
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and perspective of amorphous metal nanoparticles from laser synthesis and processing
Shun-Xing
Liang
 *a,
Lai-Chang
Zhang
b,
Sven
Reichenberger
a and
Stephan
Barcikowski
*a
*a,
Lai-Chang
Zhang
b,
Sven
Reichenberger
a and
Stephan
Barcikowski
*a
aTechnical Chemistry I and Center for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Universitaetsstrasse 7, Essen 45141, Germany. E-mail: shunxing.liang@uni-due.de; sxliang90@gmail.com; stephan.barcikowski@uni-due.de
bSchool of Engineering, Edith Cowan University, 270 Joondalup Drive, Joondalup, Perth, WA 6027, Australia
First published on 20th April 2021
Abstract
Amorphous metal nanoparticles (A-NPs) have aroused great interest in their structural disordering nature and combined downsizing strategies (e.g. nanoscaling), both of which are beneficial for highly strengthened properties compared to their crystalline counterparts. Conventional synthesis strategies easily induce product contamination and/or size limitations, which largely narrow their applications. In recent years, laser ablation in liquid (LAL) and laser fragmentation in liquid (LFL) as “green” and scalable colloid synthesis methodologies have attracted extensive enthusiasm in the production of ultrapure crystalline NPs, while they also show promising potential for the production of A-NPs. Yet, the amorphization in such methods still lacks sufficient rules to follow regarding the formation mechanism and criteria. To that end, this article reviews amorphous metal oxide and carbide NPs from LAL and LFL in terms of NP types, liquid selection, target elements, laser parameters, and possible formation mechanism, all of which play a significant role in the competitive relationship between amorphization and crystallization. Furthermore, we provide the prospect of laser-generated metallic glass nanoparticles (MG-NPs) from MG targets. The current and potential applications of A-NPs are also discussed, categorized by the attractive application fields e.g. in catalysis and magnetism. The present work aims to give possible selection rules and perspective on the design of colloidal A-NPs as well as the synthesis criteria of MG-NPs from laser-based strategies.
 Shun-Xing Liang | Shun-Xing Liang received his PhD degree in Materials Science & Engineering from Edith Cowan University (ECU), Australia, in 2020. Currently, he is working as an Alexander von Humboldt Research Fellow at the Institute of Technical Chemistry I of University of Duisburg-Essen (UDE), Germany. His research focuses on the advanced catalytic function of metallic glasses and nanostructured materials, as well as pulsed laser synthesis and processing of functional amorphous metal nanoparticles. |
 Lai-Chang Zhang | Lai-Chang Zhang is a Professor of Materials Engineering and the Program Leader-Mechanical Engineering in the School of Engineering at Edith Cowan University. After receiving his PhD in Materials Science and Engineering from the Institute of Metal Research, Chinese Academy of Sciences, he held several positions at The University of Western Australian, University of Wollongong, IFW Dresden and Technische Universität Darmstadt. His current research interests focus on the metal additive manufacturing (e.g., selective laser melting, electron beam melting), titanium alloys and composites, and processing-microstructure properties in high-performance materials. |
 Sven Reichenberger | Sven Reichenberger is the acting leader of the catalysis research group at the Institute of Technical Chemistry I of the University of Duisburg-Essen. He received his PhD in 2017 from the University of Duisburg-Essen and specialized in the field of laser-based defect engineering during a post-doctoral research. He currently conducts his habilitation on surface processes occurring during laser-based catalyst synthesis, with a focus on fuel cells, electrolyzers, and oxidation catalysts. |
 Stephan Barcikowski | Stephan Barcikowski is full professor and chair of the Institute of Technical Chemistry I at the University of Duisburg-Essen. In 2004 he received his PhD in Mechanical Engineering, followed by his habilitation in Chemistry on laser-generated nanomaterials in 2011. His research targets the nanoparticle formation mechanisms in laser ablation and fragmentation, as well as their upscaling aiming at their application in catalysis, biomedicine, and additive manufacturing. |
1. Introduction
Unlike the crystalline metallic materials with a well-defined ordered structure, amorphous/non-crystalline metallic materials with a unique short-to-medium-range ordered and long-range disordered atomic structure have a relatively short history,1–3 but they have been found to be more attractive in different fields. The atoms in the amorphous structure reside in a far-from-equilibrium state with high Gibbs free energy,4,5 and their intrinsically distinct atomic arrangement compared to their crystalline counterparts leads to a high concentration of coordinately unsaturated constituent atoms, unique electron orbital hybridization, modification, and optimization of electronic states, which are favorable to e.g. catalytic applications.6–9 Particularly, amorphous alloys, which are better known as metallic glasses (MGs), have been attractive to researchers due to their extraordinary combination of properties (e.g. high saturation magnetization and good magnetic softness,10 good plasticity, and ultrahigh specific strength,11 excellent catalytic behavior and soft magnetic properties,12 and large global ductility and very high fracture toughness),13 thereby showing promise to be advanced substitutes of crystalline alloys. Note that some extreme cases form monatomic MGs by achieving an ultrahigh liquid-quenching rate of 1014 K s−1,14 but MGs are mainly composed of two or more elements with other metal or metalloid elements to enhance the glass-forming ability (GFA). Here, GFA represents the propensity to form a glassy structure or show the glassy behavior.15,16 It is also closely related to their distinct properties compared to crystalline alloys. Over the last few decades, the processing of MGs has been developed from ribbons, wires, and powder (in micrometers) to bulk MGs (in millimeters), mainly aiming to extend their mechanical and physical applications in different fields. But for exploiting MGs in applications such as catalytic, biomedical, optical, and electronics, their size reduction especially to nanoscale would be much more attractive to maximize their potential with a unique disordered structure. In this respect, the synthesis of A-NPs or even metallic glass nanoparticles (MG-NPs) combines the features of the amorphous structure and the nanoscale size effect. However, although the gas atomization (as a rapid solidification method) and the high-energy ball milling have been widely used for manufacturing MG powders,17,18 it is still hard to achieve nano-size due to technical limitations. The chemical reduction method is also frequently used for the synthesis of MG-NPs but usually with the contamination of surfactants.6,19 Therefore, exploiting an effective and “green” strategy to successfully synthesize MG-NPs is a promising research area.In recent decades, the synthesis and processing of nanoparticles (NPs) have been extensively investigated, particularly in controlling phase, size and shape since it will highly affect their properties and correlated applications.20–23 As such, the motivation of NP synthesis and processing is usually driven by their phase regulation, size reduction and uniformity, and shape control. Many conventional chemical methods have been studied for metal NP synthesis, including thermolysis due to the breakage of chemical bonds of the substrate by heat (e.g. metallic Cu,24 Co,25 and Ag NPs26) and electrochemical synthesis relying on cathodic reduction of metal ions in an electrolyte with size adjustment by current density (e.g. metallic Ag,27 Pt,28 and Au NPs29). Other wet-chemical methods (e.g. chemical reduction,30 photo-induced reduction,31 and microemulsion techniques32) are typically and frequently used for NP synthesis with the advantages of convenient operation and moderate equipment requirements. For example, photochemical reduction has been employed for the rapid synthesis of colloidal Au NPs (i.e. in seconds to minutes) under UVA irradiation and other mild conditions,33,34 which is mainly attributed to the generation of ketyl radicals via Norrish-type-I R-cleavage for reducing Au ions to metallic Au NPs.35,36 However, metal salts or metal-organic precursors are commonly used with reducing agents to achieve NP nucleation and ripening, and surfactants (stabilizers) are also needed to control the dispersity and size of NPs in wet-chemical methods. Thus, surfactant (or reactant)-contaminated NPs resulting in toxicity or catalyst deactivation37 should be purified by post-calcination or extraction, leading to sophisticated structural modification.38,39 And even intensive reflux treatment fails to quantitatively remove the nanoparticle surface adsorbates.37 Considerable residual wastes or by-products after chemical reduction will also cause significant environmental issues compromising the sustainability of the synthesis method. As such, seeking the achievement of “naked” NPs transfers the focus on a more direct way of synthesis, where laser synthesis and processing of colloids (LSPC) including pulsed laser ablation in liquids (LAL) and pulsed laser fragmentation in liquids (LFL) has been fast developed and extensively investigated in the last two decades, with proven scalability owing to the advancement of pulsed laser systems with high output power and reduced cost.40,41
Compared to NP synthesis by chemical methods, pulsed laser synthesis of NPs is mainly a physical strategy, although chemical reactions (e.g. oxidation and carbonization) are also involved during NP synthesis, and is a more straightforward technique for the production of high purity NPs without contamination by surfactants, which thereby can be used for specific purposes with the purity requirement, such as catalysis, biomedicine, and chemo-/biosensing. They can also serve as fundamental constituents for mechanistic studies without the influence of impurity, and as perfect references for comparative studies of surfactant-functionalized NPs and immobilization of NPs on supports.42 The early research of Patil et al.43 has reported the LAL of the Fe target with the formation of metastable FeO, and Fojtik and Henglein44,45 have pioneered the laser synthesis of colloids. Subsequent investigations suggest that NPs can be obtained by ablating metal targets in a large variety of liquid environments.46–49 In combination with the availability of various laser parameters, LAL has shown a comparable and even better performance in the synthesis of colloidal NPs compared to chemical synthesis strategies. For example, the colloid stability, size control, and adsorption to supports of noble metal NPs such as gold (Au),50–52 platinum (Pt),53–55 and palladium (Pd)56 during LAL have been intrinsically studied by electrostatic stabilization using ionic strength in water (e.g. by a facile addition of inorganic saline solution or by adjusting pH). In addition, while LAL focuses on the transformation from bulk materials to nanoparticle colloids, LFL can serve as an effective way and a supplement of LAL for size reduction and narrowing the polydispersity of colloids, especially for NPs with a prominent size-dependence of characteristics to manipulate their functionality.
Indeed, there are some potential drawbacks of laser synthesis and processing of colloidal nanoparticles, including the installation and maintenance cost of various laser systems,57 the consumption of a considerable amount of energy using laser sources,58 the bimodality of nanoparticle size distribution,59 and the persistent microbubbles formed from redox reactions between the hot ablated material and the surrounding liquid.60 However, these drawbacks have been improved by extensive efforts in recent years. As for the investment and running costs, owing to the steady development of output power of the pulsed laser system, the investment cost per laser power now has seen a considerable reduction.41 Recently Dittrich et al.58 have compared the laser synthesis of NPs with different laser systems and demonstrated that less expensive, compact class lasers (<10 k€) achieved at least three times higher power-specific productivity (mg (W h)−1) compared to middle class (<100 k€) and high-end (>100 k€) laser systems. This suggests a more effective transformation of laser energy into NP formation and consequently applicability at both industrial and laboratory scale. In addition, from economic calculations it was shown that productivity directly dictates the running costs while dominating the economic aspects. When the productivity exceeds 550 mg h−1 (Au NPs), it was shown that laser synthesis becomes more economical than chemical synthesis.57 Strategies for optimizing the laser ablation setup (such as ablation cooling of the target,61,62 spatially bypassing cavitation bubbles using a flow chamber and a high-speed polygon scanner63,64) have already resulted in nanoparticle productivities of several g h−1 (e.g. 7.3 g h−1 for AuPt or 8.3 g h−1 for Pt).65 Also, optimization of size distribution to remove larger undesired nanoparticles has been established downstream using a continuously operated, tubular bowl centrifuge for economic synthesis,66 or pulsed laser fragmentation.67 Therefore, the pulsed laser synthesis is in favor of the production of “green”, economic, and functionality-tunable colloidal NPs.
Currently, most laser synthesis research has focused on laser-generated crystalline metallic materials, and noble metals (e.g. Au, Pt, and Pd) are commonly used as targets owing to not only their material density leading to higher productivity,67,68 but also the high chemical stability with high resistance to oxidation during ablation and fragmentation in a liquid environment. As such, laser-generated NPs based on noble metal targets facilitate an in-depth understanding of mechanistic studies (e.g. laser-produced plasma plume and cavitation bubble dynamics),41 minimize cross-effects from chemical reactions to obtain pure monometallic NPs, and reduce the complexity due to the involvement of other elements. Given that crystalline metal NPs have been extensively synthesized and studied via LSPC, the fundamentals and application,41,69–71 catalysis,42,72,73 and control and growth74–76 of laser-generated crystalline NPs and also crystalline oxide NPs67 have been systematically reviewed in recent years. However, to obtain A-NPs with promising properties, the LSPC technique will be a novel strategy but how to turn order into disorder and retain disorder remains as a new and important question in LSPC. To the best of our knowledge, there is still no comprehensive understanding of A-NPs by laser synthesis and processing in recent years.
In this perspective article, we attempt to discuss the recent laser-based synthesis and processing of A-NPs. The importance of understanding both long-range disordered structure and nanosized nature in metal NPs providing the fundamental aspects for synthesizing A-NPs by laser-based strategies will be highlighted. In the first part of the review, the discussion will be focused on the physical features of A-NPs, with the introduction of characterization methods specific to A-NPs, classifications, and properties primarily addressed in the subsequent sections. Then the emphasis will be placed on amorphous metal oxide and carbide NPs typically obtained by LAL. Here, the review and discussion of A-NPs will be centered on identifying possible selection rules of materials, liquids, and laser parameters. The section of amorphous metal oxide NPs by LAL will be also complemented by a discussion about amorphous metal oxide NPs gained from LFL. Discussions with respect to both LAL and LFL aim to provide a working hypothesis to successfully synthesize A-NPs with a given size and composition. The overview of these A-NPs is also extended to the prospect and potential synthesis mechanism of MG-NPs based on laser ablation of bulk MGs. Finally, their main properties and applications will be discussed.
2. Physical features of amorphous metal nanoparticles
2.1. Amorphous structure validation, amorphization criteria, and nanoparticle classification
The amorphous structure refers to a structure with long-range disordered atomic arrangements, but in principle, it is not truly a randomly arranged structure, since short-to-medium-range ordered structures have been simulated and directly observed.1,77 Even so, there are still many fundamental scientific issues related to their structural characteristics and in most cases, the structural features of the amorphous phase have to be firstly identified after the material synthesis. The simple identification of the amorphous structure can be made based on the X-ray diffraction pattern (XRD), where it gives the average diffraction feature of targets as a broad diffuse diffraction pattern (like a hump) without intensive peaks.78–80 Note that regarding the measurement of amorphous NPs, size reduction will also lead to broadening of XRD peaks and it is hard to acquire strong reflection of nanoclusters, which could lead to misinterpretation and wrong assignment of diffraction signals from small crystalline NPs as A-NPs. Wu et al.81 have found that the face-centered cubic (fcc) structure of glutathione-capped gold NPs at 2 nm and 4 nm has transformed to a non-fcc structure at nanoclusters (ca. 1 nm), where a broad diffraction peak is observed in XRD. Although NPs with the size larger than 3 nm obtained from most synthesis methods are sufficient to acquire general amorphous characteristics by the XRD pattern, which have already been extensively reported (e.g. amorphous Fe oxide NPs,82 Pd–Ni–P MG-NPs,83 and Fe–CO–Ni–B MG-NPs84), further confirmation of amorphous structure is usually needed, especially important for the single NP. As such, high-resolution transmission electron microscopy (HRTEM) proves the absence of crystal structures or lattice imperfections, and the corresponding selected area electron diffraction (SAED) of the amorphous structure exhibits the diffractive halo ring (as reported in e.g. amorphous Co@C NPs,85 amorphous Fe@C NPs,86 and Co–B MG-NPs87). In this aspect, the single-phase nature of the amorphous structure endows amorphous NPs with better corrosion resistance,88,89 a more uniform distribution of chemically active sites,90etc. compared to their crystalline counterparts due to the absence of crystallographic defects and phase segregation. In addition, differential scanning calorimetry (DSC) is considered as a complementary method for validating the amorphous structure,91 where the typical exothermic behavior during the heating process suggests the existence of phase transition or crystallization.92–94 DSC is also useful for revealing the glass transition temperature of MGs as one of their important characteristics. In fact, the out-of-equilibrium structure will spontaneously equilibrate (a phenomenon known as physical aging), although it is too slow to observe naturally. As such, the external energy input will accelerate this equilibration process. In DSC, since there is no reference point indicating the enthalpy of full transformation, the exothermic process only confirms that the materials may contain an amorphous or partially amorphous structure.95–97 Considering the unique structural feature of amorphous materials, the extended X-ray absorption fine structure (EXAFS) is also an effective technique to determine the local environment of atoms. Direct information (bonding distances, numbers, and types) can be extracted for the center atom and neighboring atoms within the first few shells,6,98,99 which is different from XRD with the essential long-range ordered structure to be detected. Complementary to EXAFS, the X-ray atomic pair distribution function (PDF) method determines the distances of paired-atoms and the number of bonding types. But the difference of PDF is the well-determined longer distances, having the advantage to distinguish the short and the medium-range ordered structure.99,100 These characterization methods are applicable for both bulk amorphous metallic materials and A-NPs.Although the aforementioned A-NPs generally have the same amorphous feature, their constituent elements will affect their formation. Rapid quenching methods usually require high purity molten alloys made by arc melting followed by a critical cooling rate of at least 105–106 K s−1 under a protection gas7,101 and oxygen-contamination will be extremely detrimental to their GFA,102 which is particularly important for bulk MGs. As such, a critical cooling rate of metallic materials to become fully amorphous could serve as one of the important indicators for their GFA. Inoue et al.103 have concluded three basic empirical rules guiding the manufacturing of bulk MGs with good GFA, which include (1) the number of constituent elements (at least three), (2) the atomic size difference of main constituent elements (over ∼12%), and (3) the significant negative heat of mixing of major elements. Under the same GFA, one material is more likely to form an amorphous structure by reducing its size in all dimensions. That is, the cooling process is much easier to “penetrate” the whole materials in low dimensions.104,105 At the same time, the dissolution of oxygen atoms into the multicomponent metallic matrix seems to lower the GFA, but in fact, some binary metal oxides show a very low crystal growth rate in silicate glasses.106 It has also been reported that the amorphous phase of oxide thin films has to be restricted to a critical thickness (normally <100 nm) and is conditionally stable due to Gibbs energy-related overcompensation by metal surface and interface energies.107 Therefore, elements of metal oxides have to be more carefully selected for amorphous structure formation and metal oxides have their limitation in expanding the size. In this aspect, MGs are relatively more flexible in element composition and dimensions, if the GFA of MGs can be improved to be high enough, where Pd40Ni10Cu30P20 bulk MG was reported to possess an extremely low critical cooling rate of 0.1 K s−1.108
Note that conventional MGs are composed of one or two principal metal elements with other metal (metal–metal MGs, e.g. Cu50Zr50,109 Ca65Mg15Zn20,110 and Al85Ni9Nd4Co2111) and/or metalloid elements (metal–metalloid MGs, e.g. Fe78Si9B13,2 Pt60Ni15P25,112 and Pd40Ni10Cu30P20108). The metalloid elements (allocated as ∼20 at%), e.g. silicon (Si), boron (B), phosphorus (P), and carbon (C), are commonly used to stabilize the metallic supercooled liquid, retard the precipitation of crystals or promote the GFA during the rapid solidification process, while there is no restriction on the composition range of metal–metal MGs. According to these examples, it can be seen that non-equiatomic compositions are typical for MGs. However, recent advances of multiple principal elements have already attracted extensive attention for the design of new alloys known as high-entropy alloys (HEAs).113–115 HEAs, which consist of five or more principal elements, possess a stabilized crystalline solid-solution phase due to the high mixing entropy, atomic size difference and mixing enthalpy, thus contributing to fascinating mechanical, physical, and chemical properties (e.g. strong fracture toughness at cryogenic temperatures,116 overcoming the low ductility while retaining high strength,117 and multi-functional activity in electrocatalysis118). Note that these concepts have already been included in the alloy design of MGs, as suggested by empirical rules from Inoue et al.103 On this basis, equiatomic MGs or high-entropy metallic glasses (HE-MGs) provide a new perspective in the alloy design. Yet, the boundary to form a solid solution phase or amorphous phase is of great interest to investigate. Guo and Liu have pointed out that the formation of a disordered phase instead of a crystalline solid solution phase in the multiple principal element system mainly originated from a more negative mixing enthalpy (−49 ≤ ΔHmix ≤ −5.5 kJ mol−1) and a larger atomic size difference (δ ≥ 9).115 These rules provide valuable guidance in the development of non-equiatomic and equiatomic MGs.
For the synthesis of most amorphous metal oxides, a suitable temperature (control) is also needed to stabilize the internal metastable structure without forming the crystals.119 On the other hand, metalloid C has been considered as an effective additive to improve the GFA in the MGs. Wu et al.120 have reported that C has a better performance to promote the GFA of Fe-based MGs than other typical metalloids such as B and P. So theoretically, amorphous metal carbides can be referred to as one group of MGs containing metals and carbon (may also include other metalloids). Here we emphasize amorphous metal carbides since they are typical laser-generated products by in situ carbonization of NPs in organic solvents (so-called reactive laser ablation in liquids41). But the discussion of MG-NPs will also highlight the formation of NPs from bulk MGs or MG ribbons by LAL. Compared to the in situ carbonization (or oxidation) approach in laser synthesis, the LAL of MG targets is much more complicated because the ablated targets usually involve more than two elements to increase GFA (as mentioned in “Inoue Criteria”).103
In order to distinguish the composition and microstructure of A-NPs in this perspective paper, we have classified and defined A-NPs into three categories with the corresponding schematic NP microstructure (as shown in Fig. 1), including amorphous metal oxide nanoparticles, amorphous metal carbide nanoparticles, and amorphous oxygen-free multicomponent nanoparticles: (1) amorphous oxide metal nanoparticles are in the form of amorphous single/multi-component metal oxide and amorphous metal@oxide core–shell NPs; (2) amorphous metal carbide nanoparticles comprise amorphous metal carbide, amorphous metal@carbon core–shell NPs and amorphous metal carbide@carbon core–shell NPs; (3) amorphous oxygen-free multicomponent nanoparticles mainly include MG-NPs with two or more oxygen-free elements and high-entropy metallic glass nanoparticles (HEMG-NPs).
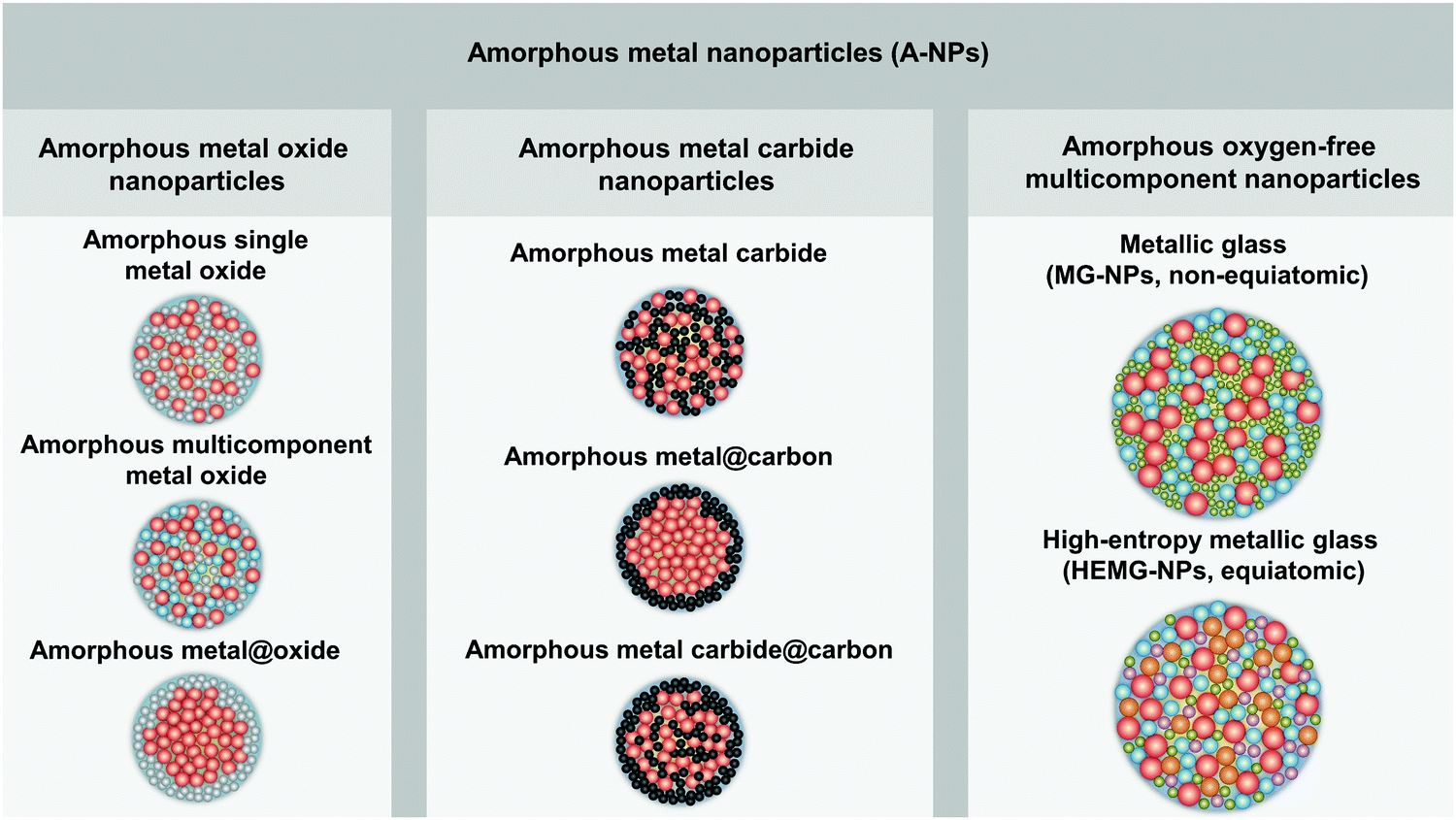 | ||
| Fig. 1 Classification of amorphous metal nanoparticles and the corresponding schematic illustration of the nanoparticle structure. | ||
2.2. Size effect and electronic structure
Given that the vitrification of single-element MGs succeeds by ultrafast cooling,14 the formation of this kind of material only exists in sufficiently small dimensions (e.g. nanoscale) rather than in bulk materials. As such, the size reduction further demonstrates its importance to form the amorphous structure.121 The most apparent size effect during the transformation from bulk materials to NPs is seen in the largely increased surface-to-volume ratio. But this behavior generally shows different defect densities in either amorphous or crystalline structure, especially for the comparison of catalytic activity. As an example, Fig. 2a not only shows the increase of crystalline TiO2 NP (i.e. anatase, anatase/rutile mixture and rutile phase) activity (regarding the generation of reactive oxygen species (ROS)) due to the size reduction, but also presents the apparently enhanced activity of amorphous Ti oxides compared to crystalline counterpart with the similar NP size, suggesting a “defect”-rich surface and a higher number of active sites during the amorphizaton.122 Note that the reduction of activity at 3–4 nm for both amorphous and crystalline structures is attributed to the significant increase of volume-specific surface area but with a relatively small increase of active sites. However, since the disordered and single-phase nature avoids the element segregation and formation of crystallographic defects, A-NPs in fact have a more uniform dispersion of the catalytic active sites on the surface of NPs leading to a more effective catalytic behavior.6,19,123 As a result, the active sites in MGs generally derived from under-coordinated metal atoms lead to the enhancement of e.g. electrochemical performance as H* adsorption/desorption in hydrogen evolution reaction (HER).84,114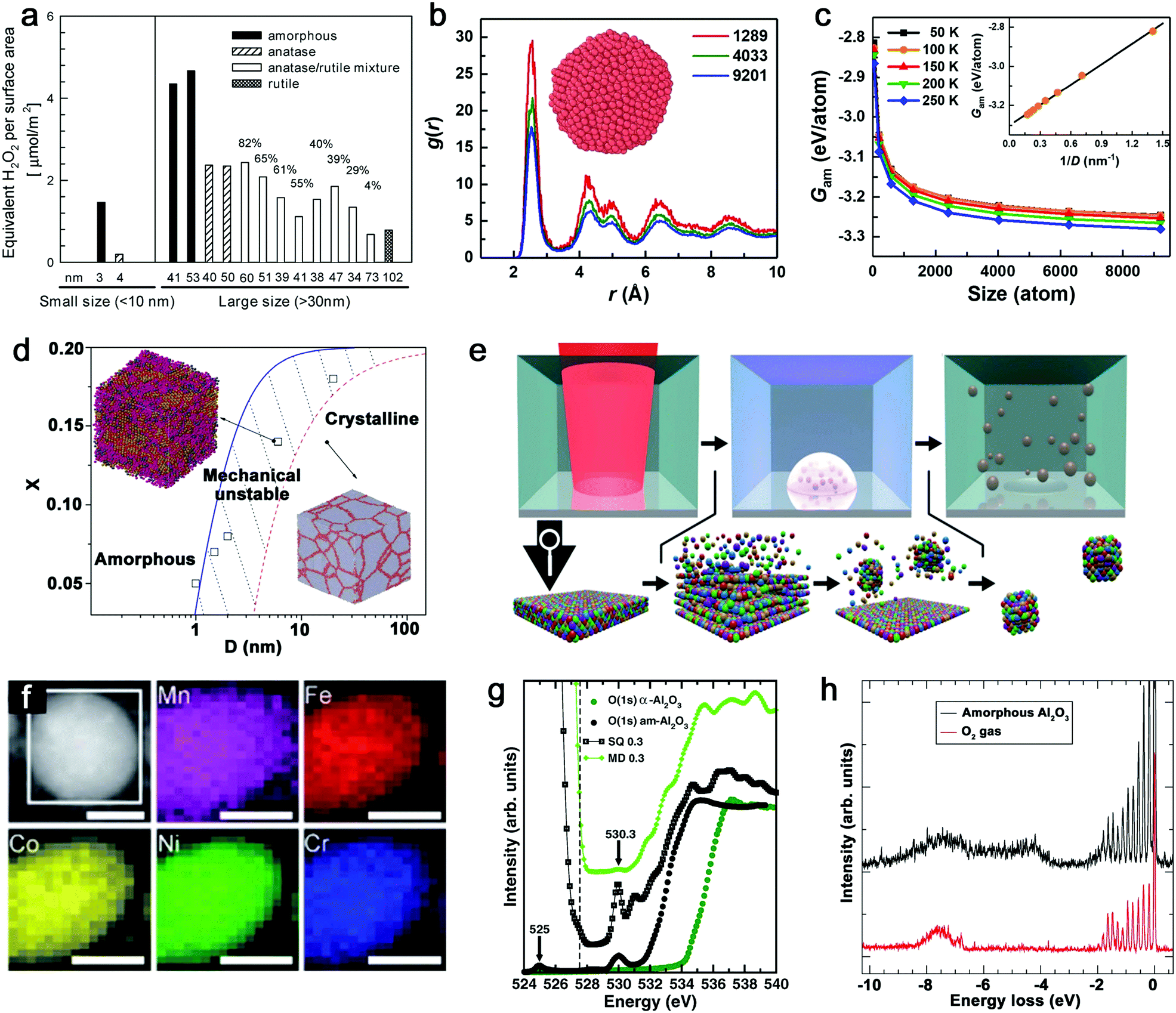 | ||
| Fig. 2 Size effect and electronic structure of amorphous metal nanoparticles. (a) The generation of reactive oxygen species per surface area by TiO2 NPs as a function of the size effect and structural effect. Reproduced with permission from ref. 122. (b) Pair distribution function of amorphous Cu NPs with three atom numbers. (c) The size dependence of Gibbs free energy of amorphous NPs. Reproduced with permission from ref. 124. (d) The phase diagram showing the relationship between the mean grain size (D) and solute concentration (X). Adapted with permission from ref. 125. (e) Pulsed laser ablation in liquid to generate HEA NPs and the formation mechanism. (f) STEM and elemental maps of Co, Cr, Fe, Mn, and Ni for a single laser-generated HEA NP. Reproduced with permission from ref. 126. (g) The oxygen K-edge NEXAFS of amorphous (black dots) and crystalline (green dots) Al2O3, compared with calculated results from MD and SQ simulation. (h) RIXS spectrum of amorphous Al2O3 and free O2 excited at 530.3 eV. Adapted with permission from ref. 127. | ||
From the aspects of thermodynamics, the Gibbs free energy difference is the driving force for crystallization and there is a competition of amorphous phase and crystalline phase during solidification.128–130 The lower free energy in the crystalline structure is correlated to its thermodynamic stability.131 However, it has been reported that size difference has an effect on the Gibbs free energy of A-NPs. An et al.124 simulated the size dependence of free energy of amorphous Cu NPs by the Frenkel–Ladd approach. They firstly verified the amorphous structure by pair distribution function (PDF) as shown in Fig. 2b, where the long-range ordered signal significantly weakens at a longer distance and the short-range ordered signal strengthens at a shorter distance, showing the characteristics of amorphous nature. The subsequent demonstration in Fig. 2c indicates the closely related increase of free energy with the decrease of the size of amorphous NPs, especially for size less than 1000 atoms (∼4 nm) due to the significant increase of free energy. Here, the size dependence of free energy in amorphous NPs also shows a similar behavior as the crystalline counterpart, but they also suggested that the slower increase of free energy of the amorphous phase compared to the crystalline phase at a sufficiently small particle size (with less thermodynamic driving force) due to the absence of facet edges (line defects) in the amorphous phase explains the propensity to form the amorphous structure in the lower dimension.124 As nanocrystals are confined in the NPs, the grain size reduction with narrowing NP size also introduced structural disorder with increased free energy in the system.125 As shown in Fig. 2d, the phase diagram indicates the transition of the crystalline to amorphous structure correlated to the mean grain size; the increased solution concentration also contributes to the larger critical size of grains for the amorphous structure.125 That is, a mixing of different atoms with size difference tends to facilitate amorphization.
Another size effect combined with amorphization presents as the regulation of electronic structure. The quantum confinement effect is generally considered to dominate the electronic properties of crystalline NPs down to a few nanometers.132 But the comparison of amorphous and crystalline Pd NPs indicates that the structural disorder of NPs significantly affects the electronic properties of NPs. The inhibited quantum confinement effect of NPs can be observed by the absence of discrete energy levels of amorphous Pd NPs in scanning tunneling microscopy/spectroscopy, which may find interesting applications in nanodevices.133 In addition, when the composition can be altered in the molten alloys, the subsequent rapid quenching achieves the effective regulation of the electronic structure of MGs due to the well-controlled and wide chemical composition range, which is not available for the crystalline form.19 Given the promise of synthesizing A-NPs by laser ablation and processing, in fact, the homogeneous distribution of elements without segregation should in future also be possible for laser-generated A-NPs to regulate their electronic structure with chosen elements. Recently, Waag et al.126 have reported a facile and scalable synthesis of high-entropy alloy (HEA) NPs by LAL, as shown in Fig. 2e. It has been shown that LAL has the ability to well retain five elements (Co, Cr, Fe, Mn, and Ni) with nearly equiatomic composition in a single laser-generated NP from the HEA bulk target observed by scanning transmission electron microscopy (STEM) and the corresponding elemental mapping (Fig. 2f),126 which has recently been advanced to the quinary alloy with varied Mn content for electrocatalytic inflection-point studies on the active sites.134 In this case, multinary NPs synthesized by LSPC, if transferrable to A-NPs, show the prospect to achieve a regulated electronic structure (e.g. electron orbital hybridization) for catalytic applications. But for the electronic structure of amorphous metal oxides, it would be more complex, which originates from the high atomic packing density. For example, the coordination of oxygen around metal atoms has been frequently focused on in the amorphous metal oxides and their performance as electronic devices mainly relies on the coordination-related defects in the optical gap.127 While Perevalov et al.135 claimed that oxygen vacancies contributed to the defect states at the conduction band of amorphous Al2O3, a recent study by Århammar et al.127 further found that the agreement of experimental results with calculated results by the stochastic quenching (SQ) method in near-edge X-ray absorption fine structure (NEXAFS) and Resonant Inelastic Scattering (RIXS) confirmed that the defect states were trapped O–O pairs, which served as hold traps (Fig. 2g and h). This feature may also be applied to other amorphous metal oxides. A typical case can be found for amorphous silica oxygen pairs.
3. Laser-based synthesis of colloidal amorphous metal nanoparticles
As shown in Fig. 3a, the general process of LAL goes through several steps, starting with the generation and rapid cooling of a plume with initially high temperature (up to 103–104 K) and pressure (up to 105–107 Pa),136,137 and initially very high cooling rates,138,139 the formation of a cavitation bubble in which the majority of NP mass is trapped,140 and the release of atom clusters and NPs in liquid after the collapse of cavitation bubbles. The clear observation of NPs inside the cavitation bubbles has also been reported by the ablation through a thin Ag film deposited on a transparent glass substrate, where this novel setup suppressed the light scattering from the bubble surface and easily obtained scattering signals by the simple inverse light scattering analysis.141 The reactive species (atoms or atomic clusters) during the laser ablation of the Zn target was also detected by NEXAFS in the vapor bubbles, as shown by Reich et al.142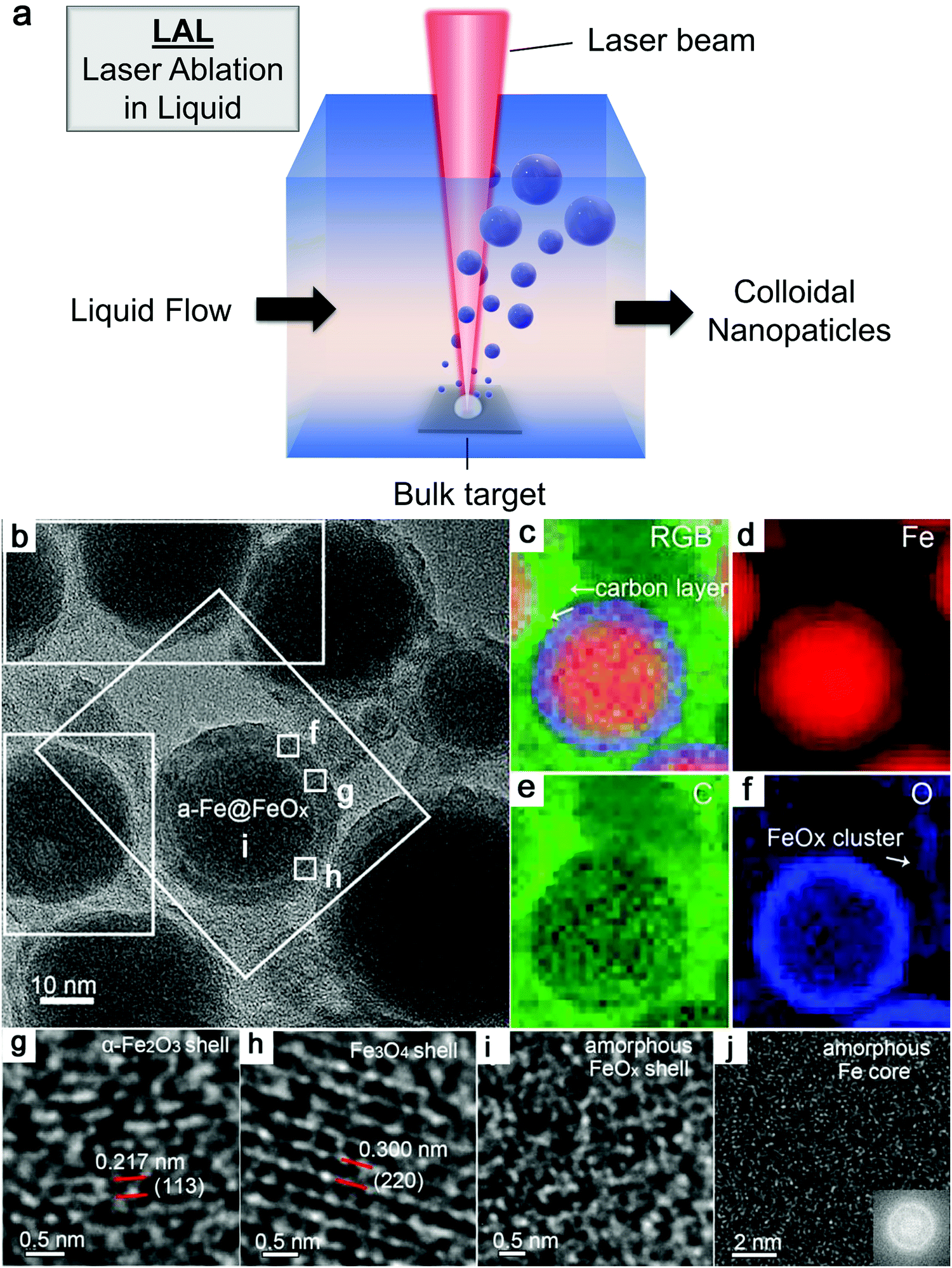 | ||
| Fig. 3 Formation of amorphous Fe oxide NPs by laser ablation in an organic solvent. (a) Schematic illustration of laser ablation in liquid. (b–f) TEM image and the corresponding EDX mapping of a Fe@FeOx core–shell NP by fs-laser ablation in acetone. (g–j) HRTEM of the selected position of the NP in (a) showing different shell structures and an amorphous core. Reproduced with permission from ref. 147. | ||
It has been known that the formation of laser-induced plasma in LAL is a far from equilibrium process and thus condensation also proceeds in nonequilibrium conditions.70,74,143 In particular, ultrashort-pulsed LAL usually provides a large temperature gradient at an initial cooling rate of 1012–1013 K s−1 when hot metal droplets are jetted into a colder and high-density liquid environment,59 facilitating the formation of metastable and defect-rich structures. But cooling rates are significantly slowed down on the time scale of a few nanoseconds, with the majority of the forming NPs being above melting temperature and in thermal equilibrium with the surrounding vapor phase.138 On the longer time scales, if the NP formation model of La Mer is considered, the time needed for critical concentration of nucleation in LAL may be shorter than in wet-chemical reduction methods,74 leading to a narrower time scale for stabilizing nonequilibrium states of ejected matter. Currently, most of the single-element noble metals (Au, Pd, Ag, Ir, Rh, etc.) with fcc structure still fail to form an amorphous structure/glassy state due to fast crystal nucleation and growth.124 Achieving amorphization of single-element noble metal nanomaterials is considered to be challenging owing to the strong metallic interactions among the atoms, the isotropic nature of metallic bonds, and the low activation barrier of the disordered structure relaxing into the crystalline state.144,145 Therefore, suppressing the nucleation in these noble metals is still harder than in other non-noble metals (e.g. Fe, Co and Ni) and the synthesis of amorphous noble metal NPs by LAL lacks sufficient evidence. But transition metals seem to be much easier to become amorphous from laser ablation, in particular when forming binary NPs via in situ reaction with the organic solvent. Accordingly, the synthesis of amorphous or hybrid amorphous–crystalline transition-metal NPs has been commonly observed by LAL (including water and organic solvents). In this section, the amorphization phenomenon and fundamentals, focusing on transition-metal oxides and carbides from LAL, will be summarized, aiming to provide a clearer clue for achieving amorphization of NPs by LAL.
3.1. Amorphous metal oxide nanoparticles
Iron. Up to now, extensive research studies have investigated the ablation of Fe or Fe oxide targets and their products by different pulsed laser durations and in different liquids.146–150 The control of size, dispersity, and phase composition of Fe oxide NPs seems to be attractive because of their excellent chemical and physical properties determining the application fields. For example, Franzel et al.148 reported the synthesis of Fe3O4 (and Fe3C) NPs by sub-ns laser ablation in ethanol; Santillán et al.149 obtained α-Fe2O3, γ-Fe2O3 and Fe3O4 NPs by femtosecond (fs) laser ablation in different liquids (i.e. HPLC water, trisodium citrate solution, acetone, and ethanol); Amendola et al.151 synthesized FeOx polycrystalline NPs with ca. 75% Fe3O4 and ca. 22% α-Fe2O3 by ns laser ablation in water; and Liu et al.152 showed the fabrication of FeO NPs by ns laser ablation in poly(vinylpyrrolidone) (PVP) solution.
However, less attention has been paid to the amorphization phenomenon of oxide NPs during LAL. This may be ascribed to insufficient evidence and synthesis limitation (appropriate selection of laser parameters, liquids, etc.) in previous reports. In a work reported by Shim et al.,153 they showed the ns laser ablation of a bulk α-Fe2O3 target in different liquids (ethanol, water, and acetone) in the batch chamber to synthesize Fe NPs. While high crystallinity was observed for the final products in both ethanol and acetone, the employment of water contributed to the formation of amorphous Fe oxide NPs, which consisted of dispersed large NPs connected by chain-like small NPs and were assigned to be hematite by Raman spectra. Yet, the identification of the determinants behind the formation of an amorphous structure by laser ablation of Fe is still lacking.
Recently, Zhang et al.147 reported a typical synthesis of amorphous Fe@FeOx (a-Fe@FeOx) core–shell NPs by fs laser ablation of a Fe bulk target in acetone. Although acetone was chosen as a liquid, the ablation products included a-Fe@FeOx NPs mainly consisting of some large NPs with the network connected by FeOx small clusters, while the energy-dispersive X-ray (EDX) mapping clearly showed Fe as a core and FeOx as a shell in the structure (Fig. 3b–f). Especially, the HRTEM with fast Fourier transform (FFT) of the Fe core presented the apparent halo ring indicating the disordered atomic (amorphous) structure in the Fe core (Fig. 3j), and a defect-rich FeOx shell composed of crystalline α-Fe2O3 and Fe3O4 (Fig. 3g–i). According to a hump-like diffuse diffraction XRD pattern, the Fe core in almost all the NPs could be considered to be amorphous and dominate the amorphous phase of laser-generated products.
Although the aforementioned network with ultrasmall FeOx clusters has been observed with certain crystallinity by laser ablation in acetone,147 the formation of small amorphous Fe hydroxides may be more favored by the pulsed laser ablation in water. In fact, a gel-like network has been commonly observed in the ablation of Fe targets in water.149,150,152,154 In addition, the investigation of the plasma–liquid interface with potentially strong reactivity is of high interest in the non-equilibrium bonding and structural arrangement.155 Using ns-LAL, Amendola et al.150 found a network of amorphous Fe hydroxide gel dispersed at a nano-scale distance. Although they also applied laser irradiation at third harmonic (355 nm) to gain a narrower size distribution of FeOx NPs (with increased particle size) after LAL, some amorphous domains were still found to be trapped in the FeOx NPs (Fig. 4a) after post-irradiation. But the formation mechanism and the influencing factors still remain to be unravelled, which will be further discussed in Section 3.1.2.
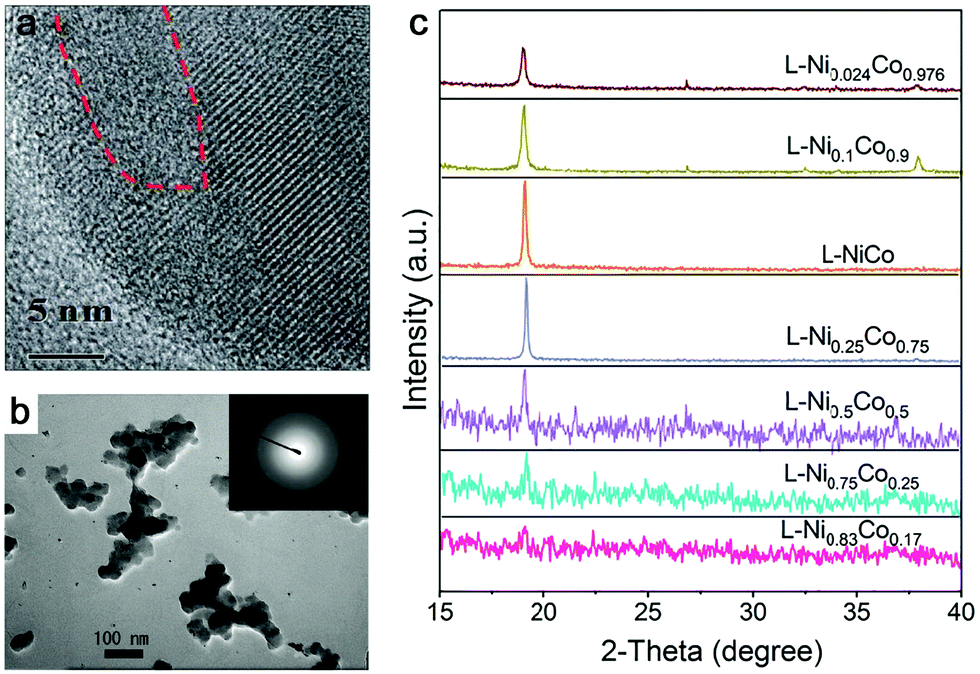 | ||
| Fig. 4 Amorphous metal oxides by laser ablation in water. (a) HRTEM of FeOx NPs after laser irradiation showing some amorphous domains. Reproduced with permission from ref. 150. (b) TEM image of amorphous Pt NPs by LAL at a wavelength of 355 nm and a laser fluence of 4 J cm−1. Reproduced with permission from ref. 155. (c) XRD patterns of products with different proportions of Co and Ni by laser ablation in 1 M KOH. Reproduced with permission from ref. 159. | ||
Other metals. Even though the amorphization behavior does not exert an effect on Au due to the strong metallic interactions among the atoms with fast nucleation, amorphous oxide NPs from dehydration of gel-like hydroxides have already been observed by ns-laser ablation of Pt at 355 nm in water (Fig. 4b),155 where at a low fluence gel-like amorphous Pt oxides formed from the oxidation of the molten Pt target, followed by direct contact (rapid quenching) with water in the absence of plasma plume and desorption from the target surface. While the formation of partially amorphous Pt/Pt-hydroxide NPs by ns-laser ablation in water has already been reported, synthesis conditions yielding amorphous Au or Ag NPs from laser synthesis and processing are yet to be discovered. Other amorphous non-noble metal oxide NPs have also been reported by the pulsed laser ablation in water. Amorphous Cu oxide particles combined with nanocrystals were obtained by ns-laser ablation of the Cu bulk target at 532 nm in water;22 amorphous Al oxide NPs (and amorphous Si oxides) were found by fs-laser ablation of aluminosilicate ceramic plates (with 29.2% Al2O3 and 59% SiO2) in water,156 and hollow amorphous Al2O3 NPs were obtained by excimer laser ablation (ns) of an Al target in water.157 Particularly, Ti seems to be in favor of the formation of the amorphous structure. Given that the main focus of the manufacturing of TiO2 is the anatase and rutile phase, common methods such as hydrolysis and condensation of molecular precursors usually produce amorphous NPs, which need post-calcination.158 This is further confirmed by laser ablation of the Ti bulk target at 355 nm in water or low concentration of sodium dodecyl sulfate (e.g. 1 mM) to produce A-NPs. But the chemical composition of produced amorphous NPs (which may include amorphous Ti or TiOx cores) has not been verified in their case although post-heat-treatment transformed the amorphous phase to anatase and rutile phases.158 In most cases, selecting oxygen- or carbon-affine transition metals instead of noble metals shows a favorable effect on the amorphization of LAL products.
In addition, amorphization will be affected by the composition range if the ablation target is composed of multiple elements. As an example in Fig. 4c, Wang et al.159 reported that the crystallinity of NiCo NPs decreased with increasing Ni content (almost full amorphization at the composition of Ni0.83Co0.17) by ns-laser ablation in a batch setup, but the increase of Co content did not have any effect on amorphization, indicating that element selection is important for amorphization in the target with a two or more element system. These products have been oxidized due to the ablation in KOH solution. On the other hand, Du and coworkers only obtained crystalline nanosheets by the ns-laser ablation of NiFe (Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe = 3:1),160 CoNi (Co:Ni = 3:1),161 and CoFe (CoFe = 3:1)162 bulk targets in water, regardless of the effect of additives. In the case of the CoNi target,161 it was shown that a high concentration of Co is not favorable for the formation of amorphous products. Unfortunately, they did not focus on the amorphization behavior by LAL. Therefore, further studies on the effects of composition on amorphization using binary alloys with common transition metals need to be conducted in the future.
:Fe = 3:1),160 CoNi (Co:Ni = 3:1),161 and CoFe (CoFe = 3:1)162 bulk targets in water, regardless of the effect of additives. In the case of the CoNi target,161 it was shown that a high concentration of Co is not favorable for the formation of amorphous products. Unfortunately, they did not focus on the amorphization behavior by LAL. Therefore, further studies on the effects of composition on amorphization using binary alloys with common transition metals need to be conducted in the future.
Note that those amorphous cores formed from reactive-LAL seem to be free of oxides and the products mainly consist of an amorphous metal@metal-oxide core–shell structure, due to either in situ or ex situ oxidation in liquid or air. It is believed that this will give rise to the scientific question of how the interaction, diffusion kinetics, and function of oxygen (or reactive oxygen species) contribute to the formation of A-NPs.
Liquid. The proper selection of solvents has been demonstrated to be necessary to synthesize NPs with a wide range of composition, in particular making benefit out of reactive-LAL, size, and shape. In organic solvents, the dissociation of molecules (e.g. acetone) containing oxygen after absorbing laser energy generates carbon, oxygen, hydrogen ions/atoms, and CO/CH3 radicals in solution.163 They subsequently react with atoms, ions, and clusters to form new NPs. But the amount of oxygen species is less than carbon and hydrogen due to the higher bond energy of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (728 kJ mol−1) than C–C (332 kJ mol−1) and C–H (414 kJ mol−1).163 As a result, the formation of amorphous metal oxide NPs is less than that of carbide NPs as shown by Zhang et al.147 However, a higher production of amorphous metal oxide NPs in LAL generally occurs in water, which is rich in oxygen (e.g. dissolved molecular O2, O2 and active O species by laser-induced splitting of H2O). In a water environment, additives as solutes are commonly applied for controlling NP size by physical or chemical interactions with ablated materials, such as Cl−, CO32−, PO43−,164 sodium dodecyl sulfate (SDS),165 polyvinylpyrrolidone54 and cetyltrimethylammonium bromide (CTAB).166 Yet, most investigations imply that solutes do not have an important role in the amorphization of metal NPs. Here, we summarize some metal NPs with laser parameters and the corresponding products by pulsed laser ablation in different liquids in Table 1. Obviously, pulsed laser ablation of active metals in water yields an amorphous metal core surrounded by an oxide shell, but the network-like amorphous metal hydroxides mostly dominate the amorphous phase in colloid NPs. Simultaneously, the crystalline NPs are also easy to be embedded into amorphous hydroxides to form a mixture. It has been proposed that the laser-generated products could be rapidly quenched by intimate contact of hot ablated droplets with water having the large heat capacity.43 As a result, the excess energy from the pulsed laser could be dissipated into water, and the metastable state in the molten metal could be rapidly trapped before the structure relaxes to thermal equilibrium, which is similar in the concept to the rapid solidification synthesis of MGs. According to the ns-laser ablation of Fe in water, Amendola et al.66,130 have proposed that a favored environment to form amorphous metal hydroxides existed at the water–plasma interface with a low temperature, a low pressure and a high water concentration due to the incomplete grain growth process and diffusion limited growth which suppressed the coalescence of nuclei and the interaction of neighbor atoms. These processes generally occur during the rapid expansion and cooling down of laser-induced plume (Fig. 5a). At stage (3), the formation of amorphous oxide NPs and hydroxide gel is more likely observed, because the pressure and temperature become low and the high water concentration can act as a quenching medium to stabilize the amorphous phase. In this situation, those quenched network-like amorphous hydroxides are released after cavitation bubble collapse. At the same time, the large NPs may also have a chance to achieve a high cooling rate. As simulated by Zhigilei and co-workers,59 larger NPs could be yielded from rapid quenching of jetted molten metal droplets by the cold and high-density region of water driven by the Rayleigh-Taylor instability, where a cooling rate as high as 1012–1013 K s−1 could also be achieved (Fig. 5b). But the effect of interaction with oxygen in water (including O species by the decomposition of water) also has to be taken into account, at least on longer time scales, not included in atomistic simulations, yet. Therefore, the final products of laser ablation of oxygen-affine transition metals (Fe, Co, Ni, etc.) in water are generally amorphous metal hydroxides with larger crystalline/amorphous NPs. But other influences of laser parameters should also be taken into consideration.
O (728 kJ mol−1) than C–C (332 kJ mol−1) and C–H (414 kJ mol−1).163 As a result, the formation of amorphous metal oxide NPs is less than that of carbide NPs as shown by Zhang et al.147 However, a higher production of amorphous metal oxide NPs in LAL generally occurs in water, which is rich in oxygen (e.g. dissolved molecular O2, O2 and active O species by laser-induced splitting of H2O). In a water environment, additives as solutes are commonly applied for controlling NP size by physical or chemical interactions with ablated materials, such as Cl−, CO32−, PO43−,164 sodium dodecyl sulfate (SDS),165 polyvinylpyrrolidone54 and cetyltrimethylammonium bromide (CTAB).166 Yet, most investigations imply that solutes do not have an important role in the amorphization of metal NPs. Here, we summarize some metal NPs with laser parameters and the corresponding products by pulsed laser ablation in different liquids in Table 1. Obviously, pulsed laser ablation of active metals in water yields an amorphous metal core surrounded by an oxide shell, but the network-like amorphous metal hydroxides mostly dominate the amorphous phase in colloid NPs. Simultaneously, the crystalline NPs are also easy to be embedded into amorphous hydroxides to form a mixture. It has been proposed that the laser-generated products could be rapidly quenched by intimate contact of hot ablated droplets with water having the large heat capacity.43 As a result, the excess energy from the pulsed laser could be dissipated into water, and the metastable state in the molten metal could be rapidly trapped before the structure relaxes to thermal equilibrium, which is similar in the concept to the rapid solidification synthesis of MGs. According to the ns-laser ablation of Fe in water, Amendola et al.66,130 have proposed that a favored environment to form amorphous metal hydroxides existed at the water–plasma interface with a low temperature, a low pressure and a high water concentration due to the incomplete grain growth process and diffusion limited growth which suppressed the coalescence of nuclei and the interaction of neighbor atoms. These processes generally occur during the rapid expansion and cooling down of laser-induced plume (Fig. 5a). At stage (3), the formation of amorphous oxide NPs and hydroxide gel is more likely observed, because the pressure and temperature become low and the high water concentration can act as a quenching medium to stabilize the amorphous phase. In this situation, those quenched network-like amorphous hydroxides are released after cavitation bubble collapse. At the same time, the large NPs may also have a chance to achieve a high cooling rate. As simulated by Zhigilei and co-workers,59 larger NPs could be yielded from rapid quenching of jetted molten metal droplets by the cold and high-density region of water driven by the Rayleigh-Taylor instability, where a cooling rate as high as 1012–1013 K s−1 could also be achieved (Fig. 5b). But the effect of interaction with oxygen in water (including O species by the decomposition of water) also has to be taken into account, at least on longer time scales, not included in atomistic simulations, yet. Therefore, the final products of laser ablation of oxygen-affine transition metals (Fe, Co, Ni, etc.) in water are generally amorphous metal hydroxides with larger crystalline/amorphous NPs. But other influences of laser parameters should also be taken into consideration.
| Target | Liquid medium (C/(C + O)) | Laser parameter | Ablation time (min) | Products | Ref. |
|---|---|---|---|---|---|
| Fe | Water (0) | 1064 nm, 9 ns, 10 Hz | — | Crystalline FeOx with an amorphous Fe hydroxide matrix | 150 |
| Ti | Water (0) | 355 nm, 10 Hz | — | Amorphous TiOx | 158 |
| Al | Water (0) | 248 nm, 30 ns, 10 Hz | 5 | Hollow amorphous Al2O3 | 157 |
| Cu | Water (0) | 532 nm, 6 ns, 1 Hz | 30 | Amorphous Cu and crystalline Cu4O3 | 22 |
| Pt | Water (0) | 355 nm, 7 ns | 15 | Amorphous Pt hydroxide | 155 and 167 |
| Ta | Water (0) | 800 nm, 100 fs, 1 kHz | 10 | Amorphous Ta oxides | 168 |
| In | Water (0) | 532 nm, 5 ns, 10 Hz | 10–120 | Crystalline In2O3 and amorphous byproducts | 169 |
| Ga | CTAB solution (0) | 1064 nm, 5 ns, 10 Hz | 20 | Amorphous Ga hydroxides | 166 |
| α-Fe2O3 | Water (0) | 355 nm, 10 ns, 30 Hz | 60 | Amorphous FeOx | 153 |
| Fe | Toluene (1) | 1064 nm, 9 ns, 10 Hz | — | Amorphous Fe@C (FeC@C) | 170 |
| Fe | Tetrahydrofuran (0.8), acetonitrile (1), dimethylformamide (0.75), dimethylsulfoxide (0.67) | 1064 nm, 9 ns, 10 Hz | — | Amorphous/polycrystalline Fe carbides | 170 |
| Fe | Methanol (0.5), ethanol (0.67), acetone (0.75), toluene (1) | 800 nm, 35 fs, 5 kHz | — | Amorphous Fe@C (FeC@C) and/or Fe crystallites | 171 |
| Fe | Acetone (0.75) | 1045 nm, 457 fs, 100 kHz | 60 | Amorphous Fe@FeOx, amorphous Fe@C, α-Fe2O3 | 147 |
| Fe | Acetone (0.75) | 1064 nm, 7 ns, 20 Hz | 30 | Crystalline FeO/Fe3O4@C | 172 |
| Fe | Hexane (1), pentane (1), decane (1) | 532 nm, 10 Hz | 1080 | Amorphous Fe7C3 and Fe(II) species | 173 |
| Co | Toluene (1) | 532 nm, 10 ns, 10 Hz | 10 | Amorphous Co@C | 85 |
| Ni, Co | Ethylene glycol (0.5) | 532 nm, 3 ns, 20 Hz | 60 | Crystalline Co/Ni@C | 174 |
| Ni, Nb, Ti | Acetone (0.75) | 1064 nm, 7 ns, 20 Hz | 30 | Crystalline Ni3C/NbC/TiC@C | 172 |
| Al | Ethanol (0.67) | 1064 nm, 30 ps, 10 Hz | 60 | Core–shell–shell metal Al-amorphous Al–Al2O3 | 175 |
| Cu | Acetone (0.75) | 1064 nm, 40 ns, 5 kHz | — | Amorphous/crystalline Cu | 176 |
| Pd | Acetonitrile (1) | 1064 nm, 3–6 ns, 10 Hz | 20 | Amorphous and crystalline Pd@C | 177 |
| GaAs | Acetone (0.75) | 532 nm, 7 ns, 10 Hz | — | Amorphous GaAs and crystalline Ga2O3 | 178 |
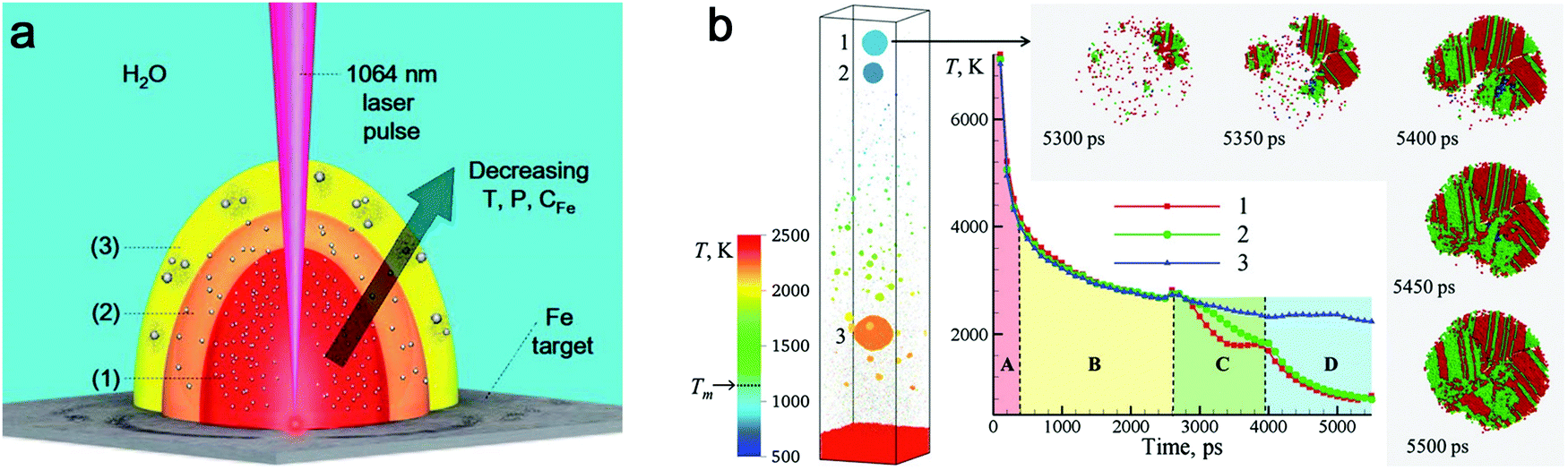 | ||
| Fig. 5 The cooling effect in water on the amorphization of oxide NPs. (a) Expansion of laser-induced plasma/plume and interaction with water. Reproduced with permission from ref. 150. (b) Atomistic simulation of 10 ps-laser ablation of the Ag target in water. Snapshot of the final configuration at 5500 ps with the evolution of temperature and the process of defect-rich crystallization of NPs. Adapted with permission from ref. 59. | ||
Laser wavelength. Besides target selection, the laser parameters (e.g. pulse duration and laser wavelength) play a role in the amorphization during laser ablation. Especially, the laser wavelength seems to have a stronger effect when the ablation is performed in water. The selection of laser wavelength is correlated to ablation efficiency. A typical analysis indicated that the laser ablation of the NiTi target in water showed increased productivity at longer wavelengths.179 Generally, one of the main reasons to choose a longer wavelength (e.g. near-infrared radiation) is to reduce the concurrent effect of photofragmentation, because the higher absorbance of already formed colloids occurs at shorter wavelengths (e.g. visible and UV light).75 The photofragmentation usually leads to size reduction of NPs. The dispersed NPs also inhibit the adsorption efficiency of laser energy for ablation if a shorter wavelength is used,41 and thus a lower productivity is observed for a batch chamber at shorter wavelengths. However, the laser-generated NPs seem to be more “active” with other species and the surrounding solution at shorter wavelengths due to the interband excitation, multiphoton absorption, and photoionization processes. These processes lead to the possibility of structural rearrangement of the already formed crystalline NPs and also break the thermodynamic equilibrium state.69
Note that most of the aforementioned ablations in water producing amorphous NPs have been carried out at the laser wavelength close to the extinction maximum, in particular within the interband region. For Pt with stronger absorption in the UV light regime, Nichols et al.155 reported higher productivity of amorphous Pt oxides due to the selection of wavelength at 355 nm instead of second harmonic (532) and 1064 nm. More importantly, a larger absorption coefficient of Pt leads to a higher surface temperature at this wavelength and the photochemical activation is enhanced between Pt and water by a large flux of UV photons.155 The former originates from a higher temperature resulting in a faster quenching efficiency in water, while the latter suggests a beneficial effect for amorphization by UV irradiation. In addition, Cu oxide NPs have a high absorbance in the visible light regime and the light absorption of Ti oxide NPs is favorable in the UV region, and both cases have produced amorphous metal oxide NPs using a pulse wavelength of 532 nm and 355 nm, respectively.22,158 All these phenomena suggest the significant role of light absorption efficiency in colloidal NPs, because these ablation experiments were conducted in the batch chamber for a long time, and thus post-irradiation (in situ LFL during LAL) has a strong effect on the produced NPs during LAL. In Fig. 6, we have summarized some data points from ablation in water according to Table 1 and marked the region which has a higher possibility to form amorphous oxide NPs. Here, the full amorphization shifts to the UV-Vis wavelength region, which is also in good agreement with the higher quantity of absorbed energy from the laser that favors the amorphization of oxide NPs in water. Therefore, a suitable wavelength (tuned to the maximal absorbance) should be considered for energy deposition into colloidal NPs when doing laser ablation in water.
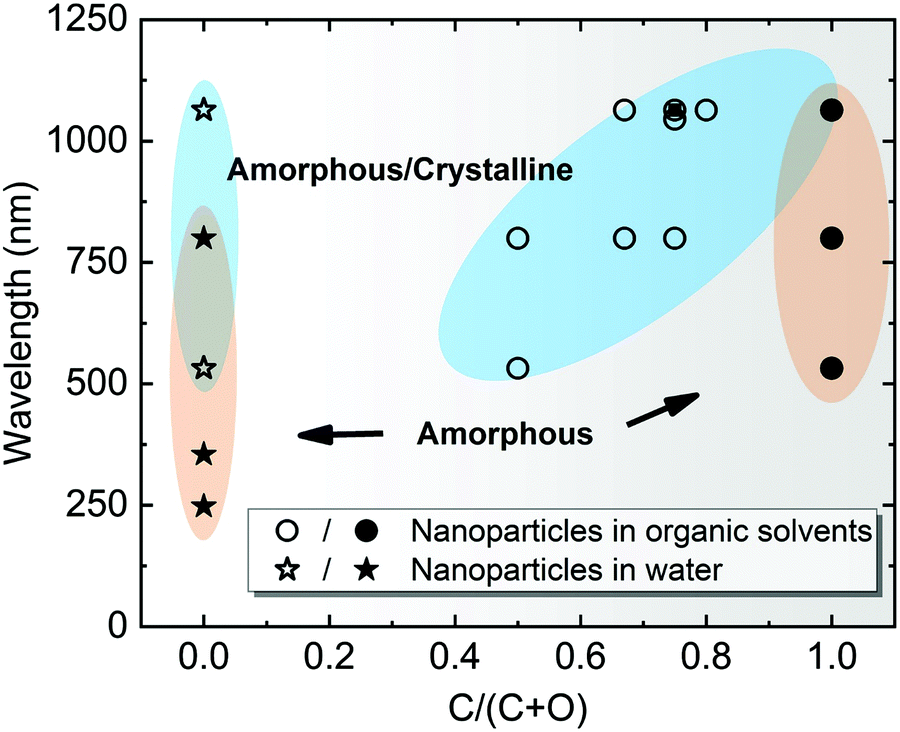 | ||
| Fig. 6 A summarization of structural characteristics of NPs by laser ablation in water (C/(C + O) = 0) and organic solvents (0 < C/(C + O) ≤ 1), showing the effect of laser wavelength as a function of C/(C + O). C/(C + O) represents the carbon to the total number of carbon and oxygen ratio in the liquid molecule. Data points are obtained from Table 1. The blue-shaded areas with hollow symbols indicate amorphous-crystalline NPs, whereas the orange-shaded areas with solid symbols indicate A-NPs. | ||
3.2. Amorphous metal carbide nanoparticles
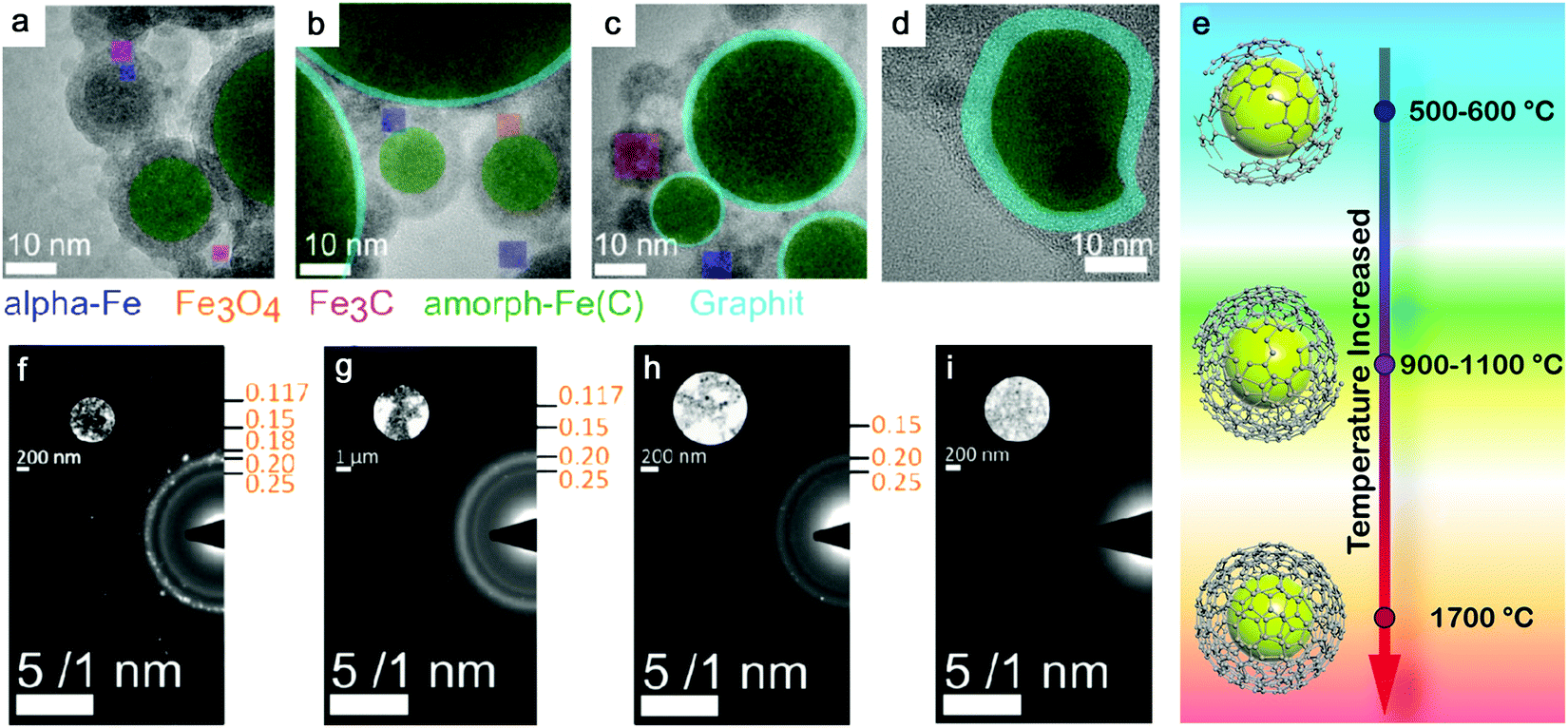 | ||
| Fig. 7 Effect of the carbon to oxygen ratio in organic solvents and carbon shell evolution. HRTEM with FFT analysis of NPs synthesized in (a) methanol (CH3OH), (b) ethanol (C2H5OH), (c) acetone (C3H6O), and (d) toluene (C7H8). Adapted with permission from ref. 171. (e) Schematic illustration of the transformation of carbon onion with increasing temperature. Reproduced with permission from ref. 172. (f)–(i) SAED of NPs synthesized in (a) methanol, (b) ethanol, (c) acetone and (d) toluene, respectively. Reproduced with permission from ref. 171. | ||
Theoretically, transitional metals having similar properties (as Fe) can achieve partial or full amorphization by pulsed laser ablation in organic solvents. Also by using toluene, amorphous Co@C core–shell NPs have been produced by ns-laser ablation at 532 nm, and C atoms possibly diffused into the Co matrix;85 amorphous Cu@C NPs were likely formed by ns-laser ablation at 1064 nm in acetone;176 a decreased crystallinity or partial amorphization of Pd encapsulated by graphitic carbon was also observed in ns-laser ablation at 1064 nm in acetonitrile.177 But the practical situation will be more complicated. One of the reasons is the competitive affinity of transition metals to carbon and oxygen, which largely determines the core composition. Zhang et al.172 have compared 16 transition metals by laser ablation in acetone and found that noble metals (Cu, Ag, Au, Pt, and Pd) only formed the metal core, whereas active metals (Ti, V, Nb, Cr, Mo, W, Ni, and Zr) formed a metal-carbon core after ablation. Those metals (Mn, Fe, and Zn) have also been found with (amorphous) oxide NPs after ablation even in organic solvents due to a higher oxygen affinity. But to maximize the function of carbonization, it is necessary to lower the effect of oxygen, where creating an oxygen-free environment during LAL could be an alternative strategy to increase the amorphization ability. On this basis, it has been reported that amorphous iron carbides could be synthesized by pulsed laser ablation of the Fe target in pentane, hexane, and decane (all without oxygen in the molecule) when Ar gas was used to isolate oxygen.173 Hence, the role of the three possible oxygen sources (the molecular oxygen dissolved in the liquid, the residual water even present in semipolar organic liquids, and the oxygen atom in the solvent molecule) in NP oxidation during LAL has to be understood. Recently, Nadarajah et al. systematically excluded the respective oxygen sources during LAL of the highly oxygen-affine transition metal alloy FeRh, where atom probe tomography could validate that both acetone and acetonitrile were capable of avoiding oxidation, if careful degassing and solvent drying was carried out prior to LAL,181 otherwise the (partial) oxidation of the NP core as well as an amorphous oxide surface layer was observed.
Selection of organic solvents. Generally, the carbon matrix surrounding NPs has a role in preventing the laser-generated NPs from undergoing oxidation and further growth or coalescence,147,170 which explains why even a higher concentration of dissolved oxygen in the organic solvents resulted in a lower oxidation of NPs after ablation.176 Regardless of the chemical structure, one of the distinguishable features among organic solvents is the oxygen content in the solvent molecule. In Fig. 7f–i, it can be seen that the amorphization of NPs has the potential to be enhanced with the increase of the carbon to the total number of carbon and oxygen ratio (C/(C + O)) in the selected methanol (0.5), ethanol (0.67), acetone (0.75), toluene (1). Selecting organic solvents which do not contain oxygen seems to favor the formation of A-NPs. It has been reported that the residual water and dissolved oxygen in organic solvents would increase the oxidation of FeRh NPs but the bonded oxygen of solvent molecules does not affect the oxidation.181 But here an analysis of the role of organic solvents can be seen in Fig. 6, which shows the effect of carbon/oxygen in solvent molecules in relation to pulse wavelength. The formation of an amorphous/crystalline structure in NPs takes place at a wide range of C/(C + O) but the full amorphization always occurs at C/(C + O) of 1. This leads to the possible conclusion that the higher content of carbon in the solvent shows a higher possibility to form new chemical bonds with metal atoms in the NPs. With the combination of metalloid carbon in metals, it increases the amorphization (or GFA) of NPs, if the transition metals have a high affinity to carbon.
However, the carbon and oxygen content in the solvent molecules is not sufficient to rationalize the phase composition (here for amorphous and crystalline phases).75 One of the reasons is the chemical structure of solvents. A competitive reaction with oxygen due to saturated chemical bonds leads to less oxidation and more carbonization of metal NPs which is favorable for the diffusion of carbon atoms. For example, tetrahydrofuran with saturated chemical bonds has a propensity to react with dissolved oxygen, thus avoiding higher oxidation of Fe atoms than in acetonitrile and dimethylformamide with unsaturated chemical bonds.170 One should also consider the complex physical and chemical reactions in the plasma plume with high-temperature and high-pressure under carbon-rich atmospheres in the organic solvents. From the current findings toluene seems to show a better performance in the amorphization of NPs. This may be due to the effect of benzylic carbon on radical stability (or radical lifetime).182 The generation of carbon-containing radicals and carbon ions/atoms usually originates from extreme conditions by laser-induced plasma.163 As a result, the stabilization of radicals facilitates the formation of carbon shells and may also have an effect on amorphization.
Given that the fast cooling of the emerging NPs from the plume provides an ideal non-equilibrium state to stabilize the amorphous feature, the subsequent cavitation bubble period (during which the matter is in thermodynamic equilibrium), however, may not further amplify the formation of amorphous NPs (at least before the bubble collapse phase). Of course, the cavitation bubble is closely related to the productivity of laser-generated colloid,183–185 and thereby could influence the ablated matter-to-solvent-ratio. But it is not clear how the cavitation bubble affects the amorphization of NPs. One can expect that the early cavitation bubble (whose precursor already appears on a sub-ns time scale, according to the above-cited simulations from the Zhigilei group) generally induces the quenching of laser-ablated matter. On the other hand, on a ns to ms time scale, the NPs will thermally equilibrate inside the bubble's vapor phase. The shielding of the colder and denser liquid region by the cavitation bubble phase boundary will largely reduce the thermal quenching efficiency, which facilitates the nucleation and growth of crystalline NPs. But in organic solvents, a higher kinematic viscosity (μ) was reported to limit the size of the cavitation bubble during the expansion and shorten the period of bubble collapse, thus reducing its lifetime (Fig. 8a) and possibly shortening the NP growth/aggregation time.186
 | ||
| Fig. 8 Bubble dynamics at liquids with different kinematic viscosities. (a) Shadowgrams of evolution of cavitation bubbles for the Au target under ns-laser ablation in water (μ = 1 mm2 s−1), polyalphaolefin (PAO) 6 (μ = 80.8 mm2 s−1), and PAO 40 (μ = 764 mm2 s−1). Adapted with permission from ref. 186. (b) Bubble collapse at D* = 0 in water with outer to inner curves at t = 57.4, 95, 105, 110, 113, 114, and 114.46 μs and (c) corresponding fast jet formation with bubble wall velocities at t = 114.2, 114.3, and 114.32 μs; jet formation at D* = 0 at a liquid viscosity (d) 20 times and (e) 40 times higher than that of water with bubble wall velocities at (d) t = 110.65, 110.67, and 110.7 μs and (e) t = 108.2, 108.3, 108.4, and 108.5 μs. Adapted with permission from ref. 187. | ||
However, it is still not clear whether the first bubble confinement plays a major role in preventing amorphization. In addition, extremely high pressure and temperature will be reached again during the bubble collapse, which may also have an effect on the confined species in the bubble. Recently, Lechner et al. have simulated the collapse of bubble shapes at a solid boundary (D* = 0, bubble expands as an initially hemispherical bubble, similar to LAL) (Fig. 8b).187 Here, D* represents the normalized distances by considering the center of the bubble to the solid boundary and the maximum radius of the bubble without any boundary. In this case, the high curvature of the bubble wall (due to kinematic viscosity of water) during bubble collapse contributes to the bell-shaped form, leading to the formation of a fast inward jet that hits the target (Fig. 8c). A standard axial-jet formation (with velocities of ∼100 m s−1) caused by axial flow focusing is found for 0.24 ≤ D* ≤ 3, but for bubbles very close to the solid boundary (0 ≤ D* ≤ 0.2), a very fast axial-jet (∼1000 m s−1) is formed by annular-liquid-flow collision. Note that viscosity plays a critical role in the speed and type of the jet. In the case of a liquid with viscosity 20 times higher than that of water at D* = 0, the super-fast jet (1000 m s−1 of bubble wall velocity) is reached (Fig. 8d), but the jet will become slower at a very high viscosity (i.e. 40 times that of water) (Fig. 8e).187 Accordingly, one may expect that this super-fast jet impinges on the target (solid boundary) with a very high kinetic energy, thus affecting the amorphization process (of trapped matter inside the bubble in case an identical jet is also formed during LAL bubble collapse, but will also contribute to target ablation). Therefore, the bubble dynamics, in particular the bubble collapse, may be also involved in the amorphization process, eventually closely related to the viscosity of the solvent. The investigation of cavitation bubbles in organic solvents (with different densities and viscosities) toward the competitive mechanism of crystallization and amorphization will be an interesting topic in the field.
Concerning the liquid selection, gaseous LAL by-products also have to be considered. A significant amount of gas is formed and accumulates in the ablation chamber's headspace or is drained by the liquid flow. For example, Kalus et al. have shown that tens of cm3 per hour permanent gas by-products were formed during LAL and LFL.60,188 For ns-ablation of Au in ethylene glycol, molecular hydrogen and carbon monoxide were identified by gas chromatographic measurements as the main decomposition products besides smaller amounts of carbon dioxide, methane, acetylene, ethylene, and ethane.188 Recently, Koshizaki et al. pointed out the positive effect of solvent pyrolysis that was useful to achieve the intended chemical composition of the particles formed by pulsed laser melting in organic liquids, with ethylene formed from ethanol playing a key role in the in situ reduction mechanism during laser melting.189
It should be noted that LAL or LFL does not ignite organic solvents in the liquid phase because of the lack of oxygen. Hence even ablation in highly flammable liquids such as acetone or acetonitrile is a safe process,181 at least at a smaller scale, at low liquid volumes, and in tight chambers. However, even ps-LAL is known to cause a significant temperature increase in the liquid,62 and during low-power ns-LAL in organic liquids, a steady temperature increase of up to 1 K min−1 has been reported that approaches the liquid's boiling point.60 This temperature increase leads to a strong increase of the solvent's vapor pressure, so that pressure-tight ablation chambers have to be used. To avoid the ignition of unintendedly leaking solvent vapor or gas by-products accumulation in the headspace (such as hydrogen), the use of flame protection housing around the ablation chamber, flooded with N2 ensures safe operation.
Carbon and carbon shell effect. Depending on the solubility of carbon in the metallic matrix (in both molten and solid states), in the case of transition metals with a higher number of unfilled d-orbitals, metal atoms will more likely attract the neighboring carbon atoms to form metal–carbon bonds with strong binding energy.172 This usually happens for the aforementioned active transition metals. In fact, considering the metalloid nature of carbon in promoting GFA of MGs, organic solvents provide a beneficial environment to form and coordinate carbon into an amorphous metallic matrix under laser ablation and related chemical reactions. In the case of pulsed laser ablation of metallocene (Fe(C5H5)2 and Co(C5H5)2), Huh et al.180 obtained amorphous Fe–C and Co–C NPs encapsulated by a graphitic carbon shell. Since a large size difference occurs in Fe (Co) and C atoms, the diffusion of C atoms into the metallic matrix is highly possible, which also inhibits the nucleation and formation of a lattice structure under fast cooling of plasma and rapid quenching by liquid. This phenomenon shows the difference of C atoms from O atoms due to different interaction and diffusion behavior, and currently there is no evidence to show the formation of amorphous metal oxide cores by laser ablation in organic solvents. Based on the homogeneous crystal nucleation theory, the high viscosity of alloy melts will largely suppress the crystal nucleation rates.190 That is, the low mobility of atoms is favorable for achieving a faster quenching than the ordering. As a high packing efficiency is closely related to the increase of GFA, an efficient atomic size ratio between solute and solvent atoms generally increases the atomic packing density of alloys to give a higher possibility of amorphization. Therefore, in the previous cases of the single amorphous metal core, the embedment of C atoms in the core is also possible, so as to increase the amorphization of metal carbides. However, the synthesis of amorphous transition-metal NPs in organic solvents does not always succeed, which is attributed to the uncontrollable composition range of carbon in the metallic matrix. As an empirical rule, a higher successful chance to achieve a full amorphous structure of metallic materials usually occurs at the metalloid element compositions located at ∼20 at%.7 Therefore, how to control the carbon content in the amorphization of NPs deserves in-depth investigation and discussion.
Given that most metal NPs ablated in organic solvents bear carbon shells, graphitic (and amorphous) carbon shells seem to be crucial for the amorphization behavior of metal NPs by laser pulses. Very recently, Sun et al.86 designed the graphitic shell encapsulated Fe and Co NPs by the modified chemical vapor deposition and the arc evaporation, which were subsequently subjected to a ns pulsed laser at 1064 nm. In Fig. 9a, the TEM image shows their crystalline Fe NPs encapsulated by graphitic shells. During the repeated laser pulses, the amorphization and recrystallization behaviors were repeatedly observed (Fig. 9b–e) due to the fast heating–cooling cycle by a short-pulsed laser. But the calculated cooling rate in their case (106–107 K s−1) was far lower than the required critical cooling rate of pure Fe or Co, which contradicted their observation of amorphization of metal NPs. As such, the formation of the amorphous phase containing metal–carbon bonds by the diffusion of C atoms seems to be satisfied with this phenomenon,86,191 which was confirmed by the electron energy-loss spectra of the Fe L2,3 edge that coincided with the shifting of Fe–C and metallic Fe. In this case, the confinement of Fe and Co NPs by graphitic shells was suggested to suppress the heterogeneous recrystallization and stabilize the disordered structure. The recrystallization of metal NPs might be due to the slow cooling, which also led to the growth of graphitic layers by carbon segregation.86 But in fact, the confinement effect of graphitic shells provides some reasonable suggestions about vitrifying laser-generated metal NPs by laser ablation in organic solvents. As the graphitic shell encapsulated metal NPs produced during laser ablation, the reirradiation by a pulsed laser leads to the remelting of solidified “trapped” metal NPs and diffusion of C atoms into the metallic matrix may decrease the mobility of molten alloys. All these phenomena promote the amorphization chance under the confinement effect of carbon shells and rapid quenching. So, this possibly becomes more obvious in the long-time irradiation by a pulsed laser in a batch chamber. But since the solubility of C atoms in metals is different, it will affect their corresponding amorphization ability, which is apparent in the noble metals. In addition, long-time irradiation will also inevitably contribute to the laser fragmentation of NPs with size reduction, especially in the batch processes, so laser fluence has to be precisely tuned below the fragmentation threshold.
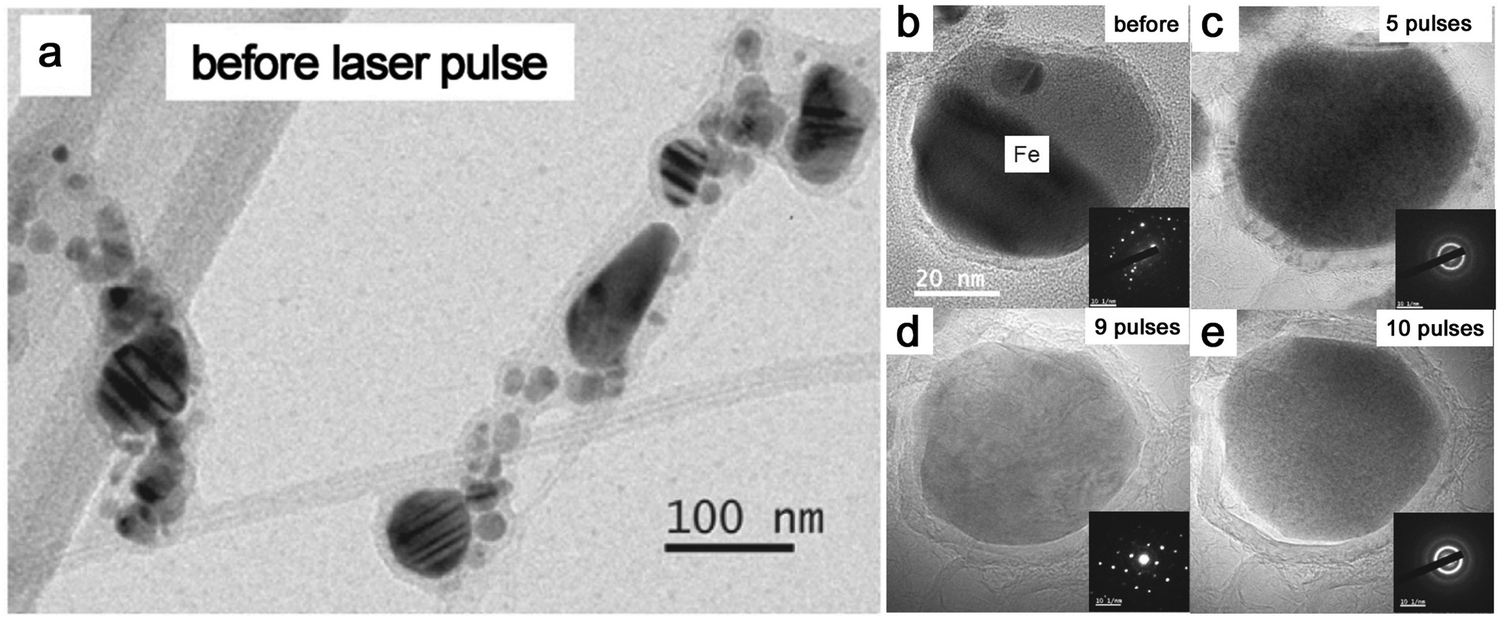 | ||
| Fig. 9 The amorphization of metal NPs under laser pulses. (a) TEM image of Fe NPs encapsulated by graphitic shells before laser pulses; (b–e) HRTEM and the corresponding SAED of an encapsulated Fe NP under different numbers of laser pulses. Adapted with permission from ref. 86. | ||
According to the aforementioned discussion, the following seem to contribute to the amorphization of NPs in organic solvents: (1) a high carbon to oxygen ratio in the organic solvents favorable to form a carbon shell and exclude the effect of oxygen, including careful removal of dissolved oxygen and residual humidity from the solvent; (2) a higher affinity of active transition metals to carbon; (3) a coordination of C atoms into the metallic matrix increasing atomic packing density; (4) the suppressed heterogeneous nucleation of metal NPs by the confinement effect of the carbon shell.
Pulse duration. Note that compared to the ablation in water, the laser wavelength has a less effect/dependence on the amorphization in organic solvents as shown in Fig. 6 (also, most organic solvents are nontransparent in the UV range, making UV-LAL quite inefficient). But choosing pulse duration is generally important to the structure and composition of NPs since it will strongly affect the LAL mechanism from bulk materials to NPs. Using ultrashort pulse durations (fs) for ablation may provide a stronger (initial) cooling rate to favor metastable structures.171 For example, nanomaterials with metastable phases such as nanodiamonds have been reported to be synthesized by the ultrashort (100 fs) laser pulsed ablation of a graphite target in water.192 By laser ablation in acetone with different pulse durations, Zhang et al.147,172 obtained Fe NPs with a good amorphous feature by fs pulses, while they obtained crystalline Fe oxides by ns pulses. The ps-laser ablation with a duration in between that of fs and ns lasers has the combined features of direct photoionization and thermal ablation processes depending on the pulse duration. As a hypothesis, this may also complicate the amorphization mechanism of NPs in the liquid, where it depends on the dominant role of direct photoionization or thermal ablation processes. By observing laser parameters in Table 1, both ns and fs pulses, to some extent, play a role in producing amorphous products, indicating that different pulse durations have the ability to form an amorphous structure based on their ablation mechanism and cooling/quenching method. However, since different targets and organic solvents have also been employed, currently they are still not fully comparable.
Regarding the multicomponent target, it is speculated that the non-congruent vaporization of ejected droplets is produced by short laser pulses, while ultrashort laser pulses have the propensity to highly retain the target stoichiometry.168 In such a case, ps-laser ablation to generate HEA NPs as discussed in Fig. 2e and f shows the good elemental homogeneity for even five elements used. On the other hand, ultrashort pulses pose far higher demands on the elemental mixing prior to ablation in liquid. Recently, Zigilei et al. predicted the quite limited mixing of target elements during ultrashort-pulsed laser ablation of a binary target, and the modeling results were backed by experiments.193 Here, single-pulse ablation of a Cu/Ag thin film sandwich did mostly lead to silver or copper-rich NPs, rather than the alloy, no matter which element was the top layer. Amendola et al. also used thin-film sandwiches to understand alloy formation, where the highest alloy yields with the intended composition and structure (in this case, iron-rich gold alloy in core–shell structure) were achieved for two scenarios: either the target was perfectly pre-mixed (no sandwich but an alloy) or longer pulse durations (ns LAL) were employed.194 Note that the thermal effects of the laser pulse in the target also contribute to alloying of targets that were segregated before the pulse arrives, as Marzun et al. showed via EDX analysis of a pressed micropowder target with Ni and Mo alloy formation only at the ablated crater region.195 Hence, ns-LAL may be a better choice to create multi-elemental NPs in case the ablation target elements are not perfectly (atomically) mixed.
Other effects may be also correlated with the polarization of the laser. It has previously been shown that a perpendicular orientation between the polarized light and a periodically structured surface leads to higher reflectivity and hence less vigorous ablation, larger particles with higher polydispersity and shorter cavitation bubble lifetime.196 For parallel orientation of both, the opposite was observed while a 10% lower pulse energy (fluence) again resulted in the same ablation rate and particle size distribution as found for the perpendicular case.196 Hence, the effect of polarization is equivalent to the effect of laser fluence, where the difference in the effective laser fluence compared for both orientations is less than 10%. If the polarization has an effect on LAL, it also depends on the capability of the ablated alloy to form laser-induced periodic surface structures (LIPSS). It has been shown that ps-LAL productivity linearly depends on the laser fluence if LIPSS is not formed, while a logarithmic trend and a decrease in productivity are evident when LIPSS is formed. Circularly polarized light cancels LIPSS formation and thereby increases the nanoparticle productivity by more than 30%.197
4. Laser-based processing of colloidal amorphous metal nanoparticles
Size control and modification aiming to achieve high fractions of monomodal ultrasmall NPs are still of great interest, which are achievable by the irradiation of raw particles (known as LFL).198 The widely accepted fragmentation mechanisms are classified by the employed laser fluence (as the driving force) and the pulse duration (as energy transfer kinetics).21 Ultrashort laser pulsed LFL (<10 ps) is correlated to electron dynamics-mediated mechanisms, where electrons escape from the confinement of the lattice structure after absorbing energy from laser pulses, leading, at sufficient laser fluence, to the charge imbalance of particles and spontaneous fission (known as Coulomb explosion/instability).6,140 On the other hand, short laser pulses (e.g. ns) with enough time for electron cooling and reaching thermal equilibrium mainly induce the photothermal evaporation processes and melting of NPs (heating–melting–evaporation mechanism).Often, LFL is performed in a batch setup with continuous (magnetic) stirring to achieve convection (Fig. 10a). As a result, the mass transport rate of educt particles to/from the irradiated volume and the repetition rate of laser pulses strongly influence the average energy and the number of laser pulses applied on the particles.67 Recently, Yang and coworkers have reported a series of amorphization behavior of transition metal oxides by second harmonic ns-laser fragmentation in a batch setup, including Fe2O3,82,199 CoO,199,200 and NiO.199,201 An interesting phenomenon is that the structures of original crystalline Fe2O3, CoO and NiO particles were rearranged into a highly disordered state after the long time irradiation in water (Fig. 10b). In their cases, the fragmentation time seems to be important for the amorphization behavior, where the initial size of crystalline particles ranges from tens to hundreds of nanometers (Fig. 10c) and 30 min is sufficient for the full amorphization of metal oxide NPs (Fig. 10d). Further irradiation by hours tends to lead to two-dimensional (2D) amorphous nanoflakes for CoO and NiO (Fig. 10e and f).200,201 This was confirmed by Liang et al.,202 who found different crystallinities of Nb2O5 NPs when particles were fragmented in water for various time durations (Fig. 10g). Since a 10 ns laser with a low repetition rate (10 Hz) was applied throughout their experiments, the fragmentation of those crystalline metal oxides probably mainly goes through the photothermal evaporation and melting. The repetitive process of photothermal processes and quenching by LFL is possible to continuously destroy the crystalline structure and suppress the nucleation of the ultrasmall structure. The high fluence used also ensures high fragmentation efficiency in order to effectively reduce the particle size.
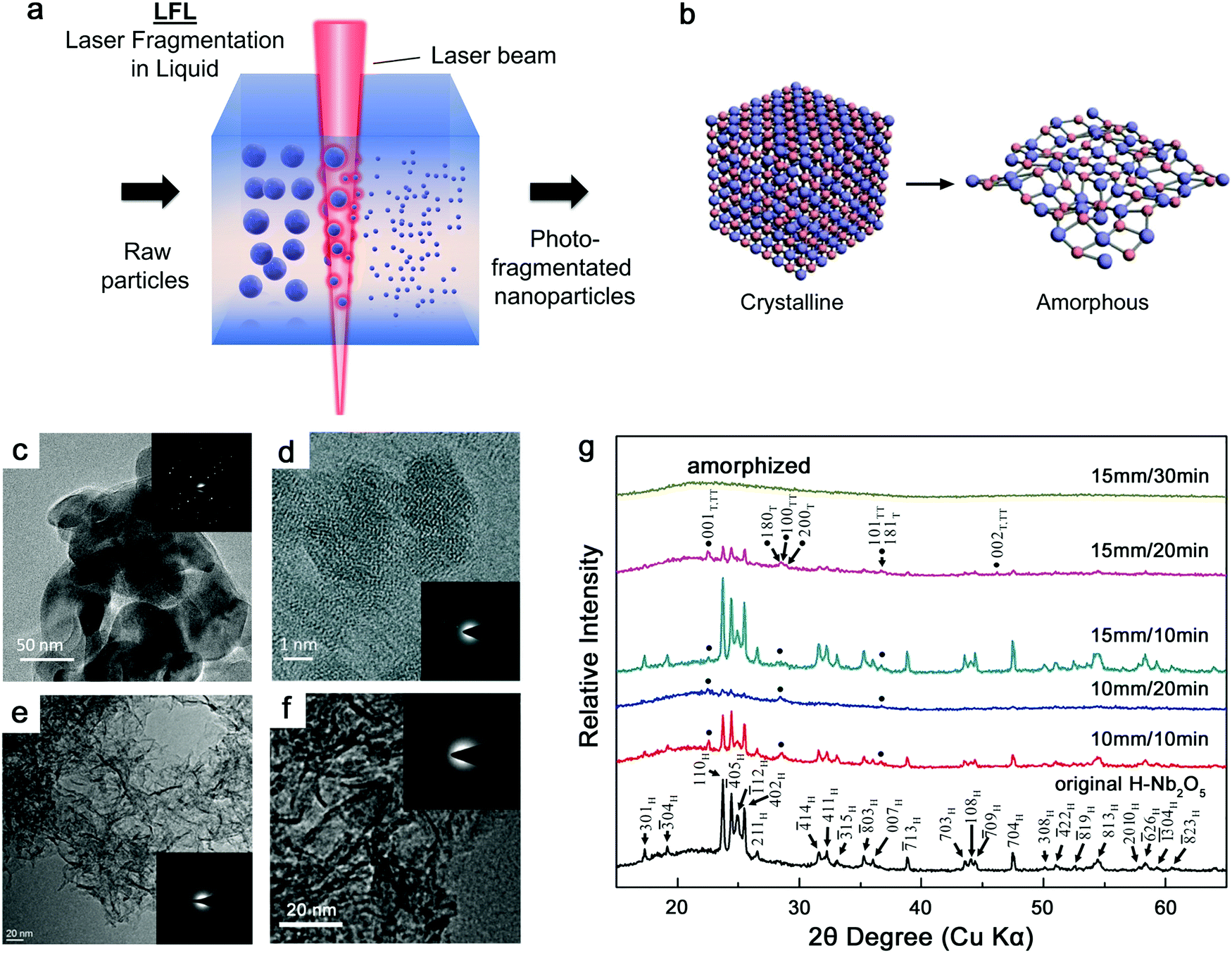 | ||
| Fig. 10 Laser fragmentation for the amorphization of metal oxide NPs. (a) Schematic illustration of laser fragmentation in liquid. (b) Amorphization process from crystalline materials. Adapted with permission from ref. 201. (c) TEM image of the crystalline hematite micropowder; (d) HRTEM image of amorphous Fe2O3 NPs after laser fragmentation of particles of (c) in water. Reproduced with permission from ref. 82. (e) Amorphous CoO nanoflakes after laser fragmentation of crystalline CoO particles in water. Reproduced with permission from ref. 200. (f) Amorphous NiO nanoflakes after laser fragmentation of crystalline NiO particles in water. Reproduced with permission from ref. 201. (g) XRD patterns of Nb2O5 particles by laser fragmentation at a specified water-to-focus distance and irradiation time. Adapted with permission from ref. 202. All insets show the corresponding SAED patterns. | ||
Although a batch setup is not favorable for increasing the productivity of LFL due to the absorption of a large amount of energy by dispersed colloidal NPs and large fluence gradients, the concurrent fragmentation behavior has a propensity to disorder the crystalline structure of the transition metal oxides. Recently, Du and coworkers showed the ns-laser fragmentation of CoNi2P particles in water to obtain amorphous CoNiPO4 NPs (Fig. 11a–c),203 where the atomic ratio was determined by inductively coupled plasma (ICP) measurements. The crystallinity was reported to be closely related to pulse energy. The amorphization tends to be achieved at a higher pulse energy (Fig. 11d). This fact further verifies the previous statement in the discussion of the laser wavelength effect in water that the light absorption efficiency of colloidal NPs favors the transformation of amorphization. Additional metalloid P seems to improve the amorphization of NPs during heating and quenching processes by fragmentation, which is in good agreement with the improvement of GFA in MG formation, but more importantly, the fragmentation seems to present a more effective way to produce the amorphous metal oxide NPs than the laser ablation in water.
 | ||
| Fig. 11 Laser fragmentation in water to synthesize amorphous multi-component metal oxide NPs. (a) TEM and (b) HRTEM images of amorphous CoNiPO4 NPs; (c) corresponding elemental mapping including Ni, P, Co and O; (d) the XRD patterns of CoNi2P particles before fragmentation and laser-fragmented CoNiPO4 NPs under different pulsed energies: 369, 480 and 541 mJ. Reproduced with permission from ref. 203. | ||
In comparison, since the ultrashort lasers have distinct fragmentation mechanisms compared to the ns laser, they may not be the perfect choice for the amorphization of NPs. For example, Waag et al.204 showed the synthesis of ultrasmall amorphous NPs and multi-crystalline particles up to 200 nm by ps-laser fragmentation of CoFe2O4 particles in water, and Siebeneicher et al.205 using ps-laser fragmentation of bismuth ferrite particles suggested that the large particles dominated the crystalline characteristics in the XRD pattern although small amorphous NPs were observed in TEM images. Since the pulse duration of ps locates between ns and fs, those investigations of the ps-laser might be involved with part of photothermal processes leading to melting processes during fragmentation. Also, the fragmentation duration was less than that of the batch setup since the free liquid jet was used to make sure that all particles went through the same number of laser pulses. In this setup, the educt particles pass through the capillary and are hit by the laser beam in a perpendicular direction by adjusting the focal spot. Repeating processes by multiple passages through the liquid jet promise the effective size reduction and homogeneous distribution of NPs. In this way, it is possible to estimate the fragmentation efficiency using “mass-specific energy input”.165
Overall, ns-LFL may be a promising tool to create A-NPs, in particular in organic solvents for in situ carbon (or even nitrogen/phosphor) doping, or in water for the creation of amorphous metal oxide NPs. However, there is still a lack of evidence that LFL is capable of synthesizing A-NPs. Not to mention, the fragmentation mechanisms are still under debate. Therefore, the amorphization behavior needs to be investigated in-depth with more understanding and should attract more interest in future studies. In addition, LFL can be possibly applied for the amorphization of metal carbide NPs due to the aforementioned confinement effect of the carbon shell on the suppression of nucleation by laser pulses. But one should also consider that the carbon shell will be destroyed during LFL, introducing more uncertain factors.
5. Prospect of metallic glass nanoparticles by laser ablation in liquid
As the counterpart of crystalline metals, MGs with amorphous structure have already been developed for decades. While crystalline single-element metal targets have been extensively applied for producing NPs by LAL, the investigation using bulk MGs to produce NPs based on laser synthesis and processing techniques is still at the infant stage. One of the main reasons for this discrepancy is not only the wider availability of crystalline metals, but also the multi-component element nature of MGs, which leads to sophisticated synthesis mechanisms and products. On the other hand, single-element NPs by LAL have been well established in recent years, but in fact, the synthesis of multicomponent metal NPs is attractive to effectively modify the properties of materials. However, the solidification of multicomponent crystalline alloys beyond solid-solution limits often leads to crystalline phases with different phase compositions. In the whole crystalline structure, elements are inhomogeneously distributed. In contrast, general methods based on rapid solidification to manufacture MGs have the ability to “quench” the atomic structure of the alloy as the molten state, thereby leading to uniform elemental distribution inside the target, rendering elemental mixing during LAL less demanding. This provides a favorable environment to generate multi-component metal NPs by LAL. Indeed, bulk MGs provide an excellent precursor with good GFA if the homogeneous element distribution can be achieved in produced NPs, which we denoted as MG-NPs. But currently, the preservation of the amorphous state of NPs after LAL of bulk MGs is still challenging. In other words, the MG structure of a target is not a prerequisite for MG-NP formation, as the LAL or LFL mechanism passes a molten stage and/or phase explosion anyway (so that laser synthesis does not “remember” the target's structure), but using an MG target is a straight-forward approach to ensure at least good starting conditions of pre-set elemental mixing and composition, and may benefit from changing ablation dynamics. In this section, the ablation phenomena of the MG surface and the bubble dynamics during LAL will be discussed, and we will provide the prospects for the synthesis of MG-NPs based on competitive mechanisms of structural changes during LAL.5.1. Surface characteristics by laser ablation
The disordered nature of MGs determines that their interaction with a pulsed laser is different from that of crystalline materials due to their structural difference, where the observation of surface morphologies usually provides important clues for the ablation mechanisms, e.g. the release of matter from the targets. In general, a short-pulsed laser, e.g. ns laser, induces the apparent thermal coupling ablation on the MG surface, thus leading to melting, vaporization, and boiling, but these thermal behaviors depending on the laser fluence and duration also result in the vertiginous change (e.g. supercooled liquid transformation, crystallization, energy state transformation, etc.) in MGs. Using a typical Zr-based MG (Zr41.2Ti13.8Cu12.5Ni10Be22.5, also known as Vitreloy 1), Williams and Brousseau investigated the relationship of crater topographies (diameter and depth) and laser fluence,206 where they found that the deposition of energy by high fluence contributed to the increase of crater diameter and depth on the MG surface (Fig. 12a), while in the ns regime, the shorter pulse duration with higher peak power resulted in a larger melting pool and more apparent melt ejection at the edge of the crater (Fig. 12b).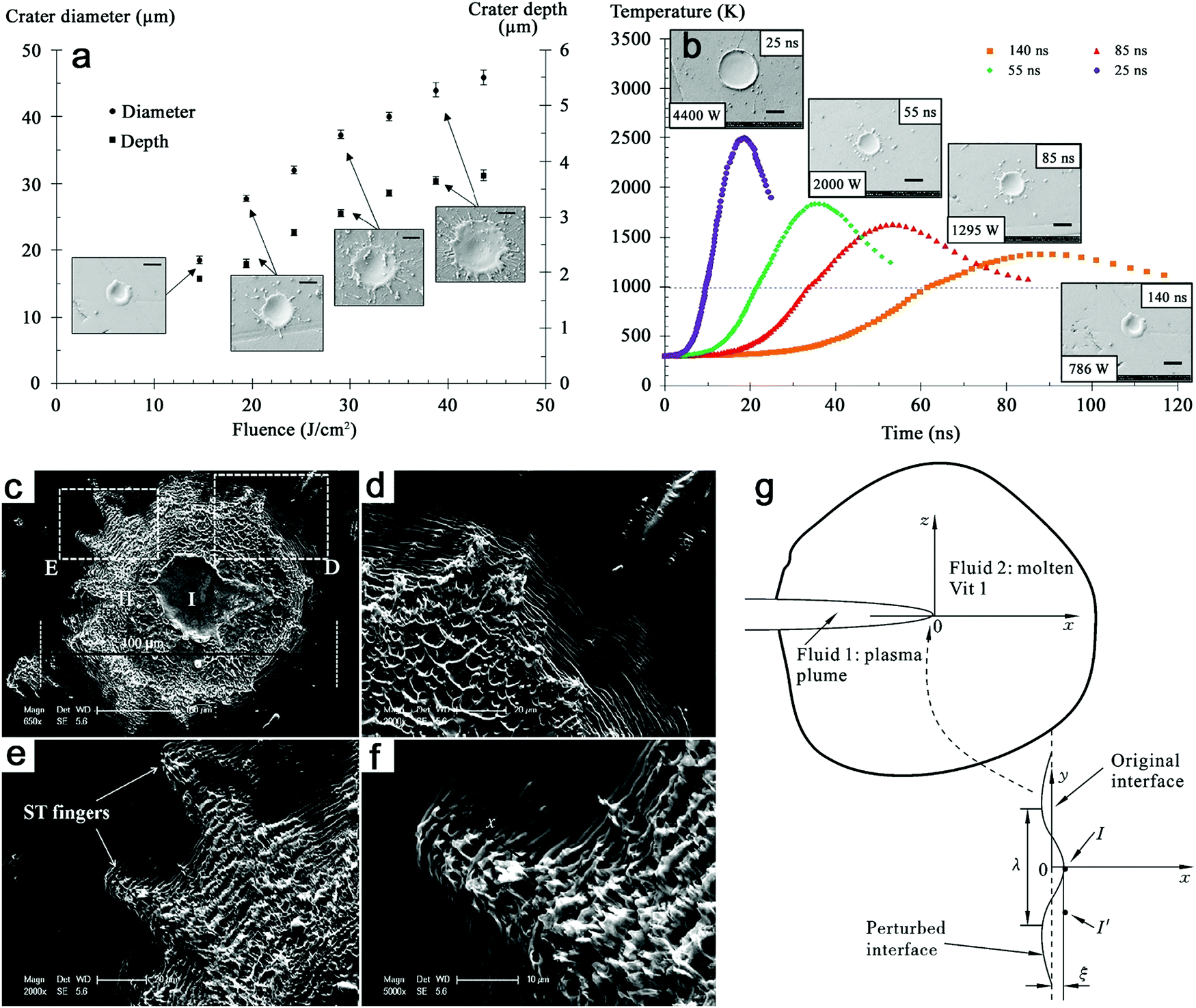 | ||
| Fig. 12 Surface characteristics of Zr-based MG after laser ablation. (a) Crater diameter and depth as a function of the fluence for a 140 ns pulse. Scale bars in SEM images: 20 μm. (b) Predicted temporal temperature variation at a fluence of 14 J cm−2 for pulse durations of 140 ns, 85 ns, 55 ns, and 25 ns. Scale bars in SEM images: 20 μm. Reproduced with permission from ref. 206. (c–f) Surface morphologies of ns-laser ablation of Vitreloy 1 in water showing surface ripples and Saffman–Taylor fingering. (g) Development of Saffman-Taylor fingering instability at a meniscus-shaped interface between the plasma plume and the molten Vitreloy 1. Adapted with permission from ref. 207. | ||
On the other hand, surface ripples are typical surface morphologies outer the carter caused by single-pulse ablation,206,208–210 where Liu et al.210 ascribed them to Kelvin–Helmholtz instability due to the interaction of the molten layer and the expanding plasma plume. The spacing of ripples could be closely related to the surface tension of the molten layer, the density, and transverse velocity of the plasma wind.210 Since MGs are usually regarded as the “frozen” liquid, the hydrodynamic instability tends to easily occur by thermal activation of the ns-laser, leaving a trace of fluid-like morphologies after the laser effect. However, such behaviors may not be consistent in the liquid environment. As such, Liu et al.207 further performed the laser ablation of Vitreloy 1 in water and found another hydrodynamic instability which was the Saffman-Taylor fingering instability phenomenon. In Fig. 12c, the central smooth region and the surrounding rough region were marked as “I” and “II”, respectively, with a starfish-shaped boundary (Fig. 12e and f). Clear surface ripples were still observed and there were melted droplets and pores in the “II” region (Fig. 12d) due to phase explosion at a temperature rise rate over 109 K s−1, indicating that the interaction of MGs with the laser under water was much more aggravated.207 Further study of Saffman-Taylor fingering instability was verified by establishing the model in an Eulerian coordinate system (x, y, z). They proposed that the flow of more viscous Vitreloy 1 liquid was initiated by the less viscous plasma plume, and according to the analysis of perturbation at the interface of two fluids, the critical wavelength (λc) satisfied the perturbation in Saffman–Taylor fingering:207
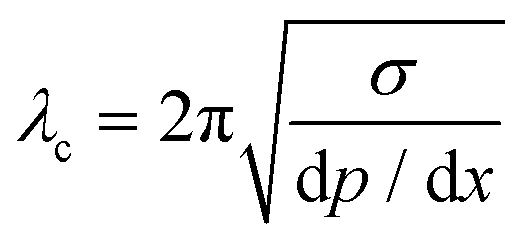 | (1) |
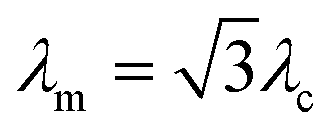 ) was consistent with the width of the experimental fingering.
) was consistent with the width of the experimental fingering.
Regarding the ultrashort pulses, Jia et al.211 have reported that single-pulse fs laser ablation of Fe73.5Cu1Nb3Si13.5B9 MG resulted in non-thermal (direct solid–vapor transition) and thermal ablation (heating and structural transformation), which was related to the incident laser fluence. The low fluence fs laser also contributed to surface ripples structure on the MG surface, which is in agreement with other reports by ns ablation. In an investigation by different laser parameters, Ma et al.212 studied the formation of concentric rings Zr55Al10Ni5Cu30 MG by multi-pulse fs lasers, suggesting that type I ring structure with nonthermal features occurred at low laser fluence due to the small laser accumulation effect, while type II ring structure with evident molten traces was found at high laser fluence. But unfortunately, surface structural changes of MG ablation by an ultrashort pulsed laser under water or other liquids are still missing.
5.2. Matter-removal and competitive mechanism
The phenomena of hydrodynamic instability have explained the melting behavior of MGs and viscous liquid dynamics under confinement in water, but ex situ surface morphologies still lack direct observation for the analysis of the ablation mechanism. In fact, pulsed laser ablation of MGs should involve vaporization and boiling of the melt. It is generally accepted that ultrashort laser pulses can induce an effective ablation in liquid by nearly instantaneous evaporation,213 where laser duration regimes from ps to fs are more favorable for the explosive boiling behavior, which is also known as phase explosion since the longer pulse duration has more apparent heat conduction within targets and plasma shielding effects lowering the ablation efficiency. However, high fluence laser ablation is also able to transfer the normal melting and vaporization into phase explosion. Yoo et al.214,215 observed the phase explosion of silicon by the dramatic increase of craters in volume when a threshold irradiance of 2.2 × 1010 W cm−2 was used in the ns-laser ablation; Bulgakova and Bulgakov indicated the dramatic increase of ablation rates of graphite, niobium and YBa2Cu3O7−δ superconductors over the threshold fluences (22, 15 and 17.5 J cm−2, respectively) using infrared ns pulses.216It is also interesting to verify whether MGs will have a different phenomenon from crystalline materials. Recently, Jiang et al.208 reported the superheating and phase explosion by ns-laser ablation of Zr-based MG at high laser fluences. In their case, the phase explosion was captured by a high-speed camera with the emission of high-temperature matter as bright sparks, where they ruled out the possibility of plasma and the shock wave by the spatiotemporal resolution of the camera.208 In addition, the formation of a liquid-gas spinodal pattern has been observed on the irradiated area. According to their modified thermal model, Fig. 13a shows the spatiotemporal profile related to MG surface temperature, where the highest temperature always occurs at the outermost surface and the temperature decreases from the surface toward the inside of the MG. Fig. 13b suggests that the ablation of MG by a single-pulse laser at high fluence causes normal heating within the pulse duration (10 ns) until the explosive boiling occurs at T1. Since the pressure at the explosive boiling of Zr-based MG is much less compared to that of crystalline metals (i.e. Ni), it is expected that MG is easier to be heated to form a highly metastable liquid with a higher degree of superheating.208 Note that the thermal conductivity of amorphous and supercooled liquid states is generally lower than that of the crystalline counterparts due to the restricted mean free path of phonons in the disordered atomic structure,206 possibly leading to (stronger) local superheating at the short laser pulses.
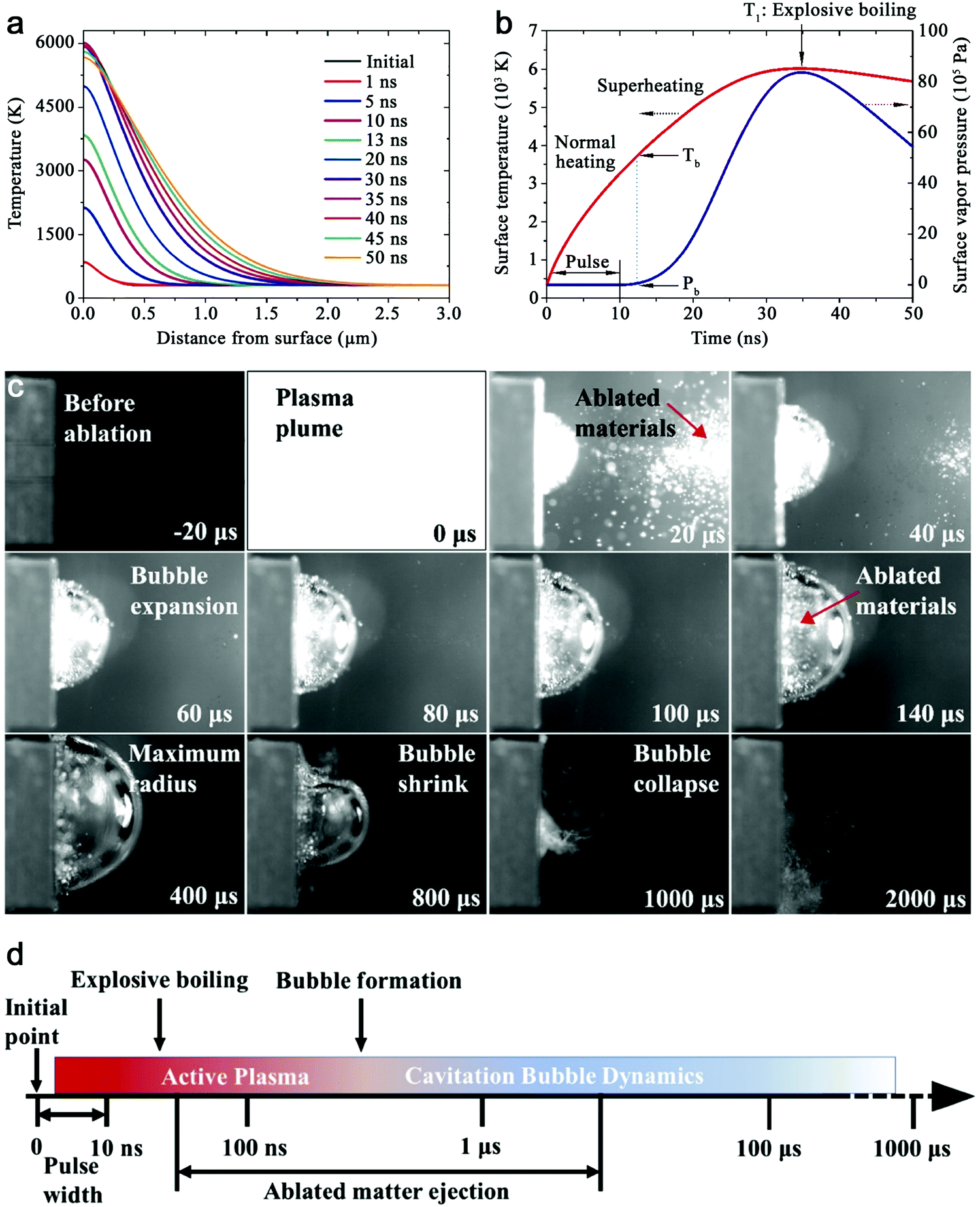 | ||
| Fig. 13 Phase explosion and bubble dynamics of MGs by ns-LAL. (a) The temperature profile as a function of distance from the surface with time evolution; (b) the surface temperature and vapor pressure as a function of time. Adapted with permission from ref. 208. (c) Snapshots of bubble evolution after a single-shot nanosecond laser ablation of the Vitreloy 1 target in water; (d) illustration of the main stage during LAL. Reproduced with permission from ref. 220. | ||
In a high time-resolved imaging study of Zr-based MG, Jiang and coworkers had further captured the formation and quenching process of laser-induced plasma in water.217 The observed plasma on the MG surface firstly expanded but was limited by the constraint of water within 10 ns, and then cooled down with the trace at around 70 ns. Given that the laser irradiance was about 3.7 × 109 W cm−2, that is 42–127 times lower than the breakdown threshold of water by infrared ns-laser pulses,218 the plasma at the target surface was likely induced by free-electron formation from ablated materials. At the same time, a peak pressure of 1.5 GPa by deposited energy into water is generally enough for explosive vaporization of water, leading to the formation of cavitation bubbles during or at the end of 10 ns laser pulse. In this case, the delayed bubble formation at ∼500 ns may indicate that the laser energy is mainly deposited into the MG target and plasma luminescence in water is caused by the ejection of electrons. The application of a single-pulse ns-laser on Vitreloy 1 led to the ejection of bright objects (bubbles or droplets) from the MG surface at the time frame of tens of μs. It is known that phase explosion of ablated matter can occur in the confined water environment.219,220 This may also lead to the conclusion that compared to crystalline materials, MGs are likely to experience superheating and phase explosion under laser ablation.208
Other works from the same group have also shown the evolution of cavitation bubbles after a single pulse ns laser.219,220Fig. 13c shows that the bubble generated from ns-laser ablation of Vitreloy 1 expands for a duration of around 400 μs, then shrinks and finally collapses at the time scale of about 1000 μs. The experimental result is reported to approximately match with the temporal variation of the bubble radius using conventional Rayleigh–Plesset theory.219 The first oscillation of the bubble generally fits the Rayleigh–Plesset equation. However, it has been reported that the Gilmore model can describe the subsequent oscillations better with consideration of liquid compressibility.221 Further consideration (e.g. vaporization, condensation, and heat conduction) is also possible but the full model will be more complicated.221Fig. 13d describes the main stages of ablation (including plasma and bubble evolution, and explosive boiling) at a time scale of 1000 μs, where Jiang et al. postulated the phase explosion to start before the bubble nucleation, and therefore part of the ablated matter is thought to be ejected directly into the colder water, which provides part of the NP formation mechanism for the ablation of bulk MGs. According to Jiang and coworkers,220 the ablation of Vitreloy 1 in water led to the formation of a fraction of small amorphous NPs (Fig. 14a) without forming crystals (SAED not shown here), while the others mainly presented as core–shell structures. The core–shell structured NPs could be divided into crystalline@amorphous core–shell type and polycrystalline core–shell type. Fig. 14a shows that Zr, Ti, Cu, Ni, and O are homogeneously distributed in the whole amorphous NP. For core–shell structures (Fig. 14b and c), it is evidenced that both core–shell structures are favorable for the elemental segregation of Cu and Ni as the crystalline core due to positive mixing enthalpy,222 which is detrimental to the GFA of MGs. Note that due to a synthesis environment in water, all NPs have been oxidized.
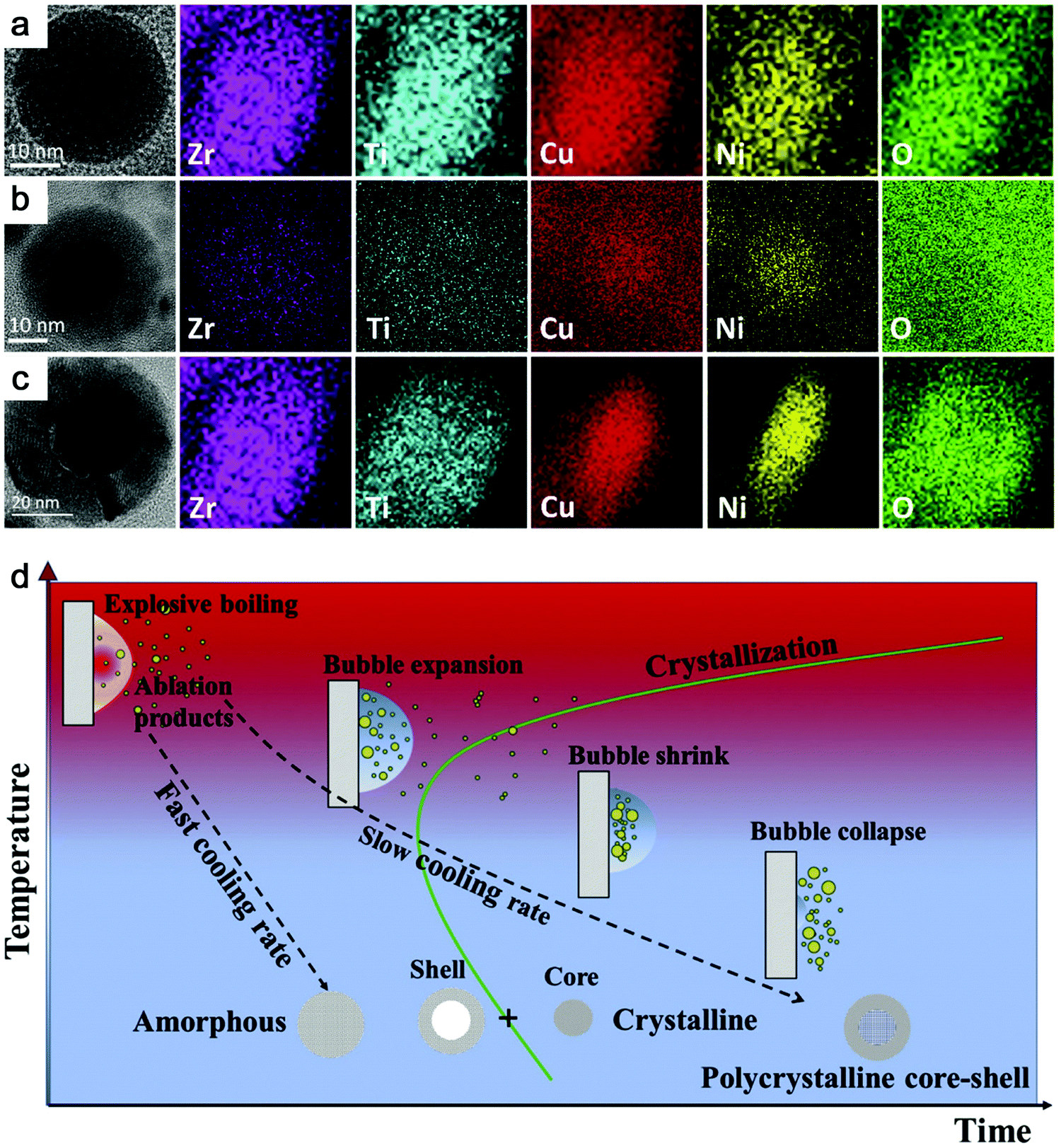 | ||
| Fig. 14 Formation mechanism of NPs during LAL of MG. HRTEM with the corresponding elemental mapping (Zr, Ti, Cu, Ni, and O) of (a) an amorphous NP, (b) a crystalline@amorphous core–shell NP and (c) a polycrystalline core–shell NP; (d) TTT diagram showing the relationship of cooling rate and formation of the NP structure. Reproduced with permission from ref. 220. | ||
Based on aforementioned different NP structures, Fig. 14d shows a time-temperature-transformation (TTT) diagram of MG-NP formation during LAL of the MG target.220 The laser energy absorption induces the atomization and ionization of the target but those active species are confined in the hot plasma plume, leading to the formation of homogeneously mixed atoms. According to the authors, the preservation of stoichiometry in the NPs most likely occurs due to the direct ejection of ablated matter by phase explosion, thus contributing to the glass-formation condition. Both factors may compensate for the side effect of oxidation, but the high cooling rate generally dominates the bypasses of the crystallization “nose” in the TTT curve. However, trapping of most of the ablated matter in the cavitation bubble is unavoidable. In a consecutive process, the cavitation bubble can survive in a microsecond timescale, which is enough for the nucleation and crystallization of NPs. Therefore, it is believed that the NPs formed in the bubble before releasing into liquid have the propensity to form a crystalline structure unless the GFA of NPs is enough to suppress the crystallization.
Since a multicomponent target in water is usually involved with complex chemical reactions, the current study is still not sufficient to reveal a general rule of the formation of MG-NPs and their competitive mechanism by the ablation of the MG target. Many other influencing factors for the formation mechanism of MG-NPs by LAL have not been studied and this would be a novel and promising area regarding NP formation by laser synthesis and target processing in liquid.
6. Properties and applications of amorphous metal nanoparticles
6.1. Heterogeneous catalysis
Currently, many investigations have focused on the application of laser-synthesized NPs as heterogeneous catalysts,223–226 due to the enrichment of catalytic active sites on the nanometer scale.227,228 In addition, amorphous alloy ribbons have also presented an attractive catalytic performance in water splitting,114,229,230 wastewater treatment,231–235 fuel cells,236,237etc., due to the ready-to-use nature with low activation energy,238,239 fast electron transfer,240,241 and abundant coordinately unsaturated atoms242,243 in the amorphous structure. Therefore, the lowering the dimension of amorphous metallic materials into NP size holds the promise to present outstanding catalytic behavior.Recently, the exploitation of ultrasmall amorphous metal oxides has been achieved by Yang and coworkers by the long-time fragmentation of the crystalline counterparts in water, where the amorphous metal oxides were subsequently employed as efficient photocatalysts for water splitting, namely solar hydrogen evolution.82,200,201 In the typical case of Ni oxide, amorphous NiO nanosheets obtained from the long-time fragmentation of crystalline NiO powders have achieved 52.02 μmol h−1 of H2 evolution in a water/methanol (as a hole scavenger) solution by simulated solar irradiation and this was increased to 1008.46 μmol h−1 by H doping on Ni atoms (Fig. 15a and b), which formed the 2D plasmonic amorphous NiO nanosheets.201 The investigation of the electronic structure found that the 2D effect and amorphization of NiO have the ability to enhance the electron doping with the compensation of intrinsic hole doping and to suppress the carrier recombination with the transformation of surface trapping sites from recombination centers to active sites.201Fig. 15d schematically illustrates the elevation of Fermi levels (Ef) by the 2D effect, amorphization and H doping in sequence to reach conduction band edge. In fact, such H doping of amorphous NiO nanosheets boosts the subsequent photocatalytic activity originating from the surface plasmon resonance (SPR) effect with high electron density (>1021 cm−3).201
 | ||
| Fig. 15 Catalytic application of laser-synthesized amorphous metal nanoparticles. Photocatalytic H2 evolution by (a) amorphous NiO nanosheets and (b) plasmonic amorphous NiO nanosheets as a function of time. Reproduced with permission from ref. 201. (c) Band structure diagram of amorphous Fe2O3 NPs (AFNs) and crystalline Fe2O3 powders (CMNs). Reproduced with permission from ref. 82. (d) Schematic illustration of band structure transformation by the 2D effect, amorphization, and H doping. Reproduced with permission from ref. 201. (e) Schematic illustration of Z-scheme photocatalytic overall water splitting by amorphous CoO nanosheet/graphene oxide. Reproduced with permission from ref. 200. Comparison of (f) overpotential values at 10 mA cm−2 and (g) Tafel plots of amorphous CoNiPO4 (L) by LFL, crystalline CoNiPO4 (E) by electrochemical oxidation, crystalline CoNi2P, and commercial RuO2 and IrO2. Adapted with permission from ref. 203. | ||
In a similar case but using Fe oxide, the application of nanoscaling by the fragmentation of large Fe2O3 particles into nano-scale Fe2O3 NPs led to more surface states and dangling bonds, which was further enhanced by the amorphization of NPs by repetitive melting and quenching processes in water.82 The amorphization can be considered as a further nanoscaling due to the disordering of the atomic structure. As a result, the up-shift of the conduction band edge over the H+/H2 potential level and the elevation of the Fermi level by additional electrons contributed to thermodynamically driven photocatalytic H2 evolution by amorphous Fe2O3 NPs (Fig. 15c).82 In general, the excitation-wavelength-dependent small polaron trapping effect of photoexcited carriers has been reported in the crystalline Fe2O3, leading to a lower photoconversion efficiency than the theoretical value.244 But the amorphization can overcome this effect, which increases the photoconversion efficiency in the visible light region, thereby achieving a higher utilization of solar irradiation. Therefore, the amorphization in metal oxides could serve as a unique function in the photocatalytic application. The coordination number of the metal–oxygen bond could also serve as a crucial factor for the superior properties of amorphous metal oxides.245 This could further demonstrate the amorphization function of oxide nanomaterials from the fragmentation of crystalline counterparts. Besides, laser-synthesized amorphous CoO nanosheets have shown efficient overall water splitting with an expected 2:1 stoichiometry by solar irradiation.200 More importantly, robust stability with continuous H2 and O2 generation of more than 120 h was presented due to the amorphization of CoO materials, and the photocatalytic activity was further promoted by a factor of 13 through the decoration of 20% graphene oxide due to the bionic Z-scheme structure (Fig. 15e), which enhanced the light absorption in both visible and near-infrared regions. This provides the new idea of incorporating amorphization and decoration to form amorphous/crystalline composites, which can further modify their unique properties in catalysis.
All these amorphous transition metal oxide nanomaterials have advantages of a very high surface-to-volume ratio and abundant coordinately unsaturated atoms, which in fact are potentially active in many other catalytic applications, such as wastewater treatment. Li et al.199 have reported the superior adsorption ability of amorphous Fe, Co, and Ni oxides by LFL in the treatment of methyl blue. The comparison study indicated that crystalline NiO needed more than 150 min for full colorization while amorphous NiO achieved that within 1 min. In fact, in addition to physical adsorption, the abundant catalytic active sites in the amorphous oxides will also induce the strong reduction of the dye leading to its fast decomposition. On the other hand, the employment of multiple elements for tailoring the electronic structure of A-NPs is expected to promote their catalytic performance. By using amorphous CoNiPO4 NPs from LFL, Sun et al.203 showed a low overpotential of 238 mV at 10 mA cm−2 (Fig. 15f) and a Tafel slope of 46 mV dec−1 in the OER performance (Fig. 15g), which were much lower than those of the electrochemically active commercial RuO2 and IrO2. They ascribed it to the existence of abundant high-valence Ni3+ in amorphous CoNiPO4 NPs.203 But it may also include the tailoring effect of the electronic structure by the P element, which has been widely reported for MGs in the studies of HER and OER.98,246
Regarding the carbon shell structure in the NPs, Kim et al.177 investigated the Pd@C core–shell structure for the reduction of nitrobenzene to aniline, where the carbon layer can facilitate hydrogenation reactions. So it is expected that the carbon-encapsulated A-NPs have the potential to be exploited as hydrogenation catalysts as well. But currently, not many amorphous metal carbides synthesized by LAL or LFL are reported in the catalytic applications.
6.2. Magnetism
Magnetic nanoparticles including elemental metals, metal oxides, and carbides are available for a wide range of applications in magnetic devices such as data storage and magnetic field sensors,170,247 and in bio-applications such as selective drug delivery and magnetic resonance imaging.248,249 Particularly, extensive interest has been focused on iron due to its easy availability and magnetic nature. For iron oxide NPs, a size range <10 nm is suitable for contrast agents in magnetic resonance imaging due to superparamagnetic behavior and good dispersion in the hematic flux, while a size range between 30 and 40 nm can be effectively applied for magnetic hyperthermia by multiple magnetic heating mechanisms.150 The size of 20 nm is suitable for both applications. As such, LAL and LFL with easy regulation of NP size and structure could serve as effective methods to modify and optimize magnetic properties of iron oxide NPs. According to the products from laser ablation of the α-Fe2O3 target in different liquids, amorphous iron oxides/hydroxides (in water) have a lower saturation magnetization (Ms) than carbon-encapsulated γ-Fe2O3 (in ethanol and acetone).153 This may be attributed to the effects of ultrasmall amorphous hydroxides and the high dispersity of NPs. In this case, Amendola et al.150 have used dilute HCl and disodium ethylenediaminetetraacetate (EDTA) to remove amorphous Fe hydroxides after LAL to increase the entire crystallinity, and the magnetic properties of the resulting products were investigated.In contrast, the formation of an amorphous Fe core in core–shell oxide NPs may have an apparent effect on the enhancement of Ms, where soft ferromagnetic amorphous Fe particles with Ms > 100 emu g−1 were compared with the smaller magnetization of α-Fe2O3 (4–5 nm) of 1 emu g−1.147,250 It has been reported that the coercivity (Hc) of large amorphous Fe@α-Fe2O3 core–shell NPs matched well with the reported Hc of amorphous Fe particles with 160 Oe at 5 K,147,250 while the Ms with 72.5 emu g−1 and 61.9 emu g−1 at 5 K and 300 K, respectively, mainly originated from amorphous cores.147 The lower Ms in amorphous Fe@α-Fe2O3 compared to amorphous elemental Fe is due to the antiferromagnetic nature of α-Fe2O3 inducing interfacial magnetic interactions and reducing the Ms.
On the other hand, the magnetism of A-NPs may be applicable as well when metal–carbon bonds are formed. Kanitz et al.171 reported that fs-laser ablation of the Fe target in methanol, ethanol, acetone, and toluene produced nanocomposites (i.e. amorphous iron, iron-oxides, and carbides) with Ms = 80, 60, 67 and 14 emu g−1 and Hc = 92, 65, 56 and 52 Oe, respectively. It is known that ferromagnetic particles give a higher Ms with decreasing NP size but this is inversed for the oxide compound.171 Given that the higher carbon to oxygen ratio of organic solvents increases the carbonization rate of entire products and vice versa, the higher Ms value in methanol is mainly attributed to the higher content of iron oxide NPs with larger NPs. But the high amount of precipitated carbon in toluene significantly reduces the magnetic effect of amorphous iron-carbide NPs. In addition, Kwong et al.85 have suggested superparamagnetic (SPM) behavior and ferromagnetic (FM) property of amorphous Co–C NPs at different temperatures, and the superparamagnetic property of metal carbide NPs could be well preserved by the graphitic shell due to the less oxidation. This superparamagnetic phenomenon of amorphous metal carbides with a carbon shell was further explained by Huh and Nakajima.180 In Fig. 16a–d, the magnetic hysteresis loops and zero-field-cooling/field-cooling (ZFC/FC) curves at Hc = 100 Oe indicate the ferromagnetism of amorphous Fe–C and Co–C NPs at room temperature and no blocking temperature below 300 K. Fig. 16e–h also shows the temperature dependence of Ms and Hc, which is different from amorphous Fe–C and Co–C without a carbon shell.251 A sharp increase of Ms and Hc at a temperature lower than 20–30 K was attributed to the superparamagnetic behavior of carbon-encapsulated amorphous NPs originating from a large increase of magnetic spin moments below the critical temperature. In Fig. 16i, a magnetic core–shell structure with carbon onion and SPM shell and FM core has been proposed to explain the anomalous magnetic behavior of amorphous Fe–C and Co–C NPs with a carbon shell at low temperatures. Accordingly, Huh and Nakajima indicated that the amorphization of Fe–C and Co–C NPs encapsulated by carbon onions would show some different features compared to common magnetic NPs, including (1) the protected surface spins, (2) the separated amorphous magnetic cores by the carbon onions, and (3) the disordered spins originating from the disordered atomic structure.180
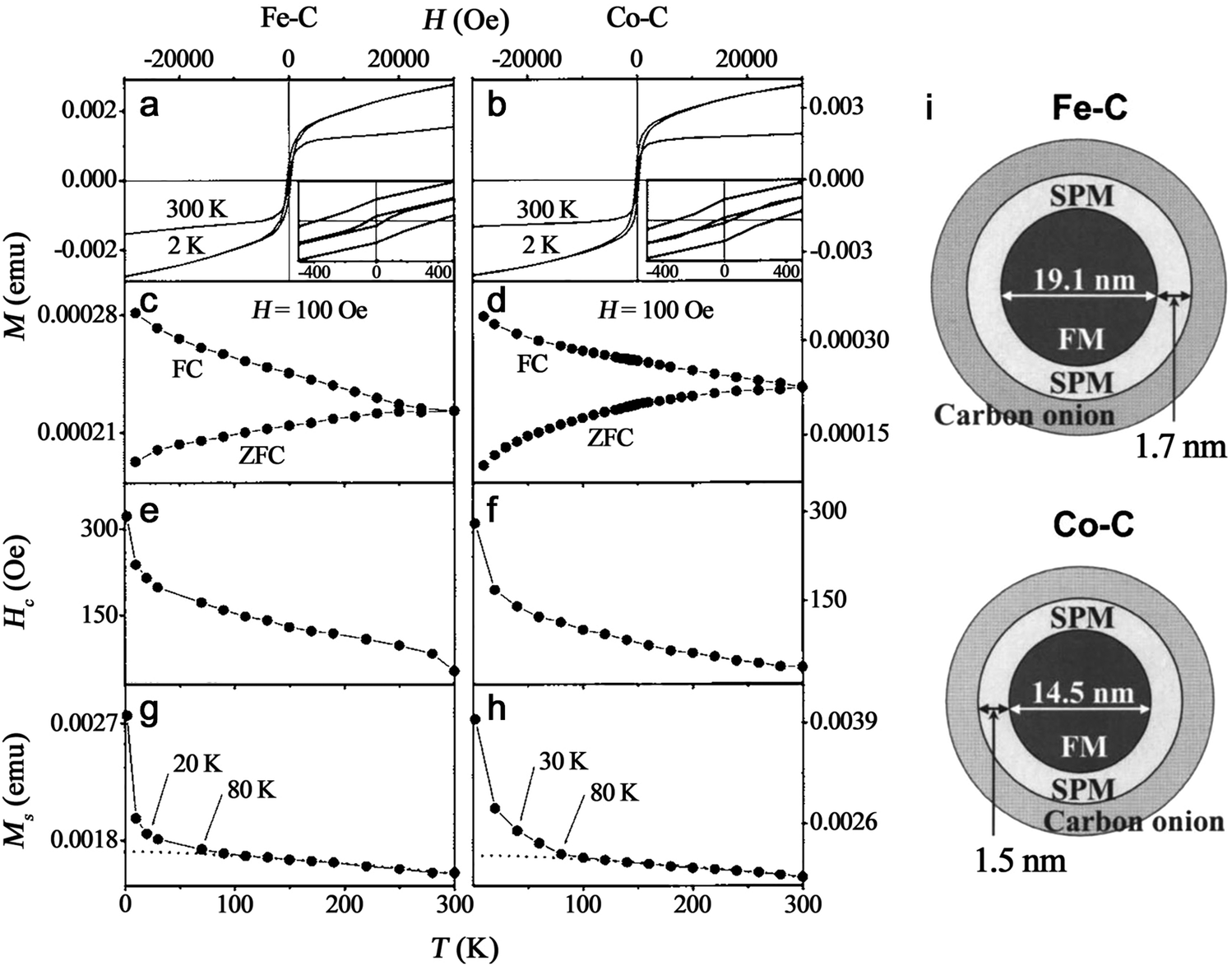 | ||
| Fig. 16 Magnetic properties of laser-generated amorphous metal carbide NPs. The hysteresis loops at 300 and 2 K of (a) Fe–C and (b) Co–C; zero-field-cooling/field-cooling (ZFC/FC) curves at H = 100 Oe of (c) Fe–C and (d) Co–C; coercivity (Hc) and saturation magnetization (Ms) as a function of temperature of (e and g) Fe–C and (f and h) Co–C. (i) The proposed magnetic core–shell structure for amorphous Fe–C and Co–C NPs. Adapted with permission from ref. 180. | ||
6.3. Other applications
Amorphous Fe NPs with excellent electromagnetic and catalytic properties have been found with the application in cancer therapy by a localized Fenton reaction by taking advantage of mild acidity and the overproduced H2O2 in a tumor microenvironment, which mainly relied on the local production of ˙OH by the on-demand release of Fe ions without the need for external energy input.248 The amorphization of Fe NPs also promotes their biodegradability and magnetism-related properties, leading to a prospective contrast agent for magnetic resonance imaging.248 Since the amorphous Fe NPs in Zhang et al.'s study were based on the hubble-bubble reduction process with involvement of surfactants, the surfactant-free amorphous Fe NPs by laser synthesis and processing in water are expected to provide a more suitable alternative due to the ultrapure requirement for bioapplications. Laser-generated amorphous/crystalline Ni60Nb40 nanocomposites have also been investigated with respect to the electrochemical nonenzymatic glucose-sensing ability in the alkaline medium.252 In fact, compared with other reports of amorphous metallic materials, the applications of laser-synthesized NPs are still very limited. But the comparable applications of amorphous metal oxide NPs from hydrothermal, pyrolysis, etc., and MG-NPs from chemical reductions have attracted interest in hydrogenation,32 hydrogenolysis,19 oxidation,6,253 supercapacitors,119 batteries, and water splitting.245 Therefore, the combination of nanoscaling and amorphization with surfactant-free nature by laser synthesis and processing should contribute to a great promise in different fields.7. Conclusions and prospects
The unique short-range ordered and long-range disordered atomic arrangement of amorphous materials leads to an excellent combination of structure and activity properties that overcomes the natural limitation of the crystalline counterparts. Extensive efforts have been devoted to developing and improving synthesis methods of amorphous materials based on hypothesized formation mechanisms. A typical approach of different amorphization processes is to suppress the crystal nucleation by employing a large temperature gradient and a fast quenching of molten metals within a short time. Currently, chemical reduction is still the main approach to achieve colloidal amorphous metal nanoparticles (A-NPs) with a large specific surface area. However, the severe contamination by surfactants from chemical reduction usually needs further calcination treatment to remove those residuals, which easily leads to structural relaxation and further crystallization of the amorphous structure. The synthesis of surfactant-free A-NPs, particularly for catalytic applications, thus encounters new challenges and requirements.Laser synthesis and processing of colloids (LSPC) has widely been used to produce colloidal crystalline NPs. Extensive investigations of their formation mechanisms and the related thermodynamics and kinetics in the liquid, phase transition, crystal nucleation, and growth were correlated to the laser-induced plasma and cavitation bubble dynamics. Yet, although also A-NPs have been synthesized in the past, the formation mechanism and related synthesis conditions are less understood. Hence, in this perspective article, we addressed previous work on laser synthesis and processing of A-NPs from the last two decades to identify related synthesis conditions and formation mechanisms.
Amorphous metal oxide NPs tend to be formed in a liquid environment enriched with oxygen when a high cooling condition is satisfied. Based on the literature survey, the oxygen “impurity” appears to be beneficial for or participates in the formation of amorphous oxide NPs during laser ablation in water. On the one hand, ns pulse duration is preferable for targets with limited elemental mixing. On the other hand, ultrashort-pulsed LAL may trigger stronger acceleration of the plume leading to jetting (and thereby stronger quenching) phenomena. Either the direct ejection of molten metals into the cold and high-density region of water or the ejection of matter into the mixing zone at the front of the expanding plume may provide sufficient quenching or oxidation condition for amorphization. However, for the matter trapped inside the cavitation bubble, the cavitation period may support the subsequent nucleation and growth of crystalline phases, at least until the bubble's collapse. During the collapse phase, high peak temperatures and pressures may again provide good starting conditions for structural changes in the nanoparticles (in the subsequent fast cooling process during release from the collapsing bubble). Multiple pulse LAL is always accompanied by LFL processes, but LFL can also be employed intendedly either on LAL-generated colloids or on other particle suspensions. The fragmentation by fs- and ps-laser pulses can provide repetitive melting and quenching processes in water and hence control the amount of amorphization with the number of laser pulses and related fluence. The significant reduction of particle size is also likely to be beneficial for amorphization, and UV LFL seems to be advantageous to yield amorphous NPs. In fact, amorphous metal oxides gained by LFL have been employed in several studies on HER and OER for water splitting, and oxygen reduction reactions (ORR) for fuel cells and supercapacitors, holding the promise of surfactant-free and ultrapure amorphous metal oxide NPs from LSPC as superior candidates compared to those from other synthesis methods.
In addition to water, organic solvents provide more choices and advantages (higher reductive potential, delivery of C, N, or P dopants to stabilize MG) as liquid medium during LAL. The enriched C environment originating from decomposition of organic solvents easily forms the carbon shell of NPs to stabilize NPs. The involvement of metalloid C satisfies the empirical rules of glass formation, where metalloid elements located at ∼20 at% induce a good glass-forming ability of metallic materials. For LAL, the simultaneous diffusion of C atoms into the metallic matrix (metal core) depends on their affinity and enthalpy to form metal–carbon bonds and largely increases the mobility of the metallic liquid, thereby supporting and promoting the amorphization ability of NPs during the quenching process. An influence can be also considered for the confinement effect of the carbon shell, which suppresses the recrystallization and stabilizes the disordered structure of the metal core. As such, regardless of the quenching rate in different liquids, the organic solvents would be more favorable for the formation of an amorphous structure due to the cooperation of C.
In general, the formation of MG-NPs can be considered as a competitive process of nucleation and amorphization. To achieve a higher fraction of the amorphous structure in the whole product, the nucleation should be suppressed to a minimum. According to the preliminary discussion of amorphous single-metal oxides and carbides by LSPC, we have summarized the key factors that affect the amorphization of MG-NPs (Fig. 17). These include, firstly, the cooling rate of the metallic liquid, where laser-generated NPs in liquids have the advantage of a high cooling rate. Secondly, the chemical composition generally dominates the critical cooling rate of MG, which results in the feasibility to form an amorphous structure under the same cooling conditions (GFA). Thus, applying a MG bulk target with a low critical cooling rate (or a high GFA) is expected to increase the chance of MG-NP product during LAL. Thirdly, the diffusion effect and chemical reaction of O and C atoms are important, where the carbon diffusion into the metallic matrix due to the decomposition of organic solvents could highly improve the GFA, while electronic properties of amorphous metal oxide NPs can be tailored by the cooperation of oxygen (mainly in water). Fourthly, preserving the target stoichiometry could maintain the GFA as in the MG target, which is feasible to obtain MG-NPs by the high cooling rate under LSPC. It has been demonstrated that LSPC can “memorize” both MGs220 and HEA126,134 target composition to synthesize NPs with the same composition. Further consideration could focus on the laser parameters (particularly the effects of laser wavelength and pulse duration) according to the current literature.
 | ||
| Fig. 17 Schematic illustration of decisive factors for amorphization of MG-NPs from LAL. | ||
Consequently, despite the progress of amorphous single-metal oxide and carbide NPs by LSPC, there are still many challenges that need to be addressed. Based on the previous list of hypotheses, general rules and mechanisms to promote the chance of A-NPs by LAL and LFL need to be developed. Given that the constituent elements and compositions largely dominate the properties and applications, future studies are expected to synthesize metallic glass nanoparticles (MG-NGs) with two or more oxygen-free elements by laser-based strategies. In this respect, bulk MGs and MG ribbons, which are mainly manufactured by casting and melt spinning respectively, with good glass-forming ability (GFA) provide a prominent precursor to manufacture MG-NPs by LAL which is barely addressed in the literature yet. The uniform elemental distribution in MG ensures an even distribution of atoms in the metallic liquid after ablation and before quenching in the solvents. In this way, MG targets provide an excellent model to study the formation of amorphous MG-NPs from LAL since the GFA in MGs is highly dependent on element compositions and their range. Additional elements can be pre-designed/alloyed in the bulk MGs to promote the corresponding property of MG-NPs after laser synthesis. Furthermore, MG powders in micro size could be also used in the LFL, which is an effective method to create ultrasmall NPs, and maybe ultrasmall MG-NPs, in future.
Accordingly, future studies on A-NPs by LSPC are expected to involve but not limited to (1) the effect of dominant laser parameters on the amorphization; (2) the correlation of bubble collapse dynamics with the amorphization and how to further suppress the nucleation of NPs with low GFA; (3) qualitative and quantitative analyses of the oxygen and carbon effect on the amorphization of different elements, including liquid degassing and dehydratisation; (4) the elemental segregation effect of A-NPs based on different target compositions; (5) the regulation of properties of A-NPs by e.g. targets with different molar fractions and atomic doping via liquid molecules; and (6) the exploitation of applications of surfactant-free A-NPs.
In conclusion, we believe that laser-generated ultrapure MG-NPs could serve as the yet overseen counterpart of crystalline nanoparticles, with promising properties, which opens a new window for the novel design of nanomaterials and substituting conventional crystalline NPs in different application fields.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
S. X. L. is grateful for the financial support by the Alexander von Humboldt Foundation Research Fellowship for postdoctoral researchers.References
- H. W. Sheng, W. K. Luo, F. M. Alamgir, J. M. Bai and E. Ma, Nature, 2006, 439, 419–425 CrossRef CAS PubMed.
- S. X. Liang, Z. Jia, Y. J. Liu, W. Zhang, W. Wang, J. Lu and L. C. Zhang, Adv. Mater., 2018, 30, 1802764 CrossRef PubMed.
- S. X. Liang, Z. Jia, W. C. Zhang, W. M. Wang and L. C. Zhang, Mater. Des., 2017, 119, 244–253 CrossRef CAS.
- L. C. Zhang, Z. Jia, F. Lyu, S. X. Liang and J. Lu, Prog. Mater. Sci., 2019, 105, 100576 CrossRef CAS.
- J. Q. Wang, Y. H. Liu, M. W. Chen, G. Q. Xie, D. V. Louzguine-Luzgin, A. Inoue and J. H. Perepezko, Adv. Funct. Mater., 2012, 22, 2567–2570 CrossRef CAS.
- Y. Pei, G. Zhou, N. Luan, B. Zong, M. Qiao and F. Tao, Chem. Soc. Rev., 2012, 41, 8140–8162 RSC.
- L. C. Zhang and S. X. Liang, Chem. – Asian J., 2018, 13, 3575–3592 CrossRef CAS PubMed.
- Z. Jia, Q. Wang, L. Sun, Q. Wang, L. C. Zhang, G. Wu, J. H. Luan, Z. B. Jiao, A. Wang, S. X. Liang, M. Gu and J. Lu, Adv. Funct. Mater., 2019, 29, 1807857 CrossRef.
- Q. Chen, Z. Yan, L. Guo, H. Zhang, L.-C. Zhang and W. Wang, J. Mol. Liq., 2020, 318, 114318 CrossRef CAS.
- A. Makino, T. Kubota, C. Chang, M. Makabe and A. Inoue, J. Magn. Magn. Mater., 2008, 320, 2499–2503 CrossRef CAS.
- B. J. Yang, J. H. Yao, J. Zhang, H. W. Yang, J. Q. Wang and E. Ma, Scr. Mater., 2009, 61, 423–426 CrossRef CAS.
- W. M. Yang, Q. Q. Wang, W. Y. Li, L. Xue, H. S. Liu, J. Zhou, J. Y. Mo and B. L. Shen, Mater. Des., 2019, 161, 136–146 CrossRef CAS.
- J. Schroers and W. L. Johnson, Phys. Rev. Lett., 2004, 93, 255506 CrossRef PubMed.
- L. Zhong, J. Wang, H. Sheng, Z. Zhang and S. X. Mao, Nature, 2014, 512, 177–180 CrossRef CAS PubMed.
- J. C. Wang, S. X. Liang, Z. Jia, W. C. Zhang, W. M. Wang, Y. J. Liu, J. Lu and L. Zhang, J. Alloys Compd., 2019, 785, 642–650 CrossRef CAS.
- J. L. Jiang, Z. Jia, Q. He, Q. Wang, F. Lyu, L. C. Zhang, S. X. Liang, J. J. Kruzic and J. Lu, J. Alloys Compd., 2020, 822, 153574 CrossRef CAS.
- S. X. Liang, Q. Zhang, Z. Jia, W. Zhang, W. Wang and L. C. Zhang, J. Colloid Interface Sci., 2021, 581, 860–873 CrossRef CAS PubMed.
- Z. Jia, L. B. T. La, W. C. Zhang, S. X. Liang, B. Jiang, S. K. Xie, D. Habibi and L. C. Zhang, J. Mater. Sci. Technol., 2017, 33, 856–863 CrossRef CAS.
- J. F. Deng, H. Li and W. Wang, Catal. Today, 1999, 51, 113–125 CrossRef CAS.
- P. Wagener, J. Jakobi, C. Rehbock, V. S. K. Chakravadhanula, C. Thede, U. Wiedwald, M. Bartsch, L. Kienle and S. Barcikowski, Sci. Rep., 2016, 6, 23352 CrossRef CAS PubMed.
- A. R. Ziefuß, S. Reichenberger, C. Rehbock, I. Chakraborty, M. Gharib, W. J. Parak and S. Barcikowski, J. Phys. Chem. C, 2018, 122, 22125–22136 CrossRef.
- K. Amikura, T. Kimura, M. Hamada, N. Yokoyama, J. Miyazaki and Y. Yamada, Appl. Surf. Sci., 2008, 254, 6976–6982 CrossRef CAS.
- S. Barcikowski and G. Compagnini, Phys. Chem. Chem. Phys., 2013, 15, 3022–3026 RSC.
- M. Salavati-Niasari, F. Davar and N. Mir, Polyhedron, 2008, 27, 3514–3518 CrossRef CAS.
- M. Salavati-Niasari, F. Davar, M. Mazaheri and M. Shaterian, J. Magn. Magn. Mater., 2008, 320, 575–578 CrossRef CAS.
- S. Navaladian, B. Viswanathan, R. P. Viswanath and T. K. Varadarajan, Nanoscale Res. Lett., 2006, 2, 44 CrossRef CAS PubMed.
- B. Yin, H. Ma, S. Wang and S. Chen, J. Phys. Chem. B, 2003, 107, 8898–8904 CrossRef CAS.
- J. Liu, W. Hu, C. Zhong and Y. F. Cheng, J. Power Sources, 2013, 223, 165–174 CrossRef CAS.
- H. Ma, B. Yin, S. Wang, Y. Jiao, W. Pan, S. Huang, S. Chen and F. Meng, ChemPhysChem, 2004, 5, 68–75 CrossRef CAS PubMed.
- N. R. Jana, L. Gearheart and C. J. Murphy, Chem. Mater., 2001, 13, 2313–2322 CrossRef CAS.
- R. Sato-Berrú, R. Redón, A. Vázquez-Olmos and J. M. Saniger, J. Raman Spectrosc., 2009, 40, 376–380 CrossRef.
- H. Li, J. Liu, S. Xie, M. Qiao, W. Dai and H. Li, J. Catal., 2008, 259, 104–110 CrossRef CAS.
- M. L. Marin, K. L. McGilvray and J. C. Scaiano, J. Am. Chem. Soc., 2008, 130, 16572–16584 CrossRef CAS PubMed.
- G. L. Hallett-Tapley, C.-O. L. Crites, M. González-Béjar, K. L. McGilvray, J. C. Netto-Ferreira and J. C. Scaiano, J. Photochem. Photobiol., A, 2011, 224, 8–15 CrossRef CAS.
- K. L. McGilvray, M. R. Decan, D. Wang and J. C. Scaiano, J. Am. Chem. Soc., 2006, 128, 15980–15981 CrossRef CAS PubMed.
- J. C. Scaiano, K. G. Stamplecoskie and G. L. Hallett-Tapley, Chem. Commun., 2012, 48, 4798–4808 RSC.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS PubMed.
- Y. Guo, D. Gu, Z. Jin, P.-P. Du, R. Si, J. Tao, W.-Q. Xu, Y.-Y. Huang, S. Senanayake, Q.-S. Song, C.-J. Jia and F. Schüth, Nanoscale, 2015, 7, 4920–4928 RSC.
- C. Streich, S. Koenen, M. Lelle, K. Peneva and S. Barcikowski, Appl. Surf. Sci., 2015, 348, 92–99 CrossRef CAS.
- S. Barcikowski, F. Devesa and K. Moldenhauer, J. Nanopart. Res., 2009, 11, 1883–1893 CrossRef.
- D. Zhang, B. Gökce and S. Barcikowski, Chem. Rev., 2017, 117, 3990–4103 CrossRef CAS PubMed.
- S. Reichenberger, G. Marzun, M. Muhler and S. Barcikowski, ChemCatChem, 2019, 11, 4489–4518 CrossRef CAS.
- P. P. Patil, D. M. Phase, S. A. Kulkarni, S. V. Ghaisas, S. K. Kulkarni, S. M. Kanetkar, S. B. Ogale and V. G. Bhide, Phys. Rev. Lett., 1987, 58, 238–241 CrossRef CAS PubMed.
- A. Fojtík and A. Henglein, Ber. Bunsen-Ges. Phys. Chem., 1993, 97, 252–254 CrossRef.
- A. Henglein, J. Phys. Chem., 1993, 97, 5457–5471 CrossRef CAS.
- J. Neddersen, G. Chumanov and T. M. Cotton, Appl. Spectrosc., 1993, 47, 1959–1964 CrossRef CAS.
- M. S. Sibbald, G. Chumanov and T. M. Cotton, J. Phys. Chem., 1996, 100, 4672–4678 CrossRef CAS.
- M. Procházka, P. Mojzeš, J. Štěpánek, B. Vlčková and P.-Y. Turpin, Anal. Chem., 1997, 69, 5103–5108 CrossRef.
- D. Zhang, B. Gökce, C. Notthoff and S. Barcikowski, Sci. Rep., 2015, 5, 13661 CrossRef PubMed.
- J. P. Sylvestre, S. Poulin, A. V. Kabashin, E. Sacher, M. Meunier and J. H. T. Luong, J. Phys. Chem. B, 2004, 108, 16864–16869 CrossRef CAS.
- V. Merk, C. Rehbock, F. Becker, U. Hagemann, H. Nienhaus and S. Barcikowski, Langmuir, 2014, 30, 4213–4222 CrossRef CAS PubMed.
- C. Rehbock, V. Merk, L. Gamrad, R. Streubel and S. Barcikowski, Phys. Chem. Chem. Phys., 2013, 15, 3057–3067 RSC.
- F. Mafuné, J. Y. Kohno, Y. Takeda and T. Kondow, J. Phys. Chem. B, 2003, 107, 4218–4223 CrossRef.
- G. Marzun, C. Streich, S. Jendrzej, S. Barcikowski and P. Wagener, Langmuir, 2014, 30, 11928–11936 CrossRef CAS PubMed.
- M. Fischer, J. Hormes, G. Marzun, P. Wagener, U. Hagemann and S. Barcikowski, Langmuir, 2016, 32, 8793–8802 CrossRef CAS PubMed.
- G. Marzun, J. Nakamura, X. Zhang, S. Barcikowski and P. Wagener, Appl. Surf. Sci., 2015, 348, 75–84 CrossRef CAS.
- S. Jendrzej, B. Gökce, M. Epple and S. Barcikowski, ChemPhysChem, 2017, 18, 1012–1019 CrossRef CAS PubMed.
- S. Dittrich, R. Streubel, C. McDonnell, H. P. Huber, S. Barcikowski and B. Gökce, Appl. Phys. A, 2019, 125, 432 CrossRef.
- C. Y. Shih, R. Streubel, J. Heberle, A. Letzel, M. V. Shugaev, C. Wu, M. Schmidt, B. Gökce, S. Barcikowski and L. V. Zhigilei, Nanoscale, 2018, 10, 6900–6910 RSC.
- M.-R. Kalus, R. Lanyumba, S. Barcikowski and B. Gökce, J. Flow Chem., 2021 DOI:10.1007/s41981-41021-00144-41987.
- C. Kerse, H. Kalaycıoğlu, P. Elahi, B. Çetin, D. K. Kesim, Ö. Akçaalan, S. Yavaş, M. D. Aşık, B. Öktem, H. Hoogland, R. Holzwarth and F. Ö. Ilday, Nature, 2016, 537, 84–88 CrossRef CAS PubMed.
- F. Waag, B. Gökce and S. Barcikowski, Appl. Surf. Sci., 2019, 466, 647–656 CrossRef CAS.
- R. Streubel, S. Barcikowski and B. Gökce, Opt. Lett., 2016, 41, 1486–1489 CrossRef CAS PubMed.
- R. Streubel, G. Bendt and B. Gökce, Nanotechnology, 2016, 27, 205602 CrossRef PubMed.
- F. Waag, R. Streubel, B. Gökce and S. Barcikowski, Appl. Nanosci., 2021, 11, 1303–1312 CrossRef CAS.
- S. Kohsakowski, F. Seiser, J.-P. Wiederrecht, S. Reichenberger, T. Vinnay, S. Barcikowski and G. Marzun, Nanotechnology, 2019, 31, 095603 CrossRef PubMed.
- V. Amendola, D. Amans, Y. Ishikawa, N. Koshizaki, S. Scirè, G. Compagnini, S. Reichenberger and S. Barcikowski, Chem. – Eur. J., 2020, 26, 9206–9242 CrossRef CAS PubMed.
- A. Neumeister, J. Jakobi, C. Rehbock, J. Moysig and S. Barcikowski, Phys. Chem. Chem. Phys., 2014, 16, 23671–23678 RSC.
- G. W. Yang, Prog. Mater. Sci., 2007, 52, 648–698 CrossRef CAS.
- J. Xiao, P. Liu, C. X. Wang and G. W. Yang, Prog. Mater. Sci., 2017, 87, 140–220 CrossRef CAS.
- C. Streich, L. Akkari, C. Decker, J. Bormann, C. Rehbock, A. Müller-Schiffmann, F. C. Niemeyer, L. Nagel-Steger, D. Willbold, B. Sacca, C. Korth, T. Schrader and S. Barcikowski, ACS Nano, 2016, 10, 7582–7597 CrossRef CAS PubMed.
- J. Zhang, M. Chaker and D. Ma, J. Colloid Interface Sci., 2017, 489, 138–149 CrossRef CAS PubMed.
- D. Zhang, J. Liu, P. Li, Z. Tian and C. Liang, ChemNanoMat, 2017, 3, 512–533 CrossRef CAS.
- D. Zhang, J. Liu and C. Liang, Sci. China: Phys., Mech. Astron., 2017, 60, 074201 Search PubMed.
- V. Amendola and M. Meneghetti, Phys. Chem. Chem. Phys., 2013, 15, 3027–3046 RSC.
- V. Amendola and M. Meneghetti, Phys. Chem. Chem. Phys., 2009, 11, 3805–3821 RSC.
- A. Hirata, P. Guan, T. Fujita, Y. Hirotsu, A. Inoue, A. R. Yavari, T. Sakurai and M. Chen, Nat. Mater., 2011, 10, 28–33 CrossRef CAS PubMed.
- Z. Jia, X. Duan, P. Qin, W. Zhang, W. Wang, C. Yang, H. Sun, S. Wang and L. C. Zhang, Adv. Funct. Mater., 2017, 27, 1702258 CrossRef.
- Z. Jia, S. X. Liang, W. C. Zhang, W. M. Wang, C. Yang and L. C. Zhang, J. Taiwan Inst. Chem. Eng., 2017, 71, 128–136 CrossRef CAS.
- X. F. Li, S. X. Liang, X. W. Xi, Z. Jia, S. K. Xie, H. C. Lin, J. P. Hu and L. C. Zhang, Metals, 2017, 7, 273 CrossRef.
- Z. Wu, J. Chen and R. Jin, Adv. Funct. Mater., 2011, 21, 177–183 CrossRef CAS.
- Z. Lin, C. Du, B. Yan and G. Yang, Catal. Sci. Technol., 2019, 9, 5582–5592 RSC.
- M. Zhao, K. Abe, S. I. Yamaura, Y. Yamamoto and N. Asao, Chem. Mater., 2014, 26, 1056–1061 CrossRef CAS.
- J. M. V. Nsanzimana, Y. Peng, Y. Y. Xu, L. Thia, C. Wang, B. Y. Xia and X. Wang, Adv. Energy Mater., 2018, 8, 1701475 CrossRef.
- H. Y. Kwong, M. H. Wong, C. W. Leung, Y. W. Wong and K. H. Wong, J. Appl. Phys., 2010, 108, 034304 CrossRef.
- J. Sun, S. K. Sinha, A. Khammari, M. Picher, M. Terrones and F. Banhart, Carbon, 2020, 161, 495–501 CrossRef CAS.
- Z. Zhu, J. Ma, L. Xu, L. Xu, H. Li and H. Li, ACS Catal., 2012, 2, 2119–2125 CrossRef CAS.
- S. Ju, J. Feng, P. Zou, W. Xu, S. Wang, W. Gao, H. J. Qiu, J. Huo and J. Q. Wang, J. Mater. Chem. A, 2020, 8, 3246–3251 RSC.
- L. C. Zhang and L. Y. Chen, Adv. Eng. Mater., 2019, 21, 1801215 CrossRef.
- Q. Q. Wang, M. X. Chen, P. H. Lin, Z. Q. Cui, C. L. Chu and B. L. Shen, J. Mater. Chem. A, 2018, 6, 10686–10699 RSC.
- N. D. Phu, D. T. Ngo, L. H. Hoang, N. H. Luong, N. Chau and N. H. Hai, J. Phys. D: Appl. Phys., 2011, 44, 345002 CrossRef.
- S. X. Liang, W. Zhang, L. Zhang, W. Wang and L. C. Zhang, Sustainable Mater. Technol., 2019, 22, e00126 CrossRef CAS.
- Z. Jia, J. C. Wang, S. X. Liang, W. C. Zhang, W. M. Wang and L. C. Zhang, J. Alloys Compd., 2017, 728, 525–533 CrossRef CAS.
- L. C. Zhang, Z. Q. Shen and J. Xu, Mater. Sci. Eng., A, 2005, 394, 204–209 CrossRef.
- L. C. Zhang, J. Xu and E. Ma, J. Mater. Res., 2002, 17, 1743–1749 CrossRef CAS.
- L. C. Zhang, J. Xu and E. Ma, Mater. Sci. Eng., A, 2006, 434, 280–288 CrossRef.
- L. Y. Chen, Y. W. Cui and L. C. Zhang, Metals, 2020, 10, 1139 CrossRef CAS.
- F. Hu, S. Zhu, S. Chen, Y. Li, L. Ma, T. Wu, Y. Zhang, C. Wang, C. Liu, X. Yang, L. Song, X. Yang and Y. Xiong, Adv. Mater., 2017, 29, 1606570 CrossRef PubMed.
- K. Kobayashi, K. Kusada, D. Wu, N. Ogiwara, H. Kobayashi, M. Haruta, H. Kurata, S. Hiroi, O. Seo, C. Song, Y. Chen, J. Kim, A. Tayal, O. Sakata, K. Ohara, T. Honma and H. Kitagawa, Chem. Commun., 2020, 56, 12941–12944 RSC.
- D. A. Dimitrov, A. L. Ankudinov, A. R. Bishop and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 14227–14237 CrossRef CAS.
- L. C. Zhang and J. Xu, J. Non-Cryst. Solids, 2004, 347, 166–172 CrossRef CAS.
- K. Pajor, T. Kozieł, G. Cios, P. Błyskun, P. Bała and A. Zielińska-Lipiec, J. Non-Cryst. Solids, 2018, 496, 42–47 CrossRef CAS.
- A. Inoue, Acta Mater., 2000, 48, 279–306 CrossRef CAS.
- L. C. Zhang, J. Xu and J. Eckert, J. Appl. Phys., 2006, 100, 033514 CrossRef.
- J. C. Wang, Z. Jia, S. X. Liang, P. Qin, W. C. Zhang, W. M. Wang, T. B. Sercombe and L. C. Zhang, Mater. Des., 2018, 140, 73–84 CrossRef CAS.
- J. Orava and A. L. Greer, J. Chem. Phys., 2014, 140, 214504 CrossRef CAS PubMed.
- F. Reichel, L. P. H. Jeurgens and E. J. Mittemeijer, Acta Mater., 2008, 56, 659–674 CrossRef CAS.
- A. Inoue and N. Nishiyama, Mater. Sci. Eng., A, 1997, 226-228, 401–405 CrossRef.
- W. H. Wang, J. J. Lewandowski and A. L. Greer, J. Mater. Res., 2011, 20, 2307–2313 CrossRef.
- E. S. Park, J. H. Na and D. H. Kim, Appl. Phys. Lett., 2007, 91, 031907 CrossRef.
- L. C. Zhang, M. Calin, M. Branzei, L. Schultz and J. Eckert, J. Mater. Res., 2007, 22, 1145–1155 CrossRef CAS.
- M. Kohda, O. Haruyama, T. Ohkubo and T. Egami, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 092203 CrossRef.
- E. P. George, D. Raabe and R. O. Ritchie, Nat. Rev. Mater., 2019, 4, 515–534 CrossRef CAS.
- Z. Jia, T. Yang, L. Sun, Y. Zhao, W. Li, J. Luan, F. Lyu, L. C. Zhang, J. J. Kruzic, J. J. Kai, J. C. Huang, J. Lu and C. T. Liu, Adv. Mater., 2020, 32, 2000385 CrossRef CAS PubMed.
- S. Guo and C. T. Liu, Prog. Nat. Sci.: Mater. Int., 2011, 21, 433–446 CrossRef.
- B. Gludovatz, A. Hohenwarter, D. Catoor, E. H. Chang, E. P. George and R. O. Ritchie, Science, 2014, 345, 1153–1158 CrossRef CAS PubMed.
- T. Yang, Y. L. Zhao, Y. Tong, Z. B. Jiao, J. Wei, J. X. Cai, X. D. Han, D. Chen, A. Hu, J. J. Kai, K. Lu, Y. Liu and C. T. Liu, Science, 2018, 362, 933–937 CrossRef CAS PubMed.
- M. W. Glasscott, A. D. Pendergast, S. Goines, A. R. Bishop, A. T. Hoang, C. Renault and J. E. Dick, Nat. Commun., 2019, 10, 2650 CrossRef.
- Q. Li, Y. Xu, S. Zheng, X. Guo, H. Xue and H. Pang, Small, 2018, 14, 1800426 CrossRef PubMed.
- Y. Wu, X. D. Hui, Z. P. Lu, Z. Y. Liu, L. Liang and G. L. Chen, J. Alloys Compd., 2009, 467, 187–190 CrossRef CAS.
- Y.-W. Kim, H.-M. Lin and T. F. Kelly, Acta Metall., 1989, 37, 247–255 CrossRef CAS.
- J. Jiang, G. Oberdörster, A. Elder, R. Gelein, P. Mercer and P. Biswas, Nanotoxicology, 2008, 2, 33–42 CrossRef CAS PubMed.
- Z. Jia, X. Duan, W. Zhang, W. Wang, H. Sun, S. Wang and L. C. Zhang, Sci. Rep., 2016, 6, 38520 CrossRef CAS PubMed.
- S. An, R. Su, S. Zhao, J. Liu, B. Liu and P. Guan, Phys. Rev. B, 2018, 98, 134101 CrossRef CAS.
- G.-P. Zheng and M. Li, Acta Mater., 2007, 55, 5464–5472 CrossRef CAS.
- F. Waag, Y. Li, A. R. Ziefuß, E. Bertin, M. Kamp, V. Duppel, G. Marzun, L. Kienle, S. Barcikowski and B. Gökce, RSC Adv., 2019, 9, 18547–18558 RSC.
- C. Århammar, A. Pietzsch, N. Bock, E. Holmström, C. M. Araujo, J. Gråsjö, S. Zhao, S. Green, T. Peery, F. Hennies, S. Amerioun, A. Föhlisch, J. Schlappa, T. Schmitt, V. N. Strocov, G. A. Niklasson, D. C. Wallace, J.-E. Rubensson, B. Johansson and R. Ahuja, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6355–6360 CrossRef.
- Y. Q. Cheng and E. Ma, Prog. Mater. Sci., 2011, 56, 379–473 CrossRef CAS.
- R. Busch, J. Schroers and W. H. Wang, MRS Bull., 2007, 32, 620–623 CrossRef CAS.
- L. C. Zhang, K. B. Kim, P. Yu, W. Y. Zhang, U. Kunz and J. Eckert, J. Alloys Compd., 2007, 428, 157–163 CrossRef CAS.
- M. Calin, L. C. Zhang and J. Eckert, Scr. Mater., 2007, 57, 1101–1104 CrossRef CAS.
- O. Millo, D. Katz, Y. Cao and U. Banin, Phys. Rev. Lett., 2001, 86, 5751–5754 CrossRef CAS.
- W. Lu, B. Wang, K. Wang, X. Wang and J. G. Hou, Langmuir, 2003, 19, 5887–5891 CrossRef CAS.
- T. Löffler, F. Waag, B. Gökce, A. Ludwig, S. Barcikowski and W. Schuhmann, ACS Catal., 2021, 11, 1014–1023 CrossRef.
- T. V. Perevalov, O. E. Tereshenko, V. A. Gritsenko, V. A. Pustovarov, A. P. Yelisseyev, C. Park, J. H. Han and C. Lee, J. Appl. Phys., 2010, 108, 013501 CrossRef.
- M. Dell’Aglio, R. Gaudiuso, O. De Pascale and A. De Giacomo, Appl. Surf. Sci., 2015, 348, 4–9 CrossRef.
- A. Kanitz, M. R. Kalus, E. L. Gurevich, A. Ostendorf, S. Barcikowski and D. Amans, Plasma Sources Sci. Technol., 2019, 28, 103001 CrossRef CAS.
- C.-Y. Shih, M. V. Shugaev, C. Wu and L. V. Zhigilei, Phys. Chem. Chem. Phys., 2020, 22, 7077–7099 RSC.
- C.-Y. Shih, M. V. Shugaev, C. Wu and L. V. Zhigilei, J. Phys. Chem. C, 2017, 121, 16549–16567 CrossRef CAS.
- S. Ibrahimkutty, P. Wagener, A. Menzel, A. Plech and S. Barcikowski, Appl. Phys. Lett., 2012, 101, 103104 CrossRef.
- K. Ando and T. Nakajima, Nanoscale, 2020, 12, 9640–9646 RSC.
- S. Reich, J. Göttlicher, A. Ziefuss, R. Streubel, A. Letzel, A. Menzel, O. Mathon, S. Pascarelli, T. Baumbach, M. Zuber, B. Gökce, S. Barcikowski and A. Plech, Nanoscale, 2020, 12, 14011–14020 RSC.
- E. Lescoute, L. Hallo, D. Hébert, B. Chimier, B. Etchessahar, V. T. Tikhonchuk, J.-M. Chevalier and P. Combis, Phys. Plasmas, 2008, 15, 063507 CrossRef.
- N. Yang, H. Cheng, X. Liu, Q. Yun, Y. Chen, B. Li, B. Chen, Z. Zhang, X. Chen, Q. Lu, J. Huang, Y. Huang, Y. Zong, Y. Yang, L. Gu and H. Zhang, Adv. Mater., 2018, 30, 1803234 CrossRef PubMed.
- G. Wu, X. Zheng, P. Cui, H. Jiang, X. Wang, Y. Qu, W. Chen, Y. Lin, H. Li, X. Han, Y. Hu, P. Liu, Q. Zhang, J. Ge, Y. Yao, R. Sun, Y. Wu, L. Gu, X. Hong and Y. Li, Nat. Commun., 2019, 10, 4855 CrossRef PubMed.
- S. Barcikowski, T. Baranowski, Y. Durmus, U. Wiedwald and B. Gökce, J. Mater. Chem. C, 2015, 3, 10699–10704 RSC.
- D. Zhang, W. Choi, Y. Oshima, U. Wiedwald, S.-H. Cho, H.-P. Lin, Y. K. Li, Y. Ito and K. Sugioka, Nanomaterials, 2018, 8, 631 CrossRef PubMed.
- L. Franzel, M. F. Bertino, Z. J. Huba and E. E. Carpenter, Appl. Surf. Sci., 2012, 261, 332–336 CrossRef CAS.
- J. M. J. Santillán, D. Muñetón Arboleda, D. F. Coral, M. B. Fernández van Raap, D. Muraca, D. C. Schinca and L. B. Scaffardi, ChemPhysChem, 2017, 18, 1192–1209 CrossRef PubMed.
- V. Amendola, P. Riello, S. Polizzi, S. Fiameni, C. Innocenti, C. Sangregorio and M. Meneghetti, J. Mater. Chem., 2011, 21, 18665–18673 RSC.
- V. Amendola, M. Meneghetti, G. Granozzi, S. Agnoli, S. Polizzi, P. Riello, A. Boscaini, C. Anselmi, G. Fracasso, M. Colombatti, C. Innocenti, D. Gatteschi and C. Sangregorio, J. Mater. Chem., 2011, 21, 3803–3813 RSC.
- P. Liu, W. Cai and H. Zeng, J. Phys. Chem. C, 2008, 112, 3261–3266 CrossRef CAS.
- P. Maneeratanasarn, T. V. Khai, S. Y. Kim, B. G. Choi and K. B. Shim, Phys. Status Solidi A, 2013, 210, 563–569 CrossRef CAS.
- A. De Bonis, T. Lovaglio, A. Galasso, A. Santagata and R. Teghil, Appl. Surf. Sci., 2015, 353, 433–438 CrossRef CAS.
- W. T. Nichols, T. Sasaki and N. Koshizaki, J. Appl. Phys., 2006, 100, 114913 CrossRef.
- M. Sivayoganathan, B. Tan and K. Venkatakrishnan, Nanoscale Res. Lett., 2012, 7, 619 CrossRef.
- Z. Yan, R. Bao, Y. Huang and D. B. Chrisey, J. Phys. Chem. C, 2010, 114, 11370–11374 CrossRef CAS.
- C. Liang, Y. Shimizu, T. Sasaki and N. Koshizaki, J. Mater. Res., 2004, 19, 1551–1557 CrossRef CAS.
- M. Wang, J.-Q. Wang, C. Xi, C.-Q. Cheng, C.-Q. Zou, R. Zhang, Y.-M. Xie, Z.-L. Guo, C.-C. Tang, C.-K. Dong, Y.-J. Chen and X.-W. Du, Angew. Chem., Int. Ed., 2020, 59, 11510–11515 CrossRef CAS PubMed.
- Z.-W. Gao, J.-Y. Liu, X.-M. Chen, X.-L. Zheng, J. Mao, H. Liu, T. Ma, L. Li, W.-C. Wang and X.-W. Du, Adv. Mater., 2019, 31, 1804769 CrossRef PubMed.
- X. Wang, Z. Li, D.-Y. Wu, G.-R. Shen, C. Zou, Y. Feng, H. Liu, C.-K. Dong and X.-W. Du, Small, 2019, 15, 1804832 CrossRef PubMed.
- Z.-W. Gao, T. Ma, X.-M. Chen, H. Liu, L. Cui, S.-Z. Qiao, J. Yang and X.-W. Du, Small, 2018, 14, 1800195 CrossRef.
- H. Zhang, C. Liang, J. Liu, Z. Tian and G. Shao, Carbon, 2013, 55, 108–115 CrossRef CAS.
- A. Letzel, B. Gökce, P. Wagener, S. Ibrahimkutty, A. Menzel, A. Plech and S. Barcikowski, J. Phys. Chem. C, 2017, 121, 5356–5365 CrossRef CAS.
- M. Lau and S. Barcikowski, Appl. Surf. Sci., 2015, 348, 22–29 CrossRef CAS.
- C.-C. Huang, C.-S. Yeh and C.-J. Ho, J. Phys. Chem. B, 2004, 108, 4940–4945 CrossRef CAS.
- W. T. Nichols, T. Sasaki and N. Koshizaki, J. Appl. Phys., 2006, 100, 114912 CrossRef.
- D. A. Bonis, M. Curcio, A. Santagata, A. Galasso and R. Teghil, Nanomaterials, 2020, 10, 145 CrossRef PubMed.
- N. Acacia, F. Barreca, E. Barletta, D. Spadaro, G. Currò and F. Neri, Appl. Surf. Sci., 2010, 256, 6918–6922 CrossRef CAS.
- V. Amendola, P. Riello and M. Meneghetti, J. Phys. Chem. C, 2011, 115, 5140–5146 CrossRef CAS.
- A. Kanitz, J. S. Hoppius, M. del Mar Sanz, M. Maicas, A. Ostendorf and E. L. Gurevich, ChemPhysChem, 2017, 18, 1155–1164 CrossRef CAS PubMed.
- D. Zhang, C. Zhang, J. Liu, Q. Chen, X. Zhu and C. Liang, ACS Appl. Nano Mater., 2019, 2, 28–39 CrossRef CAS.
- T. Matsue, Y. Yamada and Y. Kobayashi, presented in part at the ICAME 2011, 2013, pp. 179–183.
- J. Zhang and C. Q. Lan, Mater. Lett., 2008, 62, 1521–1524 CrossRef CAS.
- G. Viau, V. Collière, L. M. Lacroix and G. A. Shafeev, Chem. Phys. Lett., 2011, 501, 419–422 CrossRef CAS.
- G. Marzun, H. Bönnemann, C. Lehmann, B. Spliethoff, C. Weidenthaler and S. Barcikowski, ChemPhysChem, 2017, 18, 1175–1184 CrossRef CAS PubMed.
- Y.-j. Kim, R. Ma, D. A. Reddy and T. K. Kim, Appl. Surf. Sci., 2015, 357, 2112–2120 CrossRef CAS.
- A. De Bonis, A. Galasso, A. Santagata and R. Teghil, J. Phys. D: Appl. Phys., 2015, 49, 035301 CrossRef.
- N. Patra, K. Akash, S. Shiva, R. Gagrani, H. S. P. Rao, V. R. Anirudh, I. A. Palani and V. Singh, Appl. Surf. Sci., 2016, 366, 104–111 CrossRef CAS.
- S. H. Huh and A. Nakajima, J. Appl. Phys., 2006, 99, 064302 CrossRef.
- R. Nadarajah, S. Tahir, J. Landers, D. Koch, A. S. Semisalova, J. Wiemeler, A. El-Zoka, S.-H. Kim, D. Utzat, R. Möller, B. Gault, H. Wende, M. Farle and B. Gökce, Nanomaterials, 2020, 10, 2362 CrossRef CAS PubMed.
- J. P. Peterson and A. H. Winter, J. Am. Chem. Soc., 2019, 141, 12901–12906 CrossRef CAS PubMed.
- P. Wagener, S. Ibrahimkutty, A. Menzel, A. Plech and S. Barcikowski, Phys. Chem. Chem. Phys., 2013, 15, 3068–3074 RSC.
- S. Kohsakowski, B. Gökce, R. Tanabe, P. Wagener, A. Plech, Y. Ito and S. Barcikowski, Phys. Chem. Chem. Phys., 2016, 18, 16585–16593 RSC.
- S. Reich, P. Schönfeld, A. Letzel, S. Kohsakowski, M. Olbinado, B. Gökce, S. Barcikowski and A. Plech, ChemPhysChem, 2017, 18, 1084–1090 CrossRef CAS.
- T. Hupfeld, G. Laurens, S. Merabia, S. Barcikowski, B. Gökce and D. Amans, J. Appl. Phys., 2020, 127, 044306 CrossRef CAS.
- C. Lechner, W. Lauterborn, M. Koch and R. Mettin, Phys. Rev. Fluids, 2020, 5, 093604 CrossRef.
- M.-R. Kalus, N. Bärsch, R. Streubel, E. Gökce, S. Barcikowski and B. Gökce, Phys. Chem. Chem. Phys., 2017, 19, 7112–7123 RSC.
- K. Suehara, R. Takai, Y. Ishikawa, N. Koshizaki, K. Omura, H. Nagata and Y. Yamauchi, ChemPhysChem, 2021, 22, 675–683 CrossRef CAS PubMed.
- M. W. Chen, NPG Asia Mater., 2011, 3, 82–90 CrossRef.
- L. Sun and F. Banhart, Appl. Phys. Lett., 2006, 88, 193121 CrossRef.
- A. Santagata, A. De Bonis, A. De Giacomo, M. Dell’Aglio, A. Laurita, G. S. Senesi, R. Gaudiuso, S. Orlando, R. Teghil and G. P. Parisi, J. Phys. Chem. C, 2011, 115, 5160–5164 CrossRef CAS.
- C.-Y. Shih, C. Chen, C. Rehbock, A. Tymoczko, U. Wiedwald, M. Kamp, U. Schuermann, L. Kienle, S. Barcikowski and L. V. Zhigilei, J. Phys. Chem. C, 2021, 125, 2132–2155 CrossRef CAS.
- A. Tymoczko, M. Kamp, C. Rehbock, L. Kienle, E. Cattaruzza, S. Barcikowski and V. Amendola, Nanoscale Horiz., 2019, 4, 1326–1332 RSC.
- G. Marzun, A. Levish, V. Mackert, T. Kallio, S. Barcikowski and P. Wagener, J. Colloid Interface Sci., 2017, 489, 57–67 CrossRef CAS PubMed.
- J. A. Tomko, R. Jimenez, J. J. Naddeo, D. M. Bubb and S. M. O’Malley, Laser Phys., 2018, 28, 035602 CrossRef.
- R. Nadarajah, S. Barcikowski and B. Gökce, Opt. Express, 2020, 28, 2909–2924 CrossRef PubMed.
- A. R. Ziefuß, I. Haxhiaj, S. Müller, M. Gharib, O. Gridina, C. Rehbock, I. Chakraborty, B. Peng, M. Muhler, W. J. Parak, S. Barcikowski and S. Reichenberger, J. Phys. Chem. C, 2020, 124, 20981–20990 CrossRef.
- L. H. Li, J. Xiao, P. Liu and G. W. Yang, Sci. Rep., 2015, 5, 9028 CrossRef CAS PubMed.
- Z. Lin, C. Du, B. Yan and G. Yang, J. Catal., 2019, 372, 299–310 CrossRef CAS.
- Z. Lin, C. Du, B. Yan, C. Wang and G. Yang, Nat. Commun., 2018, 9, 4036 CrossRef PubMed.
- J. Y. Liang, C. H. Wu, Y. Zheng, P. Shen and S. Y. Chen, Appl. Phys. A: Mater. Sci. Process., 2014, 115, 1429–1438 CrossRef CAS.
- X. Sun, J. Wang, Y. Yin, H. Wang, S. Li, H. Liu, J. Mao and X. Du, Chem. – Eur. J., 2020, 26, 2793–2797 CrossRef CAS PubMed.
- F. Waag, B. Gökce, C. Kalapu, G. Bendt, S. Salamon, J. Landers, U. Hagemann, M. Heidelmann, S. Schulz, H. Wende, N. Hartmann, M. Behrens and S. Barcikowski, Sci. Rep., 2017, 7, 13161 CrossRef.
- S. Siebeneicher, F. Waag, M. Escobar Castillo, V. V. Shvartsman, D. C. Lupascu and B. Gökce, Nanomaterials, 2020, 10, 359 CrossRef CAS PubMed.
- E. Williams and E. B. Brousseau, J. Mater. Process. Technol., 2016, 232, 34–42 CrossRef CAS.
- Y. Liu, M. Q. Jiang, G. W. Yang, J. H. Chen, Y. J. Guan and L. H. Dai, Intermetallics, 2012, 31, 325–329 CrossRef CAS.
- M. Q. Jiang, Y. P. Wei, G. Wilde and L. H. Dai, Appl. Phys. Lett., 2015, 106, 021904 CrossRef.
- Y. Zhu, J. Fu, C. Zheng and Z. Ji, Opt. Laser Technol., 2016, 83, 21–27 CrossRef CAS.
- Y. Liu, M. Q. Jiang, G. W. Yang, Y. J. Guan and L. H. Dai, Appl. Phys. Lett., 2011, 99, 191902 CrossRef.
- W. Jia, Z. Peng, Z. Wang, X. Ni and C.-y. Wang, Appl. Surf. Sci., 2006, 253, 1299–1303 CrossRef CAS.
- F. Ma, J. Yang, X. Zhu, C. Liang and H. Wang, Appl. Surf. Sci., 2010, 256, 3653–3660 CrossRef CAS.
- P. Wagener, A. Schwenke, B. N. Chichkov and S. Barcikowski, J. Phys. Chem. C, 2010, 114, 7618–7625 CrossRef CAS.
- J. H. Yoo, S. H. Jeong, X. L. Mao, R. Greif and R. E. Russo, Appl. Phys. Lett., 2000, 76, 783–785 CrossRef CAS.
- J. H. Yoo, S. H. Jeong, R. Greif and R. E. Russo, J. Appl. Phys., 2000, 88, 1638–1649 CrossRef CAS.
- N. M. Bulgakova and A. V. Bulgakov, Appl. Phys. A: Mater. Sci. Process., 2001, 73, 199–208 CrossRef CAS.
- X. Song, X. Q. Wu, K. L. Xiao, C. Li, H. Y. Wang and M. Q. Jiang, Philos. Mag., 2020, 1–13 Search PubMed.
- N. Linz, S. Freidank, X.-X. Liang, H. Vogelmann, T. Trickl and A. Vogel, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 134114 CrossRef.
- M. Q. Jiang, X. Q. Wu, Y. P. Wei, G. Wilde and L. H. Dai, Extreme Mech. Lett., 2017, 11, 24–29 CrossRef.
- X. Song, K. L. Xiao, X. Q. Wu, G. Wilde and M. Q. Jiang, J. Non-Cryst. Solids, 2019, 517, 119–126 CrossRef CAS.
- S. Barcikowski, A. Plech, K. S. Suslick and A. Vogel, MRS Bull., 2019, 44, 382–391 CrossRef.
- I. Martin, T. Ohkubo, M. Ohnuma, B. Deconihout and K. Hono, Acta Mater., 2004, 52, 4427–4435 CrossRef CAS.
- M. Lau, A. Ziefuss, T. Komossa and S. Barcikowski, Phys. Chem. Chem. Phys., 2015, 17, 29311–29318 RSC.
- M. Lau, S. Reichenberger, I. Haxhiaj, S. Barcikowski and A. M. Müller, ACS Appl. Energy Mater., 2018, 1, 5366–5385 CrossRef CAS.
- P. Wagener, A. Schwenke and S. Barcikowski, Langmuir, 2012, 28, 6132–6140 CrossRef CAS PubMed.
- W. Dong, S. Reichenberger, S. Chu, P. Weide, H. Ruland, S. Barcikowski, P. Wagener and M. Muhler, J. Catal., 2015, 330, 497–506 CrossRef CAS.
- Z. Jia, F. Lyu, L. C. Zhang, S. Zeng, S. X. Liang, Y. Y. Li and J. Lu, Sci. Rep., 2019, 9, 7636 CrossRef CAS PubMed.
- Z. Jia, J. Miao, H. B. Lu, D. Habibi, W. C. Zhang and L. C. Zhang, J. Taiwan Inst. Chem. Eng., 2016, 60, 267–274 CrossRef CAS.
- Y. C. Hu, Y. Z. Wang, R. Su, C. R. Cao, F. Li, C. W. Sun, Y. Yang, P. F. Guan, D. W. Ding, Z. L. Wang and W. H. Wang, Adv. Mater., 2016, 28, 10293–10297 CrossRef CAS PubMed.
- Z.-J. Wang, M.-X. Li, J.-H. Yu, X.-B. Ge, Y.-H. Liu and W.-H. Wang, Adv. Mater., 2020, 32, 1906384 CrossRef CAS PubMed.
- J. Q. Wang, Y. H. Liu, M. W. Chen, D. V. Louzguine-Luzgin, A. Inoue and J. H. Perepezko, Sci. Rep., 2012, 2, 418 CrossRef PubMed.
- P. Wang, J. Q. Wang, H. Li, H. Yang, J. Huo, J. Wang, C. Chang, X. Wang, R. W. Li and G. Wang, J. Alloys Compd., 2017, 701, 759–767 CrossRef CAS.
- S. X. Liang, Z. Jia, W. C. Zhang, X. F. Li, W. M. Wang, H. C. Lin and L. C. Zhang, Appl. Catal., B, 2018, 221, 108–118 CrossRef CAS.
- Z. Jia, W. C. Zhang, W. M. Wang, D. Habibi and L. C. Zhang, Appl. Catal., B, 2016, 192, 46–56 CrossRef CAS.
- Q. Chen, Z. Yan, L. Guo, H. Zhang, L. Zhang, K. Kim, X. Li and W. Wang, J. Alloys Compd., 2020, 831, 154817 CrossRef CAS.
- Y. Yang, T. Adit Maark, A. Peterson and S. Kumar, Phys. Chem. Chem. Phys., 2015, 17, 1746–1754 RSC.
- G. Doubek, R. C. Sekol, J. Li, W. H. Ryu, F. S. Gittleson, S. Nejati, E. Moy, C. Reid, M. Carmo, M. Linardi, P. Bordeenithikasem, E. Kinser, Y. Liu, X. Tong, C. O. Osuji, J. Schroers, S. Mukherjee and A. D. Taylor, Adv. Mater., 2016, 28, 1940–1949 CrossRef CAS PubMed.
- S. X. Liang, X. Wang, W. Zhang, Y. J. Liu, W. Wang and L. C. Zhang, Appl. Mater. Today, 2020, 19, 100543 CrossRef.
- X. Wang, Q. Zhang, S. X. Liang, Z. Jia, W. Zhang, W. Wang and L. C. Zhang, Catalysts, 2020, 10, 48 CrossRef CAS.
- Q. Zhang, S. X. Liang, Z. Jia, W. Zhang, W. Wang and L. C. Zhang, J. Mater. Sci. Technol., 2021, 61, 159–168 CrossRef.
- S. X. Liang, W. Zhang, W. Wang, G. Jia, W. Yang and L. C. Zhang, J. Phys. Chem. Solids, 2019, 132, 89–98 CrossRef CAS.
- Z. Jia, J. L. Jiang, L. Sun, L. C. Zhang, Q. Wang, S. X. Liang, P. Qin, D.-F. Li, J. Lu and J. J. Kruzic, ACS Appl. Mater. Interfaces, 2020, 12, 44789–44797 CrossRef CAS.
- Z. Jia, J. Kang, W. C. Zhang, W. M. Wang, C. Yang, H. Sun, D. Habibi and L. C. Zhang, Appl. Catal., B, 2017, 204, 537–547 CrossRef CAS.
- L. M. Carneiro, S. K. Cushing, C. Liu, Y. Su, P. Yang, A. P. Alivisatos and S. R. Leone, Nat. Mater., 2017, 16, 819–825 CrossRef CAS PubMed.
- S. Yan, K. P. Abhilash, L. Tang, M. Yang, Y. Ma, Q. Xia, Q. Guo and H. Xia, Small, 2019, 15, 1804371 Search PubMed.
- F. Hu, H. Wang, Y. Zhang, X. Shen, G. Zhang, Y. Pan, J. T. Miller, K. Wang, S. Zhu, X. Yang, C. Wang, X. Wu, Y. Xiong and Z. Peng, Small, 2019, 15, 1901020 CrossRef PubMed.
- A. H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- C. Zhang, W. Bu, D. Ni, S. Zhang, Q. Li, Z. Yao, J. Zhang, H. Yao, Z. Wang and J. Shi, Angew. Chem., Int. Ed., 2016, 55, 2101–2106 CrossRef CAS PubMed.
- B. Thierry, F. Al-Ejeh, A. Khatri, Z. Yuan, P. J. Russell, S. Ping, M. P. Brown and P. Majewski, Chem. Commun., 2009, 7348–7350 RSC.
- M. W. Grinstaff, M. B. Salamon and K. S. Suslick, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 269–273 CrossRef CAS PubMed.
- E. P. Yelsukov, A. I. Ul’yanov, A. V. Zagainov and N. B. Arsent’yeva, J. Magn. Magn. Mater., 2003, 258-259, 513–515 CrossRef CAS.
- S. Bag, A. Baksi, S. H. Nandam, D. Wang, X. Ye, J. Ghosh, T. Pradeep and H. Hahn, ACS Nano, 2020, 14, 5543–5552 CrossRef CAS PubMed.
- L. C. Zhang, L. Y. Chen and L. Wang, Adv. Eng. Mater., 2020, 22, 1901258 CrossRef CAS.
| This journal is © the Owner Societies 2021 |