
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, in silico studies, and synthesis of new 1,8-naphthyridine-3-carboxylic acid analogues and evaluation of their H1R antagonism effects†
Vinod Kumar Gurjar and
Dilipkumar Pal *
*
Department of Pharmaceutical Sciences, Guru Ghasidas Vishwavidyalaya (A Central University), Bilaspur-495 009, CG, India. E-mail: drdilip71@gmail.com; Tel: +91-7389263761
First published on 6th April 2020
Abstract
New 1,8-naphthyridine-3-carboxylic acid derivatives were designed, synthesized and evaluated for their in vivo antihistaminic activity on guinea pig trachea by using chlorpheniramine as the standard drug. It was found that compound 5a1 displayed a promising bronchorelaxant effect in conscious guinea pigs using the in vivo model. A molecular docking study was performed to understand the molecular interaction and binding mode of the compounds in the active site of the H1 receptor. Furthermore, in silico computational studies were also performed to predict the binding modes and pharmacokinetic parameters of these derivatives. Prior to the start of experimental lab work, PASS software was used to predict the biological activities of these compounds. An in silico PASS, Swiss ADME assisted docking approach was found to be suitable to derive and synthesize effective antihistaminic agents for the present study.
1 Introduction
Heterocyclic synthesis has become a powerful technique in organic synthesis for generating new molecules for drug discovery and development.1–8 Nitrogen atom-containing heterocyclic compounds provide highly functionalized scaffolds on which pharmacophoric features can be arranged to obtain effective and selective drugs.9–17 Histamine is one of the most important chemical mediators that influences immune regulation via an acute and chronic inflammatory response through 4 different types of G-protein coupled receptors, H1, H2, H3, and H4. It is involved in the pathophysiology of allergic disorders like urticaria, rhino conjunctivitis, and asthma.18 Although antihistaminic agents belong to several chemical classes, such as ethylene-diamines, aminoethyl ethers, propyl- and propenyl-arnines, phenothiazines, piperidines and piperazines, they show remarkable chemical similarities.19 Presently, antihistamines are widely prescribed for the management of various allergic symptoms, but these drugs show complex side effect profiles, which include sedation, light-headedness, motor incoordination, cardiovascular effects, diminished alertness, concentration difficulties, fatigue and a tendency to fall asleep.20 A common structural feature for the classical first-generation antihistaminic drugs is an aryl group including phenyl, substituted phenyl and heteroaryl groups (2-pyridyl) linked to a terminal amino group via a two or three carbon chain (chlorpheniramine maleate).21 In contrast, most of the non-sedative H1R inhibitors like azelastine, levocetirizine and fexofenadine22 have a minimal muscarinic effect and show a better BRA (benefit-to-risk ratio). These drugs cannot diffuse through the blood–brain barrier and thus have a weaker sedating effect and possess higher receptor selectivity and affinity,23 and they are marked as ‘non-sedative’ H1R inhibitors. The ‘non-sedative’ second-generation drugs like terfenadine and levocetirizine also have many structural features belonging to old classic antihistamines. The condensed heterocyclic ring system of the second-generation H1-antihistamines (loratadine, fexofenadine, and azelastine) does not have the above-described pharmacophoric groups of H1-antihistamines. This concept and idea have enabled the discovery of many potent antihistaminic drugs like temelastine and mangostin. The currently used antihistaminic compounds are almost completely based on modifying the structure of some old H1 antagonists (Fig. 1).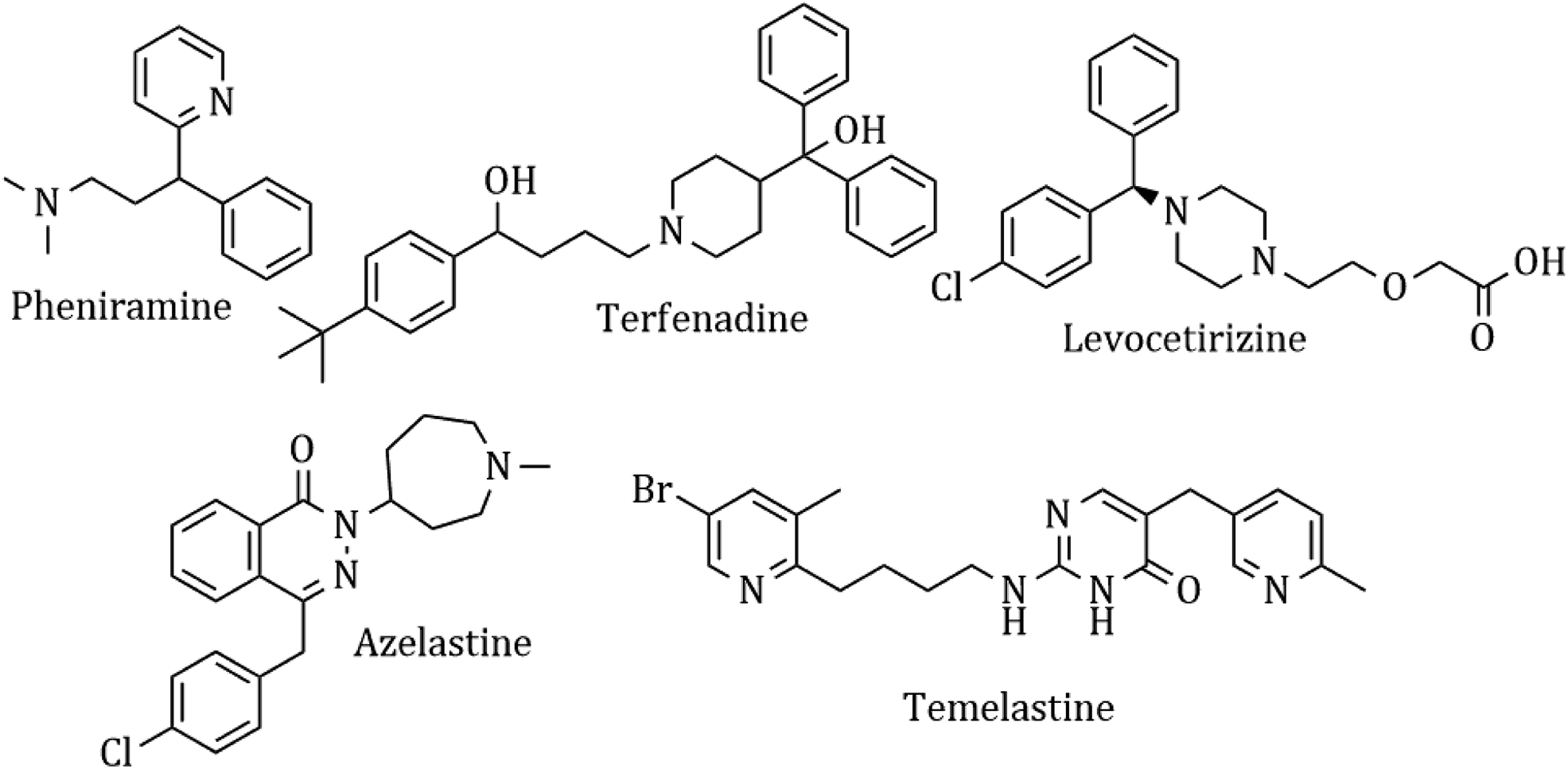 | ||
| Fig. 1 Classical antihistamines. | ||
The availability of the crystal structure of H(1)R, however, can enable new ways to study the binding of histamine and its antagonists and to find the important receptor–ligand interactions. It is also possible to compare the binding affinity of the synthesized analogues to predict their possible therapeutic activity.24,25
Azelastine is a phthalazinone derivative and a H1R antagonist.26 It is an antiallergic agent that inhibits the release of histamine and other mediators involved in the allergic response. It antagonizes histamine and leukotriene-induced bronchospasm in animal studies and reduces airway responsiveness to an inhaled antigen or distilled water and exercise challenges.27 From a comprehensive literature search of 1,8-naphthyridine, it has been found to be a potent antiallergic and antihistaminic agent.28,29 In addition, its derivatives are reported to have a broad spectrum of pharmacological activity. Notable amongst these are A1 and A2A adenosine receptor agonist,30 antibacterial,31 analgesic and anti-inflammatory32 agents, cannabinoid receptor ligands33 and phosphodiesterase (PDE 4) inhibitors.34 A review of the literature also supports that azelastine as an asthma prophylactic has three main pharmacophoric groups: (i) phthalazinone nuclei, (ii) a p-chlorobenzyl moiety and (iii) a basic amino group, N-methyl azepine. These three parts are essential to be an effective H1R antagonistic compound.
In reference to our continuous interest in establishing potential non-sedating, nonclassical histaminergic (H1) blockers with low toxicity profiles, we have previously published a research paper where the synthesis of some substituted 1,8-naphthyridine derivatives with different cyclic amine substituents of 1,8-naphthyridine-3-carboxylic acid35 has been described. Consequently, in this work, the key compound 1,8-naphthyridine-3-carboxylic acid is selected, which is susceptible to forming amide derivatives. In the present paper, we have reported the design, synthesis and in vitro antihistaminic activity of 1,8-naphthyridine-3-carboxylic acid derivatives that are modified at the terminal 3-carboxylic acid end (Fig. 2). In the current drug discovery process, the potential of a novel compound is frequently studied initially through virtual tools. The possibility of a compound to exhibit useful therapeutic activity (sometimes called ‘drug-likeness’) is predicted from its molecular structure.36,37 Prediction of bioavailability and bioavailability-related properties, such as solubility and lipophilicity, is significant before synthesis. This could be the best method to avoid depletion of chemicals, valuable time and above all possible environmental problems. Furthermore, we also performed in silico ADMET profile and PASS analyses of all compounds.
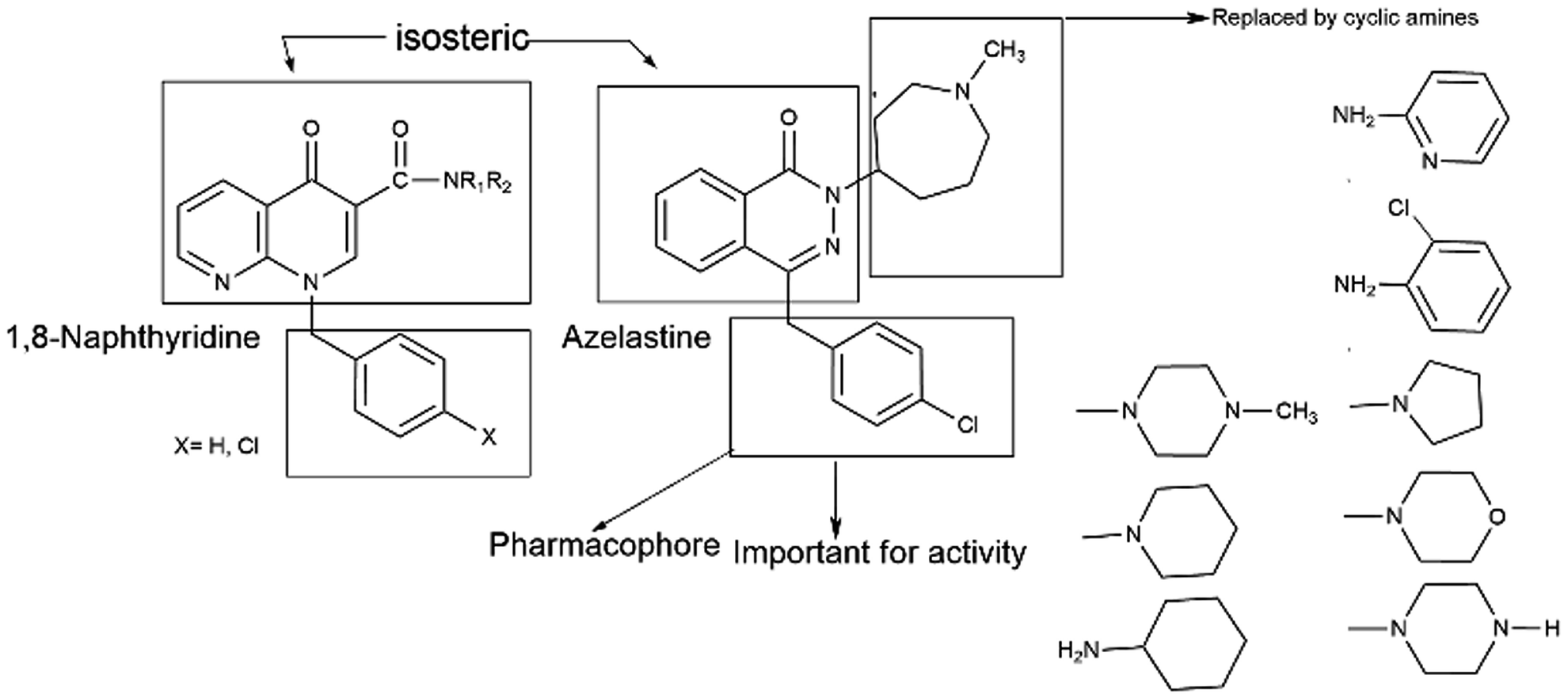 | ||
| Fig. 2 Design of 1,8-naphthyridine derivatives. | ||
2 Materials and methods
2.1 General
All chemicals and reagents were purchased from commercial sources (Himedia, Sigma-Aldrich) having the best analytical-grade and used without further purification unless otherwise stated. Melting points were checked using open capillary methods on a Macro scientific apparatus and were uncorrected. All reactions were routinely checked by thin-layer chromatography (TLC) analysis on silica gel G plates eluted with a mixture of chloroform![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol (4:1) as a solvent system. The IR spectra were recorded using a Shimadzu FTIR spectrophotometer. 1HNMR spectra were recorded using a Bruker (Avance III) 400 MHz system spectrometer with tetramethyl silane (TMS) as the internal standard and DMSO as the solvent. Chemical shifts were reported as parts per million (ppm). Signals were represented as follows: s: singlet, d: doublet, m: multiplet, and t: triplet. High-resolution mass spectra (HRMS) were obtained using the Electrospray Ionization (ESI) technique on a Bruker Fourier Transform Ion Cyclotron Resonance Mass Spectrometer.
:methanol (4:1) as a solvent system. The IR spectra were recorded using a Shimadzu FTIR spectrophotometer. 1HNMR spectra were recorded using a Bruker (Avance III) 400 MHz system spectrometer with tetramethyl silane (TMS) as the internal standard and DMSO as the solvent. Chemical shifts were reported as parts per million (ppm). Signals were represented as follows: s: singlet, d: doublet, m: multiplet, and t: triplet. High-resolution mass spectra (HRMS) were obtained using the Electrospray Ionization (ESI) technique on a Bruker Fourier Transform Ion Cyclotron Resonance Mass Spectrometer.
The compounds were synthesized in a sequence of reactions from the starting material 2-aminopyridine (1). Diethyl ethoxy methylene malonate (2), p-chloro benzyl chloride/benzyl chloride (3) and 1,8-naphthyridine-3-carboxylic acid (4) were obtained using reported methods. The synthetic route to the title compound is illustrated in the following Scheme 1.35,38
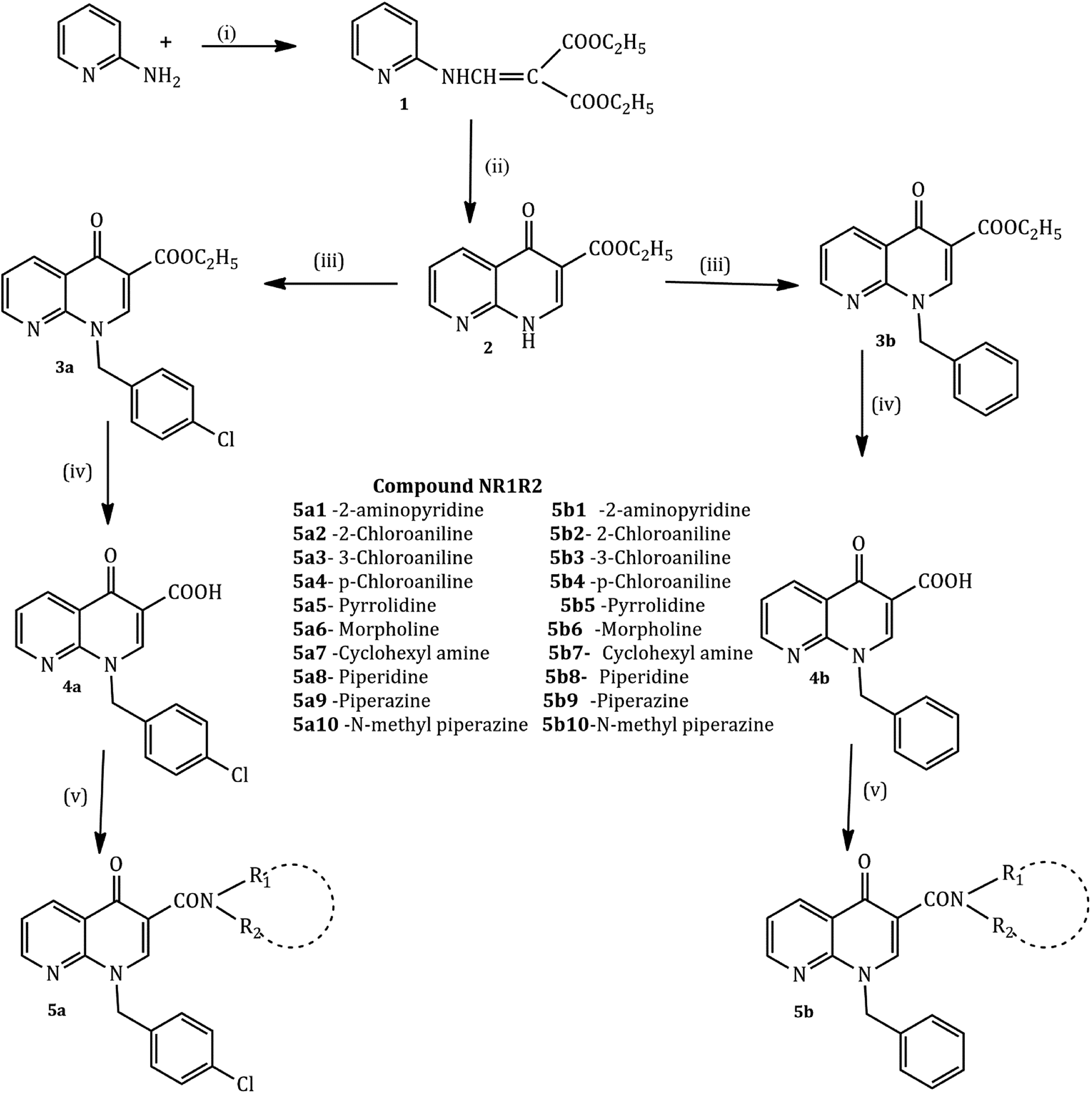 | ||
| Scheme 1 Preparation of 1,4-dihydro-4-oxo-1,8-naphthyridine-3-carboxylic acid derivatives. Reagents and conditions: (i) diethyl ethoxy methylene malonate, 120 °C, 1 h, (ii) diphenyl ether, 250 °C reflux, 4 h, (iii) p-chlorobenzylchloride/benzyl chloride, NaH, DMF, 90 °C reflux, 24 h, (iv) NaOH, EtOH, 100 °C reflux, 2 h and (v) R–NH R–NH2, heat 25–100 °C in a sealed tube for 24 h. | ||
2-Aminopyridine (1 mmol) and diethyl ethoxy methylene malonate (1 mmol) were heated at 120–130 °C for 2 h producing crude malonate (1) that was purified by recrystallization from light petroleum ether. The crude ester (1, 0.017 mol) and diphenyl ether in excess were heated at 240–250 °C for one hour, cooled at room temperature and washed with petroleum ether. The resulting white powder of malonate (2) was recrystallized from dimethylformamide. Sodium hydride (NaH, 1.81 mmol, 50% in mineral oil) and a solution of malonate (2) (1.5 mmol) in 10 mL of dry DMF were stirred at room temperature. After one hour, p-chlorobenzyl chloride (for 3a) and chlorobenzyl chloride (for 3b) (1 mmol each) were added and the mixture was stirred for 24 h. The resultant solution was evaporated and the addition of ethyl ether caused the precipitation of carboxylate 3a and 3b as pure solids. The 1,8-naphthyridine-3-carboxylic acid ethyl esters 3a and 3b (4.13 mmol) and a mixture of 10% sodium hydroxide (5 mL) and ethyl alcohol (5 mL) were refluxed for 2 h. After cooling, the solution was adjusted to pH 4 with aqueous 10% hydrochloric acid. The resulting precipitate of 4a and 4b was collected. A mixture of the 1,8-naphthyridine acids 4a and 4b (1 mmol) and appropriate amines (2-aminopyridine, 2-chloro aniline, cyclohexane amine, and n-phenyl piperazine) (10 mmol each) was heated in a sealed tube at 120° for 24 h. After cooling, the reaction mixtures were treated with ethyl ether to give the title compounds as a pure solid.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O keto), 1651.1 (CO amide), 734 (C–Cl); mass (m/z): 390 (M+), chemical formula: C21H15ClN4O2, MW 390.82, anal calc.: C, 64.54; H, 3.87; N, 14.34; found: C, 64.44; H, 3.78; N, 14.30%.O amide), 1651.1 (CO ring), 797.70 (C–Cl); mass m/z 423 (M+), chemical formula: C22H15Cl2N3O2, MW 423.29, anal calc.: C, 62.28; H, 3.56; N, 9.90; found: C, 62.21; H, 3.72; N, 9.81%.O keto), 1651.1 (CO amide), 780 (C–Cl); mass (m/z): 423 (M+), chemical formula: C22H15Cl2N3O2, MW 424.28, anal calc.: C, 62.28; H, 3.56; N, 9.90; found: C, 62.21; H, 3.72; N, 9.81%.O keto), 1651.1 (CO amide), 797.7 (C–Cl); mass m/z 423 (M+), chemical formula: C22H15Cl2N3O2, MW 424.28, anal calc.: C, 62.28; H, 3.56; N, 9.90; found: C, 62.28; H, 16.71; N, 9.90%.O keto), 1692.1 (CO amide), 781 (C–Cl); mass (m/z): 367 (M+), chemical formula: C20H18ClN3O2, MW 367.84, anal calc.: C, 65.31; H, 4.93; N, 11.42; found: C, 65.22; H, 4.84; N, 11.41%.O keto), 1692.1 (CO amide), 781.00 (C–Cl); mass (m/z): 383 (M+); chemical formula: C20H18ClN3O3, MW 383.84, anal calc.: C, 62.58; H, 4.73; N, 10.95; found: C, 62.51; H, 4.70; N, 10.86%.O keto), 1692.1 (CO amide), 781.0 (C–Cl); mass (m/z): 395 (M+), chemical formula: C22H22ClN3O2, MW 395.87, anal calc.: C, 66.75; H, 5.60; N, 10.61; found: C, 66.62; H, 5.42; N, 10.52%.O keto), 1651.1 (CO), 780.10 (C–Cl); mass (m/z): 381 (M+), chemical formula: C21H20ClN3O2, MW 381.87, anal calc.: C, 66.05; H, 5.28; N, 11.00; found: C, 66.01; H, 5.22; N, 10.87%.O amide), 1651.1 (CO ring), 797.8 (C–Cl); mass (m/z): 382 (M+), chemical formula: C20H19ClN4O2, MW 382.85, anal calc.: C, 62.74; H, 5.00; N, 14.63; found: C, 62.65; H, 5.01; N, 14.54%.O keto), 1692.1 (CO), 781.0 (C–Cl); mass (m/z): 396 (M+), chemical formula: C21H21ClN4O2, MW 396.87, anal calc.: C, 63.55; H, 5.33; N, 14.12; found: C, 63.48; H, 5.24; N, 14.09%.O keto), 1651.1 (CO ring), 780.10 (C–Cl); mass (m/z): 356 (M+), chemical formula: C21H16N4O2, MW 356.38, anal calc.: C, 70.77; H, 4.53; N, 15.72; found: C, 70.70; H, 4.46; N, 15.65%.O amide), 1651.1 (CO amide), 797.8 (C–Cl); mass (m/z): 389 (M+), chemical formula: C22H16ClN3O2, MW 389.84, anal calc.: C, 67.78; H, 4.14; N, 10.78; found: C, 67.71; H, 4.05; N, 10.70%.O keto), 1651.1 (CO amide), 780 (C–Cl); mass (m/z): 389 (M+), chemical formula: C22H16ClN3O2, MW 389.83, anal calc.: C, 67.78; H, 4.14; N, 10.78; found: C, 67.71; H, 4.05; N, 10.70%.O keto), 1651.1 (CO amide), 797.71 (C–Cl); mass (m/z): 389 (M+), chemical formula: C22H16ClN3O2, MW 389.83, anal calc.: C, 67.78; H, 4.14; N, 10.78; found: C, 67.71; H, 4.05; N, 10.70%.O keto), 1692.1 (CO) cm−1. Mass (m/z): 333 (M+), chemical formula: C20H19N3O2, MW 333.39, anal calc.: C, 72.05; H, 5.74; N, 12.60; found: C, 72.06; H, 5.75; N, 12.61%.O keto), 1692.1 (CO); mass (m/z): 349 (M+); chemical formula: C20H19N3O3, MW 349.37, anal calc.: C, 68.75; H, 5.48; N, 12.03; found: C, 68.62; H, 5.41; N, 12.00%.O keto), 1692.1 (CO amide), 781 (C–Cl). Mass (m/z): 361 (M+), chemical formula: C22H23N3O2, MW 361.44, anal calc.: C, 73.11; H, 6.41; N, 11.63; found: C, 73.03; H, 6.32; N, 11.53%.O keto), 1651.1 (CO); mass (m/z): 347 (M+), chemical formula: C21H21N3O2, MW 347.41, anal calc.: C, 72.60; H, 6.09; N, 12.10; found: C, 72.54; H, 6.01; N, 12.04%.O keto), 1651.2 (CO); mass (m/z): 348 (M+), chemical formula: C20H20N4O2, MW 348.41, anal calc.: C, 68.95; H, 5.79; N, 16.08; found: C, 68.86; H, 5.69; N, 16.02%.O keto), 1692.1 (CO); mass (m/z): 362 (M+), chemical formula: C21H22N4O2, MW 362.43, anal calc.: C, 69.59; H, 6.12; N, 15.46; found: C, 69.51; H, 6.03; N, 15.37%.
O keto), 1651.1 (CO amide), 734 (C–Cl); mass (m/z): 390 (M+), chemical formula: C21H15ClN4O2, MW 390.82, anal calc.: C, 64.54; H, 3.87; N, 14.34; found: C, 64.44; H, 3.78; N, 14.30%.O amide), 1651.1 (CO ring), 797.70 (C–Cl); mass m/z 423 (M+), chemical formula: C22H15Cl2N3O2, MW 423.29, anal calc.: C, 62.28; H, 3.56; N, 9.90; found: C, 62.21; H, 3.72; N, 9.81%.O keto), 1651.1 (CO amide), 780 (C–Cl); mass (m/z): 423 (M+), chemical formula: C22H15Cl2N3O2, MW 424.28, anal calc.: C, 62.28; H, 3.56; N, 9.90; found: C, 62.21; H, 3.72; N, 9.81%.O keto), 1651.1 (CO amide), 797.7 (C–Cl); mass m/z 423 (M+), chemical formula: C22H15Cl2N3O2, MW 424.28, anal calc.: C, 62.28; H, 3.56; N, 9.90; found: C, 62.28; H, 16.71; N, 9.90%.O keto), 1692.1 (CO amide), 781 (C–Cl); mass (m/z): 367 (M+), chemical formula: C20H18ClN3O2, MW 367.84, anal calc.: C, 65.31; H, 4.93; N, 11.42; found: C, 65.22; H, 4.84; N, 11.41%.O keto), 1692.1 (CO amide), 781.00 (C–Cl); mass (m/z): 383 (M+); chemical formula: C20H18ClN3O3, MW 383.84, anal calc.: C, 62.58; H, 4.73; N, 10.95; found: C, 62.51; H, 4.70; N, 10.86%.O keto), 1692.1 (CO amide), 781.0 (C–Cl); mass (m/z): 395 (M+), chemical formula: C22H22ClN3O2, MW 395.87, anal calc.: C, 66.75; H, 5.60; N, 10.61; found: C, 66.62; H, 5.42; N, 10.52%.O keto), 1651.1 (CO), 780.10 (C–Cl); mass (m/z): 381 (M+), chemical formula: C21H20ClN3O2, MW 381.87, anal calc.: C, 66.05; H, 5.28; N, 11.00; found: C, 66.01; H, 5.22; N, 10.87%.O amide), 1651.1 (CO ring), 797.8 (C–Cl); mass (m/z): 382 (M+), chemical formula: C20H19ClN4O2, MW 382.85, anal calc.: C, 62.74; H, 5.00; N, 14.63; found: C, 62.65; H, 5.01; N, 14.54%.O keto), 1692.1 (CO), 781.0 (C–Cl); mass (m/z): 396 (M+), chemical formula: C21H21ClN4O2, MW 396.87, anal calc.: C, 63.55; H, 5.33; N, 14.12; found: C, 63.48; H, 5.24; N, 14.09%.O keto), 1651.1 (CO ring), 780.10 (C–Cl); mass (m/z): 356 (M+), chemical formula: C21H16N4O2, MW 356.38, anal calc.: C, 70.77; H, 4.53; N, 15.72; found: C, 70.70; H, 4.46; N, 15.65%.O amide), 1651.1 (CO amide), 797.8 (C–Cl); mass (m/z): 389 (M+), chemical formula: C22H16ClN3O2, MW 389.84, anal calc.: C, 67.78; H, 4.14; N, 10.78; found: C, 67.71; H, 4.05; N, 10.70%.O keto), 1651.1 (CO amide), 780 (C–Cl); mass (m/z): 389 (M+), chemical formula: C22H16ClN3O2, MW 389.83, anal calc.: C, 67.78; H, 4.14; N, 10.78; found: C, 67.71; H, 4.05; N, 10.70%.O keto), 1651.1 (CO amide), 797.71 (C–Cl); mass (m/z): 389 (M+), chemical formula: C22H16ClN3O2, MW 389.83, anal calc.: C, 67.78; H, 4.14; N, 10.78; found: C, 67.71; H, 4.05; N, 10.70%.O keto), 1692.1 (CO) cm−1. Mass (m/z): 333 (M+), chemical formula: C20H19N3O2, MW 333.39, anal calc.: C, 72.05; H, 5.74; N, 12.60; found: C, 72.06; H, 5.75; N, 12.61%.O keto), 1692.1 (CO); mass (m/z): 349 (M+); chemical formula: C20H19N3O3, MW 349.37, anal calc.: C, 68.75; H, 5.48; N, 12.03; found: C, 68.62; H, 5.41; N, 12.00%.O keto), 1692.1 (CO amide), 781 (C–Cl). Mass (m/z): 361 (M+), chemical formula: C22H23N3O2, MW 361.44, anal calc.: C, 73.11; H, 6.41; N, 11.63; found: C, 73.03; H, 6.32; N, 11.53%.O keto), 1651.1 (CO); mass (m/z): 347 (M+), chemical formula: C21H21N3O2, MW 347.41, anal calc.: C, 72.60; H, 6.09; N, 12.10; found: C, 72.54; H, 6.01; N, 12.04%.O keto), 1651.2 (CO); mass (m/z): 348 (M+), chemical formula: C20H20N4O2, MW 348.41, anal calc.: C, 68.95; H, 5.79; N, 16.08; found: C, 68.86; H, 5.69; N, 16.02%.O keto), 1692.1 (CO); mass (m/z): 362 (M+), chemical formula: C21H22N4O2, MW 362.43, anal calc.: C, 69.59; H, 6.12; N, 15.46; found: C, 69.51; H, 6.03; N, 15.37%.2.2 Antihistaminic activity study
The antihistaminic screening of the synthesized compounds was carried out through the process of protection from histamine-induced constriction on guinea pig trachea. The animals were maintained in a fully controlled animal facility at 23 ± 2 °C, a relative humidity (RH) of 45–55%, and under a 12 h light–dark cycle; and they had sufficient standard nutritional animal feed and purified water. All the animals were acclimatized to their environment for one week before carrying out the experiments. The Institutional Animal Ethics Committee approved the protocol adopted for the experimentation on animals. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Pinnacle Biomedical Research Institute (PBRI), Bhopal, India and approved by the Animal Ethics Committee of the Institute (Approval reference number PBRI/IAEC/PN-17082). In all experimental procedures, efforts were made to minimize pain and suffering.A modified Van Arman technique was adopted to determine the in vivo antihistaminic potential of the synthesized derivatives.39 Male Dunkin Hartley Guinea pigs of either sex (250–300 g) were fasted prior to the experiment for 24 h. Three animals were chosen per individual group. The test compounds and reference drug (chlorpheniramine maleate) were given by oral administration at the dose level of 10 mg kg−1 of body weight suspended in 1% CMC and combined with histamine hydrochloride aerosol (aqueous solution 0.2%) in a vaponephrine Pocket Nebulizer and drizzled into a closed transparent chamber for 15 seconds. Proconvulsive (time for onset of convulsions) time in seconds, during the aerosol experiment, was measured. The delitescence of convulsion and tumble numbers for each animal were noted within a 6 min interval of exposure. The test animals remaining balanced (behaving normally) for over 6 min were treated as guarded against histamine-induced bronchoconstriction. Chlorpheniramine maleate was given intraperitoneal (Ip) at a dose level of 25 mg kg−1 of body weight for the restoration of the experimental animals. Bronchial challenge by histamine forced a bronchospastic response in all sensitive animals. The delay in the rise of the bronchospastic response was measured as a bronchoprotective effect and expressed in terms of percent protection compared to the control group.
| Percent protection = [1 − (T1/T2)] × 100 |
2.3 Sedative-hypnotic exercise
The sedative potential of the compounds was determined by measuring the decrease in locomotor activity using an astrophotometer (INCO, Ambala, India).40–46 Swiss albino mice of either sex (20–25 g) were chosen as test animals where six animals were kept per individual group in polypropylene cages in a controlled environment, with free access to food and water and maintained on a 12 h/12 h day/night cycle. The basal activity score for all animals was noted. Then, test compounds and the standard drug at the dose of 5 mg kg−1 of body weight were given by the oral route in a 1% CMC suspension. The activity score was noted at 1, 2 and 3 h after drug application. Sedation was determined by a substantial decline in locomotor activity. The percentage fall in the count was taken as the measure of sedative activity. The percent reduced locomotor activity was noted and expressed in terms of percentage fall in locomotor exercise determined by the formula:| Percent fall in locomotor activity = [(A − B)/A] × 100 |
The percentage decrease in photoactometer reading is reported as the degree of sedation produced.
2.4 Statistical analysis
Statistical evaluation of the biological activities of the test admixtures on various animals was performed by one-way (ANOVA) followed by Dunnett's test (manually). In all studies, the significance level of the means of each test group was calculated and compared with the control group. A level of p < 0.05 was considered significant in all cases.2.5 Molecular docking study
Molecular docking was performed using Auto Dock Tools (ADT) (version 1.5.4) and Auto Dock Vina (version 1_1_2 docking programs) (interactive molecular graphics programs) to understand the drug molecule interaction with the protein, the potential binding mode, and energy. The structures relating to the human histamine H1 receptor in complex with doxepin (PDB ID: 3RZE) retrieved from protein data bank (PDB).The ligands were drawn with Chem Draw Ultra version 12.0 (Cambridge Soft) followed by subsequent molecular mechanics (MM2) energy minimization of ligands using ChemBio3D Ultra version 12.0 with GAMESS Interface by assuring connection error in the bonds. These energy-minimized ligands (structures) were employed for molecular docking study. The torsions of the ligands were set by detecting the roots in Auto Dock Vina 1.1.2. Ligand preparation was done by adding Gasteiger charges.
Redundant chains, non-essential ions, water molecules, and ligands were discarded. The only exception was made for one structural water molecule that mediates a salt-bridge for the co-crystal ligand in both crystal structures. The PDBQT formats of ligands and protein were prepared by the AutoDock Tools 1.5.4 package (http://mgltools.scripps.edu).47 Protein and Grid preparation was done using Auto dock tools, and Auto dock Vina 1.1.2 (ref. 48) was used to perform molecular docking. The search grid of the histamine H1 receptor was identified as centre: 15.386, centre: 32.27 and centre: 20.835 with dimensions of size: 30, size: 28, and size: 28. The active site was defined to include all atoms within 6.0 Å radius of the native ligand. The first ten top-ranked docking poses were saved for each docking run. To validate the molecular docking protocol, the respective reference ligands were initially docked into the crystal structure of the enzymes.
2.6 ADME analysis
The success of a drug candidate is determined not only by its good potential but also by a satisfactory ADME profile. Because a wide variety of experimental media and high throughput in vitro ADME screens are available, it has the capacity to predict some important properties in silico and is valuable for analysis of the good qualities of the molecules. Nowadays, it is recommended that the employment of computational ADME, in combination with in vivo and in vitro predictions, should be done as early as possible in the drug discovery process so as to reduce the number of safety issues.49 Lipinski's rule of five is helpful in describing molecular properties of a drug candidate, which is necessary to evaluate the important pharmacokinetic parameters such as ADME. The rule is helpful in drug design and development of a potential drug molecule.50,51 The ADME study was carried out using the SWISS ADME predictor for the present investigation. This is a free web tool to evaluate the ADMET properties, like aqueous solubility (logS), skin permeability (logKp), synthetic accessibility score (SA), percentage absorption, pharmacokinetics, drug-likeness, and medicinal chemistry friendliness properties of small molecules.52 The criteria of molecular weight ≤500, ≤5 hydrogen bond donors (HBDs), 10 hydrogen bond acceptors (HBAs), and ≤10 rotatable bonds (RBs) were selected for the present study.53
The search engine further gave a compiled result on lipophilicity and hydrophilicity of these molecules by integrating results obtained from various logP and S prediction programs called ILOGP, XLOGP3, WLOGP, ESOL, and SILICOS-IT. logP, a measure of lipophilicity of a molecule, is the logarithm of the ratio of the concentration of a drug substance in two solvents in a unionized form. The lower the logP value, the stronger the lipophilicity of the chemical substance. The aqueous solubility of a compound significantly affects its absorption and distribution characteristics.54 On the other hand, low water solubility often leads to bad absorption, and, therefore, the general aim is to avoid poorly soluble compounds. logS is a unit expressing solubility, and it is the 10-based logarithm of the solubility measured in mol L−1. The distribution of logS in traded drugs reveals a value somewhere between −1 and −4 and will be optimized for better absorption and distribution of drugs in the body.
2.7 Prediction of activity spectra for substances (PASS)
We used PASS (Prediction of Activity Spectra for Substances, available at http://www.pharmaexpert.ru/PASSonline/predict.php) for computational screening of possible biological effects55 like antihistaminic and other related activities, i.e., antiallergic, anti-asthmatic, histamine release inhibition, rhinitis treatment, IgE antagonism, bronchodilation, immunomodulation, IL antagonism, PDE inhibition, 5HT3 antagonism, anti-inflammatory activities, etc. This software program designed a tool for evaluating the general biological potential of an organic drug-like candidate. PASS provides simultaneous predictions of a wide range of biological activity based on the structure of organic compounds. Thus, PASS can be used to estimate the biological activity profiles for virtual molecules, prior to their chemical synthesis and biological testing. It predicts the required pharmacological effect, as well as molecular mechanisms of action and generation of unnecessary side effects such as mutagenicity, teratogenicity, carcinogenicity, and embryotoxicity. This tool provided quantitative structure–activity relationships based on the decomposition of chemical structures using 2D and/or 3D descriptors, followed by the generation of models obtained from bioactive ligands.56 The activity was estimated in terms of Pa (probable activity) and Pi (probable inactivity). Structures with Pa greater than Pi were the only compounds considered for a particular pharmacological activity.573 Results and discussion
3.1 Synthetic chemistry
A series of cyclic amine substituted 1,8-naphthyridine-3-carboxylic acid analogues was efficiently synthesized based on the methods we had developed in our previous schemes.35The key compounds, 1-(4-chlorobenzyl)-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylic acid 4a and 1-benzyl-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylic acid 4b, were prepared as outlined via a four-step methodology in excellent yield.
The condensation reaction of 2-aminopyridine with ethoxy methylene malonate by Gould–Jacobs reaction yielded diethyl 2-((pyridine-2-ylamino)methylene) malonate 2 that was cyclized during refluxing with phenoxy ether to give ethyl 4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylate 3.58 N-alkylation of 3 in anhydrous DMF with appropriate alkyl chloride in the presence of sodium hydride gave the 1-alkyl-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylic acid ethyl esters. Hydrolysis in 10% aqueous NaOH solution yielded the corresponding 1-alkyl-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylic acids 4a and 4b. The coupling step was achieved in 24 h by heating the corresponding acids with the appropriate amines (2-aminopyridine, chloroaniline, morpholine, pyrrolidine, piperazine, etc.) in a dry DMF solution in a sealed tube. The target compounds 5a1–10 and 5b1–10 were purified by recrystallization from appropriate solvents.
The synthesized compounds were purified via column chromatography using a methanol:chloroform (10:40) mixture as the eluent. Characterization of the synthesized molecules was performed using FTIR spectroscopy, 1HNMR spectroscopy, elemental analysis and mass spectrometry, which confirmed the formation of the structure. Further, the structures were established by mass spectral data in accordance with their molecular formula. Compound 5a, for example, exhibited a molecular ion peak at m/z 390.2 (M+) in its mass spectrum. Its 1H NMR spectrum included the characteristic signals at 8.02 (d, 1H, Ar–H), 9.11 (d, 1H, Ar–H), 7.66 (d, 1H, Ar–H), and 7.76 (d, 2H Ar–H), corresponding to the naphthyridine ring structure.
3.2 Antihistaminic activity study
A series of compounds containing the 1,8-naphthyridine-3-carboxylic acid scaffold was evaluated for their in vivo antihistaminic activity. All the test compounds were found to exhibit comparable antihistaminic activity (Table 1). The percentage protection result showed that all the test compounds of the series exhibited comparable protection in the range of 52–61%. The pharmacological studies of the test compounds indicated that different substituents present at the third position of 1,8-naphthyridine-3-carboxylic acid exert varied pharmacological activity. Replacement of the hydroxyl group of the acid with alicyclic amine (pyrrolidinyl and piperidinyl compounds 5a5, 5a8, 5b5, and 5b8, respectively) showed good antihistaminic activity. Replacement of the heterocyclic amine (morpholinyl, piperazinyl and N-methyl piperazinyl compounds 5a6, 5a9, 5a10, 5b6, 5b9 and 5a10, respectively) showed comparable antihistaminic activity. Furthermore, replacement with aromatic amine (amino pyridinyl and cyclohexylamine compounds 5a1, 5a7, 5b1 and 5b7, respectively) led to a further increase in activity. Amongst the series, compound 5a1 (pyridinyl derivative) was the most potent with the percentage protection of 61.45 that is comparable to that of the standard drug (65.20%).| Antihistaminic activity | Percent CNS depression | ||||
|---|---|---|---|---|---|
| Compd | Time of onset of convulsion (s) | % Protection | 1 h | 2 h | 3 h |
| a Each value represents the mean ± SEM (n = 6). Significance levels *p < 0.001, **p > 0.05. | |||||
| 5a1 | 371 ± 6.53* | 61.45 ± 0.15* | 13.02 ± 0.22** | 13.60 ± 1.26** | 9.81 ± 1.52** |
| 5a2 | 352 ± 2.46* | 59.38 ± 0.19* | 9.12 ± 1.02** | 9.15 ± 1.02** | 6.00 ± 0.58** |
| 5a3 | 352 ± 1.82* | 59.37 ± 0.14* | 10.14 ± 0.56** | 11.16 ± 0.77** | 8.03 ± 0.91** |
| 5a4 | 341 ± 2.40* | 58.06 ± 0.16* | 7.11 ± 0.88** | 8.16 ± 0.73** | 5.01 ± 0.43** |
| 5a5 | 311 ± 2.43* | 54.01 ± 0.14* | 8.16 ± 0.73** | 8.19 ± 0.76** | 6.00 ± 0.58** |
| 5a6 | 328 ± 2.86* | 56.40 ± 0.19* | 7.11 ± 0.88** | 7.17 ± 0.87** | 5.01 ± 0.43** |
| 5a7 | 305 ± 1.52* | 53.11 ± 0.13* | 7.11 ± 0.88** | 9.81 ± 1.52** | 6.00 ± 0.58** |
| 5a8 | 340 ± 4.29* | 57.94 ± 0.36* | 6.00 ± 0.58** | 7.17 ± 0.87** | 5.18 ± 0.4** |
| 5a9 | 316 ± 4.56* | 54.74 ± 0.42* | 8.18 ± 0.74** | 10.13 ± 0.55** | 6.07 ± 0.59** |
| 5a10 | 311 ± 2.46* | 54.01 ± 0.23* | 9.61 ± 1.42** | 10.11 ± 0.56** | 7.11 ± 0.88** |
| 5b1 | 354 ± 5.33* | 59.60 ± 0.352* | 8.58 ± 0.09** | 11.17 ± 0.10** | 5.29 ± 0.11** |
| 5b2 | 353 ± 3.87* | 59.49 ± 0.284* | 9.53 ± 0.13** | 11.2 ± 0.09** | 5.16 ± 0.08** |
| 5b3 | 366 ± 3.94* | 60.92 ± 0.315* | 6.43 ± 0.26** | 8.10 ± 0.09** | 4.67 ± 0.13** |
| 5b4 | 354 ± 4.23* | 59.60 ± 0.265* | 8.28 ± 0.10** | 9.42 ± 0.17** | 5.09 ± 0.10** |
| 5b5 | 355 ± 6.42* | 59.71 ± 1.53* | 8.23 ± 1.23** | 12.1 ± 1.51** | 6.1 ± 1.62** |
| 5b6 | 304 ± 6.69* | 52.96 ± 1.42* | 9.23 ± 1.72** | 13.1 ± 1.60** | 9.1 ± 1.54** |
| 5b7 | 305 ± 6.38* | 53.11 ± 1.28* | 11.10 ± 1.52** | 14.1 ± 1.62** | 7.2 ± 1.93** |
| 5b8 | 359 ± 5.41* | 60.16 ± 1.39* | 12.9 ± 1.72** | 15.1 ± 1.83** | 9.1 ± 1.7** |
| 5b9 | 301 ± 5.87* | 52.49 ± 1.72* | 6.21 ± 1.94** | 12.1 ± 1.87** | 6.1 ± 1.92** |
| 5b10 | 361 ± 6.72* | 60.38 ± 1.40* | 8.29 ± 1.93** | 10.1 ± 1.04** | 8.1 ± 1.87** |
| Control | 143 ± 3.29* | — | 6.10 ± 0.49** | 4.1 ± 0.59** | 4 ± 0.91** |
| Chlorpheniramine | 411 ± 4.43* | 65.20 ± 0.33* | 38.80 ± 0.32** | 34.80 ± 0.72** | 29.58 ± 0.72** |
3.3 Sedative-hypnotic exercise
Sedation is the most common side effect associated with more than 20% of antihistamines used by human beings. Hence, in vivo sedative–hypnotic activity was also evaluated for the test compounds. The percentage fall in photoactometer count was measured as the degree of sedation produced. The results of this study shown in Table 1 demonstrate that most of the tested compounds exhibited insignificant sedation (less than 10%). Chlorpheniramine maleate (first-generation H1R inhibitor) and Cetirizine (second-generation H1R inhibitor) were used as reference drugs for this study. Chlorpheniramine maleate showed approximately 30% sedation and cetirizine showed around 10–12% sedation. All the test compounds showed lower sedative potentials; while compared to the reference drug, the test compounds exhibited equipotent activity. Thus, this series can be developed as a clinically useful novel class of non-sedative antihistamines.3.4 Molecular docking study
Molecular docking is a valuable technique in computational chemistry to deeply analyse ligand recognition and it has led to important breakthroughs in drug discovery and design in the field of medicinal chemistry.59 Molecular docking methodology explores the binding mode and affinity of a small molecule within the binding site of the receptor target protein. The docked ligands were ranked according to their binding affinity in ligand–receptor complexes (Fig. 3 and 4a–f).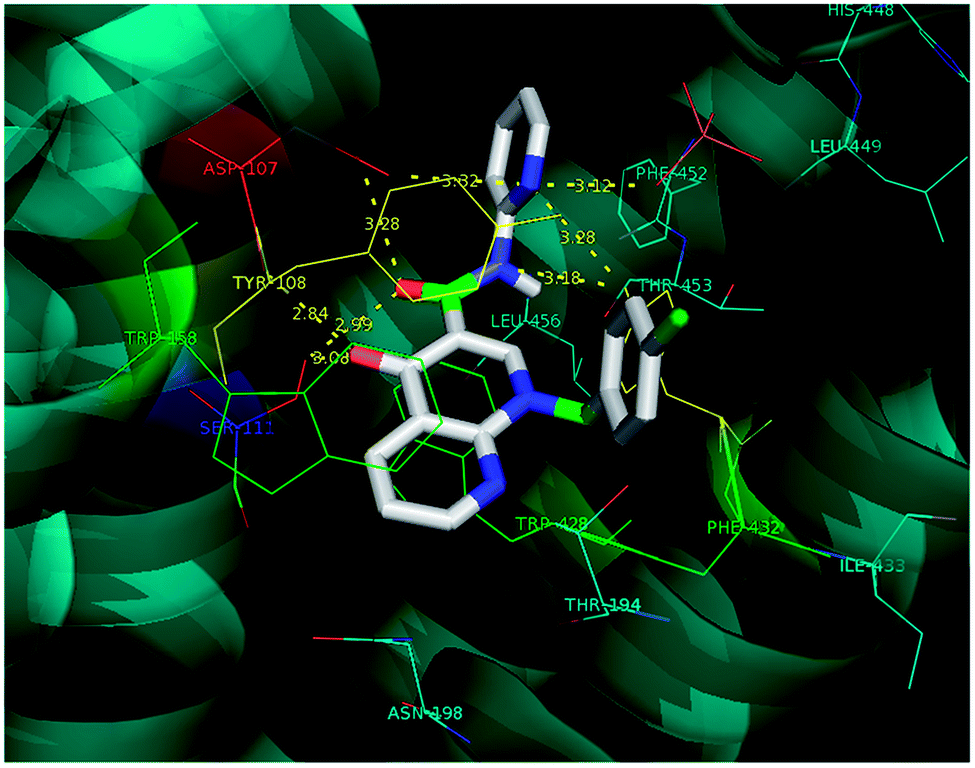 | ||
| Fig. 3 The docking poses of 5a1 in the binding site of the human histamine H1 receptor in the complex (PDB ID: 3RZE). | ||
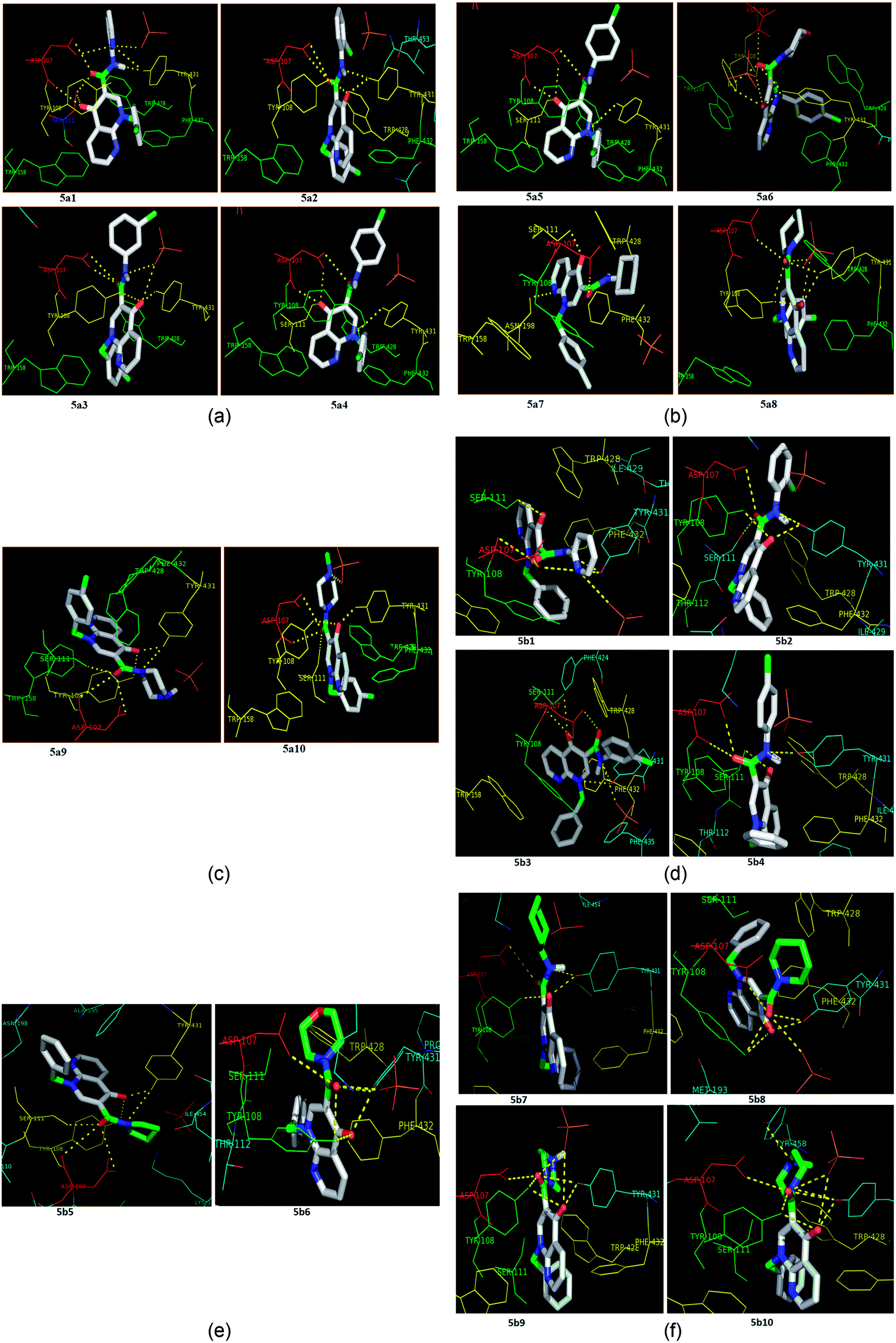 | ||
| Fig. 4 (a) The docking poses of 5a1–5a4 in the binding site of the human histamine H1 receptor in the complex (PDB ID: 3RZE). (b) The docking poses of 5a5–5a8 in the binding site of the human histamine H1 receptor in the complex (PDB ID: 3RZE). (c) The docking poses of 5a9 and 5a10 in the binding site of the human histamine H1 receptor in the complex (PDB ID: 3RZE). (d) The docking poses of 5b1–5b4 in the binding site of the human histamine H1 receptor in the complex (PDB ID: 3RZE). (e) The docking poses of 5b5 and 5b6 in the binding site of the human histamine H1 receptor in the complex (PDB ID: 3RZE). (f) The docking poses of 5b7–5b10 in the binding site of the human histamine H1 receptor in the complex (PDB ID: 3RZE). | ||
Molecular docking was performed on a set of test compounds, viz. 5a1–10 and 5b1–10, against the H1 receptor in order to identify the critical ligand–protein interactions. The scores due to docking for the tested ligands determined that all the prepared admixtures possessed potential for interaction with one or more amino acids in the active site (binding pocket) of the receptor. Compound 5a1 was the most potent inhibitor of the receptor. In the docking, the binding of 5a1 was supported by three hydrophilic interactions between residues Asp107, Ser111, and Tyr431 and the ligand at bond distances of 2.84 and 3.08 and 3.18 Å. According to the dockings, compounds 5a2–10 and 5b1–10 shared a similar binding profile with 5a and also showed two hydrophilic interactions with residue Asp107. The docked ligands were found to have a similar binding capacity to the co-crystallized ligands. Theoretically, the entire synthesized compounds showed moderate to good docking scores and binding energy to the selected protein target ranged from −7.1 to −10.6 kcal mol−1, which is in good agreement with the observed antihistaminic activity. Most of the test ligands showed comparable docking scores in comparison to the reference drug azelastine, which was used as a reference for interpretations of biological study and analysis. Amongst these compounds, the complex of 5b2 (o-chloroaniline derivative) showed high binding affinities during the docking study in the active sites of the H1R receptor (PDB ID: 3RZE). The results are shown in Table 2.
| Comd | No. of H bonds | Docking score kcal mol−1 | H-bond with amino acid |
|---|---|---|---|
| 5a1 | 7 | −7.9 | A:Asp-107, Ser111, Tyr431 |
| 5a2 | 5 | −9.5 | A:Asp-107, Tyr431, Tyr108 |
| 5a3 | 5 | −7.1 | A:Asp-107, Tyr431, Tyr108 |
| 5a4 | 5 | −7.5 | A:Asp-107, Tyr431, Ser111 |
| 5a5 | 8 | −8.2 | A:Asp-107, Tyr431, Tyr108 |
| 5a6 | 7 | −7.2 | A:Asp-107, Tyr431, Tyr108 |
| 5a7 | 3 | −7.4 | A:Asp-107, Ser111, Asb198 |
| 5a8 | 7 | −7.4 | A:Asp-107, Tyr431, Tyr108 |
| 5a9 | 5 | −7.6 | A:Asp-107, Tyr431, Tyr108, Ser111 |
| 5a10 | 6 | −7.1 | A:Asp-107, Tyr431, Tyr108, Ser111 |
| 5b1 | 7 | −8.0 | A:Asp-107, Tyr431, Ser111 |
| 5b2 | 5 | −9.8 | A:Asp-107, Tyr431, Tyr108, Ser111 |
| 5b3 | 7 | −7.4 | A:Asp-107, Tyr431, Ser111 |
| 5b4 | 4 | −7.7 | A:Asp-107, Tyr431, Ser111 |
| 5b5 | 6 | −8.4 | A:Asp-107, Tyr431, Tyr108, Ser111 |
| 5b6 | 6 | −7.2 | A:Asp-107, Tyr431, Tyr108, Ser111 |
| 5b7 | 5 | −8.2 | A:Asp-107, Tyr431, Tyr108 |
| 5b8 | 6 | −8.6 | A:Asp-107, Tyr431, Tyr108 |
| 5b9 | 7 | −7.4 | A:Asp-107, Tyr431, Tyr108 |
| 5b10 | 8 | −8.2 | A:Asp-107, Tyr431, Tyr108, Tyr458 |
| Azelastine | 3 | −10.6 | A:Asp-107, Tyr431, Ser111 |
3.5 ADME analysis
The results obtained from in silico studies clearly indicate that the compounds had drug-like candidate properties with no violation of any of the drug-likeness rules discussed above. It was interesting to note that the results of the SWISS ADME predictor values of logP, molar refractivity and the total polar surface area in these molecules were in excellent agreement with the most important rules of drug-likeness.
Though these compounds exhibited a good hydrophilic–lipophilic balance and the same predicted bioavailability, the halogen derivative with high lipophilicity was expected to show decent GI absorption. In addition, we calculated the total polar surface area (TPSA) since it is another key property that is related to drug bioavailability. Thus, passively absorbed molecules with TPSA >140 are thought to have low oral bioavailability. The results obtained from the Swiss ADME search engine are listed in Table 3.
| Compd | MW | R-bonds | H-A | H-D | TPSA | MR | WlogP (lipophilicity) |
ESOL logS |
GI absorption | BBB permeant | logKp (cm s−1) |
Lipinski violations | PAINS alerts | Synthetic accessibility |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a R bond = rotatable bond, H-A = hydrogen bond acceptor, H-D = hydrogen bond donor, TPSA = topological polar surface area, logP = lipophilicity, logS = water solubility, logKp = permeability coefficient, PAINS = pan-assay interference structure, CLPH = chlorpheniramine, AZT = azelastine. |
||||||||||||||
| 5a1 | 390.8 | 5 | 4 | 1 | 76.88 | 108.77 | 3.55 | −4.65 | High | Yes | −6.28 | 0 | 0 | 2.67 |
| 5a2 | 424.3 | 5 | 3 | 1 | 63.99 | 115.99 | 4.81 | −5.69 | High | Yes | −5.52 | 0 | 0 | 2.59 |
| 5a3 | 424.3 | 5 | 3 | 1 | 63.99 | 115.99 | 4.81 | −5.69 | High | Yes | −5.52 | 0 | 0 | 2.58 |
| 5a4 | 424.3 | 5 | 3 | 1 | 63.99 | 115.99 | 4.81 | −5.69 | High | Yes | −5.52 | 0 | 0 | 2.56 |
| 5a5 | 367.8 | 4 | 3 | 0 | 55.2 | 106.07 | 2.95 | −4.35 | High | Yes | −6.24 | 0 | 0 | 2.46 |
| 5a6 | 383.8 | 4 | 4 | 0 | 64.43 | 107.16 | 2.19 | −3.89 | High | Yes | −6.95 | 0 | 0 | 2.58 |
| 5a7 | 395.9 | 5 | 3 | 1 | 63.99 | 111.68 | 4.16 | −5.15 | High | Yes | −5.61 | 0 | 0 | 2.69 |
| 5a8 | 381.9 | 4 | 3 | 0 | 55.2 | 110.88 | 3.34 | −4.65 | High | Yes | −6.07 | 0 | 0 | 2.57 |
| 5a9 | 382.8 | 4 | 4 | 1 | 67.23 | 112.79 | 1.38 | −3.71 | High | Yes | −7.14 | 0 | 0 | 2.61 |
| 5a10 | 396.9 | 4 | 4 | 0 | 58.44 | 117.69 | 1.72 | −4.07 | High | Yes | −6.9 | 0 | 0 | 2.72 |
| 5b1 | 356.4 | 5 | 4 | 1 | 76.88 | 103.76 | 2.9 | −4.06 | High | Yes | −6.51 | 0 | 0 | 2.65 |
| 5b2 | 389.8 | 5 | 3 | 1 | 63.99 | 110.98 | 4.16 | −5.11 | High | Yes | −5.75 | 0 | 0 | 2.56 |
| 5b3 | 389.8 | 5 | 3 | 1 | 63.99 | 110.98 | 4.16 | −5.11 | High | Yes | −5.75 | 0 | 0 | 2.55 |
| 5b4 | 389.8 | 5 | 3 | 1 | 63.99 | 110.98 | 4.16 | −5.11 | High | Yes | −5.75 | 0 | 0 | 2.53 |
| 5b5 | 333.4 | 4 | 3 | 0 | 55.2 | 101.06 | 2.3 | −3.76 | High | Yes | −6.48 | 0 | 0 | 2.41 |
| 5b6 | 349.4 | 4 | 4 | 0 | 64.43 | 102.15 | 1.54 | −3.3 | High | Yes | −7.19 | 0 | 0 | 2.54 |
| 5b7 | 361.4 | 5 | 3 | 1 | 63.99 | 106.67 | 3.51 | −4.55 | High | Yes | −5.84 | 0 | 0 | 2.65 |
| 5b8 | 347.4 | 4 | 3 | 0 | 55.2 | 105.87 | 2.69 | −4.06 | High | Yes | −6.31 | 0 | 0 | 2.52 |
| 5b9 | 348.4 | 4 | 4 | 1 | 67.23 | 107.78 | 0.73 | −3.12 | High | No | −7.38 | 0 | 0 | 2.57 |
| 5b10 | 362.4 | 4 | 4 | 0 | 58.44 | 112.68 | 1.07 | −3.48 | High | Yes | −7.13 | 0 | 0 | 2.68 |
| CLPH | 274.8 | 5 | 2 | 0 | 16.13 | 108.77 | 3.82 | −3.82 | High | Yes | −5.57 | 0 | 0 | 2.7 |
| Azelastine | 381.9 | 3 | 3 | 0 | 38.13 | 115.99 | 3.92 | −5.20 | High | Yes | −5.53 | 1 | 0 | 3.62 |
The analysis indicates that the derivatives fell within the permissible range of standard drugs, as is evident from the boiled-egg diagram (Fig. 5a–c and 6).
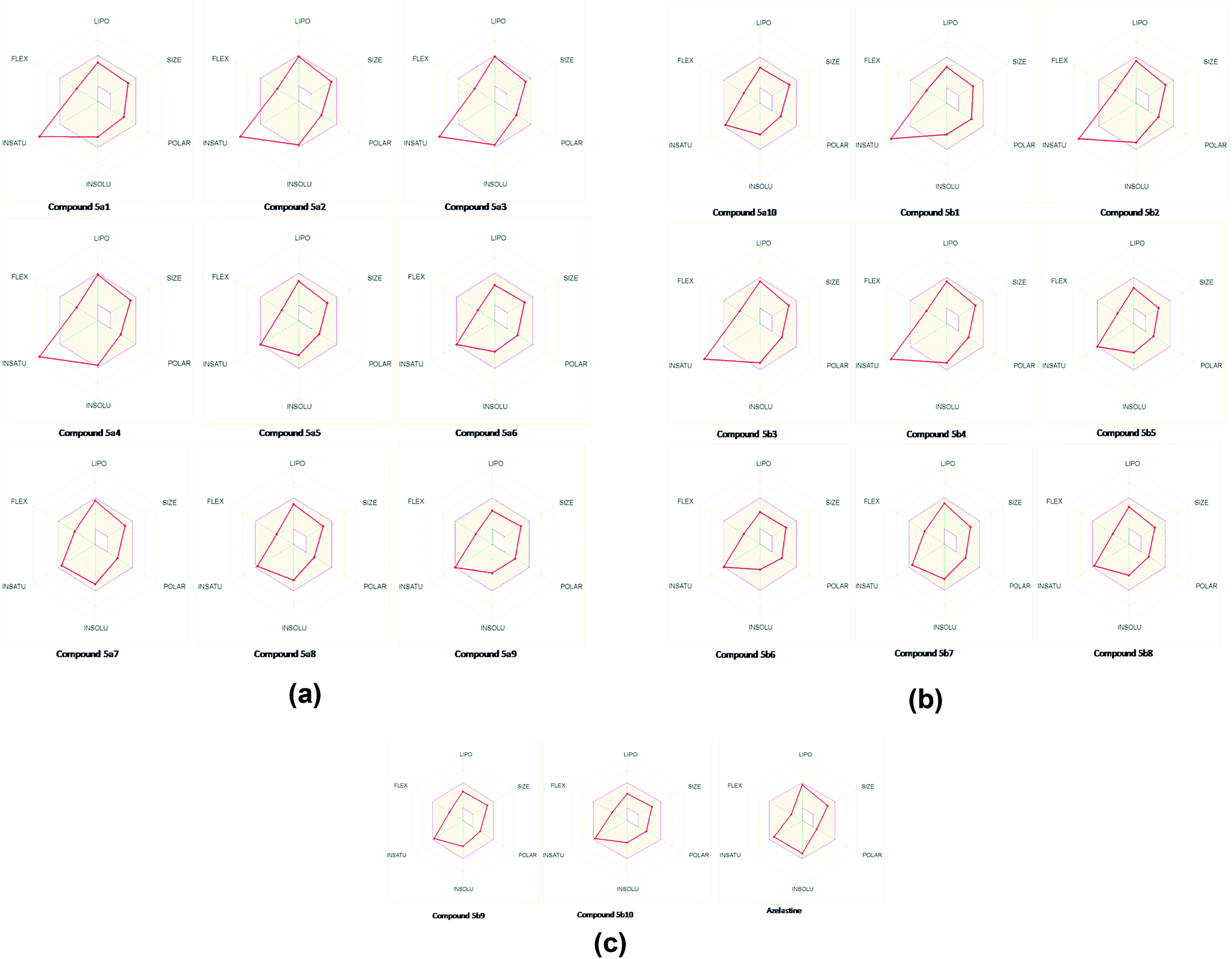 | ||
| Fig. 5 (a) Bioavailability radar graph of 1,8-naphthyridine derivatives 5a1–a9 (pink area reflects the allowed values of drug likeness properties of the molecule). (b) Bioavailability radar graph of 1,8-naphthyridine derivatives 5a10–5b8 (pink area reflects the allowed values of drug-likeness properties of the molecule). (c) Bioavailability radar graph of 1,8-naphthyridine derivatives 5b9–5b10 (pink area reflects the allowed values of drug-likeness properties of the molecule). | ||
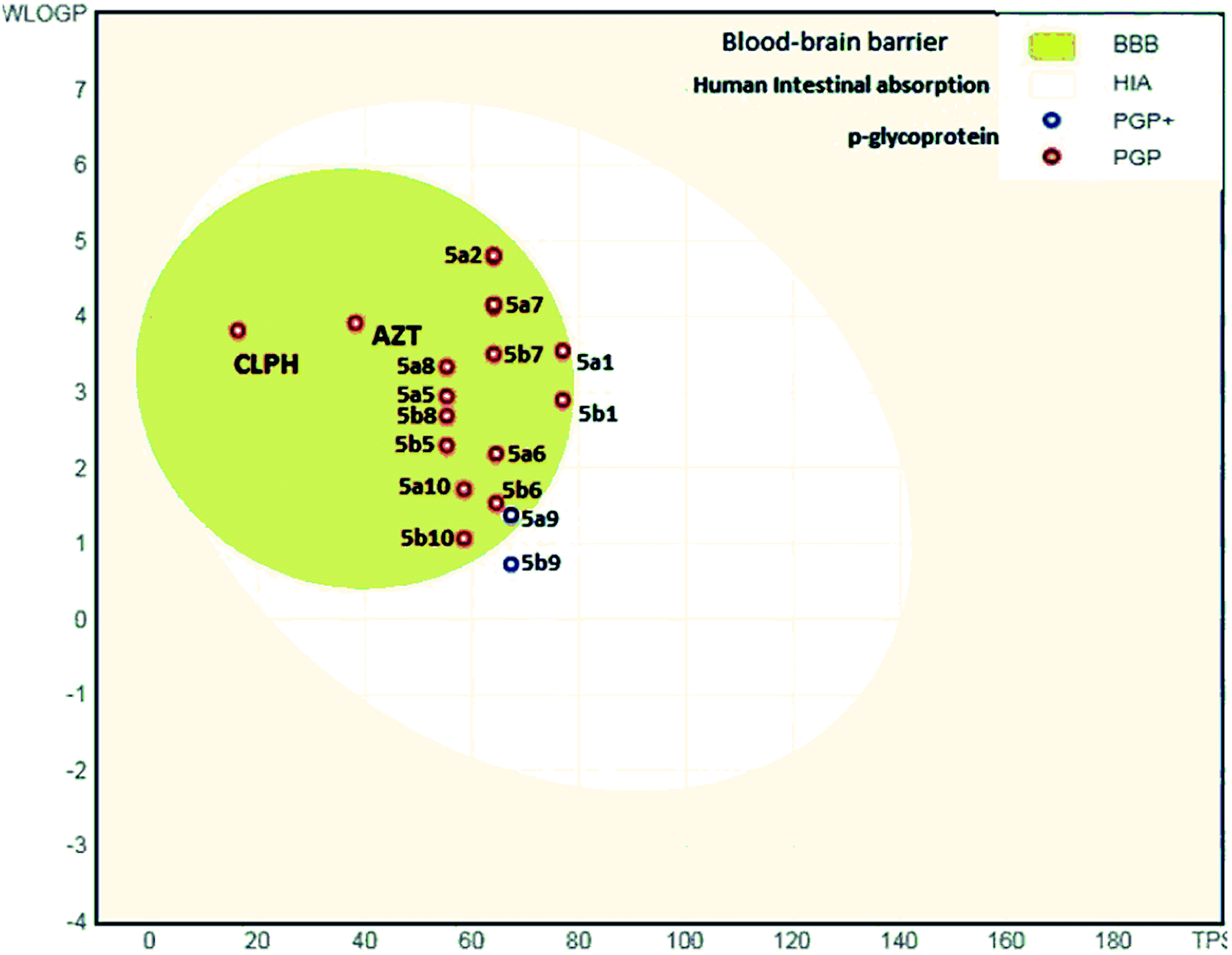 | ||
| Fig. 6 ADME properties of compounds 5a1–a10 and 5b1–b10 and azelastine by graphical representation (boiled-egg) (predict gastrointestinal absorption and brain penetration of small molecules). | ||
It is also clear that compounds 5a1–5a10 and 5b1–5b10 cannot be affected by the P-glycoprotein of the CNS system. Compounds present in the yellow zone except 5a9 and 5b9 can permeate through the blood–brain barrier (BBB). All compounds present in the white area can be absorbed very easily by the gastrointestinal tract. In the present study, the synthesized ligand and its complexes were found to be in good agreement with the given criteria and can be said to possess good oral bioavailability.
3.6 Biological activity spectrum PASS analysis
The biological activity spectra of the synthesized compounds were determined by using an online version of PASS software. The results obtained (Tables 4 and 5) were interpreted and used in a flexible manner. The antihistaminic activities of the derivatives were only slightly changed by amidation at position 3. However, an additional chloro-substituent increased the activity remarkably, as was observed for compound 5a3. Compound 5a3 showed the highest Pa for H1R inhibitory activity (0.359), whereas compound 5b1 showed the highest Pa for antiallergic and antiasthmatic activity (0.575 and 0.520, respectively), compound 5a2 showed the highest Pa for histamine release inhibitor (0.373), compound 5a9 showed the highest Pa for rhinitis treatment (0.479), and compound 5b6 showed the highest Pa for IL antagonist activity (0.300).| Activity name | 5a1 | 5a2 | 5a3 | 5a4 | 5a5 | 5a6 | 5a7 | 5a8 | 5a9 | 5a10 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | |
| a Hks – histidine kinase, PDE – phosphodiesterase, IgE – immunoglobulin E, H1R – histamine H1 receptor, HRI – histamine release inhibitor, IL – interleukin, Pa (probability “to be active”), Pi (probability “to be inactive”), NA – not available, AIFML – anti-inflammatory, IMM – immunomodulatory. | ||||||||||||||||||||
| Antiallergic | 0.556 | 0.020 | 0.528 | 0.024 | 0.521 | 0.024 | 0.544 | 0.022 | 0.294 | 0.096 | 0.297 | 0.095 | 0.500 | 0.028 | 0.311 | 0.088 | 0.256 | 0.125 | 0.284 | 0.103 |
| Antiasthmatic | 0.490 | 0.029 | 0.479 | 0.030 | 0.450 | 0.035 | 0.468 | 0.032 | 0.193 | 0.180 | 0.204 | 0.167 | 0.447 | 0.036 | 0.209 | 0.162 | 0.192 | 0.180 | NA | NA |
| Antihistaminic | NA | NA | NA | NA | 0.237 | 0.210 | NA | NA | 0.171 | 0.053 | 0.140 | 0.079 | NA | NA | 0.185 | 0.045 | 0.145 | 0.073 | 0.188 | 0.043 |
| AIFML | 0.259 | 0.202 | 0.254 | 0.167 | 0.113 | 0.071 | 0.255 | 0.164 | NA | NA | NA | NA | 0.240 | 0.201 | NA | NA | NA | NA | NA | NA |
| Histamine antagonist | NA | NA | NA | NA | NA | NA | NA | NA | 0.112 | 0.071 | NA | NA | NA | NA | 0.122 | 0.062 | 0.097 | 0.094 | 0.123 | 0.061 |
| Bronchodilator | 0.127 | 0.056 | 0.138 | 0.047 | 0.166 | 0.098 | 0.143 | 0.044 | NA | NA | NA | NA | 0.187 | 0.024 | NA | NA | NA | NA | NA | NA |
| H1R agonist | 0.161 | 0.104 | 0.166 | 0.098 | 0.359 | 0.115 | 0.165 | 0.099 | 0.179 | 0.083 | 0.078 | 0.070 | 0.238 | 0.040 | 0.171 | 0.092 | 0.208 | 0.057 | 0.165 | 0.100 |
| H1R antagonist | NA | NA | NA | NA | NA | NA | NA | NA | 0.100 | 0.045 | NA | NA | NA | NA | 0.094 | 0.051 | 0.092 | 0.053 | 0.153 | 0.025 |
| HRI | NA | NA | 0.373 | 0.107 | 0.310 | 0.096 | 0.353 | 0.118 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Hks | 0.321 | 0.088 | 0.310 | 0.096 | 0.080 | 0.038 | 0.320 | 0.089 | 0.333 | 0.081 | 0.318 | 0.090 | 0.288 | 0.111 | 0.321 | 0.088 | 0.309 | 0.096 | 0.310 | 0.096 |
| IgE antagonist | 0.092 | 0.018 | NA | NA | 0.140 | 0.023 | 0.084 | 0.029 | NA | NA | NA | NA | 0.092 | 0.017 | NA | NA | NA | NA | NA | NA |
| IMM | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | 0.198 | 0.161 | NA | NA | NA | NA | NA | NA |
| IL antagonist | 0.141 | 0.139 | 0.158 | 0.127 | 0.152 | 0.124 | 0.152 | 0.135 | NA | NA | 0.225 | 0.044 | NA | NA | NA | NA | NA | NA | NA | NA |
| PDE inhibitor | 0.150 | 0.026 | 0.159 | 0.023 | 0.149 | 0.026 | 0.168 | 0.020 | 0.084 | 0.066 | 0.093 | 0.057 | 0.158 | 0.023 | 0.094 | 0.056 | 0.082 | 0.067 | 0.094 | 0.052 |
| Rhinitis treatment | 0.317 | 0.093 | 0.282 | 0.125 | 0.311 | 0.098 | 0.312 | 0.097 | 0.333 | 0.081 | 0.350 | 0.072 | 0.269 | 0.140 | 0.326 | 0.087 | 0.479 | 0.023 | 0.363 | 0.066 |
| Activity name | 5b1 | 5b2 | 5b3 | 5b4 | 5b5 | 5b6 | 5b7 | 5b8 | 5b9 | 5b10 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | |
| a Hks – histidine kinase, PDE – phosphodiesterase, IgE – immunoglobulin E, H1R – histamine H1 receptor Pa (probability “to be active”), Pi (probability “to be inactive”), NA – not available. | ||||||||||||||||||||
| Antiallergic | 0.575 | 0.018 | 0.536 | 0.023 | 0.521 | 0.024 | 0.534 | 0.023 | 0.280 | 0.106 | 0.283 | 0.104 | 0.505 | 0.027 | 0.296 | 0.095 | 0.240 | 0.141 | 0.269 | 0.114 |
| Antiasthmatic | 0.520 | 0.025 | 0.490 | 0.029 | 0.450 | 0.035 | 0.458 | 0.034 | 0.176 | 0.050 | 0.195 | 0.177 | 0.461 | 0.033 | 0.200 | 0.172 | 0.149 | 0.069 | NA | NA |
| Antihistaminic | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | 0.143 | 0.075 | NA | NA | 0.189 | 0.043 | NA | NA | 0.193 | 0.041 |
| AIFML | 0.245 | 0.218 | NA | NA | 0.237 | 0.210 | 0.255 | 0.165 | NA | NA | NA | NA | 0.257 | 0.161 | NA | NA | NA | NA | NA | NA |
| Histamine antagonist | NA | NA | NA | NA | NA | NA | NA | NA | 0.116 | 0.067 | 0.096 | 0.095 | NA | NA | 0.128 | 0.058 | 0.100 | 0.089 | 0.129 | 0.057 |
| Bronchodilator | 0.156 | 0.037 | NA | 0.041 | 0.113 | 0.071 | 0.140 | 0.046 | NA | NA | NA | NA | 0.219 | 0.017 | NA | NA | NA | NA | NA | NA |
| H1R agonist | NA | NA | 0.148 | 0.113 | 0.166 | 0.098 | 0.173 | 0.089 | 0.151 | 0.121 | NA | NA | 0.218 | 0.050 | 0.143 | 0.137 | 0.183 | 0.078 | NA | NA |
| H1R antagonist | NA | NA | NA | NA | NA | NA | NA | NA | 0.095 | 0.050 | NA | NA | NA | NA | 0.089 | 0.056 | 0.087 | 0.059 | 0.139 | 0.028 |
| HRI | 0.256 | 0.180 | 0.155 | 0.106 | 0.359 | 0.115 | 0.355 | 0.118 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Hks | 0.261 | 0.138 | 0.375 | 0.107 | 0.310 | 0.096 | 0.321 | 0.088 | 0.271 | 0.126 | 0.260 | 0.139 | 0.232 | 0.175 | 0.261 | 0.138 | 0.250 | 0.151 | 0.250 | 0.151 |
| IgE antagonist | 0.105 | 0.008 | 0.294 | 0.057 | 0.080 | 0.038 | 0.085 | 0.028 | NA | NA | NA | NA | 0.107 | 0.008 | NA | NA | NA | NA | NA | NA |
| IMM | 0.222 | 0.132 | NA | NA | NA | NA | 0.196 | 0.164 | NA | NA | 0.194 | 0.167 | 0.236 | 0.115 | NA | NA | NA | NA | NA | NA |
| IL antagonist | 0.166 | 0.011 | 0.181 | 0.028 | 0.140 | 0.023 | 0.138 | 0.025 | NA | NA | 0.300 | 0.015 | 0.270 | 0.028 | 0.250 | 0.041 | NA | NA | NA | NA |
| PDE inhibitor | 0.194 | 0.015 | 0.127 | 0.020 | 0.149 | 0.026 | 0.069 | 0.034 | 0.109 | 0.046 | 0.118 | 0.040 | 0.120 | 0.039 | 0.068 | 0.037 | 0.106 | 0.048 | 0.113 | 0.043 |
| Rhinitis treatment | 0.294 | 0.113 | 0.057 | 0.133 | 0.311 | 0.098 | 0.309 | 0.099 | NA | NA | 0.241 | 0.176 | 0.300 | 0.107 | 0.474 | 0.025 | 0.336 | 0.080 | 0.336 | 0.080 |
4 Conclusions
The synthesis, characterization, in silico ADME and in vivo antihistaminic activity of a new series of 1,8-naphthyridine-3-carboxylic acid derivatives have been described in this manuscript. A combination of cyclic amines with a naphthyridine ring and possession of sufficient hydrogen donor–acceptor sites resulted in good antihistaminic activity.These compounds showed significant H1R inhibitory (antihistaminic) activity through histamine-induced bronchoconstriction on conscious guinea pigs by the in vivo model. Out of the synthesized compounds, the pyridyl derivative 5a1 showed maximum antihistaminic activity with 61.45% protection. Further, the average sedative properties of the synthesised compounds were found to be very small (<16%) when compared to the standard drug chlorpheniramine maleate (38%). The molecules showed promising in silico results, as indicated by their significant scoring functions and high protein–ligand interaction energy, which simultaneously predicted the activity of the test compounds. In conjugation with the in silico results, the compounds showed very promising antihistaminic activities. The in silico ADME profiling, toxicity, drug-likeness, drug-scoring results, PASS analysis and in vitro antihistaminic activities suggested that the compounds are promising leads for the development of selective, safe and potent antihistamines. The noteworthy conclusion of the study is the development of 1,8-napthyridine derivatives with pyrazolines, morpholine, piperidine and piperazine like cyclic amines as remarkable anti-histaminic agents in general. Further study is in progress with these compounds and related heterocyclic scaffolds to get information about the chemical structure that efficiently modulates histamine release to discover new small molecule(s) endowed with noteworthy antihistaminic activities.
Conflicts of interest
The authors declare that there are no conflicts of interest in this manuscript.Abbreviations
| RPMC | Rat peritoneal mast cells |
| PDE 4 | Phosphodiesterase 4 |
| ADMET | Absorption distribution, metabolism, execration, toxicity |
| PASS | Prediction of activity spectra for substances |
| TPSA | Total polar surface area |
Acknowledgements
The authors acknowledge the SLT Institute of Pharmaceutical Sciences, Guru Ghasidas University, Bilaspur Chhattisgarh and Punjab University Chandigarh for performing IR, NMR and Mass spectroscopy and PBRI, Bhopal, M. P. for the evaluation of the biological activities of the compounds.References
- D. Pal and S. Saha, J. Adv. Pharm. Technol. Res., 2012, 4, 98–104 Search PubMed.
- P. Rani, D. Pal, R. R. Hegde and S. R. Hashim, Adv. Anticancer Agents Med. Chem., 2016, 16, 898–906 CrossRef PubMed.
- P. Rani, D. Pal, R. R. Hegde and S. R. Hashim, J. Chemother., 2016, 28, 255–265 CrossRef PubMed.
- P. E. Silva Junior, L. C. D. Rezende, J. P. Gimenes, V. G. Maltarollo, J. Dale, G. H. G. Trossini, F. S. Emery and A. Ganesand, RSC Adv., 2016, 6, 22777–22780 RSC.
- D. Pal, R. Tripathy, D. D. Pandey and P. Mishra, J. Adv. Pharm. Technol. Res., 2014, 5, 196–201 CrossRef PubMed.
- S. Saha, D. Pal and S. Kumar, Trop. J. Pharm. Res., 2016, 15, 1401–1411 CrossRef CAS.
- D. Pal and S. Saha, RSC Adv., 2019, 9, 28061–28077 RSC.
- S. B. Nimse and D. Pal, RSC Adv., 2015, 5, 27986–28006 RSC.
- S. Haider, Journal of Phytochemistry and Biochemistry, 2017, 1, e101 Search PubMed.
- S. Saha, D. Pal and S. Kumar, Inven. Impact Med. Chem., 2017, 1, 1–8 Search PubMed.
- S. Saha, D. Pal and S. Kumar, Indian J. Exp. Biol., 2017, 55, 831–837 Search PubMed.
- S. Saha, D. Pal and S. Kumar, Int. J. Pharmacol. Pharm. Sci., 2017, 9, 247–251 Search PubMed.
- B. Eftekhari–Sis, M. Zirak and A. Akbari, Chem. Rev., 2013, 113, 2958–3043 CrossRef.
- Y. Ju and R. S. Varma, J. Org. Chem., 2006, 71, 135–141 CrossRef.
- E. M. Gordon, R. W. Barrett, W. J. Dower, S. P. A. Fodor and M. A. Gallop, J. Med. Chem., 1994, 37, 1385–1401 CrossRef PubMed.
- D. Pal and S. Saha, J. Adv. Pharm. Technol. Res., 2012, 3, 92–99 CrossRef CAS PubMed.
- P. Rani, D. Pal, R. R. Hegde and S. R. Hashim, BioMed Res. Int., 2014, 386473, 1–9 Search PubMed.
- S. Zhou and G. Huang, RSC Adv., 2020, 10, 5874–5885 RSC.
- P. Kuna, D. Jurkiewicz, M. M. Czarnecka-Operacz, R. Pawliczak, J. Woroń, M. Moniuszko and A. Emeryk, Alergol, 2016, 33, 397–410 Search PubMed.
- L. M. Du Buske, J. Allergy Clin. Immunol., 1996, 98, S307–S318 CrossRef.
- M. Gobinath, N. Subramanian and V. Alagarsamy, J. Saudi Chem. Soc., 2015, 19, 282–286 CrossRef.
- N. Chairungsrilerd, K. Furukawa, T. Ohta, S. Nozoe and Y. Ohizumi, Eur. J. Pharmacol., 1996, 314, 351–356 CrossRef.
- R. J. Hopp, A. Bewtra, N. M. Nair and R. G. Townley, J. Allergy Clin. Immunol., 1985, 76, 609–613 CrossRef.
- R. Kiss, Z. Kovári and G. Keseru, Eur. J. Med. Chem., 2004, 39, 959–967 CrossRef.
- S. Thangapandian, N. Krishnamoorthy, J. Shalini, S. Sakkiah, P. Lazar, Y. Lee and K. Woo Lee, Bull. Korean Chem. Soc., 2010, 31, 52–58 CrossRef.
- K. Tasaka and M. Akagi, Arzneimittelforschung, 1979, 29, 488–493 Search PubMed.
- D. McTavish and E. M. Sorkin, Drugs, 1989, 38, 778–800 CrossRef.
- M. H. Sherlock, J. J. Kaminski, W. C. Tom, J. F. Lee, S. F. Wong, W. Kreutner, R. W. Bryant and A. T. McPhail, J. Med. Chem., 1988, 31, 2108–2121 CrossRef.
- Y. Nishikawa, T. Shind, K. Ishi, H. Nakamura, T. Kon, H. Uno and J. Matsumoto, Chem. Pharm. Bull., 1989, 37, 1256–1259 CrossRef PubMed.
- C. Manera, L. Betti, T. Cavallini, G. Giannaccini, A. Martinelli, G. Ortore, G. Saccomanni, L. Trincavelli, T. Tuccinardia and P. L. Ferrarini, Bioorg. Med. Chem. Lett., 2005, 15, 4604–4610 CrossRef PubMed.
- J. D. Gohil, H. B. Patel and M. P Patel, RSC Adv., 2016, 6, 74726–74733 RSC.
- M. Di Braccio, G. Grossi, S. Alfei, V. Ballabeni, M. Tognolini, L. Flammini, C. Giorgio, S. Bertoni and E. Barocelli, Eur. J. Med. Chem., 2014, 86, 394–405 CrossRef.
- P. L. Ferrarini, V. Calderone, T. Cavallini, C. Manera, G. Saccomanni, L. Pani, S. Ruiu and G. L. Gessa, Bioorg. Med. Chem., 2004, 15, 1921–1933 CrossRef.
- K. Takayama, M. Iwata, H. Hisamichi, Y. Okamoto, M. Aoki and A. Niwa, Chem. Pharm. Bull., 2002, 50, 1050–1059 CrossRef.
- V. K. Gurjar and D. Pal, Int. J. Pharm. Biol Arch., 2018, 9, 265–273 Search PubMed.
- W. P. Walters and M. A. Murcko, Adv. Drug Delivery Rev., 2002, 54, 255–271 CrossRef PubMed.
- M. S. Lajiness, M. Vieth and J. Erickson, Curr. Opin. Drug Discovery Dev., 2004, 7, 470–477 Search PubMed.
- C. G. Van Arman, L. M. Miller and M. P. O'Malley, J. Pharmacol. Exp. Ther., 1961, 133, 90–97 Search PubMed.
- P. B. Dews, Br. J. Pharmacol., 1953, 8, 46–48 CrossRef.
- D. Pal, S. Dutta and A. Sarkar, Acta Pol. Pharm., 2009, 66, 535–541 Search PubMed.
- D. Pal, S. Sannigrahi and U. K. Mazumder, Indian J. Exp. Biol., 2009, 47, 743–747 Search PubMed.
- D. Pal, S. K. Pahari and A. K. Pathak, Asian J. Chem., 2007, 19, 4452–4458 Search PubMed.
- D. Pal, S. B. Nimse, S. Khatun and P. Bandyopadhyaya, Nat. Prod. Sci., 2006, 12, 44–49 Search PubMed.
- D. Pal, M. Sahoo and A. Mishra, European Bulletin of Drug Research, 2005, 13, 91–97 Search PubMed.
- D. Pal, C. Panda, S. Sinhababu, A. Dutta and S. Bhattacharya, Acta Pol. Pharm., 2003, 60, 481–486 Search PubMed.
- M. F. Sanner, J. Mol. Graphics Modell., 1999, 17, 57–61 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 Search PubMed.
- M. T. Khan, Curr. Drug Metab., 2010, 11, 285–295 CrossRef.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717, DOI:10.1038/srep42717.
- B. C. Doak, B. Over, F. Giordanetto and J. Kihlberg, Chem. Biol., 2014, 21, 1115–1142 CrossRef.
- G. Moroy, V. Y. Martiny, P. Vayer, B. O. Villoutreix and M. A. Miteva, Drug Discovery Today, 2012, 17, 44–55 CrossRef.
- A. Lagunin, A. Stepanchikova, D. Filimonov and V. Poroikov, Bioinformatics, 2000, 16, 747–748 CrossRef.
- M. N. Drwal and R. Griffith, Drug Discovery Today: Technol., 2013, 10, e395–e401 CrossRef.
- P. G. Jamkhande, S. K. Pathan and S. J. Wadher, Int. J. Mycobact., 2016, 5, 417–425 CrossRef.
- G. Y. Leshere, E. J. Froelich, M. D. Gruett, J. H. Bailey and R. P. Brundage, J. Med. Chem., 1962, 5, 1063–1065 CrossRef.
- S. Vilar, E. Sobarzo-Sanchez, L. Santana and E. Uriarte, Curr. Med. Chem., 2017, 24, 4340–4359 CrossRef CAS.
- N. S. Pagadala, K. Syed and J. Tuszynski, Biophys. Rev., 2017, 9, 91–102 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00746c |
| This journal is © The Royal Society of Chemistry 2020 |