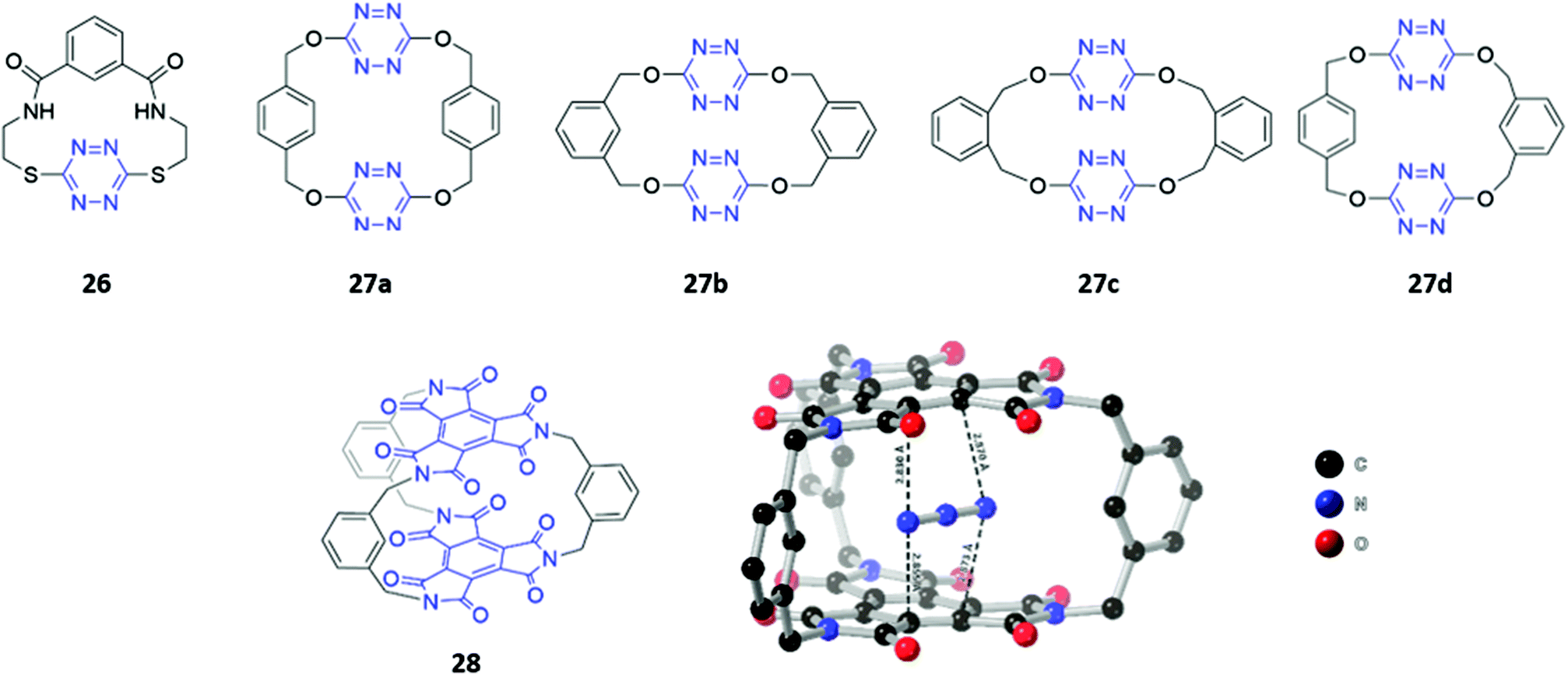 | ||
| Fig. 13 Structures of macrocycles that form anion–π interactions with guests. (Top) Structures of 26 and 27a–d with tetrazine units highlighted in blue. 27a–d showed weak binding to azide and thiocyanate with Ka values ranging from 23–166 M−1 and 3–22 M−1 respectively. (Bottom) Structure of 28 with BTI groups shown in blue and crystal structure of 28·N3−. Hydrogen atoms have been omitted for clarity. 28 bound N3− with Ka = 1.54 × 104 M−1 in acetone. | ||
Recently, the same group synthesised a C3-symmetric cage 28 based on benzene triimide (BTI) (Fig. 13), which was found to bind N3−, SCN− and I−via anion–π interactions, with a remarkable affinity for N3− in particular (Ka = 15372 M−1 in acetone). The formation of charge-transfer interactions between the electron-deficient BTI moieties and azide resulted in a naked-eye colour change of the solution from colourless to bright orange. A co-crystal of 28 with azide showed that the anion is sandwiched between the two BTI planes with intermolecular distances of 2.830–2.873 Å, providing evidence for strong anion–π interactions.57
Photoswitchable anion receptors that can undergo reversible conformational changes have the potential to demonstrate altered affinities for different guests upon light irradiation. Bandyopadhyay and co-workers reported a fluorescent azobenzene-containing macrocycle 29 which resides predominantly in the ‘open’ E form, where it binds preferentially ATP over GTP in aqueous tris buffer (1 mM, pH 6.8) (Fig. 14). This is seen in a 7-fold fluorescence enhancement when ATP is added to 29, whereas GTP only induces a 3-fold enhancement. Crystallographic and computational studies suggested that the protonated pyridinium group of 29 forms charged interactions with both GTP and ATP, but only the adenosine moiety of ATP contains an appropriately-positioned amine group to form an additional hydrogen bond with the azobenzene lone pair. However, upon irradiation with light at 366 nm, 29 photoisomerises to the ‘closed’ Z form, which is unable to form a hydrogen bond with ATP due to geometrical constraints. The reduced affinity of the Z form for ATP is reflected in the low fluorescence enhancement (1.5-fold), which is slightly lower than the GTP-induced enhancement (1.9-fold).58 While the findings are somewhat preliminary, this work demonstrates the potential of photoswitchable macrocycles with tunable affinities for ostensibly similar anionic guests.
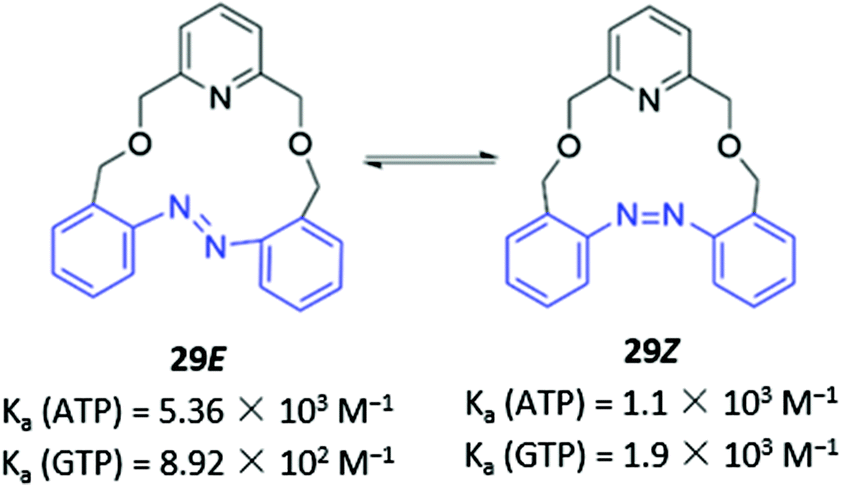 | ||
| Fig. 14 Photoswitchable azobenzene-based anion receptor 29 with photoisomerisable azobenzene groups shown in blue. | ||
Unlike the majority of optical anion sensors, which possess colorimetric and fluorescent signalling units, Pasini and co-workers designed a chiroptical probe 30 that responds to halide binding via changes in its circular dichroism spectrum in methylcyclohexane/dichloromethane (9:1, v/v) (Fig. 15). The halide guests docked in the cleft generated by the triazole units and phenyl spacer, forming weak CH–X− hydrogen bonds with the triazole groups and the phenyl ring. The inclusion of halides in the macrocyclic cavity changed the dihedral angle of the BINOL group, which altered the intensity of its characteristic CD signal. Notably, these changes were not observed in the acyclic analogue, which is inherently conformationally flexible.59
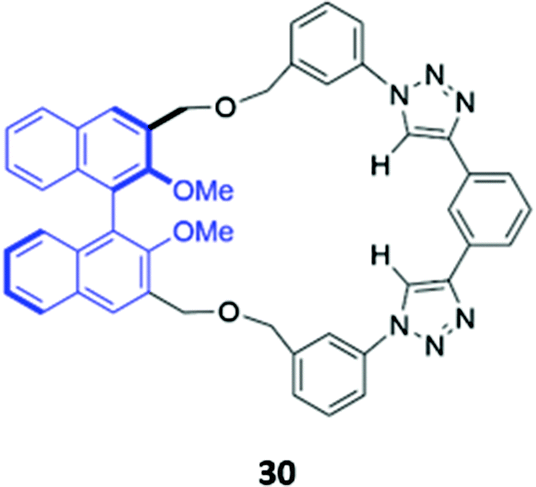 | ||
| Fig. 15 Structure of a chiroptical probe for halide sensing with BINOL shown in blue. The Ka values for binding of halides to 30 range from 8.9–18.1 M−1 in CDCl3. | ||
Another example of atypical anion sensor design comes in the form of four trans-homoporphodimethenes 31a–d synthesised by Ravikanth and co-workers (Fig. 16). These receptors are irreversible chemodosimeter sensors that selectively recognise cyanide via covalent bond formation. Addition of cyanide to a THF solution of the 31a led to significant fluorescence enhancement, with NMR studies suggesting this was due to nucleophilic attack of cyanide on the meso-carbon of the dipyrrin moiety, converting it to a cyanodipyrromethane.60
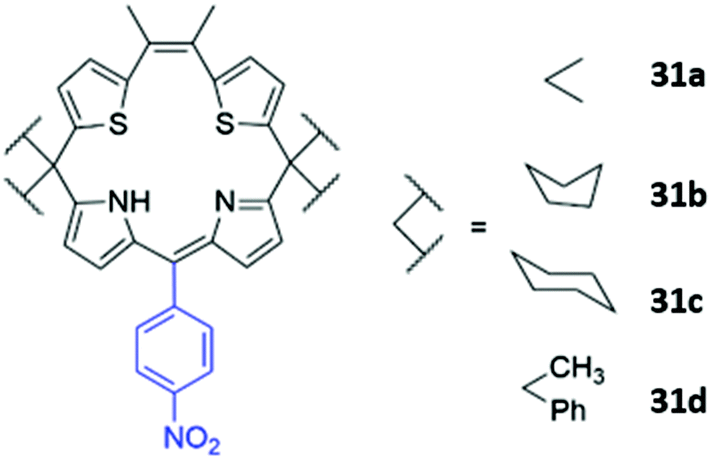 | ||
| Fig. 16 Structure of homoporphodimethenes for selective cyanide sensing with nitrophenyl chromophore highlighted in blue. | ||
3. Charged macrocycles
3.1. Macrocycles with ionisable groups
The incorporation of cationic groups capable of forming strong electrostatic interactions with anions is another common approach to enhance the potency of macrocyclic anion receptors, particularly when these electrostatic forces are combined with more directional interactions like hydrogen or halogen bonding. Amines, in particular, facilitate reversible guest-binding as they can be protonated in acidic media to ‘turn-on’ the anion recognition capabilities of the host. In some cases, the binding of a basic anionic guest leads to deprotonation of the amine group, which quenches the emission of adjacent fluorophores via enhanced PET. Therefore, amines are useful and versatile groups that can assist in binding anions as well as inducing spectral changes in the receptor.A handful of optical anion sensors rely predominantly on charged interactions established between multiple protonated amines and the anionic guest. Senge and co-workers employed a tetrakis(2-aminophenyl)porphyrin 32 as a selective host for pyrophosphate (Fig. 17). The hexaprotonated form of 32, formed in the presence of excess TFA, exhibited exceptionally high affinity for pyrophosphate, with an association constant of 2.26 × 109 M−1 in CH2Cl2. Pyrophosphate binding was accompanied by notable changes in the UV–Vis spectrum, including shifts in the Soret band, reduced intensity of the main porphyrin Q band and the appearance of two new Q bands, which resulted in a ‘naked eye’ colour change from yellow-brown to green. Interestingly, upon addition of excess of pyrophosphate (16 equiv.), the increased basicity of the solution causes deprotonation of the amino groups and regeneration of the free receptor. Addition of TFA reverses these changes, demonstrating switchable behaviour that could be repeated for at least eight cycles.61
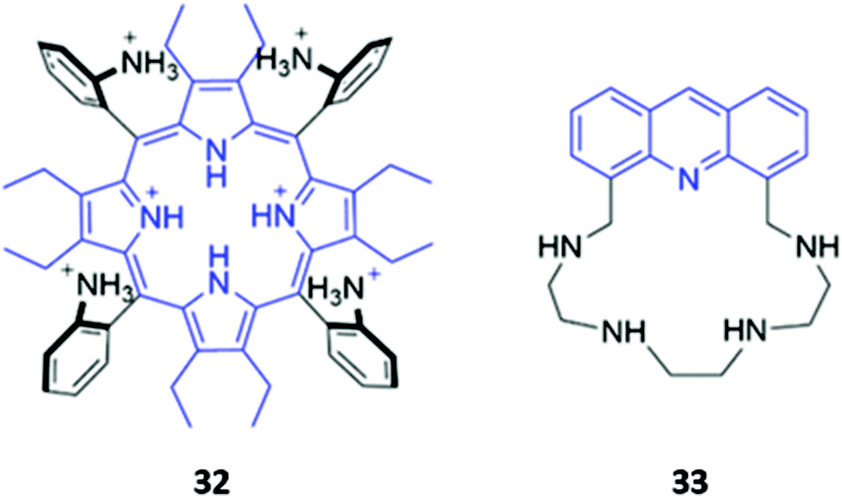 | ||
| Fig. 17 Structure of hexaprotonated tetrakis(2-aminophenyl)porphyrin host 32 (Ka (P2O74−) = 2.26 × 109 M−1 in CH2Cl2) and tetra-aza macrocycle 33 (Ks (F−) = 3.98 × 109 M−1 and Ks (Cl−) = 1.58 × 1011 M−1 in water). | ||
Gellini and co-workers synthesised a tetra-aza macrocycle 33 containing an acridine fluorophore (Fig. 17). At low-to-intermediate pH, the free receptor exists predominantly in the +2 or +3 protonation state and can undergo a proton migration from one of the protonated ammonium groups to the acridine moiety, forming an acridinium-like fragment that fluoresces intensely at 450 nm. Upon binding of fluoride or chloride in water, the emission is enhanced, with calculations suggesting that the F− and Cl− complexes of 33 preferentially undergo the proton transfer upon excitation to the S2 state due to a reduction of the energy barrier for this reaction. In contrast, addition of bromide or iodide to 33 results in fluorescence quenching, which was attributed to the heavy atom effect.62
Hybrid macrocycles that utilise both electrostatic interactions and hydrogen bonding interactions have been shown to act as potent anion sensors. Kataev and co-workers synthesised a series of hybrid amido-amine macrocycles 34a–c of varying sizes (Fig. 18). 34a showed an increase in fluorescence intensity upon addition of fluoride, whereas other halides and oxyanions induced aggregation and fluorescence quenching in aqueous MES buffer (10 mM, pH 6.2). This interesting optical response was attributed to the size of the macrocyclic cavity in 34a, which was suitable for encapsulating up to three fluoride ions, encouraging protonation of the tertiary amine groups and hindering PET from the amine to the naphthalimide fluorophore. The larger ionic radii of other anions prevent more than one anion from fitting inside the binding cavity, with the second anion bound on the periphery of the macrocycle, thereby resulting in aggregation by intermolecular π–π and electrostatic interactions.63 The same group synthesised a similar hybrid amide-amine macrocycle 35 (Fig. 18), which was able to bind various nucleoside monophosphates, of which cGMP was most strongly bound (K = 8.1 × 104 M−1 in aqueous acetate buffer with 2% DMSO). The binding of cGMP by the dicationic macrocycle led to fluorescence quenching due to π–π stacking of the guanine base with the pendant naphthalimide groups of the macrocycle.64 Subsequently, a cage-like amido-amine receptor 36 was reported, which contains an anthracene fluorophore held in a ‘strap’ over the macrocyclic cavity (Fig. 18). The receptor formed a 1:2 receptor–anion complex with phosphate in aqueous solution. Initial binding of phosphate leads to fluorescence quenching due to deprotonation of the benzylic amines by the basic phosphate anion, enhancing PET to the anthracene moiety. In excess phosphate, a fluorescence enhancement is observed, attributed to the protonation of the tertiary amines to maintain charge balance when additional anions coordinate to the outside of the receptor cavity.65
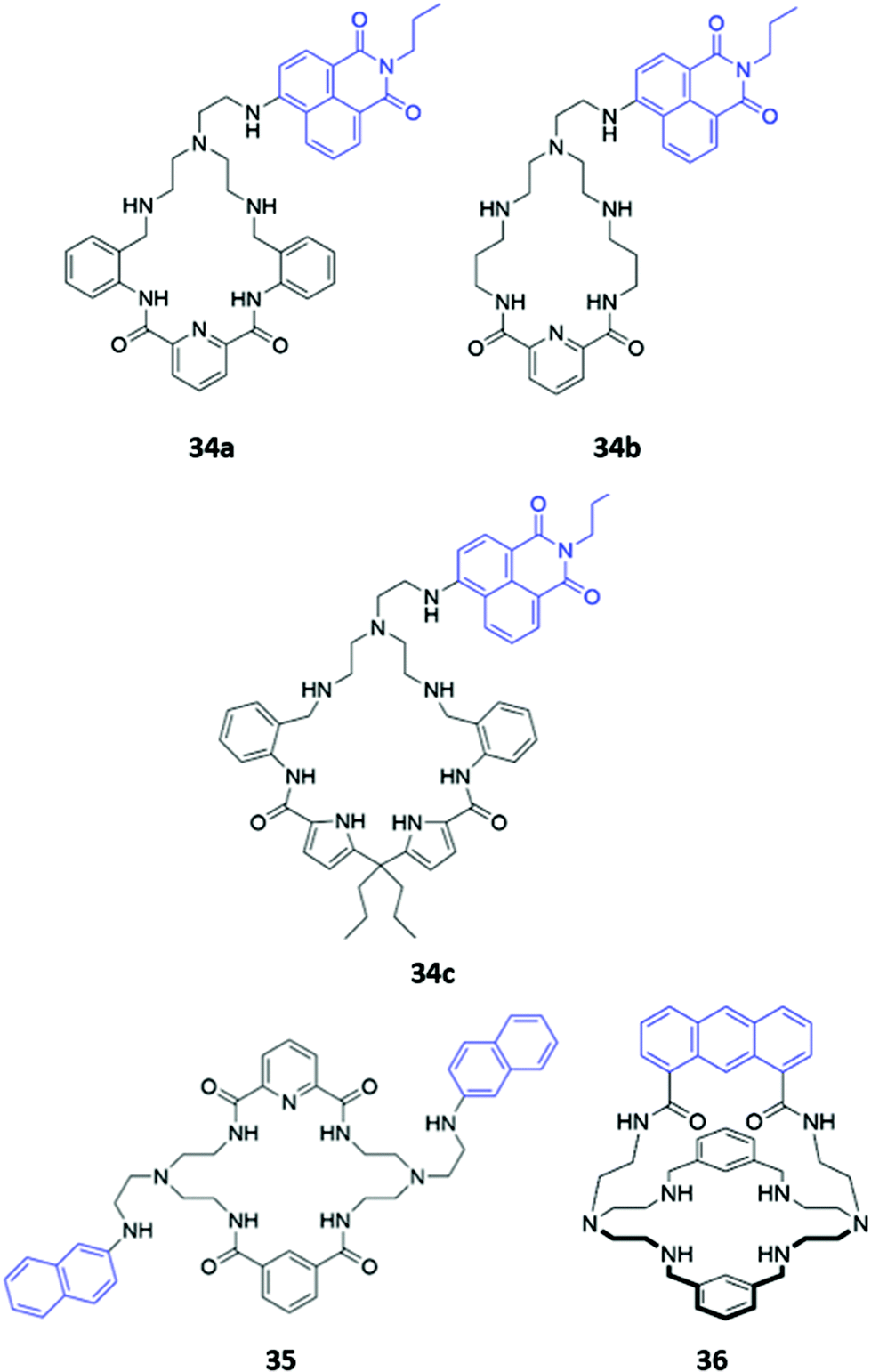 | ||
| Fig. 18 Structures of hybrid amine-containing macrocycles 34–36 with various fluorophores shown in blue. | ||
The versatility of amine groups is further demonstrated in their ability to bind both anionic and cationic guests. Chehri and co-workers synthesised a hybrid macrocycle 37 containing both ether and amine linkers, which served as a dual sensor for Fe(III) and iodide in aqueous solution (Fig. 19). Fe(III) was coordinated by the lone pairs of the ether and amine groups, leading to fluorescence quenching of the naphthalene groups via ligand-to-metal charge transfer, which opens a new non-radiative deactivation channel for electrons in the excited state. The addition of I− to the naked receptor induces a change in the UV–vis absorbance spectrum, with the appearance of a new band at 360 nm, accompanied by a change from colourless to pale-pink. The fluorescence intensity also decreases due to the heavy atom effect.66 Another dual sensor for Hg(II) and HSO4− consisted of a tetraazamacrocycle attached to a bromosalicylaldehyde pendant arm 38 (Fig. 19). Coordination of Hg(II) led to fluorescence quenching in CH3CN/HEPES buffer (2:8, v/v, 20 μM, pH 7.4). In contrast, the binding of HSO4− in the same solvent mixture, which was postulated to occur via multiple hydrogen bonds and/or charged interactions with the amine and hydroxyl groups of 38, induced chelation-enhanced fluorescence by restricting the free rotation of the sensor.67
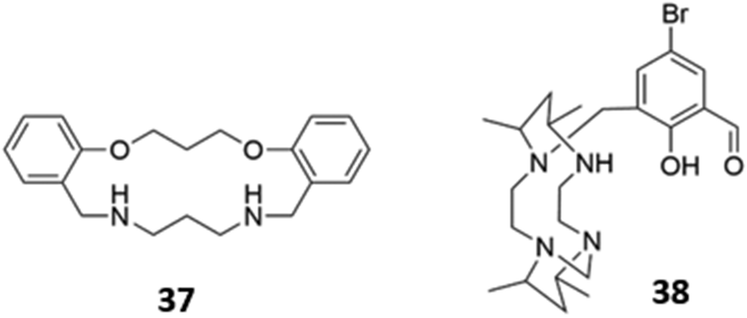 | ||
| Fig. 19 Structures of amine-based dual cation/anion receptors. | ||
Ionisable pyridine groups have also been used to enhanced the binding of receptors to anions. Johnson and co-workers reported two pyridine-containing macrocycles 39a–b (Fig. 20), which exhibit chloride-selective binding via a combination of hydrogen bonding from the amide/urea groups as well as charge-assisted hydrogen bonding from the protonated pyridinium moiety. The receptors were found to be unstable in aqueous solution, but could be incorporated into a thin film. In the solid state, intermolecular interactions between the fluorophore backbones induced a redshift of the emission peak when the receptors were exposed to an acidic solution of chloride.68
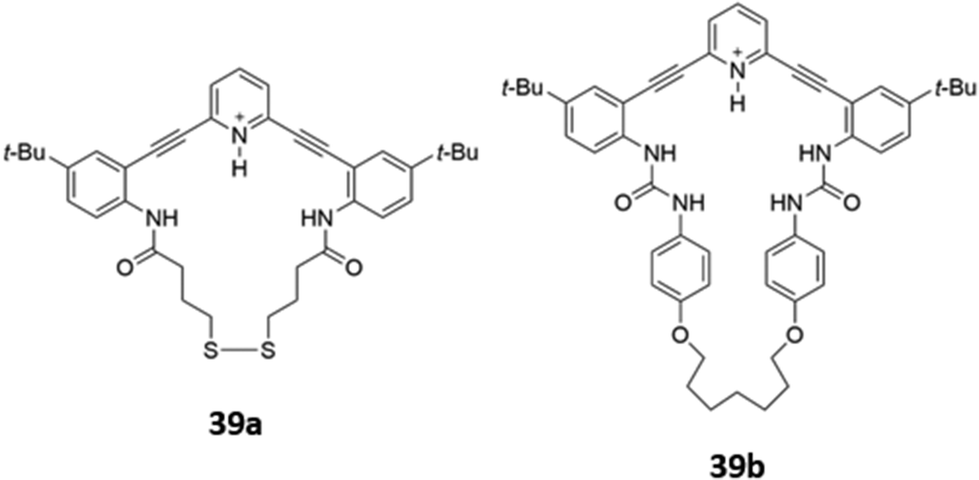 | ||
| Fig. 20 Structures of pyridine-based macrocyclic receptors for chloride sensing. | ||
3.2. Macrocycles with intrinsically charged groups
Cationic groups such as imidazolium and benzimidazolium units are capable of forming both electrostatic interactions and charge-assisted hydrogen bonds with anionic guests, and have been widely employed in a range of macrocyclic optical anion sensors. The majority of these hosts are cyclophane-like macrocycles consisting of multiple charged binding groups and signalling units connected by methylene spacers. Weakly-associated counterions such as PF6− are typically utilised in the preparation of such hosts to minimise competition for binding sites between the counterion and the target anion.Some early examples of cyclophane-like benzimidazolium receptors were reported by Gao and co-workers.69,70 A series of acridine-containing hosts 40a–b (Fig. 21), containing linkers of varying length and flexibility, were synthesised and found to bind H2PO4−via charge-assisted hydrogen bonds between the two benzimidazolium CH groups and the anion. This resulted in a significant redshift of the acridine fluorescence from 430 nm to 556 nm in acetonitrile, which was attributed to the anion-induced assembly of excimers between the acridine rings of two macrocycles. The degree of redshift increased as ring size decreased, which likely indicated differing extents of excimer formation; in smaller rings, there was greater repulsion energy between the benzimidazolium rings in the ground state and largest stabilisation energy in the excited state, leading to the lowest-energy excimer.69 Liu and co-workers recently reported a related pair of cyclophanes 41a–b, possessing two dibenzimidazolium units and an anthraquinone fluorophore (Fig. 21). The smaller macrocycle 41a exhibited fluorescence enhancement in the presence of acetate while the larger macrocycle 41b was a sensor for nitrate in H2O/CH3CN (1:1, v/v).71
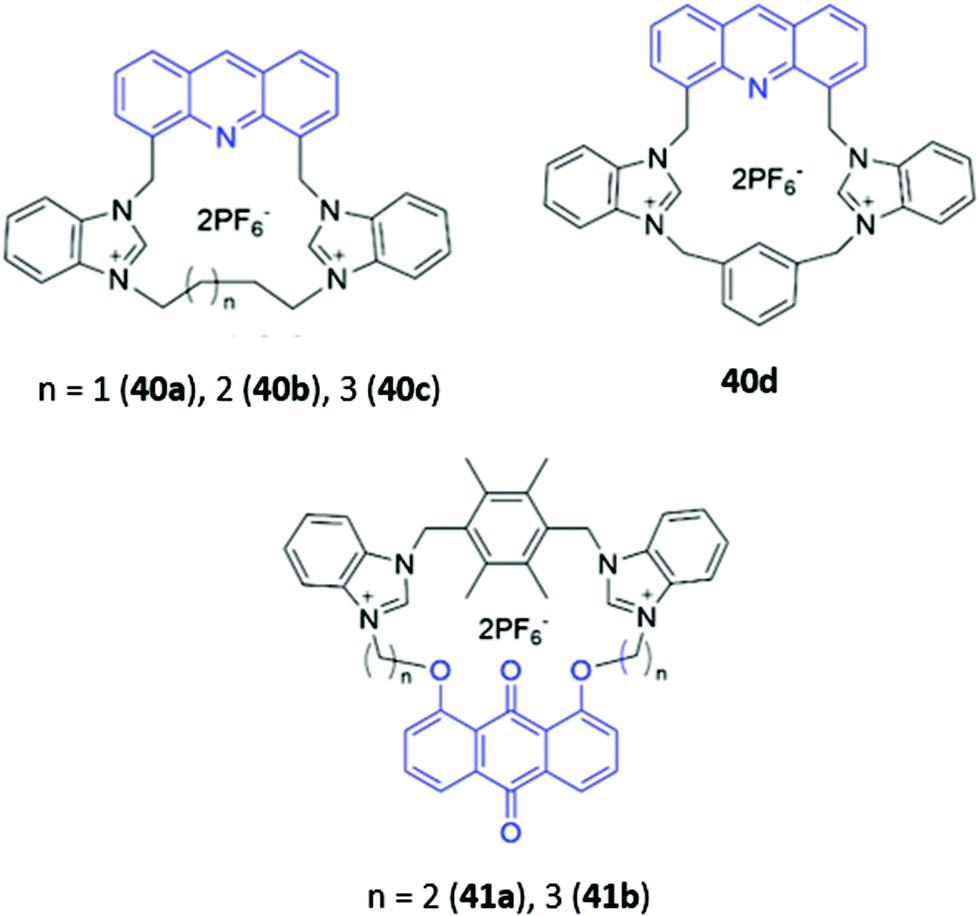 | ||
| Fig. 21 Structures of cyclophane-like receptors with benzimidazolium groups and various fluorophores (shown in blue). | ||
Zheng and co-workers synthesised a dicationic imidazolium-based macrocycle 42 with tetraphenylethylene fluorophores (Fig. 22), which are known to exhibit aggregation induced fluorescence (AIE). The receptor initially showed no emission when subjected to a range of inorganic anions; however, when 42 was mixed with one equivalent of zinc(II) acetate in a H2O/DMSO solution (99.5:0.5 v/v), blue fluorescence was observed upon the addition of P2O74−. The authors suggested that Zn(II) promoted aggregation of the pyrophosphate-bound complexes, resulting in AIE.72 Another dicationic imidazolium-based macrocycle 43 (Fig. 22), reported by Gong and co-workers, exhibited turn-on fluorescence in response to HP2O73−. However, unlike in 42, this fluorescence enhancement was attributed to the anion-induced disaggregation of non-fluorescent excimers present in concentrated acetonitrile solutions of the free receptor 43. A weaker fluorescence enhancement was also observed in the presence of H2PO4− and HCO3−.73
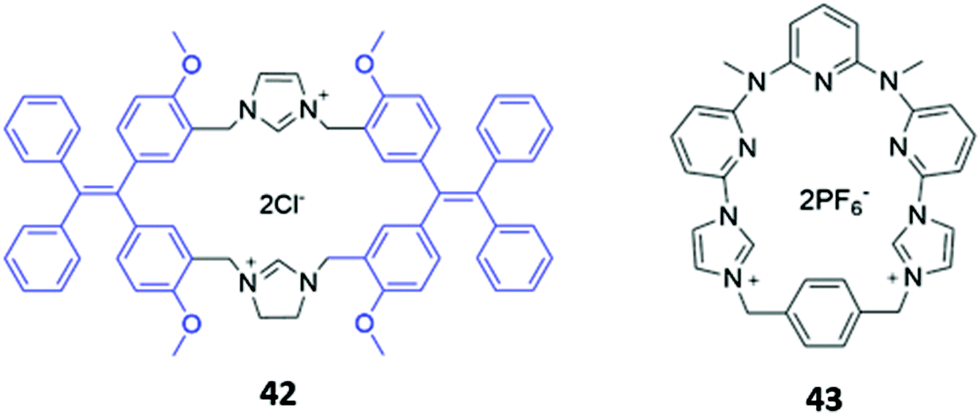 | ||
| Fig. 22 Structures of imidazolium-based macrocycles for sensing pyrophosphate, with Ka = 1.41 × 104 M−1 for 42 in H2O/DMSO and 4.7 × 106 M−1 for 43 in CH3CN. | ||
Metal-based chromophores have also been employed in conjunction with cationic imidazolium binding units. A simple ferrocene-containing macrocycle 44 with two imidazolium binding units exhibited optical responses to fluoride, acetate and dihydrogen phosphate in acetonitrile (Fig. 23). The free receptor is non-fluorescent due to quenching of the excited state by ferrocene, but the binding of fluoride and acetate appears to inhibit the quenching process, leading to weak fluorescence enhancement. Interestingly, the addition of dihydrogen phosphate to 44 did not elicit a fluorescence response, but instead caused a redshift and reduction in intensity of the main absorption band.74 Another imidazolium-containing macrocycle 45 (Fig. 23), which adopts a bowl-like conformation in solution and incorporates an Ru-arene as a fluorophore, was found to bind halides via CH–X hydrogen bonds, with iodide in particular inducing changes in the UV–Vis absorption spectrum of 45 in methanol.75
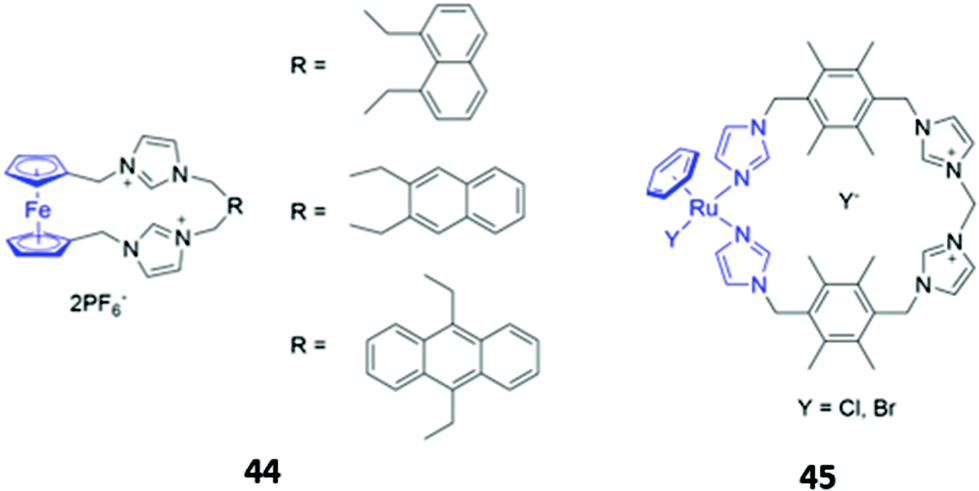 | ||
| Fig. 23 Cationic macrocycles containing metal-based chromophores (shown in blue). | ||
Cationic haloimidazolium groups can bind anions via charge-assisted halogen bonding interactions, which are highly directional interactions between the anion and an electron-deficient σ-hole present at the pole of a halogen atom covalently bound to an electron-withdrawing group such as imidazolium.76 Halogen bonding interactions were exploited for anion sensing in a series of cyclophanes consisting of haloimidazolium binding groups linked by naphthalene spacer groups (46a–c) (Fig. 24). The bromo- and iodo-imidazoliophane receptors displayed high affinities for iodide (Ka = 6.31 × 105 M−1) and bromide (Ka = 9.55 × 105 M−1) respectively. The binding of these ions was accompanied by a redshift and enhancement of the naphthalene emission band. Notably, the fluorescence response was observed even in a competitive CD3OD/D2O (9:1) solvent mixture. In contrast, a hydrogen bonding analogue 46d displayed no fluorescence response in the same solvent mixture, providing evidence for the greater strength and selectivity of halogen bonding for anion recognition in aqueous media.77
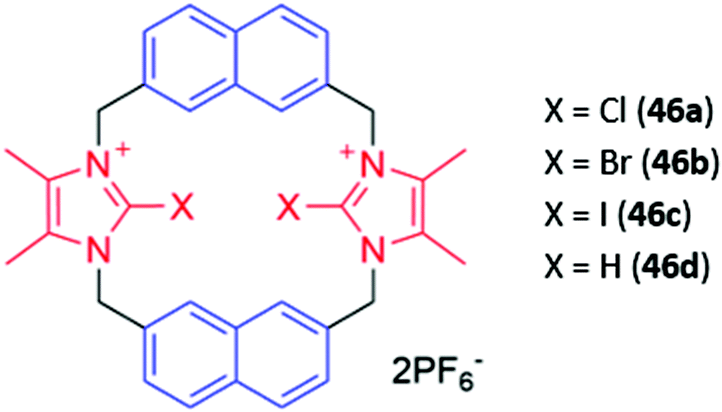 | ||
| Fig. 24 Structures of proto- or halo-imidazolium-containing cyclophanes (44a–d) with imidazolium shown in red and naphthalene fluorophores in blue. 46b bound I− with Ka = 6.31 × 105 M−1 and 46d bound Br− with Ka = 9.55 × 105 M−1 in 9:1 CH3OH/H2O. | ||
While imidazolium-based motifs are commonly used in macrocyclic anion receptors, charged pyridinium groups have also been employed. A hybrid macrocycle 47, containing four cationic pyridinium groups and four amide groups (Fig. 25), was found to preferentially bind 1,4-phenylenediacetate, which exhibits size complementarity for the large cavity. The dicarboxylate binds at the charged pyridinium sites and forms hydrogen bonds with the nearby amide groups, as well as weak π-stacking interactions with the aromatic anthracene spacer. Fluorescence enhancement was observed upon anion addition to an acetonitrile solution of 47, attributed to inhibition of PET from the binding sites to the anthracene moieties.78 Recently, Cao and co-workers synthesised a octacationic cage 48 based on eight pyridinium moieties and two tetraphenylethylene (TPE) units. 48 was shown to bind the fluorescent dye sulforhodamine 101 (SR101), which contains two sulfonate groups that form electrostatic interactions with the pyridinium sites as well as an aromatic core that can form π-stacking interactions with the TPE moieties. Binding of SR101 to 48 led to quenching of the fluorescence emission at 545 nm as well as the appearance of a sharp emission peak at 621 nm, resulting in a fluorescence colour change from yellow to red under UV illumination.79
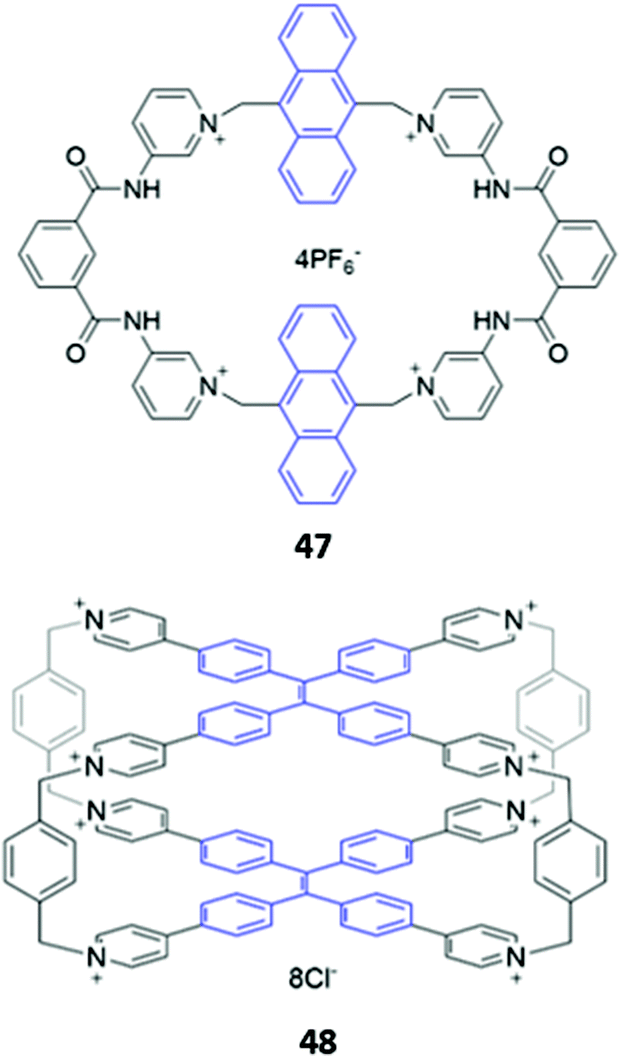 | ||
| Fig. 25 Structure of pyridinium-based macrocycles with fluorophores shown in blue. | ||
4. Metallomacrocycles
The cationic and Lewis acidic nature of metal ions, as well as the interesting photophysical properties exhibited by some transition metal and lanthanide ions, has prompted the development of many metal-based systems for anion sensing.15 Combining metal ions with macrocyclic ligands, usually decorated with additional chromophores or anion-binding sites, can enhance the sensing capabilities of these systems.Homodimetallic complexes of cryptand-like ligands have been used to selectively encapsulate anions that are of suitable size and geometry to bridge the metal centres. An early example was reported by Ugozzoli and co-workers in 2000, in which three di-copper bis-tren cryptands with linkers of varying length and flexibility (49a–c) demonstrated preferential binding for different anions (Fig. 26). 49a exhibited a preference for linear anions like N3− and NCO−, which are long enough to bridge the two Cu(II) centres. In contrast, the more flexible ether and 2,5-dimethylfuran spacers in 49b and 49c respectively enabled the receptors to rearrange into geometries suitable for binding various halide ions. The coordination of chloride to the copper centres of 49c in particular gave rise to an intense absorption band at 410 nm in aqueous solution, which was attributed to ligand-to-metal charge transfer.80
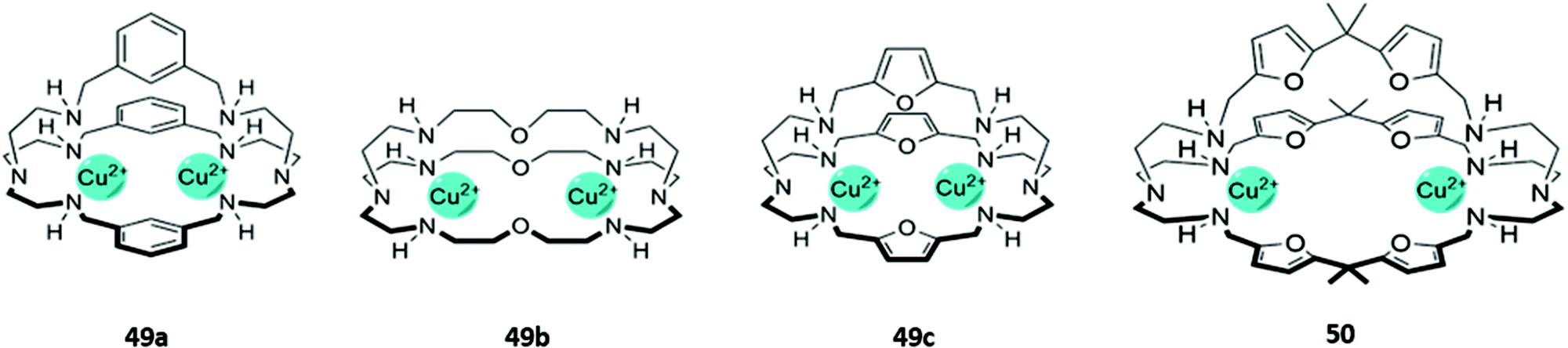 | ||
| Fig. 26 Structures of dicopper cryptand-like anion receptors. 50 binds GTP as a 1:2 host–guest complex with K1 = 5.01 × 104 M−1 and K2 = 1.58 × 104 M−1 in 1:1 CH3OH/H2O. | ||
A dicopper complex of an extended cryptand-like ligand 50 was found to bind nucleoside monophosphates in H2O/CH3OH (1:1, v/v) (Fig. 26). The anionic guests bridge the two Cu(II) ions in the cryptand, binding through the phosphate group and a donor atom on the nucleobase. In the absence of a nucleoside monophosphate, 50 could form a non-fluorescent complex with the anionic dye 6-carboxyfluorescein. The dye was displaced upon binding of GMP to the receptor, switching on the yellow fluorescence emission of the free dye.81 This indicator–displacement method, in which the displacement of a dye (the indicator) from the receptor by the target anion switches on the fluorescence of the free dye, was later used extensively by Hossain and co-workers for the optical sensing of dicarboxylates. Over a series of publications, the authors demonstrated that dinuclear complexes of foldamer-like macrocyclic ligands could form non-fluorescent ion pairs with eyosin Y (an anionic dye). The dye was readily displaced by dicarboxylates such as oxalate and citrate, which were bound more strongly by the receptors. Anion binding and the concomitant release of eyosin Y was signalled by an increase in the characteristic fluorescence of the free dye.82–88
In a twist on the indicator–displacement method, Zhang and co-workers synthesised a host a 2-(hydroxy)-naphthyl imino-functionalised pillar[5]arene 51 (Fig. 27), which coordinated to Fe(III) through its imine nitrogen and hydroxyl groups in H2O/DMSO (1:9, v/v). In the presence of Fe(III), the blue fluorescence of 51 was quenched. However, upon addition of fluoride, Fe(III) is displaced to form [FeF6]3−, restoring the fluorescence of the free receptor. Therefore, 51 could be used as either a ‘turn-off’ fluorescence sensor for Fe(III) or a ‘turn-on’ fluorescence sensor for fluoride.89
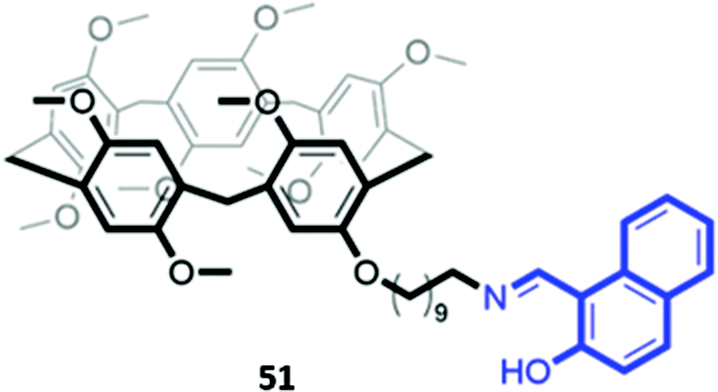 | ||
| Fig. 27 Structure of pillar[5]arene receptor 51 functionalised with a 2-(hydroxy)-naphthyl imino fluorophore (in blue). | ||
Macrocyclic complexes of zinc have also been explored as anion sensors. A mononuclear Zn(II) complex of a bipyridine crown ether 52 (Fig. 28) was shown to undergo a 3-fold fluorescence enhancement in the presence of acetate, with smaller enhancements for larger carboxylates in H2O/CH3OH (1:4, v/v, buffered at pH 7.4). The carboxylates were postulated to chelate the Zn(II) centre as well as form hydrophobic interactions with the crown ether ring; interestingly, an acyclic analogue of 52 showed no fluorescence response to carboxylates under the same conditions, suggesting at the importance of the crown ether.90 A tetranuclear Zn(II)–Schiff base complex 53, which contained multiple imine and amine groups (Fig. 28), underwent a blueshift of its absorption band upon addition of fluoride or acetate in aqueous methanolic solvent mixtures. Computational studies suggested this was due to a deprotonation of the iminium groups by the basic anions, making the iminium substituents less electron-withdrawing and widening the HOMO–LUMO gap of the adjacent aromatic ring. The spectral changes could be reversed by addition of a protic acid to the receptor–anion solutions.91
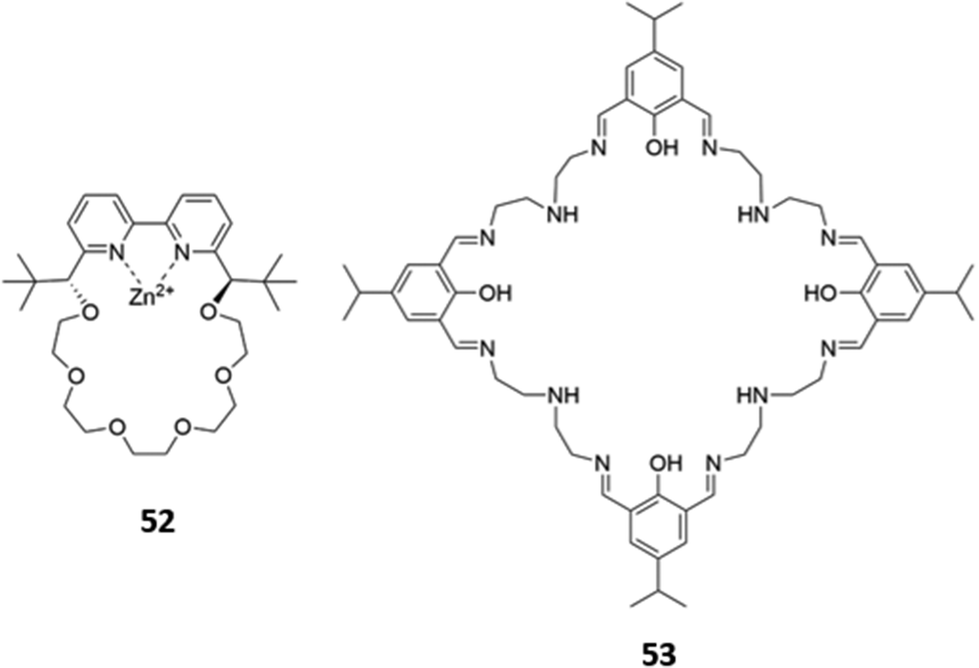 | ||
| Fig. 28 Structures of Zn-based metallomacrocyclic anion receptors 52 (Ka (CH3COO−) = 880 M−1 in 1:4 H2O/MeOH) and 53. | ||
A handful of metallomacrocyclic anion sensors have been synthesised containing metal ions that do not directly coordinate to the anionic guests, but instead serve as fluorogenic or chromogenic signalling units, whilst providing a positively-charged environment to enhance anion binding. Ru(II) polypyridyl complexes have been widely used in chemosensing due to their favourable photophysical properties, including high photostability, long emission lifetimes and emission in the visible region.92 It is therefore unsurprising that Ru complexes are amongst the most common metal-based chromophores used in optical anion sensors.93–95 An early example reported in 1996 was a series of macrocyclic and calix[4]arene-based receptors 54a–d containing bipyridyl units to which ruthenium(II) could be appended (Fig. 29). 1H NMR studies in DMSO-d6 indicated that receptors preferably bound H2PO4− over Cl−via convergent hydrogen bonds. A comparison of the two calix[4]arene receptors showed that 54c, which contains shorter linkers and therefore a smaller binding cavity, exhibited stronger binding to all anions tested. The interaction of 54c with H2PO4− resulted in a blueshift of the Ru(II) MLCT emission band, accompanied by an increase in emission intensity. The latter was attributed to the rigidification of the macrocycle upon anion binding, which decreases the rate of non-radiative decay.96 A series of hetero-dinuclear macrocycles containing ruthenium(II) tris(bipyridyl) alongside ferrocene, cobaltocenium and osmium(II) tris(bipyridyl) were subsequently synthesised and found to bind chloride strongly in acetonitrile with a concomitant enhancement in emission intensity.96,97
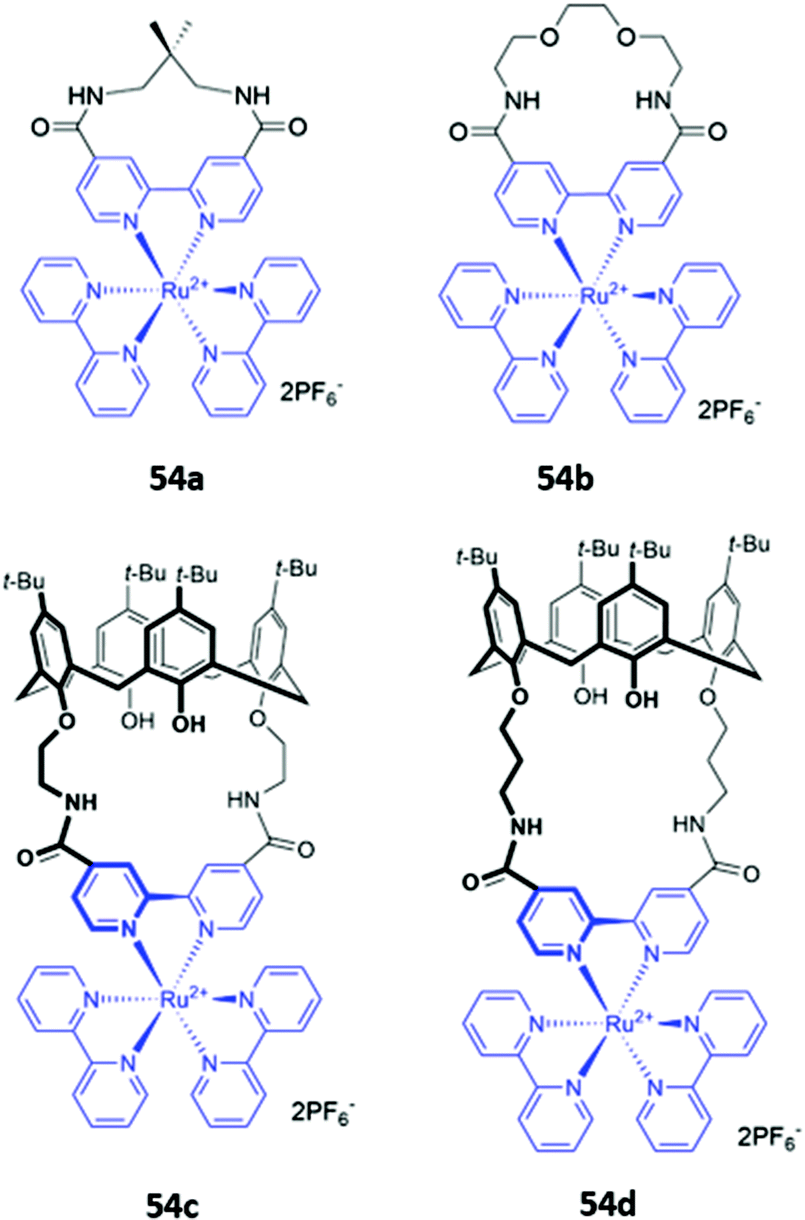 | ||
| Fig. 29 Macrocycles incorporating ruthenium(II) bipyridyl complexes (shown in blue) for sensing of various anions. | ||
Mukherjee and co-workers synthesised platinum-based square macrocycles 55a–c which contain a conjugated Pt-ethynyl-carbazole fluorophore and amide groups for anion binding (Fig. 30). The Pt(II) centres were not involved in anion binding; instead, they served the dual purposes of directing the formation of square macrocycles and facilitating PET from the amide groups to the carbazole, quenching fluorescence from the excited state of the carbazole in the free receptor. Binding of pyrophosphate to the amide groups suppressed electronic transfer, leading to significant fluorescence enhancement in DMF.98
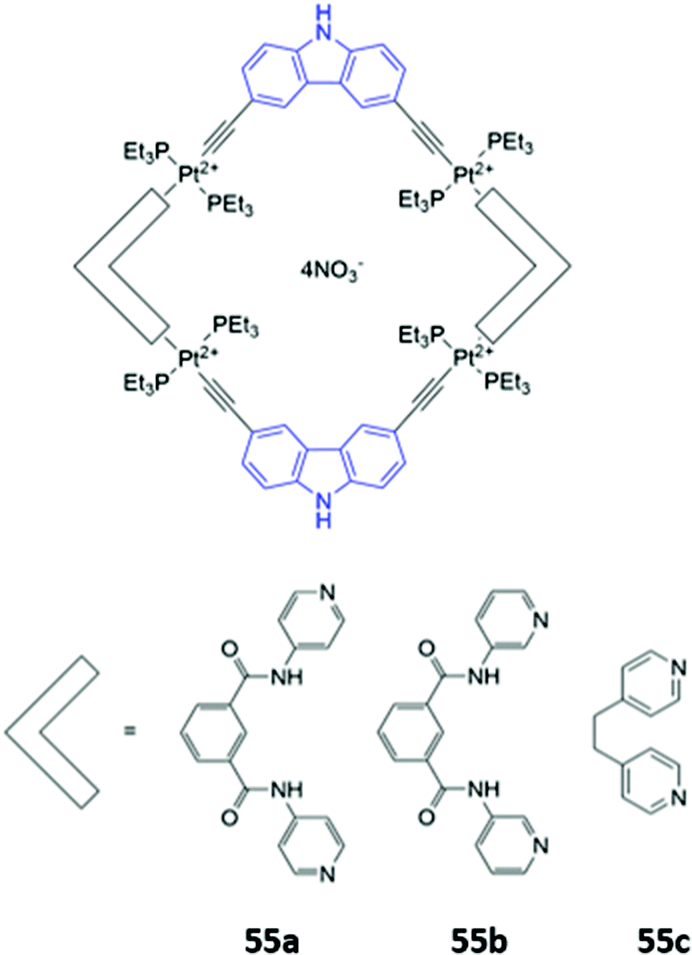 | ||
| Fig. 30 Structures of Pt-containing square macrocycles with carbazole fluorophores shown in blue. 55a binds P2O74− with Ka = 2.4 × 104 M−1 in DMF. | ||
5. Mechanically interlocked structures
Mechanically interlocked molecules (MIMs), such as rotaxanes and catenanes, possess topologically unique cavities that mimic the three-dimensional binding sites of proteins. These binding cavities provide a shielded hydrophobic environment to reduce the thermodynamic cost of binding, particularly in competitive solvents. The cavities can also be decorated with multiple pre-organised binding motifs that are complementary in geometry and size to the target guest. Building upon the seminal MIM metal template synthesis work of Sauvage in particular,99–103 in the past two decades, significant progress has been made in the development of complementary template-based approaches for MIM synthesis, including anion templation104 and active metal templation105–108 strategies. These powerful synthetic methods have enabled the preparation of complex interlocked structures in greatly improved yields, making MIMs more than synthetic curiosities and unlocking the vast potential of these molecules as platforms for anion sensing.5.1. MIMs containing organic luminophores
In an analogous manner to macrocyclic anion hosts, optical sensing capabilities can be introduced to MIMs by the incorporation of fluorogenic or chromophoric units that exhibit anion-induced spectral changes. The use of aromatic luminophores such as naphthalene, which also serve as spacer groups to adjust the size of the macrocycles, was a natural starting point.A halogen bonding [2]catenane 56 was constructed from two identical macrocycles, each of which contained a cationic bromoimidazolium for anion binding as well as naphthalene groups as signalling units (Fig. 31). The [2]catenane selectively bound Br− (Ka = 1.48 × 105 M−1) and Cl− (Ka = 3.76 × 106 M−1) in acetonitrile solution to form 1:1 stoichiometric host–guest complexes, with molecular dynamics (MD) simulations indicating that the halide anions were held in the binding cavity by convergent halogen bonds. Anion binding was accompanied by a decrease in the emission band at 309 nm, alongside the appearance of a new broad band at 445 nm.109 Another example of a [2]catenane-based anion sensor was the hetero-[2]catenane 57, which bound a range of anions via a combination of hydrogen bonding from isophthalamide group of one macrocycle and charge-assisted halogen bonding from the iodotriazolium of the second macrocycle (Fig. 31). Upon addition of anions, in particular acetate and H2PO4−, an enhancement of the naphthalene emission bands was observed in acetonitrile, attributed to an increase in the rigidity of 57, which reduces the available non-radiative decay pathways. In contrast, no observable changes in the UV–vis absorption spectrum occurred under analogous conditions. This reflects the lower sensitivity of UV–vis spectroscopy compared to fluorescence spectroscopy, with the former unable to detect the subtle electronic and structural changes that occur in the highly pre-organised [2]catenane host.110
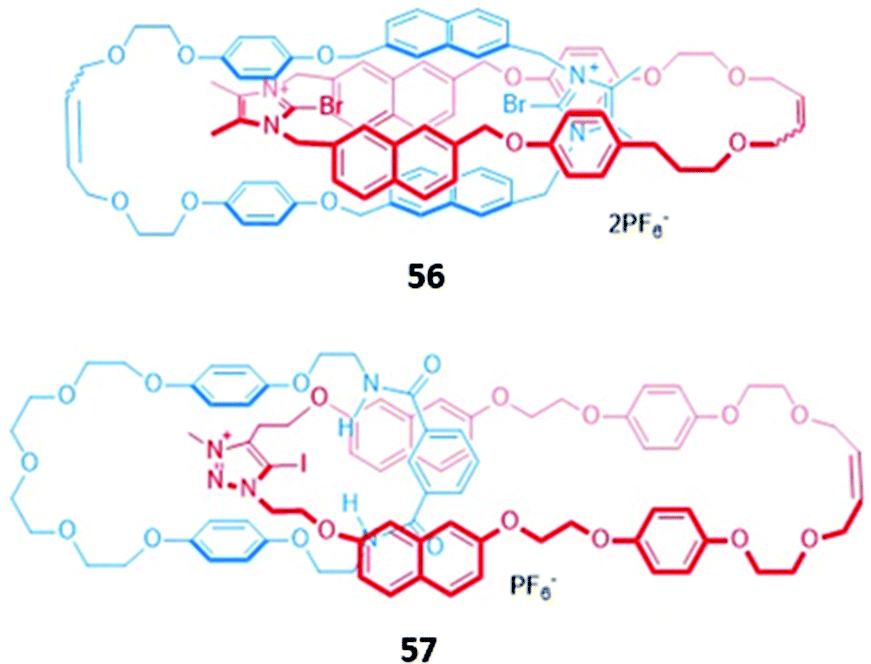 | ||
| Fig. 31 A halogen bonding [2]catenane 56 selective for Br− (Ka = 1.48 × 105 M−1) and Cl− (Ka = 3.76 × 106 M−1) in CH3CN and mixed halogen/hydrogen bonding [2]catenane 57 which binds AcO− (Ka = 1.5 × 105 M−1) and H2PO4− (Ka = 4.5 × 104 M−1) in CH3CN. | ||
An indolocarbazole-containing [2]rotaxane 58, synthesised by active metal templation, displayed an unusual preference for oxyanions (CH3COO− and H2PO4−) over halide ions in acetone-d6/D2O (95:5, v/v) (Fig. 32). A strong affinity for sulfate was also observed, possibly due to the complementary tetrahedral geometry of the dianion and the arrangement of the hydrogen bond donor groups in the binding cavity of 58. The addition of anions induced a redshift in the emission band of the indolocarbazole fluorophore. In addition, chloride binding resulted in fluorescence enhancement, while fluoride and acetate led to quenching.111
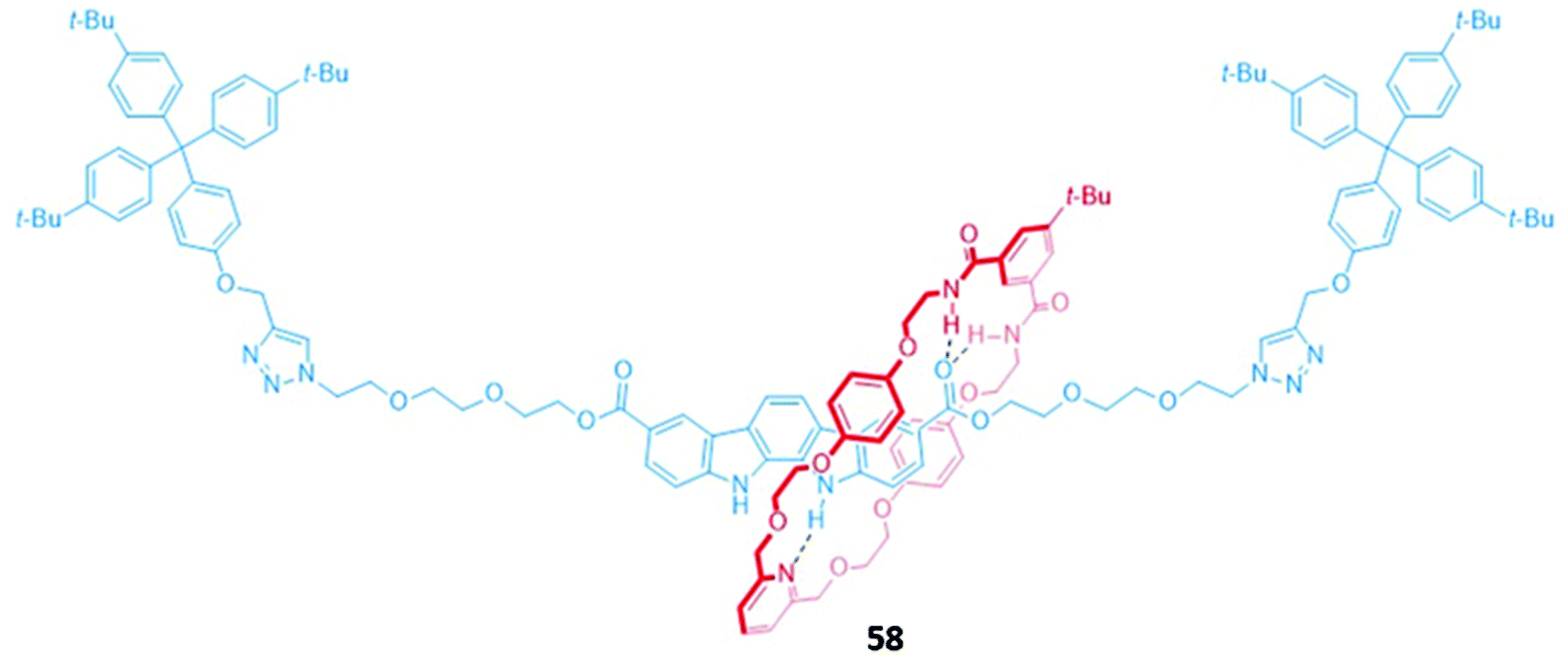 | ||
| Fig. 32 A neutral indolocarbazole-containing [2]rotaxane which binds AcO− (Ka = 2.36 × 103 M−1) and H2PO4− (Ka = 2.05 × 103 M−1 in 95:5 acetone-d6/D2O). | ||
Higher-order interlocked structures have been shown to produce interesting optical responses upon the binding of anions. A dicationic [3]rotaxane 59 (Fig. 33) containing a naphthalene reporter group was synthesised by chloride anion templation and formed 1:2 host–guest complexes with the monoanions Cl−, Br− and AcO−, producing modest enhancements of fluorescence intensity arising from the increased rigidity of the receptor. In contrast, the addition of sulfate to a CDCl3/CD3OD (1:1, v/v) solution of 59 initially resulted in a 1:1 stoichiometric complex with the anion bridging the two macrocycles. The rotary flexibility of the naphthalene linker allowed the rotaxane to fold around the sulfate guest to form a sandwich complex, quenching the naphthalene fluorescence through the formation of close contacts. The second equivalent of sulfate induced a conformational change in 59 to a more open form, forming a 1:1 host–guest complex in which each sulfate is associated with one macrocycle. This conformational change restored the fluorescence of the naphthalene units.112
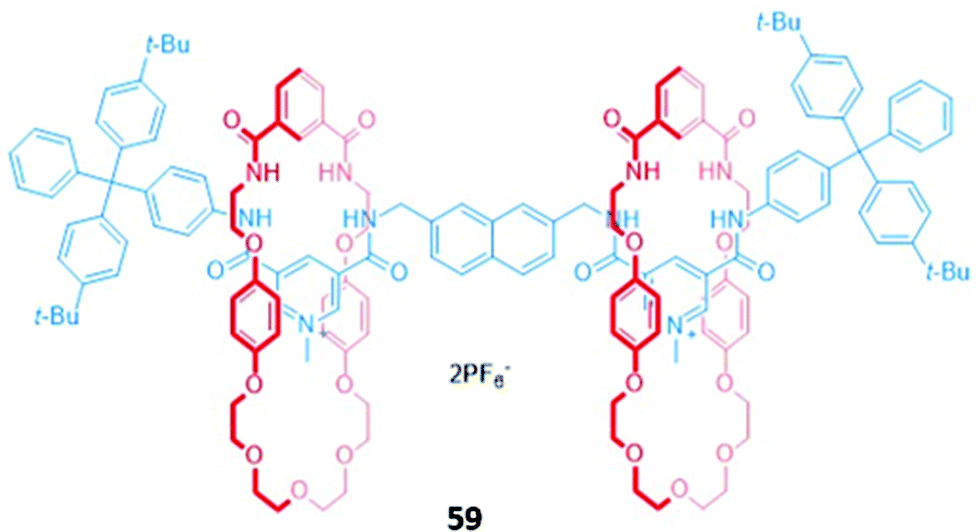 | ||
| Fig. 33 A dicationic [3]rotaxane which displays an unusual optical response to SO42− (K1 > 104 M−1 in 45:45:10 CDCl3/CD3OD/D2O). | ||
A chiral [3]rotaxane 60, which possessed a central (S)-BINOL unit flanked by two halogen bonding 3,5-bis(iodotriazole)pyridinium motifs in the axle (Fig. 34), was employed as a sensor for dicarboxylates. The dicarboxylate guests resided in the binding pocket between the two macrocycles, spanning the gap between the halogen bonding sites of the axle to form 1:1 stoichiometric sandwich complexes. Significant fluorescence quenching was observed in the presence of dicarboxylates, presumably due to the formation of close contacts between the bridging anion and the (S)-BINOL fluorophore, whereas the binding of smaller anions like chloride elicited much weaker responses. 60 exhibited an impressive ability to discriminate between both enantiomers (Ks/KR for glutamate = 5.7 ± 0.3) and geometric isomers (Kfumarate/Kmalonate = 4.4 ± 0.3) in CHCl3/CH3OH/H2O (60:39:1 v/v/v). MD simulations suggested the observed selectivity stemmed from differences in the number of synergistic halogen and hydrogen bonding interactions that could be formed between 60 and the respective stereo- or geometric isomers, alongside important contributions from anion-solvent interactions.113
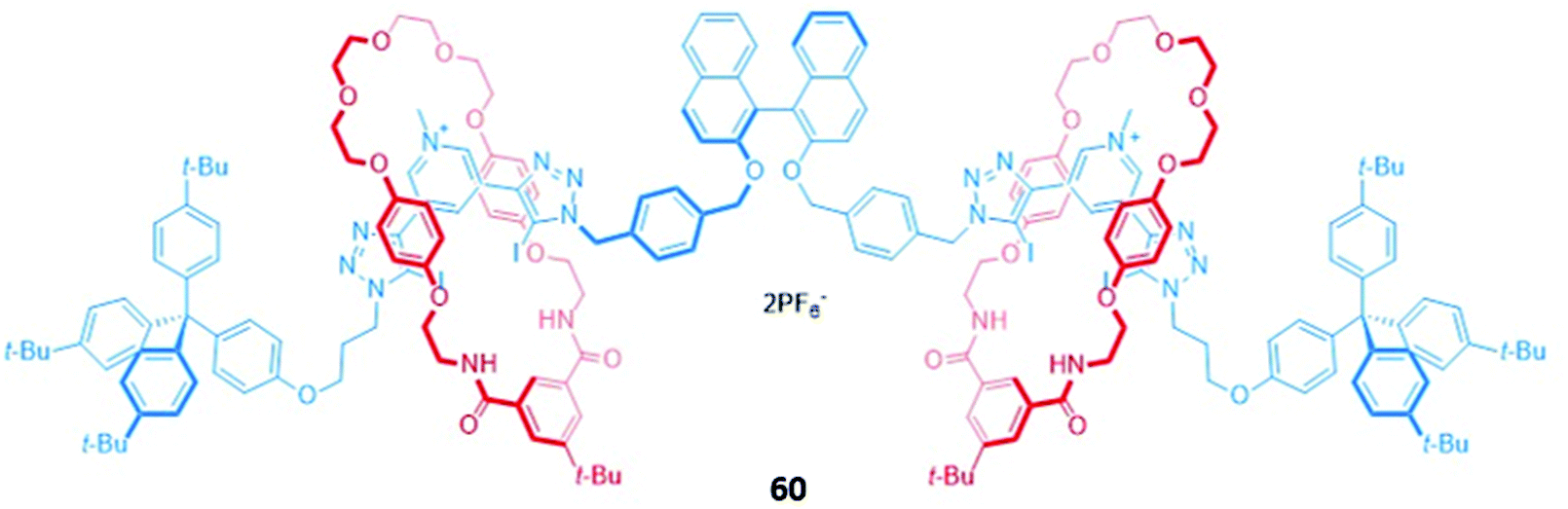 | ||
| Fig. 34 A chiral (S)-BINOL-containing [3]rotaxane with two bis(iodotriazolium) stations, which displays stereoselective binding of (S)-/(R)-glutamate (Ks/KR = 5.7 ± 0.3) and fumarate/malonate (Kfumarate/Kmalonate = 4.4 ± 0.3) in 60:39:1 CHCl3/CH3OH/H2O. | ||
5.2. MIMs containing metal-based luminophores
Luminescent metal complexes have also been incorporated into the structure of MIM-based anion receptors to produce effective optical sensors. An early example was a pair of cationic [2]rotaxanes 61a–b with rhenium(I) or ruthenium(II) bipyridyl complexes incorporated into the macrocyclic components (Fig. 35). The rotaxanes preferentially bound chloride over larger oxyanions, which were unable to penetrate the binding cavity. This preference may have been a consequence of using a chloride template to construct the interlocked structure, producing a cavity that was complementary to the anion template. The addition of chloride or H2PO4− induced an increase in fluorescence intensity in both rotaxanes, with the optical response persisting even in competitive solvent systems containing up to 10% water.114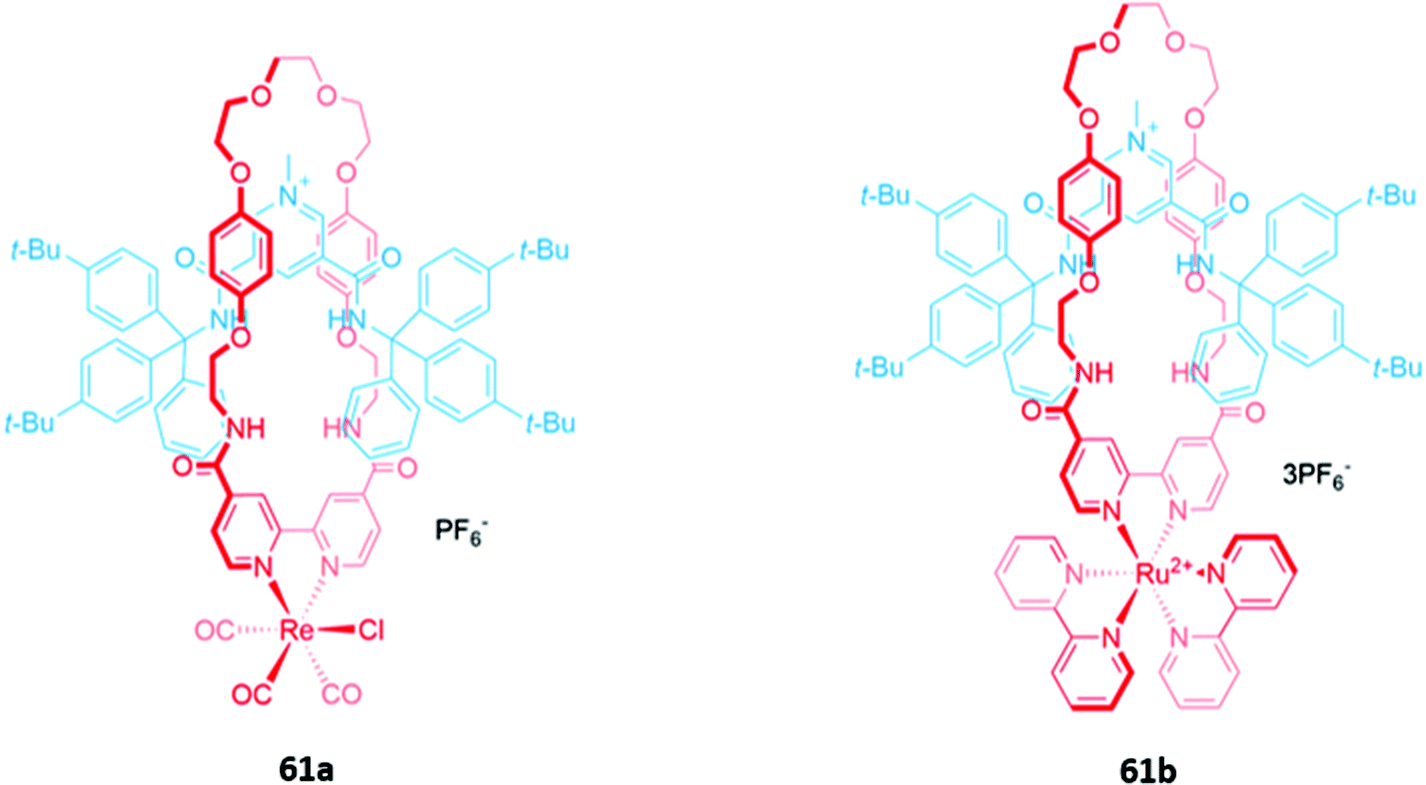 | ||
| Fig. 35 [2]Rotaxanes with a ruthenium(II) (61a) or rhenium(I) (61b) complex integrated into the macrocycle which preferentially bind chloride (Ka > 104 M−1 for 61a in 9:1 acetone/D2O and Ka = 1500 M−1 for 61b in 7:3 acetone/D2O). | ||
A ruthenium(II) bipyridyl complex was once again integrated into the macrocyclic component of a [2]rotaxane 62, which contained permethylated β-cyclodextrin stoppers for water solubility and exhibited a fluorescence response to anions in pure water (Fig. 36). NMR studies suggested that 62 formed 1:1 receptor–anion complexes in which the anionic guest resides in the cavity formed between the macrocycle and axle, where it is held by convergent hydrogen and halogen bonds. The receptor demonstrated a remarkably high affinity for iodide (Ka = 6300 M−1 in water), as well as moderate affinities for chloride (Ka = 190 M−1), bromide (Ka = 1020 M−1) and sulfate (Ka = 450 M−1). The addition of iodide and sulfate induced modest enhancements of the ruthenium(II) MLCT emission, attributed to rigidification of the receptor upon complexation with the anions, allowing the rotaxane to be employed as a luminescent sensor for these anions in water.115
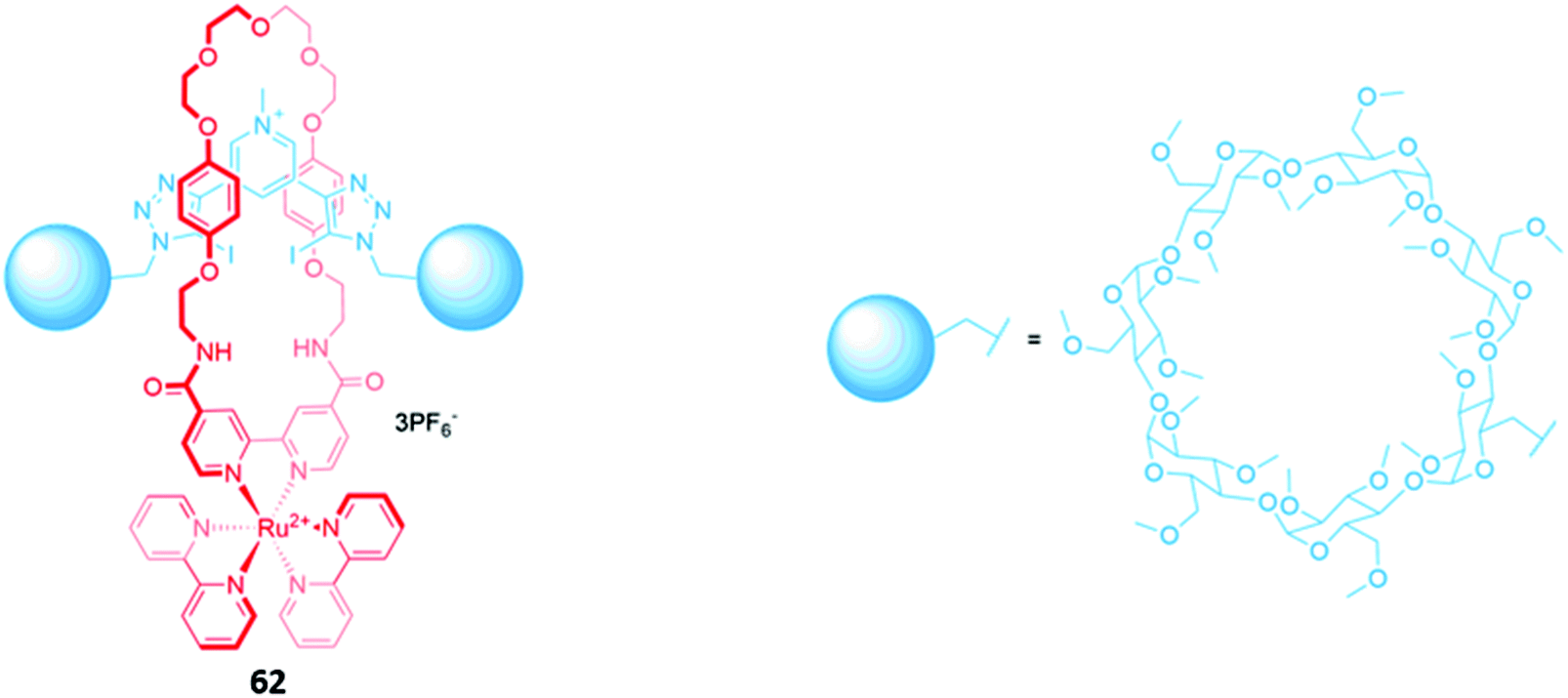 | ||
| Fig. 36 A water-soluble ruthenium(II) bipyridyl-functionalised [2]rotaxane which binds iodide (Ka = 6300 M−1 in water). | ||
Recently, luminescent 1,3-di(2-pyridyl)benzene complexes of Pt(II) and Ru(II) were incorporated into a range of acyclic, macrocyclic and interlocked anion hosts (63a–f) (Fig. 37). The acyclic Pt receptor 63a bound anions in a 1:1 stoichiometry and underwent significant fluorescence enhancement of 60% upon addition of chloride or sulfate, attributed to rigidification of the receptor upon anion coordination. On the other hand, the macrocyclic analogue 63c showed a smaller fluorescence enhancement due to the higher pre-organisation and rigidity of the free receptor. Promisingly, preliminary luminescent titrations of the cationic [2]rotaxane 63f with chloride suggested that the interlocked host exhibited a significantly higher affinity for the halide than that of the acyclic or macrocyclic analogues in a competitive solvent mixture of acetone-d6/D2O (7:3, v/v).116
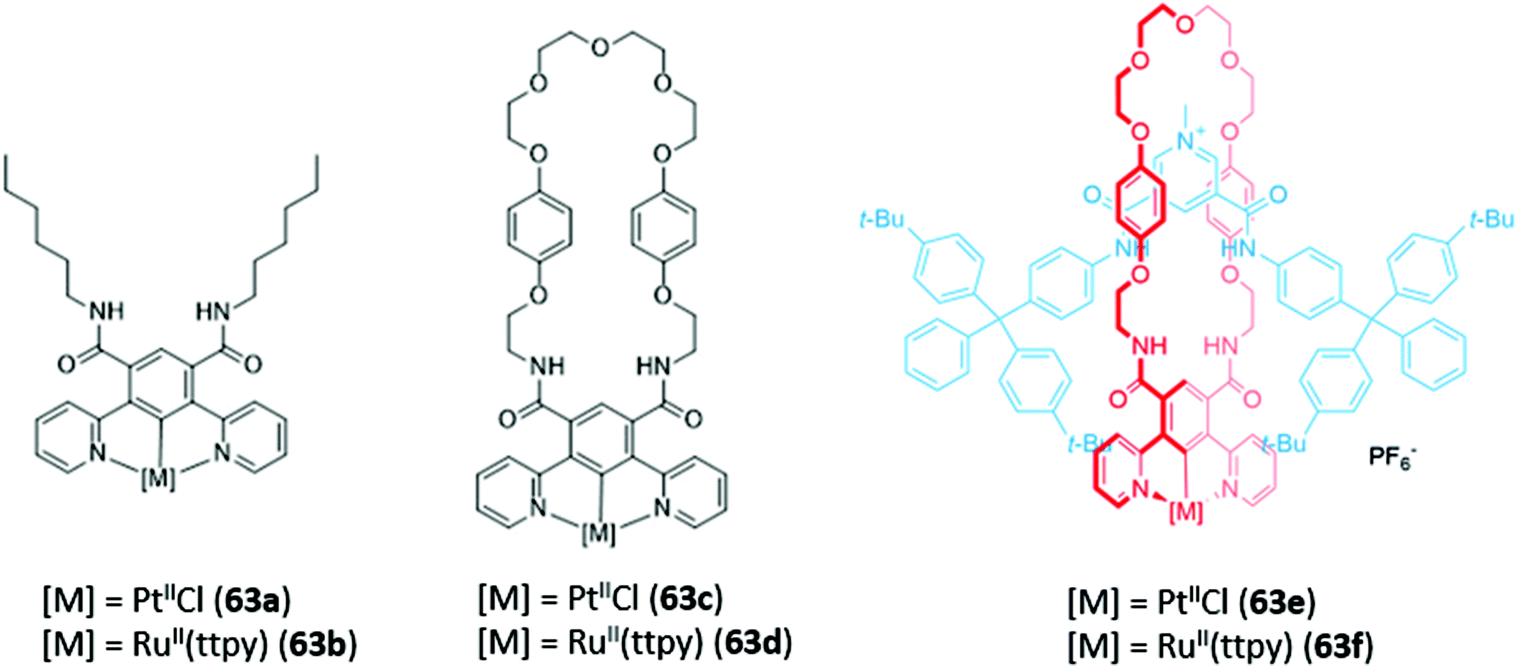 | ||
| Fig. 37 Anion receptors incorporating 1,3-di(2-pyridyl)benzene fluorophores 63a–f. | ||
An all-halogen bonding [2]rotaxane 64, which contains a rhenium(I) complex integrated into the macrocyclic component (Fig. 38), was shown to selectively bind halide ions over a range of oxyanions. The preference for halides was attributed to complementarity in size and shape between the spherical anions and the binding cavity, with X-ray crystallographic studies confirming the presence of halogen bonds between the chloride anion and the iodotriazole/iodotriazolium groups. Luminescence titrations in solvent systems containing up to 50% water showed an initial increase in the intensity of the rhenium(I) MLCT band in the presence of halides, followed by fluorescence quenching when a large excess of the anions was added.117
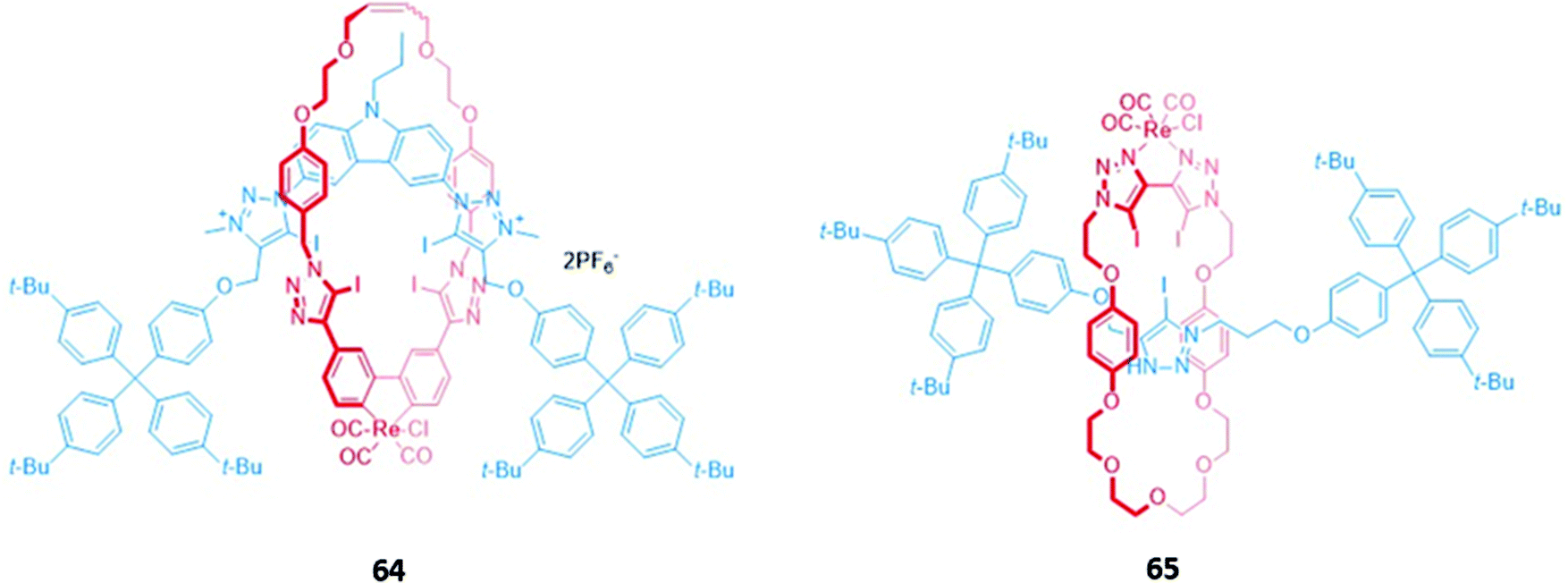 | ||
| Fig. 38 [2]rotaxanes with a rhenium(I) complex integrated into the macrocycle. 64 binds various halides while 65 binds chloride preferentially with Ka = 560 M−1 in CHCl3. | ||
A related rhenium-containing [2]rotaxane 65 (Fig. 38) was prepared via an elegant synthetic route involving an active template Cu-mediated alkyne–azide cycloaddition reaction (AT-CuAAC).118 A macrocycle containing a flexible bis-iodotriazole motif was used to bind the copper(I) template, which promoted a Cu–AAC reaction between the two stopper precursors to yield the interlocked host. After removal of the metal template, introduction of a more sterically-demanding rhenium(I) complex, which coordinated to the bis-iodotriazole group in an exotopic manner, inverted the bis-iodotriazole group so that the halogen bond donors were directed into the macrocyclic cavity. 65 preferentially bound chloride over larger halides and acetates, leading to a decrease in the broad rhenium(I) MLCT absorption band in CHCl3.
Surprisingly, despite the useful luminescent properties of lanthanide ions in sensing and imaging,119 the only report of a lanthanide-containing MIM used in optical anion sensing was a europium-containing [2]rotaxane 66, which was synthesised via nitrite anion templation (Fig. 39).120 Upon removal of the nitrite template, it was found that fluoride addition caused substantial quenching of the Eu(III) emission in acetone/H2O (99:1 v/v). The increase in fluorescence lifetime and changes in the intensity of the hypersensitive ΔJ = 2 bands in fluoride-bound 66 suggested that fluoride was coordinated directly to the Eu(III) centre. Charge-assisted hydrogen bonds from the axle also enhanced the affinity of 66 for fluoride, as shown by the binding strength of the macrocyclic component alone. Nitrite and acetate led to weaker fluorescence quenching, which was attributed to a different mode of binding, wherein these anions did not directly coordinate to the emissive Eu(III) ion but were instead associated with the organic groups in the binding cavity.
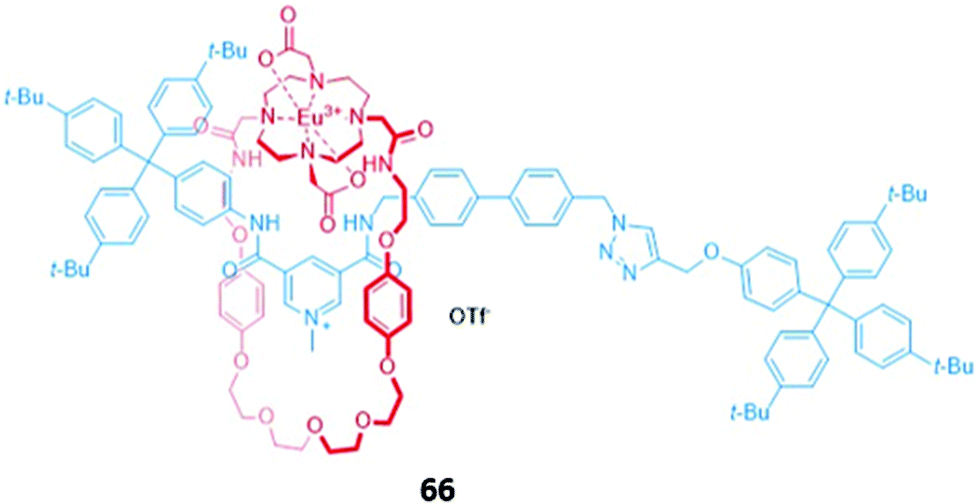 | ||
| Fig. 39 A [2]rotaxane with a europium(III) complex integrated into the macrocycle which binds fluoride (Ka = 2.42 × 105 M−1 in 99:1 acetone/water). | ||
A zinc metalloporphyrin was used as a signalling group in a hetero-[2]catenane 67, which was synthesised via chloride anion templation, with auxiliary contributions from aromatic donor–acceptor interactions and pyridyl coordination to the apical site of Zn(II) centre (Fig. 40). These interactions further served to pre-organise the interlocked cavity for chloride recognition. Crystallographic studies showed a high degree of complementarity between the binding cavity and the spherical halide, which was bound via six convergent CH–Cl and NH–Cl hydrogen bonds in DMSO. The encapsulation of chloride by 67 led to a gradual blueshift of the Soret band absorbance as well as slight increases in emission intensity.121
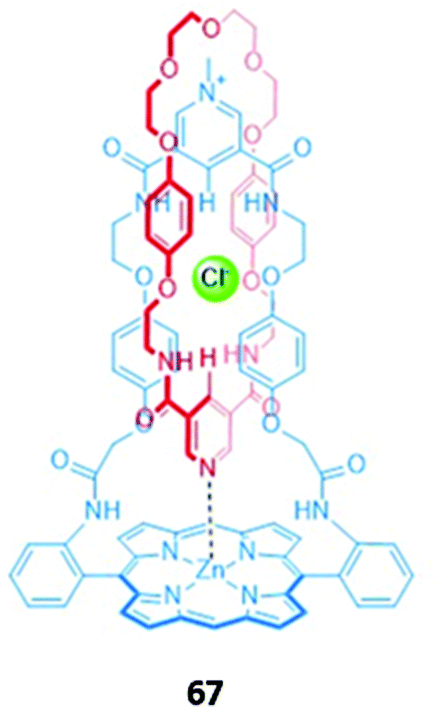 | ||
| Fig. 40 A hetero-[2]catenane incorporating a zinc porphyrin chromophore which selectively binds Cl− (Ka = 2144 M−1 in DMSO-d6). | ||
5.3. MIMs that undergo anion-induced shuttling
One of the most fascinating and useful properties of mechanically interlocked systems is the ability of the components to undergo reversible co-conformational changes in response to a stimulus. Due to this dynamic behaviour, MIMs are regarded as highly promising candidates for design of molecular machines and switches.122,123 A number of MIMs that undergo anion-induced conformational changes have been constructed and show promise as optical anion sensors. The majority of these systems are based on multi-station rotaxanes, which produce a photophysical response when the macrocycles translocate from a photoactive station to an anion binding station.A simple chloride-selective [2]rotaxane 68 was synthesised by Goldup and co-workers using AT-CuAAC (Fig. 41). In the free receptor, strong intramolecular hydrogen bonds between the urea station of the axle and the bipyridine group of the macrocycle precluded the binding of anions. However, protonation of the bipyridine group in acidic solutions forced the macrocycle to move away from the urea station and onto the adjacent triazole station. This displacement was inferred from 1H NMR experiments and confirmed by crystallographic analysis, which additionally showed the formation of a new hydrogen bond between the protonated bipyridine and the triazole as well as π–π interactions between the bipyridine and the naphthalimide stopper. The ‘freed’ urea group in the protonated receptor was able to form hydrogen bonds with various anions, exhibiting a marked preference for chloride (Ka > 104 in 1:1 CDCl3/CD3CN) and inducing a fluorescence enhancement in 68. The authors postulated this was due to a combination of receptor rigidification as well as the formation of π–π interactions with the naphthalimide group in the conformation adopted upon protonation of the [2]rotaxane.124
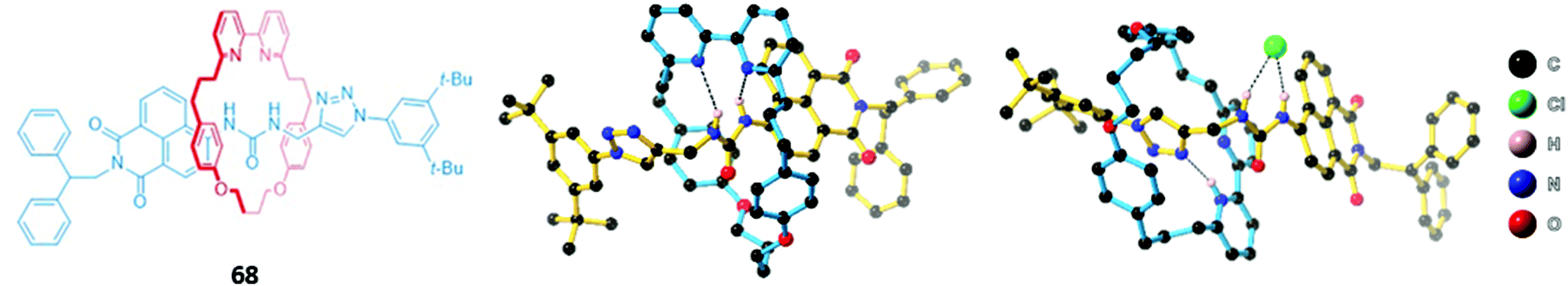 | ||
| Fig. 41 (Left) Structure of urea-containing [2]rotaxane 68; (middle) crystal structure of apo-receptor 68; (right) crystal structure of 68·HCl. Hydrogen bonds are shown in dashed lines and all other hydrogen atoms have been omitted for clarity. 68 binds Cl− strongly with Ka > 104 in 1:1 CDCl3/CD3CN. | ||
Lin and co-workers prepared a BODIPY-stoppered three-station [2]rotaxane 69, containing a central dibenzylammonium group and two peripheral triazolium stations (Fig. 42).125,126 In acidic solution, the crown ether-based macrocycle preferentially resides over the ammonium group, a co-conformation stabilised by hydrogen bonds between the ammonium NH groups and the oxygen atoms of the crown ether. The addition of OH− resulted in deprotonation of the ammonium group, allowing the macrocycle to instead shuttle between the triazolium stations. The reduced distance between the macrocycle and the BODIPY stoppers resulted in quenching of the BODIPY fluorescence at 516 nm due to enhanced PET from the macrocycle to the fluorophore. 69 also underwent quenching in the presence of H2PO4− in acetonitrile. However, this optical response was not a result of anion-induced shuttling; instead, computational calculations indicated that electrostatic interactions between the peripheral triazolium motifs and H2PO4− caused the axle component to fold around the anion, thereby activating PET. This was confirmed by quenching studies conducted on the free axle, which possessed greater flexibility and accordingly displayed a higher binding constant (K = 4.80 × 104 M−1) than the [2]rotaxane (K = 3.75 × 104 M−1).
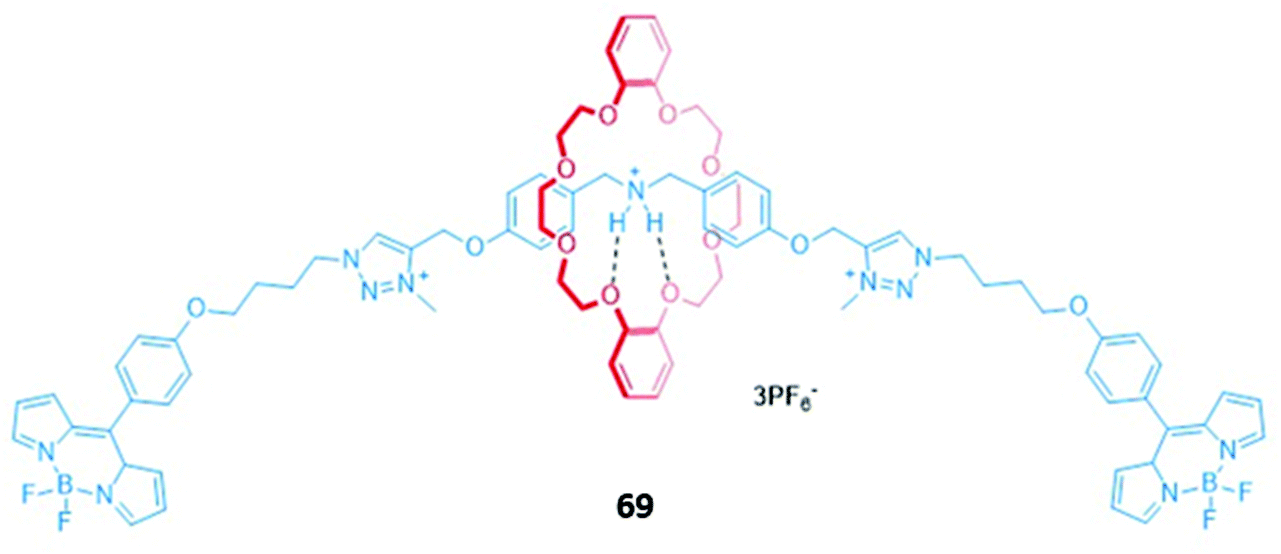 | ||
| Fig. 42 A BODIPY-stoppered [2]rotaxane with a central ammonium station and peripheral imidazolium anion binding stations which binds H2PO4− (K = 3.75 × 104 M−1 in CH3CN). | ||
The first true example of an optical anion sensor that produces a colorimetric response through the controlled, anion-induced translocation of a macrocycle along a multi-station axle is a pair of naphthalene diimide (NDI)-containing [2]rotaxanes 70a–b (Fig. 43).127 The rotaxanes differ in the binding motif present in the axle, with 70a containing a triazolium station for hydrogen bonding, while 70b contains an iodotriazolium for halogen bonding. In the presence of the non-coordinating anion PF6−, the macrocycle resides predominantly on the NDI station due to the formation of stable charge transfer interactions between the electron-poor NDI and the electron rich hydroquinone groups of the macrocycle. The addition of halides favours a translocation of the macrocycle to the triazolium or iodotriazolium station, which allows the isophthalimide group to participate in encapsulation of the guest. The loss of the donor–acceptor charge transfer interactions between the NDI units and the macrocycles in the resulting complex leads to a naked eye colour change from orange to colourless. NMR studies were used to estimate the percentage occupancies of the stations in the presence of different anions. When iodide was added, the halogen bonding rotaxane 70b exhibited significantly higher positional integrity in 1:1 CDCl3/CD3OD, with an iodotriazolium occupancy of 63%, compared to only 37% for the proto-triazolium in 70a, thereby demonstrating the superiority of halogen bonding for anion binding.
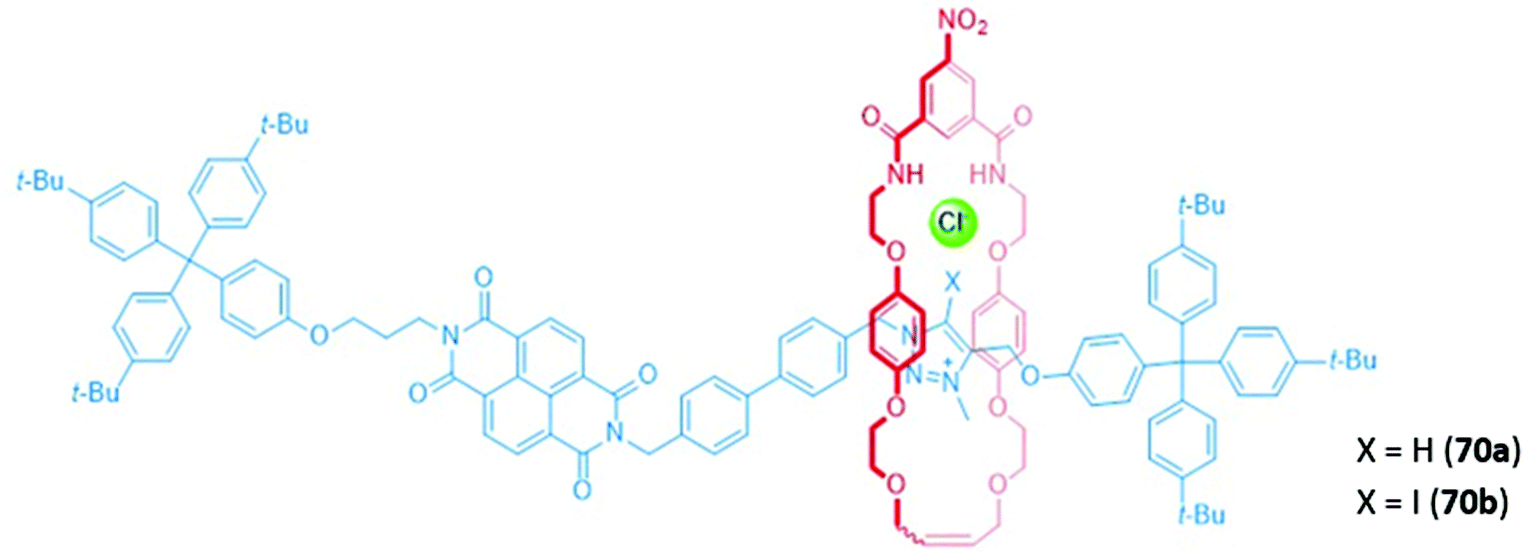 | ||
| Fig. 43 NDI-containing [2]rotaxanes with a prototriazolium (70a) or iodotriazolium (70b) anion binding station. | ||
The use of dynamic higher-order interlocked structures has proven to be a fruitful avenue of research, yielding a series of sophisticated and elegant anion sensors. One example is a four-station [3]rotaxane 71 that exhibited selectivity for nitrate (Fig. 44), which is challenging to target due to its lower affinity for hydrogen bonds and high degree of solvation. In the free receptor, the two macrocycles symmetrically occupied the naphthalene diimide (NDI) stations of the axle, as shown by 1H NMR and 2D 1H ROESY NMR studies in CDCl3/CD3OD (1:1, v/v). Upon addition of nitrate, the macrocycles undergo a concerted pincer motion from the peripheral NDI stations to the central halogen bonding iodotriazolium stations, with an accompanying discolouration of the solution in CHCl3. The preference for nitrate over halides and more basic oxyanions such HCO3− and H2PO4− was attributed to complementarity between the triangular geometry of the nitrate anion and the arrangement of the binding motifs in the three-dimensional cavity of 71. Once again, halogen bonding interactions proved to be superior for anion recognition, with the hydrogen bonding analogue of 71 exhibiting a lower affinity for nitrate. This suggests that the stricter preference for a linear geometry in the halogen bonding interaction was instrumental in creating a geometrically-defined binding site.128
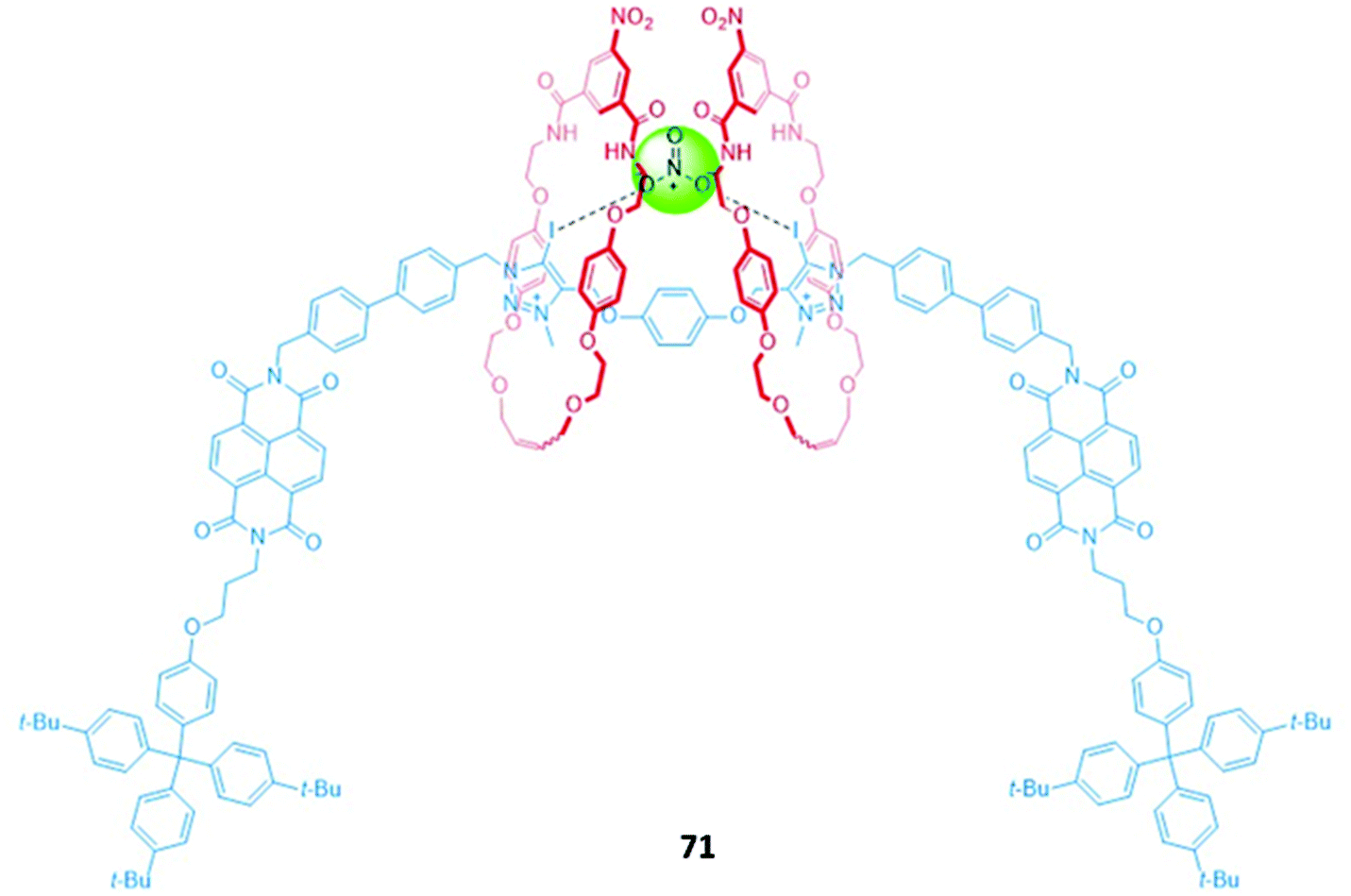 | ||
| Fig. 44 A nitrate-selective [3]rotaxane with iodotriazolium anion binding stations and NDI chromophores. 71 binds NO3− with Ka = 1782 M−1 in 1:1 CDCl3/CD3OD while the hydrogen bonding analogue exhibits a lower Ka of 653 M−1. | ||
A hetero-[3]catenane 72, comprising a large four-station central macrocycle and two smaller isophthalamide-containing macrocycles (Fig. 45), was constructed by chloride anion templation. The central macrocycle contains a tetrachloro-functionalised perylene diimide (PDI) unit as well as two triazolium anion-binding stations. The free receptor exhibited solvent-dependent co-conformational changes; in aprotic solvents like CHCl3, the smaller macrocycles reside predominantly on the triazolium stations, while the introduction of a polar protic solvent like CH3OH induces circumrotatory motion of the small macrocycles to the two naphthalimide subunits of the PDI. This change in co-conformation was accompanied by a naked eye colour change from orange to red. The anion binding properties of 72 were thus investigated in 3:1 CHCl3/CH3OH, where the small macrocycles preferentially reside on the PDI unit. 72 was found to bind chloride in a 1:2 host–guest stoichiometry, with an accompanying colour change from red to orange. This is consistent with the loss of intramolecular interactions between the PDI stations and the hydroquinone groups of the small macrocycles, indicating that chloride binding to the triazolium units induced the concomitant movement of the small macrocycles to the triazolium binding sites. A significant enhancement of the PDI fluorescence was also observed upon chloride binding in a solvent mixture of 45:45:10 CHCl3/CH3OH/H2O, allowing 72 to be used as a fluorescent anion sensor in competitive media.129
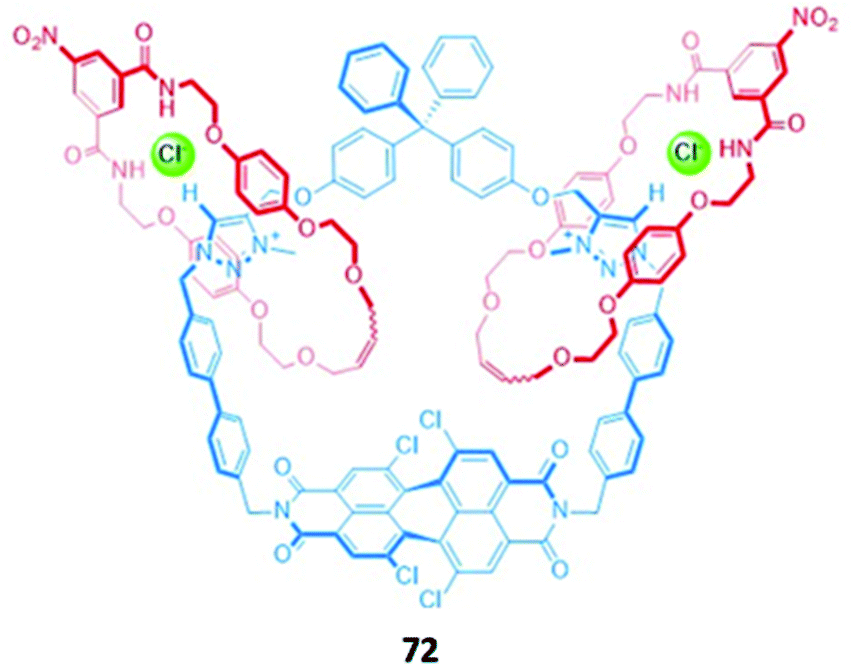 | ||
| Fig. 45 A hetero-[3]catenane incorporating a perylene diimide chromophore. 72 binds various anions as a 1:2 host–guest complex with highest affinity for Cl− (K1 = 1.55 × 104 M−1 and K2 = 1.58 × 102 M−1 in 3:1 CHCl3/CH3OH). | ||
An exotic fullerene-containing four-station [3]rotaxane 73 was shown to undergo dynamic shuttling and photophysical changes in the presence of chloride (Fig. 46). The axle component comprised a central C60 fullerene bis-triazolium motif flanked by two peripheral NDI stations, while the two macrocycles contained isophthalamide binding motifs tethered to ferrocene groups. The [3]rotaxane adopted two distinct co-conformations mediated by anion binding; in the presence of the non-coordinating anion PF6−, the ferrocenyl macrocycles reside predominantly on the NDI stations, while addition of chloride induced a translational motion of the macrocycles to the central bis-triazolium stations. In the co-conformation adopted in the PF6− salt of 73 in CHCl3 solution, NDI emission is quenched due to PET from the ferrocenyl groups to the NDI stations. In the chloride-bound [3]rotaxane, the shuttling of the macrocycles to the central fullerene unit results in the formation of a C60 fullerene-based charge-separated state and also precludes ferrocenyl-NDI electron transfer, thereby switching on NDI emission.130
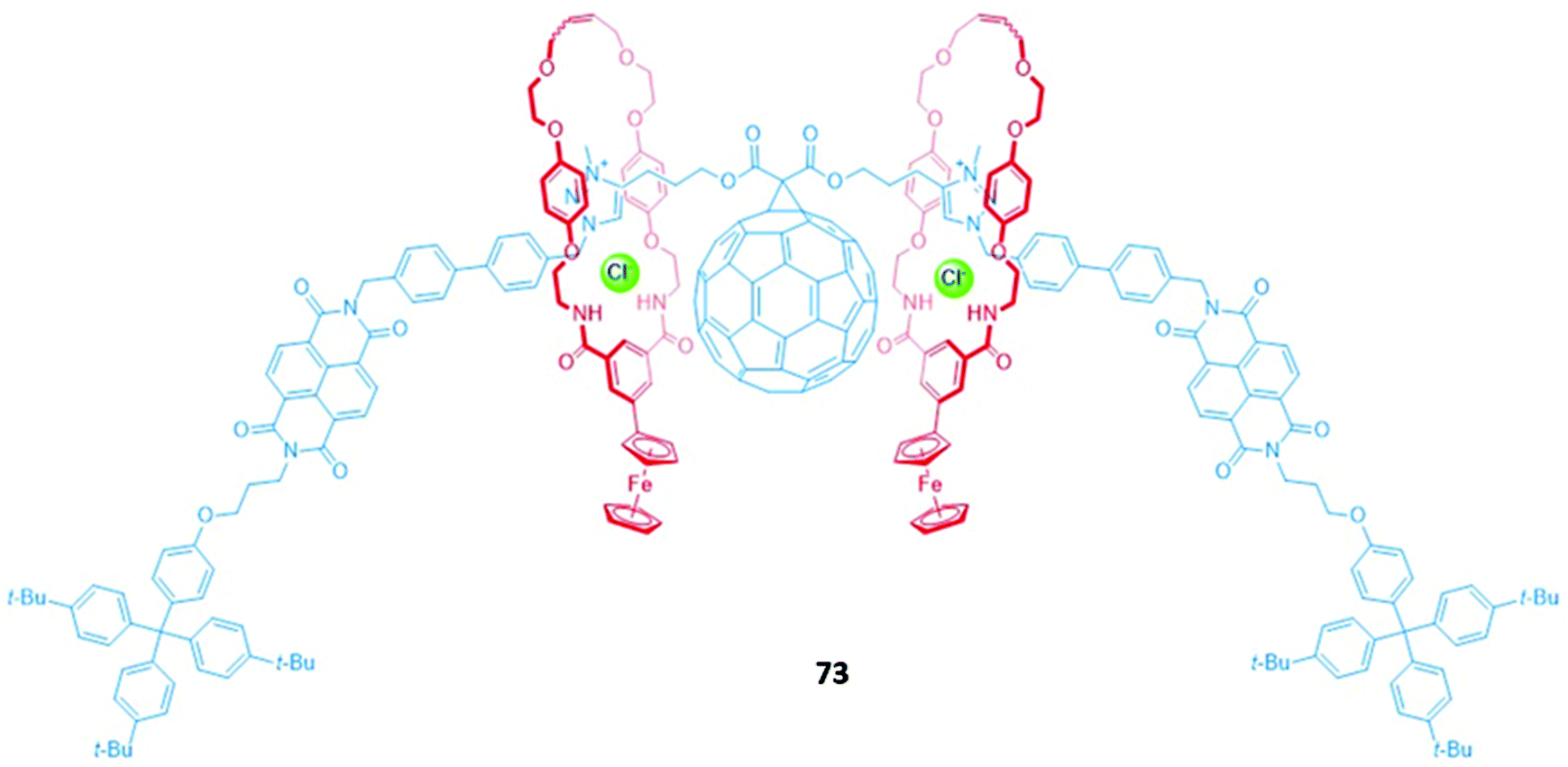 | ||
| Fig. 46 A fullerene-containing [3]rotaxane with appended ferrocene units. | ||
5.4. MIMs that undergo anion-induced dissociation
The anion-triggered dissociation of MIMs into the respective axle and macrocyclic components can lead to a turn-on of fluorescence arising from the separation of a fluorogenic unit from a quencher. Reversible dissociation was first demonstrated using a pseudorotaxane 74 comprising an NDI-based axle with no stoppers and an isophthalamide naphthohydroquinone-containing macrocycle (Fig. 47). Although 74 is not strictly a MIM due to the lack of mechanical bonds in the structure, in the absence of an anion, the axle was threaded through the macrocycle to form a coloured pseudorotaxane stabilised by charge transfer interactions between the NDI unit and the naphthohydroquinone groups. Addition of various anions, including sulfate, benzoate, fluoride, chloride and H2PO4−, displaces the NDI axle by formation of hydrogen bonding interactions with the isophthalamide group, resulting in a discoloration of the solution in CHCl3.131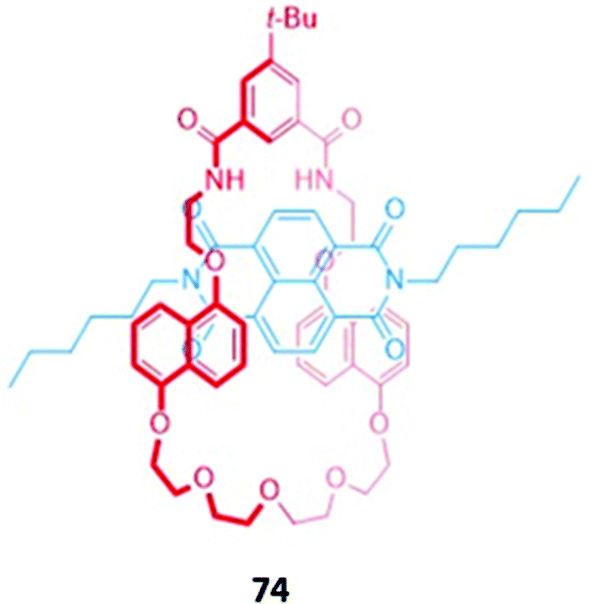 | ||
| Fig. 47 Pseudo-rotaxane assembly that undergoes anion-triggered dissociation. | ||
Jasti and co-workers synthesised a self-immolating [2]rotaxane 75 capable of selectively sensing fluoride (Fig. 48).132 The macrocyclic component of 75 was a fluorescent carbon ‘nanohoop’ while the asymmetrical axle is stoppered by a fluorescence-quenching 3,5-dinitrobenzyl unit and a fluoride-cleavable triisopropylsilane (TIPS). The intact [2]rotaxane is non-fluorescent in CHCl3 due to the proximity of the quencher to the macrocyclic fluorophore. The addition of fluoride promoted cleavage of the TIPS stopper, allowing rapid de-threading of the axle from the macrocycle. The separation of the macrocycle from the quencher resulted in a dramatic 123-fold enhancement of fluorescence. A similar concept was employed by Zhu and co-workers using synthesised a [2]rotaxane 76 with a pillar[5]arene-based macrocycle and a silane-stoppered axle (Fig. 48). As before, disassembly of the rotaxane in the presence of fluoride restored the fluorescence of the pillar[5]arene in DMSO/THF (1:1, v/v). A simple test kit was made by adsorbing 76 onto strips of filter paper, which exhibited fluorescence turn-on when dipped in THF solutions containing fluoride.133
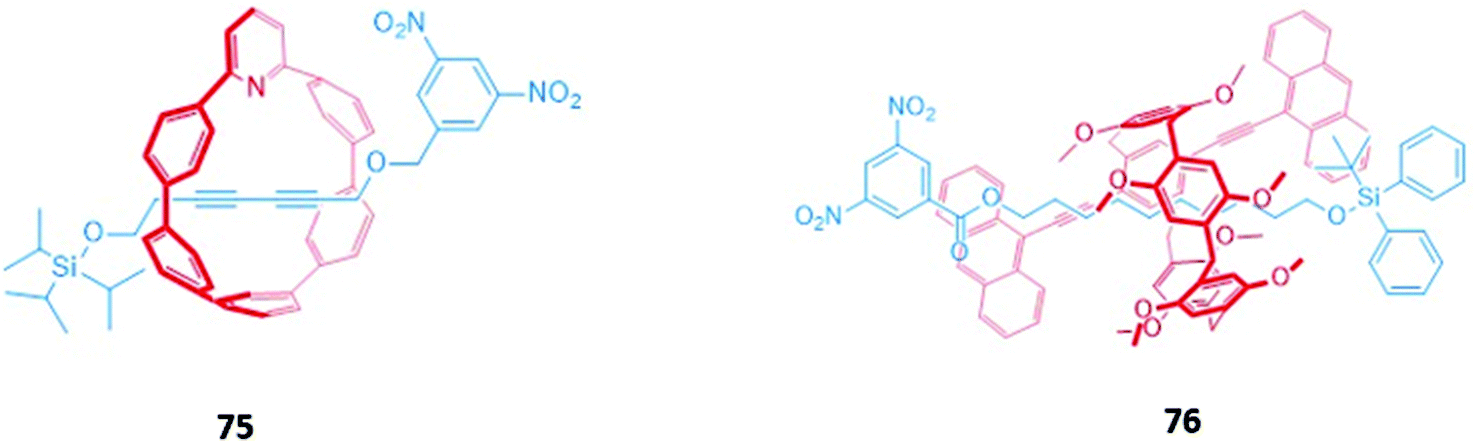 | ||
| Fig. 48 Fluoride-selective self-immolating rotaxanes. | ||
6. Conclusions
This review highlights the significant progress that has been achieved in the development of optical anion sensors based on macrocyclic and interlocked hosts over the past two decades. A plethora of fluorogenic and chromogenic signalling units, both organic and metal-based, have been integrated into macrocyclic hosts to produce a range of optical responses to anion binding, including ‘naked eye’ colour changes and fluorescence enhancement/quenching. Sensors capable of distinguishing between different anions have also been developed; while the majority of these systems rely on multi-sensor arrays, a notable bis-calix[4]pyrrole-appended phenazine that uses vibration induced emission to differentiate dicarboxylates of varying length has also been reported.42While the majority of macrocyclic hosts rely on hydrogen bonding or electrostatic interactions for the recognition of anions, recent years have seen increased use of other non-covalent interactions, such as halogen bonding and anion–π interactions. Halogen bonding motifs have been shown to significantly enhance the strength and selectivity of anion binding in competitive media. Moving forward, we foresee the development of potent halogen bonding motifs and their incorporation into anion receptors to tackle the challenge of achieving strong anion binding in water.
Mechanically interlocked molecules have emerged as highly promising candidates for optical anion sensing due to the presence of three-dimensional binding cavities that can be exploited to provide shape and size complementarity to target anions. Future developments in MIM-based anion sensors may exploit the three-dimensional nature of the binding cavities to selectively detect environmentally-relevant oxyanions such as nitrate, which are challenging targets due to their high solvation and complex geometries. The propensity of interlocked systems to undergo anion-induced shuttling also provides a novel means of producing an optical response, as exemplified in a series of sophisticated anion sensors based on [3]rotaxanes and [3]catenanes. There is great scope to exploit the switchable nature of MIM-based molecular shuttles in the design of complex sensors and devices, and we foresee exciting developments in this direction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. M. T. acknowledges the Clarendon Fund and the Oxford Australia Scholarships Fund for a postgraduate research scholarship.Notes and references
- M. P. Anderson, R. J. Gregory, S. Thompson, D. W. Souza, S. Paul, R. C. Mulligan, A. E. Smith and M. J. Welsh, Science, 1991, 253, 202 CrossRef CAS PubMed.
- F. Delange, Thyroid, 1994, 4, 107–128 CrossRef CAS PubMed.
- J. L. Way, Annu. Rev. Pharmacol. Toxicol., 1984, 24, 451–481 CrossRef CAS PubMed.
- V. H. Smith and D. W. Schindler, Trends Ecol. Evol., 2009, 24, 201–207 CrossRef PubMed.
- J. H. P. Watson and D. C. Ellwood, Nucl. Eng. Des., 2003, 226, 375–385 CrossRef CAS.
- P. K. Dasgupta, J. V. Dyke, A. B. Kirk and W. A. Jackson, Environ. Sci. Technol., 2006, 40, 6608–6614 CrossRef CAS PubMed.
- J. L. Sessler, P. A. Gale and W.-S. Cho, in Anion Receptor Chemistry, The Royal Society of Chemistry, 2006, pp. 320–369 Search PubMed.
- P. Prados and R. Quesada, Supramol. Chem., 2008, 20, 201–216 CrossRef CAS.
- C. H. Park and H. E. Simmons, J. Am. Chem. Soc., 1968, 90, 2431–2432 CrossRef CAS.
- G. T. Spence and P. D. Beer, Acc. Chem. Res., 2013, 46, 571–586 CrossRef CAS PubMed.
- M. J. Chmielewski, J. J. Davis and P. D. Beer, Org. Biomol. Chem., 2009, 7, 415–424 RSC.
- P. A. Gale and C. Caltagirone, Coord. Chem. Rev., 2018, 354, 2–27 CrossRef CAS.
- D. A. McNaughton, M. Fares, G. Picci, P. A. Gale and C. Caltagirone, Coord. Chem. Rev., 2021, 427, 213573 CrossRef CAS.
- S. K. Sahoo, G.-D. Kim and H.-J. Choi, J. Photochem. Photobiol., C, 2016, 27, 30–53 CrossRef CAS.
- A. B. Aletti, D. M. Gillen and T. Gunnlaugsson, Coord. Chem. Rev., 2018, 354, 98–120 CrossRef CAS.
- R. Kumar, A. Sharma, H. Singh, P. Suating, H. S. Kim, K. Sunwoo, I. Shim, B. C. Gibb and J. S. Kim, Chem. Rev., 2019, 119, 9657–9721 CrossRef CAS PubMed.
- D. M. Homden and C. Redshaw, Chem. Rev., 2008, 108, 5086–5130 CrossRef CAS PubMed.
- F. Sansone, L. Baldini, A. Casnati and R. Ungaro, New J. Chem., 2010, 34, 2715–2728 RSC.
- S. K. Kim, J. H. Bok, R. A. Bartsch, J. Y. Lee and J. S. Kim, Org. Lett., 2005, 7, 4839–4842 CrossRef CAS PubMed.
- E. Brunetti, J.-F. Picron, K. Flidrova, G. Bruylants, K. Bartik and I. Jabin, J. Org. Chem., 2014, 79, 6179–6188 CrossRef CAS PubMed.
- B.-Y. Hou, D.-X. Wang, H.-B. Yang, Q.-Y. Zheng and M.-X. Wang, J. Org. Chem., 2007, 72, 5218–5226 CrossRef CAS PubMed.
- P. G. Sutariya, H. Soni, S. A. Gandhi and A. Pandya, New J. Chem., 2019, 43, 737–747 RSC.
- H.-F. Xie, C. Wu, J. Zou, Y.-X. Yang, H. Xu, Q.-L. Zhang, C. Redshaw and T. Yamato, Dyes Pigm., 2020, 178, 108340 CrossRef CAS.
- M. Nemati, R. Hosseinzadeh, R. Zadmard and M. Mohadjerani, Sens. Actuators, B, 2017, 241, 690–697 CrossRef CAS.
- B. Uttam, R. Kandi, M. A. Hussain and C. P. Rao, J. Org. Chem., 2018, 83, 11850 CrossRef CAS PubMed.
- A. S. Miranda, P. M. Marcos, J. R. Ascenso, M. N. Berberan-Santos, R. Schurhammer, N. Hickey and S. Geremia, Molecules, 2020, 25, 4708 CrossRef CAS PubMed.
- A. A. Vavilova, R. V. Nosov and I. I. Stoikov, Mendeleev Commun., 2016, 26, 508–510 CrossRef CAS.
- E. Quinlan, S. E. Matthews and T. Gunnlaugsson, J. Org. Chem., 2007, 72, 7497–7503 CrossRef CAS PubMed.
- Y. Chen, X. Liu, H. Huang, W. Wu and Y. Zheng, Sci. China: Chem., 2010, 53, 569–575 CrossRef CAS.
- Z.-Y. Li, H.-K. Su, H.-X. Tong, Y. Yin, T. Xiao, X.-Q. Sun, J. Jiang and L. Wang, Spectrochim. Acta, Part A, 2018, 200, 307–312 CrossRef CAS PubMed.
- S. Arimori, M. G. Davidson, T. M. Fyles, T. G. Hibbert, T. D. James and G. I. Kociok-köhn, Chem. Commun., 2004, 4, 1640–1641 RSC.
- J. Jin, J. Y. Park and Y. S. Lee, J. Phys. Chem. C, 2016, 120, 24324–24334 CrossRef CAS.
- P. R. Sharma, S. Pandey, V. K. Soni, G. Choudhary and R. K. Sharma, Supramol. Chem., 2019, 31, 634–644 CrossRef CAS.
- M. Singh, N. Singh, J. R. Ascenso and P. M. Marcos, Supramol. Chem., 2019, 31, 313–321 CrossRef.
- S. Li, D.-X. Wang and M.-X. Wang, Tetrahedron Lett., 2012, 53, 6226–6229 CrossRef CAS.
- A. Baeyer, Ber. Dtsch. Chem. Ges., 1886, 19, 2184–2185 CrossRef.
- P. A. Gale, J. L. Sessler, V. Král and V. Lynch, J. Am. Chem. Soc., 1996, 118, 5140–5141 CrossRef CAS.
- J. Sessler, P. Anzenbacher, H. Miyaji, K. Jursikova, E. Bleasdale and P. A. Gale, Ind. Eng. Chem. Res., 2000, 39, 3471–3478 CrossRef CAS.
- M. Pushina, P. Koutnik, R. Nishiyabu, T. Minami, P. Savechenkov and P. Anzenbacher, Chem. – Eur. J., 2018, 24, 4879–4884 CrossRef CAS PubMed.
- J. Yoo, M.-S. Kim, S.-J. Hong, J. L. Sessler and C.-H. Lee, J. Org. Chem., 2009, 74, 1065 CrossRef CAS PubMed.
- W. Huang, L. Sun, Z. Zheng, J. Su and H. Tian, Chem. Commun., 2015, 51, 4462–4464 RSC.
- W. Chen, C. Guo, Q. He, X. Chi, V. M. Lynch, Z. Zhang, J. Su, H. Tian and J. L. Sessler, J. Am. Chem. Soc., 2019, 141, 14798–14806 CrossRef CAS PubMed.
- W.-T. Gong, K. Hiratani and S. S. Lee, Tetrahedron, 2008, 64, 11007–11011 CrossRef CAS.
- A. Costero, M. Bañuls, M. Aurell and M. Arellano, J. Inclusion Phenom. Macrocyclic Chem., 2006, 54, 61–66 CrossRef CAS.
- P. Piatek and J. Jurczak, Chem. Commun., 2002, 2, 2450–2451 RSC.
- F. Wang, R. Nandhakumar, Y. Hu, D. Kim, K. M. Kim and J. Yoon, J. Org. Chem., 2013, 78, 11571–11576 CrossRef CAS PubMed.
- T. Ema, K. Okuda, S. Watanabe, T. Yamasaki, T. Minami, N. A. Esipenko and P. Anzenbacher, Org. Lett., 2014, 16, 1302 CrossRef CAS PubMed.
- D. Lichosyt, P. Dydio and J. Jurczak, Chem. – Eur. J., 2016, 22, 17673–17680 CrossRef CAS PubMed.
- S. N. Berry, L. Qin, W. Lewis and K. A. Jolliffe, Chem. Sci., 2020, 11, 7015–7022 RSC.
- A. Aldrey, V. García, C. Lodeiro, A. Macías, P. Pérez-Lourido, L. Valencia, R. Bastida and C. Núñez, Tetrahedron, 2013, 69, 4578–4585 CrossRef CAS.
- A. Aldrey, C. Núñez, V. García, R. Bastida, C. Lodeiro and A. Macías, Tetrahedron, 2010, 66, 9223–9230 CrossRef CAS.
- C. Núñez, A. Aldrey, V. García, R. Bastida, A. Macías and C. Lodeiro, Inorg. Chim. Acta, 2012, 381, 85–94 CrossRef.
- I. A. Rather, S. A. Wagay and R. Ali, Coord. Chem. Rev., 2020, 415, 213327 CrossRef CAS.
- P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907–9972 CrossRef CAS PubMed.
- Y. Zhao, Y. Li, Z. Qin, R. Jiang, H. Liu and Y. Li, Dalton Trans., 2012, 41, 13338–13342 RSC.
- H.-B. Liu, Q. Zhang and M.-X. Wang, Angew. Chem., Int. Ed., 2018, 57, 6536–6540 CrossRef CAS PubMed.
- D.-H. Tuo, Y.-F. Ao, Q.-Q. Wang and D.-X. Wang, Org. Lett., 2019, 21, 7158–7162 CrossRef CAS PubMed.
- M. S. Hossain, S. A. Rahaman, J. Hatai, M. Saha and S. Bandyopadhyay, Chem. Commun., 2020, 56, 4172–4175 RSC.
- M. Caricato, A. Olmo, C. Gargiulli, G. Gattuso and D. Pasini, Tetrahedron, 2012, 68, 7861–7866 CrossRef CAS.
- P. Isar, M. Rajeswara Rao and M. Ravikanth, Eur. J. Org. Chem., 2018, 3095–3104 CrossRef CAS.
- K. Norvaiša, K. J. Flanagan, D. Gibbons and M. O. Senge, Angew. Chem., Int. Ed., 2019, 58, 16553–16557 CrossRef PubMed.
- R. Chelli, G. Pietraperzia, A. Bencini, C. Giorgi, V. Lippolis, P. R. Salvi and C. Gellini, Phys. Chem. Chem. Phys., 2015, 17, 10813–10822 RSC.
- A. S. Oshchepkov, T. A. Shumilova, S. R. Namashivaya, O. A. Fedorova, P. V. Dorovatovskii, V. N. Khrustalev and E. A. Kataev, J. Org. Chem., 2018, 83, 2145–2153 CrossRef CAS PubMed.
- A. S. Oshchepkov, T. A. Shumilova, M. Zerson, R. Magerle, V. N. Khrustalev and E. A. Kataev, J. Org. Chem., 2019, 84, 9034–9043 CrossRef CAS PubMed.
- B. S. Morozov, S. S. R. Namashivaya, M. A. Zakharko, A. S. Oshchepkov and E. A. Kataev, ChemistryOpen, 2020, 9, 171–175 CrossRef CAS PubMed.
- R. Azadbakht and N. Chehri, New J. Chem., 2018, 42, 17690–17699 RSC.
- M. Sethupathi, G. Muthusankar, V. Thamilarasan, N. Sengottuvelan, G. Gopu, N. M. Vinita, P. Kumar and F. Perdih, J. Photochem. Photobiol., B, 2020, 203, 111739 CrossRef CAS PubMed.
- C. L. Vonnegut, A. M. Shonkwiler, L. N. Zakharov, M. M. Haley and D. W. Johnson, Chem. Commun., 2016, 52, 9506–9509 RSC.
- D. Zhang, X. Jiang, H. Yang, A. Martinez, M. Feng, Z. Dong and G. Gao, Org. Biomol. Chem., 2013, 11, 3375–3381 RSC.
- D. Zhang, X. Jiang, H. Yang, Z. Su, E. Gao, A. Martinez and G. Gao, Chem. Commun., 2013, 49, 6149–6151 RSC.
- Y. Liu, Z. Zhao, R. Huo and Q. Liu, Sci. Rep., 2019, 9, 502 CrossRef PubMed.
- J.-H. Wang, J.-B. Xiong, X. Zhang, S. Song, Z.-H. Zhu and Y.-S. Zheng, RSC Adv., 2015, 5, 60096–60100 RSC.
- J. Yang, C.-C. Dong, X.-L. Chen, X. Sun, J.-Y. Wei, J.-F. Xiang, J. L. Sessler and H.-Y. Gong, J. Am. Chem. Soc., 2019, 141, 4597–4612 CrossRef CAS PubMed.
- H.-T. Niu, Z. Yin, D. Su, D. Niu, J. He and J.-P. Cheng, Dalton Trans., 2008, 3694 RSC.
- J. Li, P. Zhang, Y. Xu, Z. Su, Y. Qian, S. Li, T. Yu, P. J. Sadler and H.-K. Liu, Dalton Trans., 2017, 46, 16205 RSC.
- J. Y. C. Lim and P. D. Beer, Chem, 2018, 4, 731–783 CAS.
- F. Zapata, A. Caballero, N. G. White, T. D. W. Claridge, P. J. Costa, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2012, 134, 11533 CrossRef CAS PubMed.
- K. Ghosh and A. R. Sarkar, Tetrahedron Lett., 2009, 50, 85–88 CrossRef CAS.
- H. Duan, Y. Li, Q. Li, P. Wang, X. Liu, L. Cheng, Y. Yu and L. Cao, Angew. Chem., 2020, 59, 10101–10110 CrossRef CAS.
- V. Amendola, E. Bastianello, L. Fabbrizzi, C. Mangano, P. Pallavicini, A. Perotti, A. Manotti Lanfredi and F. Ugozzoli, Angew. Chem., Int. Ed., 2000, 39, 2917–2920 CrossRef CAS.
- V. Amendola, G. Bergamaschi, A. Buttafava, L. Fabbrizzi and E. Monzani, J. Am. Chem. Soc., 2010, 132, 147–156 CrossRef CAS PubMed.
- J. S. Mendy, M. A. Saeed, F. R. Fronczek, D. R. Powell and M. A. Hossain, Inorg. Chem., 2010, 49, 7223–7225 CrossRef CAS PubMed.
- M. Rhaman, M. Hasan, A. Alamgir, L. Xu, D. Powell, B. Wong, R. Tandon and M. Hossain, Sci. Rep., 2018, 8, 286–286 CrossRef PubMed.
- M. M. Rhaman, A. Alamgir, B. M. Wong, D. R. Powell and M. A. Hossain, RSC Adv., 2014, 4, 54263–54267 RSC.
- M. M. Rhaman, F. R. Fronczek, D. R. Powell and M. A. Hossain, Dalton Trans., 2014, 43, 4618–4621 RSC.
- M. A. Saeed, D. R. Powell and M. A. Hossain, Tetrahedron Lett., 2010, 51, 4904–4907 CrossRef CAS.
- M. Hu and G. Feng, Chem. Commun., 2012, 48, 6951–6953 RSC.
- D. Hontz, J. Hensley, K. Hiryak, J. Lee, J. Luchetta, M. Torsiello, M. Venditto, D. Lucent, W. Terzaghi, D. Mencer, A. Bommareddy and A. L. Vanwert, ACS Omega, 2020, 5, 19469 CrossRef CAS PubMed.
- W. Zhu, H. Fang, J.-X. He, W.-H. Jia, H. Yao, T.-B. Wei, Q. Lin and Y.-M. Zhang, New J. Chem., 2018, 42, 11548–11554 RSC.
- P.-L. Ng, C.-S. Lee, H.-L. Kwong and A. S. C. Chan, Inorg. Chem. Commun., 2005, 8, 769–772 CrossRef CAS.
- T. Chakraborty, S. Dasgupta, A. Bhattacharyya, E. Zangrando, D. Escudero and D. Das, New J. Chem., 2019, 43, 13152–13161 RSC.
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- M. D. Pratt and P. D. Beer, Tetrahedron, 2004, 60, 11227–11238 CrossRef CAS.
- A. Mishra, V. Vajpayee, H. Kim, M. H. Lee, H. Jung, M. Wang, P. J. Stang and K.-w. Chi, Dalton Trans., 2012, 41, 1195–1201 RSC.
- P. De Wolf, P. Waywell, M. Hanson, S. L. Heath, A. J. H. M. Meijer, S. J. Teat and J. A. Thomas, Chem. – Eur. J., 2006, 12, 2188–2195 CrossRef CAS PubMed.
- F. Szemes, D. Hesek, Z. Chen, S. W. Dent, M. G. B. Drew, A. J. Goulden, A. R. Graydon, A. Grieve, R. J. Mortimer, T. Wear, J. S. Weightman and P. D. Beer, Inorg. Chem., 1996, 35, 5868–5879 CrossRef CAS.
- P. D. Beer, F. Szemes, V. Balzani, C. M. Salà, M. G. B. Drew, S. W. Dent and M. Maestri, J. Am. Chem. Soc., 1997, 119, 11864–11875 CrossRef CAS.
- S. Shanmugaraju, A. Bar, K. Chi and P. S. Mukherjee, Organometallics, 2010, 29, 2971–2980 CrossRef CAS.
- C. O. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1989, 28, 189–192 CrossRef.
- P. Gaviña and J.-P. Sauvage, Tetrahedron Lett., 1997, 38, 3521–3524 CrossRef.
- M. C. Jiménez, C. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284–3287 CrossRef.
- J. F. Nierengarten, C. O. Dietrich-Buchecker and J. P. Sauvage, J. Am. Chem. Soc., 1994, 116, 375–376 CrossRef CAS PubMed.
- N. Solladié, J.-C. Chambron and J.-P. Sauvage, J. Am. Chem. Soc., 1999, 121, 3684–3692 CrossRef.
- K. M. Bąk, K. Porfyrakis, J. J. Davis and P. D. Beer, Mater. Chem. Front., 2020, 4, 1052–1073 RSC.
- J. E. M. Lewis, P. D. Beer, S. J. Loeb and S. M. Goldup, Chem. Soc. Rev., 2017, 46, 2577–2591 RSC.
- J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed.
- K. D. Hänni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240–1251 RSC.
- J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC.
- A. Caballero, F. Zapata, N. G. White, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 1876–1880 CrossRef CAS.
- J. M. Mercurio, A. Caballero, J. Cookson and P. D. Beer, RSC Adv., 2015, 20, 9298–9306 RSC.
- A. Brown, T. Lang, K. M. Mullen and P. D. Beer, Org. Biomol. Chem., 2017, 15, 4587–4594 RSC.
- M. J. Langton and P. D. Beer, Chem. – Eur. J., 2012, 18, 14406–14412 CrossRef CAS PubMed.
- J. Y. C. Lim, I. Marques, V. Félix and P. D. Beer, Chem. Commun., 2018, 54, 10851–10854 RSC.
- L. M. Hancock, E. Marchi, P. Ceroni and P. D. Beer, Chem. – Eur. J., 2012, 18, 11277–11283 CrossRef CAS.
- M. J. Langton, I. Marques, S. W. Robinson, V. Félix and P. D. Beer, Chem. – Eur. J., 2016, 22, 185–192 CrossRef CAS.
- R. C. Knighton, S. Dapin and P. D. Beer, Chem. – Eur. J., 2020, 26, 5288–5296 CrossRef CAS.
- B. R. Mullaney, A. L. Thompson and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11458–11462 CrossRef CAS PubMed.
- M. J. Langton, Y. Xiong and P. D. Beer, Chem. – Eur. J., 2015, 21, 18910–18914 CrossRef CAS.
- C. Allain and S. Faulkner, Future Med. Chem., 2010, 2, 339–350 CrossRef CAS.
- M. J. Langton, O. A. Blackburn, T. Lang, S. Faulkner and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11463–11466 CrossRef CAS.
- A. Brown, M. J. Langton, N. L. Kilah, A. L. Thompson and P. D. Beer, Chem. – Eur. J., 2015, 21, 17664–17675 CrossRef CAS.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- A. W. Heard and S. M. Goldup, ACS Cent. Sci., 2020, 6, 117–128 CrossRef CAS PubMed.
- M. Denis, L. Qin, P. Turner, K. A. Jolliffe and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 5315–5319 CrossRef CAS.
- R. Arumugaperumal, V. Parthiban, T. Shukla, R. Putikam, R. Singh, S.-P. Wu, M.-C. Lin and H.-C. Lin, Sens. Actuators, B, 2018, 270, 382–395 CrossRef CAS.
- R. Arumugaperumal, V. Srinivasadesikan, M. V. Ramakrishnam Raju, M.-C. Lin, T. Shukla, R. Singh and H.-C. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 26491–26503 CrossRef CAS PubMed.
- T. A. Barendt, S. W. Robinson and P. D. Beer, Chem. Sci., 2016, 7, 5171–5180 RSC.
- T. A. Barendt, A. Docker, I. Marques, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 11069–11076 CrossRef CAS PubMed.
- T. A. Barendt, L. Ferreira, I. Marques, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 9026–9037 CrossRef CAS PubMed.
- T. A. Barendt, I. Rašović, M. A. Lebedeva, G. A. Farrow, A. Auty, D. Chekulaev, I. V. Sazanovich, J. A. Weinstein, K. Porfyrakis and P. D. Beer, J. Am. Chem. Soc., 2018, 140, 1924–1936 CrossRef CAS PubMed.
- K. M. Mullen, J. J. Davis and P. D. Beer, New J. Chem., 2009, 33, 769–776 RSC.
- J. M. Van Raden, B. M. White, L. N. Zakharov and R. Jasti, Angew. Chem., Int. Ed., 2019, 58, 7341–7345 CrossRef CAS PubMed.
- Q. Li, Y. Wu, Y. Liu, L. Shangguan, B. Shi and H. Zhu, Org. Lett., 2020, 22, 6662 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |