
Rh-Catalyzed intramolecular decarbonylative cyclization of ortho-formyl group tethered alkylidenecyclopropanes (ACPs) for the construction of 2-methylindenes†
Xing
Fan
,
Ruixing
Liu
,
Yin
Wei
 and
Min
Shi
*
and
Min
Shi
*
State Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, University of Chinese Academy of Sciences, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: mshi@mail.sioc.ac.cn
First published on 10th June 2019
Abstract
A Rh-catalyzed intramolecular cascade decarbonylative cyclization reaction of ortho-formyl group-tethered alkylidenecyclopropanes (ACPs) has been developed, affording 2-methylindenes in moderate to good yields. The reaction proceeded through a decarbonylative generation of Rh–H species, intramolecular migratory insertion, β-carbon elimination, and a reductive elimination from a π-allylic rhodium intermediate on the basis of a deuterium-labeling experiment as well as other control experiments.
Indene derivatives are important structural motifs found in some medicines.1 For example, fenistil and its derivatives have been used as antihistamine drugs, aldosterone synthase inhibitors and antituberculosis agents (see Fig. S1 in the ESI†).1a–d They can also be used as ligands by deprotonation to form metallocene complexes with metals such as Au, Rh, Ru, Cr, Mo, Zr, etc. for various catalytic reactions.1e–h Moreover, indene derivatives have important applications in solar cells, optical materials and so on.1i,j
Recently, decarbonylative coupling reactions catalyzed by transition metals for substrates containing a carbonyl group have emerged as a new research direction.2 These reactions rely on a typical process using a ketone or an aldehyde as a substrate such as Tsuji–Wilkinson decarbonylation3 to realize decarbonylative coupling reactions (Scheme 1a). In 1994, the group of Ito reported the Rh(I)-catalyzed decarbonylative coupling of cyclobutanone derivatives to produce cyclopropane derivatives (Scheme 1b).4 Since then, Rh(I)-catalyzed decarbonylative coupling reactions have been extensively investigated,5 such as decarbonylation of diynones or diones for the synthesis of conjugated diynes and ynones.5a–c In recent years, other metals such as Pd6 and Ni7 have also been utilized for the decarbonylative couplings of carbonyl group containing compounds.
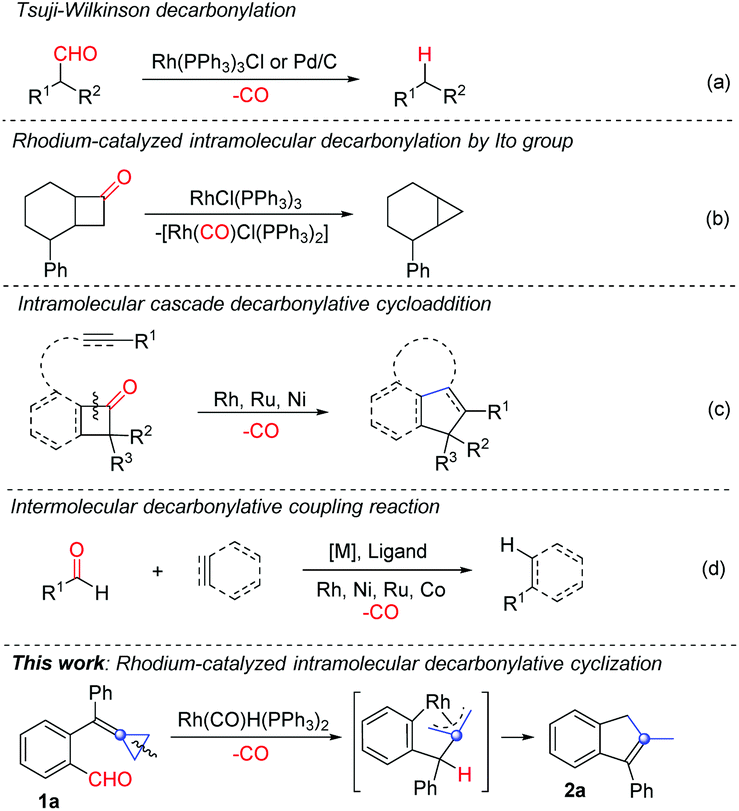 | ||
| Scheme 1 Strategies for decarbonylation or cascade decarbonylative coupling of carbonyl group containing compounds and our synthetic strategy for decarbonylative cyclization of ortho-formyl group tethered alkylidenecyclopropanes. | ||
Besides the direct decarbonylative coupling reactions, other types of cyclization reactions can also be realized in combination with decarbonylative coupling. For example, Rh(I)-catalyzed intramolecular cascade decarbonylative cyclization of cyclobutanones,8a,b benzocyclobutenones8c or isatins8d with unsaturated functional groups to produce the corresponding cyclized products has been disclosed (Scheme 1c).8 In addition, the intermolecular decarbonylative coupling reactions of carbonyl compounds with alkenes, alkynes or arenes have also been developed (Scheme 1d).9 Furthermore, it should be noted that many of these decarbonylative coupling reactions with unsaturated functional groups could be carried out under the catalysis of other transition metals such as Ni,10 Mn,11 Ru,12 Co13 and so on14 (Scheme 1c and d).
Recently, the use of alkylidenecyclopropanes (ACPs) as a special class of olefinic substances for cascade cyclization reactions in an intra- or intermolecular manner using transition metal catalysis has witnessed significant progress.15 In 2007, Fürstner and co-workers16 reported a cascade cyclization comprising a rhodium-catalyzed C–H activation followed by a hydrometalation of adjacent ACP and a regioselective C–C bond activation of the cyclopropane ring as well as a reductive elimination of the resulting Rh ring intermediate to afford functionalized cycloheptene derivatives in good yields. In 2011, the group of Aïssa also disclosed a Rh-catalyzed chemoselective intramolecular hydroformylation of α,α-disubstituted 4-alkylidenecyclopropanals under mild conditions, affording cycloheptenones in good yields.17
Among the reported examples of Rh catalyzed hydroformylation of ACPs, we were wondering whether the intramolecular decarbonylative cyclization reaction could be realized when similar ACP substrates were used in the Rh(I)-catalyzed transformations. Accordingly, ortho-formyl group-substituted biphenylalkylidenecyclopropane 1a was prepared and used as a template substrate for the examination of our working hypothesis (Scheme 1, this work).
We initially investigated the ligand effect on the intramolecular decarbonylative cyclization reaction of 1a with Wilkinson's catalyst and found that using dppp as a ligand afforded the desired product 2a in 19% yield in toluene at 80 °C overnight (Table 1, entry 1). Raising the reaction temperature from 80 °C to 110 °C gave the corresponding cyclized product 2a in 42% yield, however, when the reaction was carried out at 130 °C in a sealed tube, 2a was still given in 42% yield (Table 1, entry 2 vs. 3). Thus, we set up the reaction temperature at 110 °C for the further reaction condition screening.
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Cat. (mol%) | Ligand | T/°C | Solvent | Yieldb (%) |
| a All reactions were carried out with 1a (0.1 mmol), Rh cat. (2.5 mol%), and ligand (10 mol%) in 1.0 mL of solvent for 12 h. b 1H NMR yields using 1,3,5-trimethoxybenzene as an internal standard. c Isolated yields. d This reaction was carried out in a sealed tube. acac = acetylacetone. | |||||
| 1 | Rh(PPh3)3Cl | dppp | 80 | PhMe | 19 |
| 2 | Rh(PPh3)3Cl | dppp | 110 | PhMe | 42c |
| 3 | Rh(PPh3)3Cl | dppp | 130 | PhMe | 42d |
| 4 | Rh(PPh3)3Cl | Xantphos | 110 | PhMe | 36 |
| 5 | Rh(PPh3)3Cl | P(C6F5)3 | 110 | PhMe | 56 |
| 6 | [Rh(cod)Cl]2 | P(C6F5)3 | 110 | PhMe | 28 |
| 7 | [Rh(cod)Cl]2 | — | 110 | PhMe | 29 |
| 8 | [Rh(cod)OH]2 | P(C6F5)3 | 110 | PhMe | 32 |
| 9 | [Rh(cod)OH]2 | — | 110 | PhMe | 33 |
| 10 | Rh(PPh3)3Cl | — | 110 | PhMe | 48 |
| 11 | Rh(cod)BF4 | — | 110 | PhMe | — |
| 12 | Rh(CO)Cl(PPh3)2 | — | 110 | PhMe | 77c |
| 13 | Ir(CO)Cl(PPh3)2 | — | 110 | PhMe | 8 |
| 14 | Rh(CO)Cl(acac) | — | 110 | PhMe | 49 |
| 15 | Rh(CO)Cl[P(C6F5)3]2 | — | 110 | PhMe | 16 |
| 16 | Rh(CO)H(PPh3)2 | — | 110 | PhMe | 83c |
| 17 | Rh(CO)H(PPh3)2 | — | 110 | DCE | 24 |
| 18 | Rh(CO)H(PPh3)2 | — | 110 | MeCN | 7 |
| 19 | Rh(CO)H(PPh3)2 | — | 110 | DMF | — |
| 20 | Rh(CO)H(PPh3)2 | — | 110 | Xylene | 60 |
| 21 | Rh(CO)H(PPh3)2 | — | 110 | PhCl | 39 |
| 22 | Rh(CO)H(PPh3)2 | — | 110 | PhCF3 | 42 |
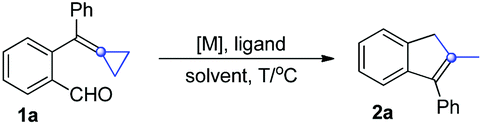
Next, several other commercially available phosphine ligands were used in the reaction. We found that the use of Xantphos as an external ligand did not improve the reaction outcome (Table 1, entry 4), while when the electron deficient P(C6F5)3 was employed as the ligand, 2a was produced in a higher yield of 56% under otherwise identical conditions (Table 1, entry 5). Subsequently, we performed the reactions using [Rh(cod)Cl]2 and [Rh(cod)OH]2, in which no phosphine ligand was coordinated to the rhodium metal center and found that the yields of 2a were almost identical in the presence or absence of the P(C6F5)3 ligand (Table 1, entry 6 vs. 7, 8 vs. 9). Afterwards, we realized that the yield of 2a could reach 48% when using Wilkinson's catalyst alone even without any external phosphine ligand (Table 1, entry 10). These results suggested that the addition of an extra phosphine ligand did not significantly improve the yield of 2a when using Wilkinson's type of Rh catalyst. Moreover, a cationic rhodium catalyst such as Rh(cod)BF4 had no catalytic activity for this reaction (Table 1, entry 11). Therefore, we started to seek out other catalysts for this reaction rather than the ligand.
Gratifyingly, when Rh(CO)Cl(PPh3)2 was used as the catalyst, the yield of 2a was up to 77% isolated yield (Table 1, entry 12). However, the use of Ir(CO)Cl(PPh3)2 as the catalyst gave 2a in only 8% yield and most of the starting materials 1a were recovered (Table 1, entry 13). When the coordinated PPh3 ligand was changed with other ligands such as acac (acetylacetone) or P(C6F5)3, the yields of 2a were 49% and 16%, respectively (Table 1, entries 14 and 15). Using Rh(CO)H(PPh3)2 as the catalyst could produce 2a in 83% isolated yield (Table 1, entry 16). Finally, the examination of the solvent effect in DCE, MeCN, DMF, xylene, PhCl and PhCF3 revealed that toluene was the best choice (Table 1, entries 17–22) (for more details on the optimization of reaction conditions, see Tables S1–S3 in the ESI†).
With the optimized conditions in hand, we next investigated the scope and limitations of this reaction, and the results are summarized in Scheme 2. When a chlorine atom or a methoxy substituent was introduced at the benzene ring bearing a formyl group, the reaction could take place smoothly, giving the desired products 2b and 2c in good yields. When the substituents were introduced at another aromatic ring, the reactions also proceeded smoothly, giving the desired products 2d–2j in moderate to good yields regardless of whether an electron-withdrawing group such as a fluorine atom and other halogen atoms or electron-donating groups such as Me, OMe, OBn and Ph were introduced at the aromatic ring. In addition, we also investigated substrates 1k and 1l, having different substituents on both aromatic rings, and found that the reactions proceeded smoothly, furnishing the desired products 2k and 2l with yields of 61% and 79%, respectively. As for the bromine atom substituted substrates 1m and 1n, the yields of the desired products 2m and 2n were only 22% and 26%, respectively, presumably due to the fact that the oxidative addition of Ar–Br to the Rh(I) center consumed the substrate 1m or 1n, resulting in the formation of 2m or 2n in lower yields. A similar phenomenon was also observed in the case of using substrate 1f in the reaction. For substrate 1o, in which the benzene ring is replaced by a naphthalene moiety, the desired product 2o was obtained in 78% yield. Substrate 1p containing a heteroaromatic ring was also tolerated, giving the corresponding product 2p in 33% yield.
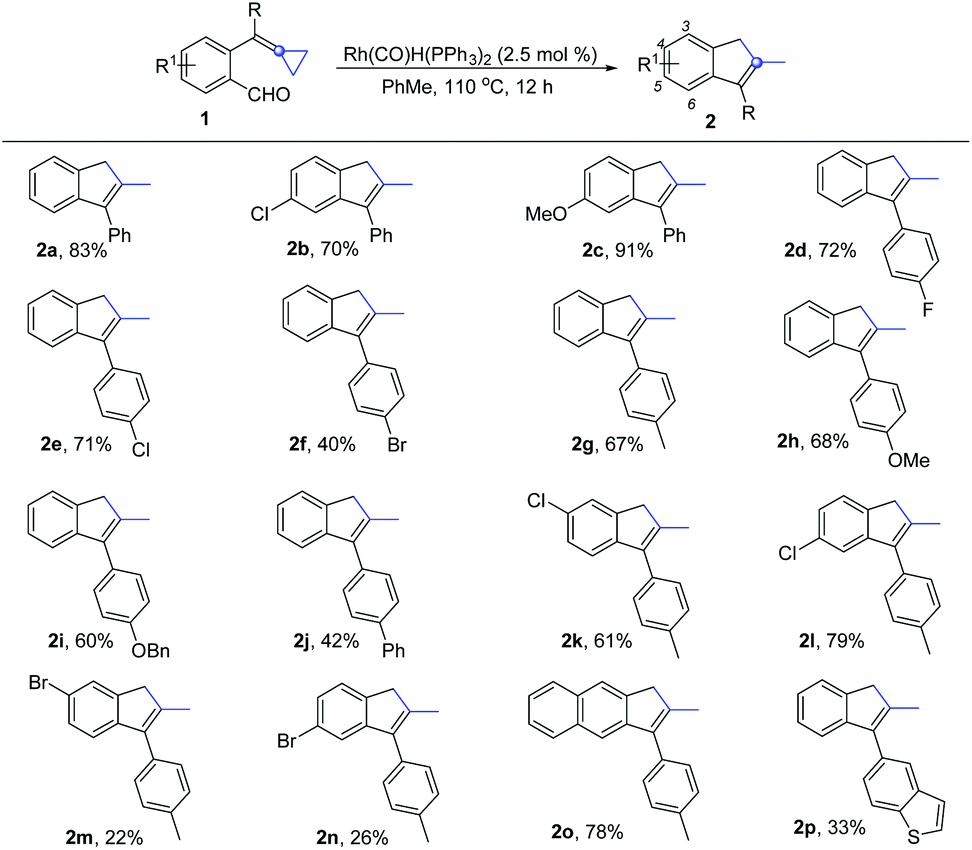 | ||
| Scheme 2 Substrate scope for the synthesis of 2. All reactions were carried out with 1 (0.2 mmol) and Rh(PPh3)2(CO)H (2.5 mol%) in toluene (2.0 mL) at 110 °C for 12 h. Isolated yields. | ||
The structure of product 2k was determined by X-ray diffraction. The ORTEP drawing is shown in Fig. 1 and the CIF data are presented in the ESI.†18
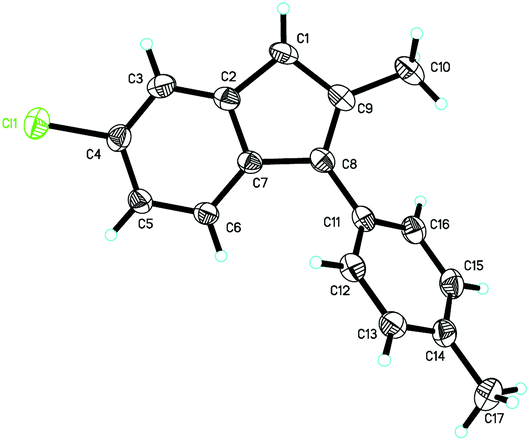 | ||
| Fig. 1 X-ray crystal structure of compound 2k. | ||
To further clarify the substrate scope and the reaction mechanism of this intramolecular cascade cyclization process, we performed several control experiments and a deuterium labeling experiment. When 2-(1-phenylprop-1-en-1-yl)benzaldehyde 1q was used as a substrate, no reaction occurred under the standard conditions (Scheme 3a). When using alkylidenecyclobutane 1r as a substrate, no reaction could take place as well (Scheme 3b). These results suggested that the methylenecyclopropane moiety is essential for this transformation. Meanwhile, we also prepared a deuterium labeled substrate 1a-d (>99% D content) as a substrate for this transformation under the standard conditions, furnishing the corresponding product 2a-d in 47% yield along with >95% deuterium incorporation at the terminal methyl group (Scheme 3c). This result indicated that one hydrogen atom in the methyl group of product 2 is derived from the formyl group in substrate 1.
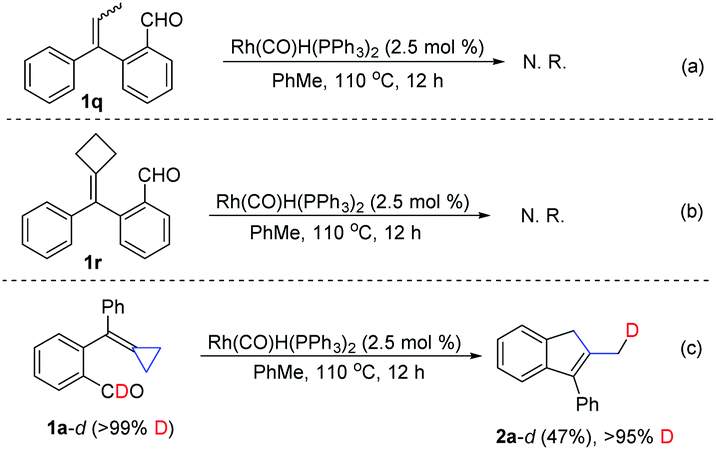 | ||
| Scheme 3 Control experiments and a labeling experiment. | ||
On the basis of the control and deuterium labeling experiments and previous literature,19 a plausible mechanism for this reaction is outlined in Scheme 4. Initially, oxidative addition of the aldehyde moiety of substrate 1a-d with Rh(I) along with a decarbonylative process produces Rh-D species A, which undergoes insertion of the double bond of the ACP moiety to afford intermediate B. β-Carbon elimination affords the intermediate C or D. Afterward, reductive elimination from intermediate D affords intermediate E, which undergoes a [1,3-H] migration to give the thermodynamically stable product 2a-d.20
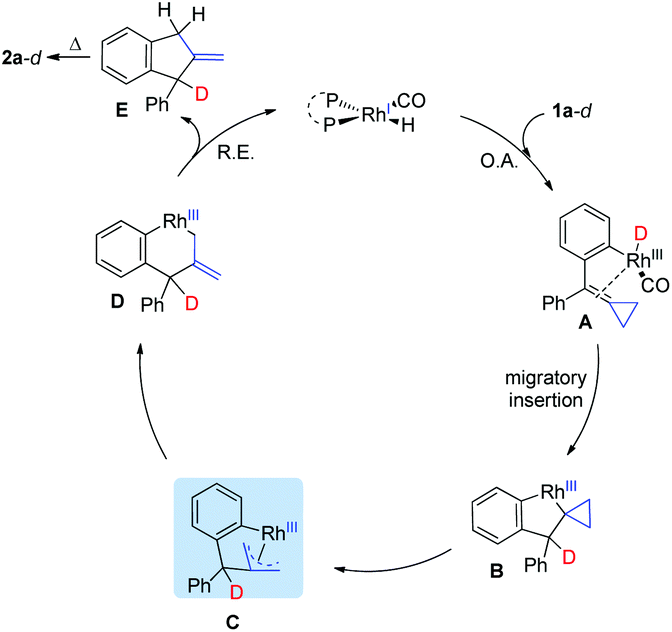 | ||
| Scheme 4 A plausible reaction mechanism. | ||
In summary, we have developed a novel Rh-catalyzed decarbonylative cascade cyclization reaction of ortho-formyl group-tethered alkylidenecyclopropanes (ACPs) for the preparation of 2-methylindene derivatives. The reaction was initiated by a decarbonylative oxidative addition of the carbonyl group with Rh(I) to generate a Rh–H species. Next, the Rh–H species underwent a migratory insertion to the double bond of the ACP moiety, leading to the distal C–C bond cleavage to give the allylic rhodium complex. The subsequent reductive elimination and a 1,3-hydrogen migration would give the desired cyclized indene derivatives. The reaction mechanism has been proposed on the basis of the control and deuterium labeling experiments. Further investigations towards the application of this methodology to synthesize more practicable compounds are underway in our lab.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful for the financial support from the National Basic Research Program of China [(973)-2015CB856603], the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000 and sioczz201808), and the National Natural Science Foundation of China (No. 20472096, 21372241, 21572052, 20672127, 21421091, 21372250, 21121062, 21302203, 20732008, 21772037, 21772226 and 21861132014).Notes and references
- (a) D. Rehn, U. Gundert-Remy, E. Weber and G. Hennings, Biopharm. Drug Dispos., 1984, 5, 291 CrossRef CAS PubMed; (b) D. Rehn, H. Geissler, O. Schuster, H. Lukas and G. Hennings, Fundam. Clin. Pharmacol., 1990, 4, 673 CrossRef CAS PubMed; (c) P. H. Reggio, S. Basu-Dutt, J. Barnett-Norris, M. T. Castro, D. P. Hurst, H. H. Seltzman, M. J. Roche, A. F. Gilliam, B. F. Thomas, L. A. Stevenson, R. G. Pertwee and M. E. Abood, J. Med. Chem., 1998, 41, 517 CrossRef PubMed; (d) E. Alcalde, N. Mesquida, J. Frigola, S. López-Pérez and R. Mercè, Org. Biol. Chem., 2008, 6, 3795 RSC; (e) P. A. Deck, Coord. Chem. Rev., 2006, 250, 1032 CrossRef CAS; (f) B. M. Wile, R. McDonald, M. J. Ferguson and M. Stradiotto, Organometallics, 2007, 26, 1069 CrossRef CAS; (g) V. V. Izmer, A. Y. Lebedev, M. V. Nikulin, A. N. Ryabov, A. F. Asachenko, A. V. Lygin, D. A. Sorokin and A. Z. Voskoboynikov, Organometallics, 2006, 25, 1217 CrossRef CAS; (h) J. Eppinger, M. Spiegler, W. Hieringer, W. A. Herrmann and R. Anwander, J. Am. Chem. Soc., 2000, 122, 3080 CrossRef CAS; (i) Y. He, H.-Y. Chen, J. Hou and Y. Li, J. Am. Chem. Soc., 2010, 132, 1377 CrossRef CAS PubMed; (j) K. Nikitin, C. Fleming, H. Müller-Bunz, Y. Ortin and M. J. McGlinchey, Eur. J. Org. Chem., 2010, 5203 CrossRef CAS.
- For selected review articles, see: (a) L. Guo and M. Rueping, Chem. – Eur. J., 2018, 24, 7794 CrossRef CAS PubMed; (b) R. J. Somerville and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 6708 CrossRef CAS PubMed; (c) A. Dermenci and G. Dong, Sci. China: Chem., 2013, 56, 685 CrossRef CAS; (d) A. Dermenci, J. W. Coe and G. Dong, Org. Chem. Front., 2014, 1, 567 RSC.
- (a) J. Tsuji and K. Ohno, Tetrahedron Lett., 1965, 6, 3969 CrossRef; (b) K. Ohno and J. Tsuji, J. Am. Chem. Soc., 1968, 90, 99 CrossRef CAS; (c) J. Tsuji and K. Ohno, Synthesis, 1969, 157 CAS.
- (a) K. Kiyotomi, A. Hiromichi, W. Masami and T. Shiichiro, Chem. Lett., 1974, 3, 215 CrossRef; (b) M. Murakami, H. Amii, K. Shigeto and Y. Ito, J. Am. Chem. Soc., 1996, 118, 8285 CrossRef CAS.
- (a) A. Dermenci, R. E. Whittaker and G. Dong, Org. Lett., 2013, 15, 2242 CrossRef CAS PubMed; (b) A. Dermenci, R. E. Whittaker, Y. Gao, F. A. Cruz, Z.-X. Yu and G. Dong, Chem. Sci., 2015, 6, 3201 RSC; (c) R. E. Whittaker and G. Dong, Org. Lett., 2015, 17, 5504 CrossRef CAS PubMed; (d) L. Yang, T. Zeng, Q. Shuai, X. Guo and C.-J. Li, Chem. Commun., 2011, 47, 2161 RSC.
- (a) A. Maleckis and M. S. Sanford, Organometallics, 2014, 33, 2653 CrossRef CAS; (b) D. Shiro, H. Nagai, S.-i. Fujiwara, S. Tsuda, T. Iwasaki, H. Kuniyasu and N. Kambe, Heteroat. Chem., 2014, 25, 518 CrossRef CAS; (c) C. Liu and M. Szostak, Angew. Chem., 2017, 129, 12892 CrossRef; (d) Y. Ogiwara, Y. Sakurai, H. Hattori and N. Sakai, Org. Lett., 2018, 20, 4204 CrossRef CAS PubMed.
- (a) P. Mi, P. Liao, T. Tu and X. Bi, Chem. – Eur. J., 2015, 21, 5332 CrossRef CAS PubMed; (b) K. Ding, S. Xu, R. Alotaibi, K. Paudel, E. W. Reinheimer and J. Weatherly, J. Org. Chem., 2017, 82, 4924 CrossRef CAS PubMed; (c) C. Liu and M. Szostak, Chem. Commun., 2018, 54, 2130 RSC; (d) T.-T. Zhao, W.-H. Xu, Z.-J. Zheng, P.-F. Xu and H. Wei, J. Am. Chem. Soc., 2018, 140, 586 CrossRef CAS PubMed.
- (a) P. A. Wender, A. G. Correa, Y. Sato and R. Sun, J. Am. Chem. Soc., 2000, 122, 7815 CrossRef CAS; (b) T. Xu, N. A. Savage and G. Dong, Angew. Chem., Int. Ed., 2014, 53, 1891 CrossRef CAS PubMed; (c) X. Zhou, H. M. Ko and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 1386 Search PubMed; (d) R. Zeng and G. Dong, J. Am. Chem. Soc., 2015, 137, 1408 CrossRef CAS PubMed.
- (a) L. Yang, C. A. Correia, X. Guo and C.-J. Li, Tetrahedron Lett., 2010, 51, 5486 CrossRef CAS; (b) L. Yang, X. Guo and C.-J. Li, Adv. Synth. Catal., 2010, 352, 2899 CrossRef CAS; (c) Q. Shuai, L. Yang, X. Guo, O. Baslé and C.-J. Li, J. Am. Chem. Soc., 2010, 132, 12212 CrossRef CAS PubMed.
- P.-h. Chen, T. Xu and G. Dong, Angew. Chem., Int. Ed., 2014, 53, 1674 CrossRef CAS PubMed.
- Z. Zong, W. Wang, X. Bai, H. Xi and Z. Li, Asian J. Org. Chem., 2015, 4, 622 CrossRef CAS.
- (a) X. Guo, J. Wang and C.-J. Li, J. Am. Chem. Soc., 2009, 131, 15092 CrossRef CAS PubMed; (b) X. Guo, J. Wang and C.-J. Li, Org. Lett., 2010, 12, 3176 CrossRef CAS PubMed; (c) T. Kondo, A. Nakamura, T. Okada, N. Suzuki, K. Wada and T.-a. Mitsudo, J. Am. Chem. Soc., 2000, 122, 6319 CrossRef CAS; (d) T. Kondo, Y. Taguchi, Y. Kaneko, M. Niimi and T.-a. Mitsudo, Angew. Chem., Int. Ed., 2004, 43, 5369 CrossRef CAS PubMed.
- E. Watanabe, A. Kaiho, H. Kusama and N. Iwasawa, J. Am. Chem. Soc., 2013, 135, 11744 CrossRef CAS PubMed.
- (a) R.-J. Tang, Q. He and L. Yang, Chem. Commun., 2015, 51, 5925 RSC; (b) Y.-J. Jang, M.-C. Yan, Y.-F. Lin and C.-F. Yao, J. Org. Chem., 2004, 69, 3961 CrossRef CAS PubMed.
- For selected review articles, see: (a) F. Wang, S. Yu and X. Li, Chem. Soc. Rev., 2016, 45, 6462 RSC; (b) H. Taniguchi, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2009, 131, 11298 CrossRef CAS PubMed; (c) L. Ackermann, S. I. Kozhushkov and D. S. Yufit, Chem. – Eur. J., 2012, 18, 12068 CrossRef CAS PubMed.
- C. Aïssa and A. Fürstner, J. Am. Chem. Soc., 2007, 129, 14836 CrossRef PubMed.
- D. Crépin, C. Tugny, J. H. Murray and C. Aïssa, Chem. Commun., 2011, 47, 10957 RSC.
- The structure of 2k has been confirmed by X-ray diffraction and the CIF data are shown in the ESI.†.
- I. F. D. Hyatt, H. K. Anderson, A. T. Morehead and A. L. Sargent, Organometallics, 2008, 27, 135 CrossRef CAS.
- Q. Zhou, S. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 16013 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and copies of NMR spectra of new compounds. CCDC 1888696. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qo00614a |
| This journal is © the Partner Organisations 2019 |