
Resolving protein mixtures using microfluidic diffusional sizing combined with synchrotron radiation circular dichroism†
Christian
Bortolini
 ab,
Tadas
Kartanas
a,
Davor
Copic
c,
Itzel
Condado Morales
a,
Yuewen
Zhang
a,
Pavan K.
Challa
a,
Quentin
Peter
a,
Tamás
Jávorfi
d,
Rohanah
Hussain
d,
Mingdong
Dong
b,
Giuliano
Siligardi
*d,
Tuomas P. J.
Knowles
*ae and
Jérôme
Charmet
*f
ab,
Tadas
Kartanas
a,
Davor
Copic
c,
Itzel
Condado Morales
a,
Yuewen
Zhang
a,
Pavan K.
Challa
a,
Quentin
Peter
a,
Tamás
Jávorfi
d,
Rohanah
Hussain
d,
Mingdong
Dong
b,
Giuliano
Siligardi
*d,
Tuomas P. J.
Knowles
*ae and
Jérôme
Charmet
*f
aChemistry Department, University of Cambridge, Lensfield Road, Cambridge, CB3 0FF, UK
bInterdisciplinary Nanoscience Center, Aarhus University, Aarhus, 8000, Denmark
cInstitute for Manufacturing, Engineering Department, University of Cambridge, Charles Babbage Road, Cambridge, CB3 0FS, UK
dDiamond Light Source, Harwell Science and Innovation Campus, Didcot, OX11 0DE, UK. E-mail: giuliano.siligardi@diamond.ac.uk
eCavendish Laboratory, University of Cambridge, J. J. Thomson Avenue, Cambridge, CB3 0HE, UK. E-mail: tpjk2@cam.ac.uk
fInstitute of Digital Healthcare, WMG, University of Warwick, Coventry, CV4 7AL, UK. E-mail: j.charmet@warwick.ac.uk
First published on 22nd November 2018
Abstract
Circular dichroism spectroscopy has become a powerful tool to characterise proteins and other biomolecules. For heterogeneous samples such as those present for interacting proteins, typically only average spectroscopic features can be resolved. Here we overcome this limitation by using free-flow microfluidic size separation in-line with synchrotron radiation circular dichroism to resolve the secondary structure of each component of a model protein mixture containing monomers and fibrils. To enable this objective, we have integrated far-UV compatible measurement chambers into PDMS-based microfluidic devices. Two architectures are proposed so as to accommodate for a wide range of concentrations. The approach, which can be used in combination with other bulk measurement techniques, paves the way to the study of complex mixtures such as the ones associated with protein misfolding and aggregation diseases including Alzheimer's and Parkinson's diseases.
1 Introduction
A range of biophysical tools are available to study homogeneous biomolecule mixtures at the molecular level, but it remains extremely challenging to study heterogeneous mixtures. A particularly striking example is that of proteins associated with misfolding and aggregation diseases, including Alzheimer's and Parkinson's diseases, that aggregate into molecular species of different sizes and solubilities from the very early stages of the disease.1 Despite significant progress made in recent years, the misfolding pathway remains difficult to fully characterise due in large part to a lack of biophysical tools and methods to study, in molecular detail, such mixtures in their native environment without losing the temporal information of the various molecular changes and interactions.Bulk measurement techniques such as circular dichroism (CD),2,3 infrared (IR) spectroscopy,4,5 nuclear magnetic resonance (NMR)6,7 or thioflavin T (ThT) fluorescence8–11 have been used to study protein folding and aggregation. However, such techniques only report on the ensemble average and therefore do not allow for a precise resolution of interactions and changes at the molecular level.
Single molecule techniques, such as atomic force microscopy (AFM),12,13 electron microscopy (EM),14,15 infrared nanospectroscopy16 and high resolution imaging17,18 have received a considerable interest in recent years to study protein folding and aggregates at the molecular level. For example AFM and transmission electron microscopy (TEM) are commonly used to study peptide self-assembly through the mapping of sample morphology at different aggregation stages.19–21 To date, however, the level of structural information available from such approaches has been more limited than from bulk spectroscopy. Besides, such measurement often require operation in non-native environment.
Even though conventional separation techniques, such as liquid chromatography, have been combined with bulk measurement techniques to resolve complex mixtures22–26 their operation usually results in a loss of the temporal information and native environment. For example, it has been acknowledged that the interaction with the stationary phase and the dilution of the samples in size exclusion chromatography may modify the state of the sample.27,28
In recent years, microfluidic approaches have opened up new opportunities to study complex biological processes.29–32 Here, we take advantage of the laminar flow properties inherent to operation in microfluidic devices to separate a complex mixture into well-resolved size-dependent fractions, using an H-filter configuration.33 In brief, we flow the solution of interest containing the protein mixture, alongside a buffer solution. The different components of the mixture diffuse in a size-dependent manner, into the buffer solution and the free-flowing solution is then separated into well resolved fractions (see Fig. 1). The concentration of the isolated fraction is obtained based on the concentration profile (Fig. 1c) calculated by numerically solving the problem of particles diffusing in a fully developed Poiseuille flow in a rectangular microfluidics channel (see Calculation of the diffusion profile section). The fractions are then studied with a label-free bulk measurement technique, in this case synchrotron radiation circular dichroism (SRCD), a powerful technique to study the secondary structure of chiral molecules (see Synchrotron radiation circular dichroism section). In particular, we use the highly collimated microbeam generated at Diamond B23 beamline for SRCD,34 which enables on-chip measurement. We show that this combination gives information that could not be obtained by studying the complex mixture without separation. In particular, we demonstrate that by excluding larger species from one fraction (fraction 2 in Fig. 1), it is possible to resolve precisely its structure and reconstruct the structure of the other species (fraction 2) by subtracting the spectrum of the resolved fraction from the that of the overall mixture. Even though the concept proposed herein is used with SRCD, it is can also be adapted to other bulk measurement techniques, such as UV/vis, IR absorption and fluorescence microscopy.
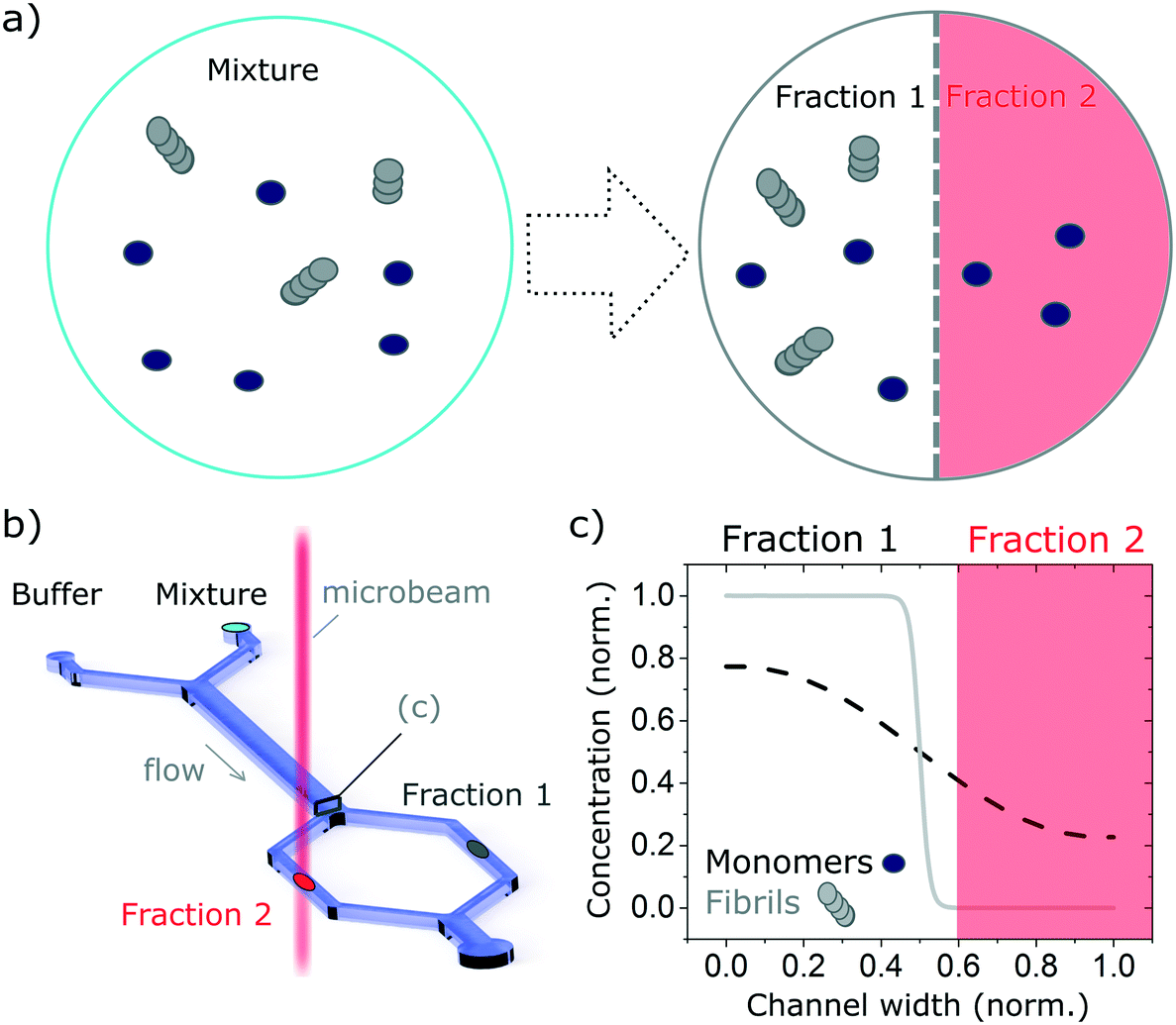 | ||
| Fig. 1 Schematic of the measurement principle. The combination of microfluidics-based free-flow separation with bulk measurement technique enables the study, in molecular detail, of heterogeneous mixtures in their native environment while retaining the temporal information. In this manuscript we combine diffusion-based separation with the ability to measure SRCD using the highly collimated microbeam of B23 beamline to study proteins mixtures. (a) The complex mixture, here made up of monomers and fibrils, is flown alongside a buffer solution. The monomers, with a smaller hydrodynamic radius, diffuse faster than the fibres, enabling the separation of the mixture into well-defined fractions that can be studied separately. (b) Sketch of the diffusion-based separation microfluidic device and microbeam light to probe each fraction. (c) Diffusion profile of the monomers and fibrils normalised concentrations at the end of the microfludics device diffusion length. The fractions collected depend on the hydrodynamic resistance of the separation channels (normalised channel width). | ||
A challenge encountered in interfacing microfluidics with CD is the incompatibility of conventional polydimethylsiloxane (PDMS) based microfluidic devices with Dd-UV measurement (see Fig. S1†). Even though the combination of SRCD and microfluidics has been reported previously to study protein refolding kinetics of cytochrome C from 4 M to 0.8 M GuHCL,35 the mixing devices used in these studies were made of fused silica with the beamlight focused on a masked slit of 60 μm × 15 mm. The fabrication of such devices requires access to specialised microfabrication equipment and expertise, which is usually not readily available in conventional biophysical laboratories. Since soft-lithography is one of the most widely used technique to fabricate microfluidic devices,36,37 and in an effort to make our finding available to a broad scientific community, we propose here microfluidic devices fabricated using conventional PDMS-based soft-lithography, compatible with far-UV measurement, including SRCD. In this manuscript, we describe proof-of-principle microfluidic devices based on two architectures enabling the integration of measurement chambers of different height, thus allowing the measurement of a wide range of concentration (see Fig. 2).
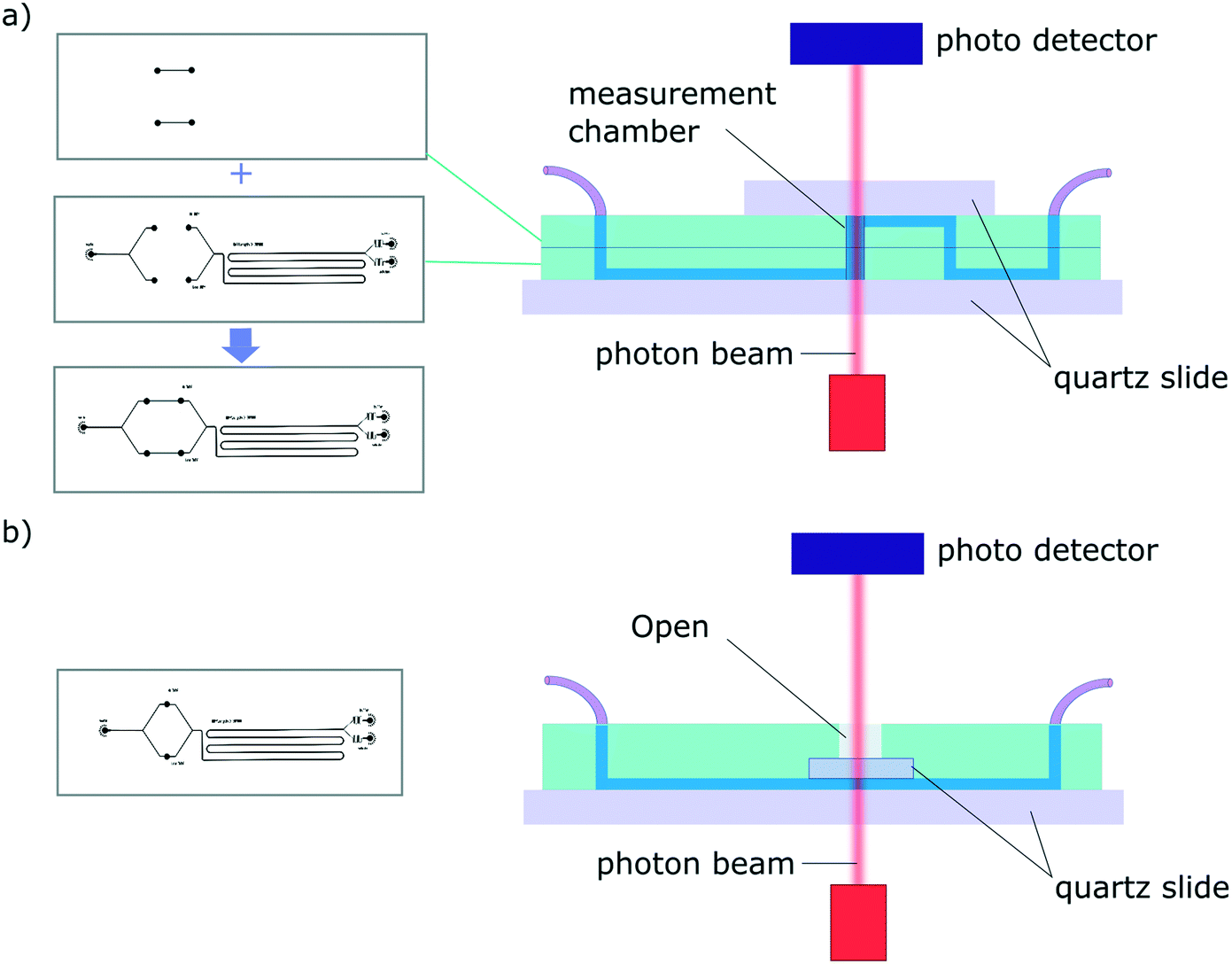 | ||
| Fig. 2 Schematic of a typical diffusion devices for the 2 architectures presented. In each case the measurement chambers are confined between quartz windows. The masters are represented on the left and the cross-section of the resulting device is show on the right (a) architecture 1 consists in two-layer devices. The height of the measurement chamber (equivalent to the path length, l) can be tuned by adapting the height of each PDMS layer. (b) Architecture 2 is based on one-master, one-layer devices. The path length, l, is defined by the height of the channels. | ||
2 Materials and methods
Sample preparation
Insulin fibrils were assembled in vitro by incubating 348 μM (2 mg ml−1) of bovine insulin (Sigma Aldrich) in HCl (pH 1.4) for 8 hours at 60 °C on non-binding plates (half-volume, CORNING 3881). The fibril formation was evaluated using controls insulin were incubated under the same conditions with ThT (40 μM) as shown in Fig. S2.† The unlabelled fibrils were then sonicated to homogenise the mixture into approximately 100 nm long fibres (Fig. S3† for TEM images) and diluted in water to the desired concentration. Insulin and BSA monomers (Sigma Aldrich) were dissolved in HCl (pH 1.4) and phosphate buffer (pH 2.7) respectively and directly diluted to the desired concentration in water.Synchrotron radiation circular dichroism
Circular dichroism (CD), a spectroscopic technique used to probe conformational changes of chiral molecules23–38 is particularly well suited to study protein folding and aggregation. In the case of proteins, CD is mainly employed to assess their secondary structure and conformation by measuring the absorption of circularly polarised light in the protein backbone. If the contribution to the CD signal of the side chains is neglected, which is usually a good approximation, information about protein folding can be accurately estimated, even when analysing samples at very low concentration and with composite α-helical and β-sheet domains.2 The far-UV region of CD spectra (from 180 to 240 nm) exhibits typical absorption characteristics of α-helices (190 and 208 for π → π*, 222 nm for n → π*) and β-sheets (195 nm for π → π*, 218 nm for the n → π*). CD spectra, therefore, contain detailed information about dissymmetric characteristics of the peptide backbone, the challenge resides in the extraction of this information. An excellent way to improve the CD signal-to-noise ratio, and therefore observe even small differences in these aggregates, is to employ synchrotron radiation as the source of UV light. Here, we use the highly collimated micro-beam light with high photon flux, available at Diamond B23 beamline for SRCD, since it is possible to focus such a beam to a spot area down to 45 μm × 15 μm, using an objective lens, onto the microfluidic channels. Besides, the SRCD spectra can be measured at different positions along the microfluidic channels using a motorized XY-stage.Fabrication of the devices
Due to the strong absorption of many materials in the far-UV range, the fabrication of microfluidic devices for SRCD measurements has, to-date, been limited to fused silica devices that can only be fabricated in specialised laboratories35 (see Fig. S1† for a SRCD graph obtained using a conventional PDMS device). We have overcome this problem by developing 2 new microfluidic devices architectures fabricated using conventional PDMS-based soft-lithography, one of the most widely used fabrication technique for microfluidic devices. We have achieved this by integrating, within the microfluidic devices, measurement chambers confined between 2 quartz slides. In order to accommodate for measurement in a wide range of concentrations, C, we have developed 2 device architectures enabling the tuning of the path length, l so that the absorption, A, as defined the Beer–Lambert law: A = εlC, where ε is the molar extinction coefficient, stays between 0.4–1.4, ideally 0.8. Typically, for a 100 μm path length, the protein concentration should be between 0.4–0.8 mg ml−1.The first architecture consists of a two-layer device that comprise, at appropriate positions along the microfluidic circuit, measurement chambers confined vertically between two quartz slide windows (Fig. 2a). The fabrication process consists in aligning the 2 complementary PDMS devices, with channels facing out, before plasma bonding. The connections between the 2 layers and to the inlet and outlet ports are made using a biopsy punch. Finally, quartz windows are plasma bonded on each side of the device to seal the channels. The second set of devices is fabricated using a single layer architecture and a one-mould process (Fig. 2b). The fabrication steps consist in pouring uncured PDMS in the master mold and pressing a 5 × 5 mm quartz window (cut from a quartz slide using a diamond scriber) onto the measurement chamber area. Once the device is cured, the structured PDMS is peeled gently, making sure the quartz window stays in place, and the ports are punched. Finally, a quartz slide is plasma bonded onto the device to seal the channels (see Fig. S4†). The second architecture was also tested successfully using a 3D printed master (see Fig. S5.a†).
Calculation of the diffusion profile
We consider particles diffusing in a fully developed flow in a rectangular microfluidic channel, so the problem can be simplified by time independence and a translationally invariant flow in the channel direction (x axis). The flow in such a channel is given by the Poiseuille flow. From the incompressible Navier–Stokes equation: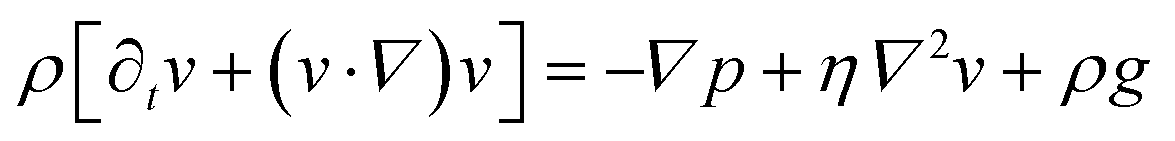 | (1) |
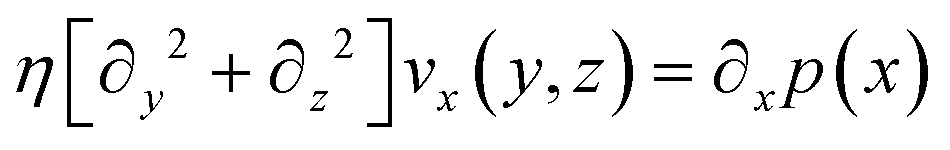 | (2) |
This equation can be solved analytically. The following formulation of the solution is symmetrical and converges quickly:39
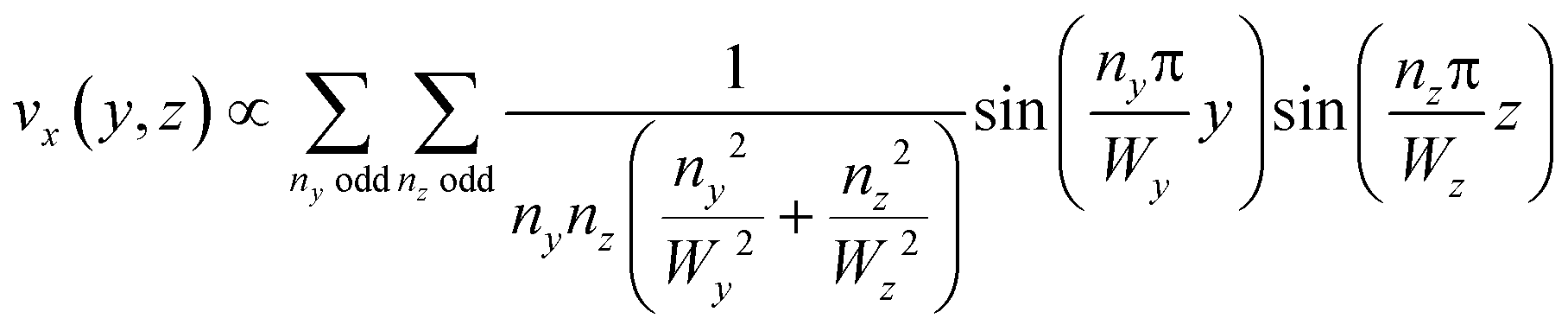 | (3) |
The general convection–diffusion equation for the local concentration c(x, y, z) and diffusion coefficient D is given by:
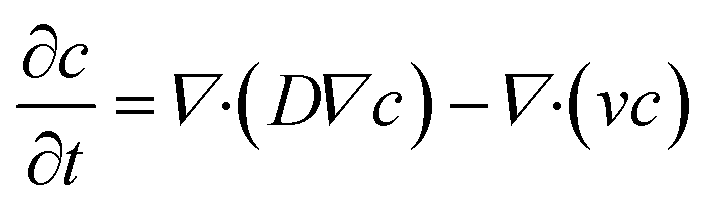 | (4) |
It is similarly simplified by translational invariance of the flow and time invariance:
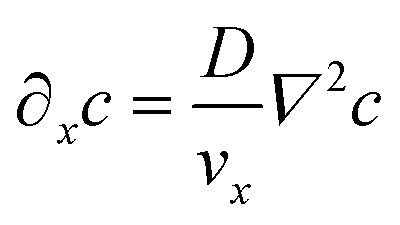 | (5) |
Assuming that diffusion in the x direction is negligible (D∂x2c ≪ vx∂xc):
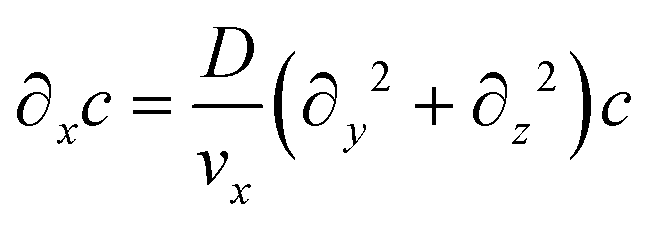 | (6) |
This equation is numerically integrated with a trapezoid method and Neumann boundary conditions. The space step dx is chosen with the Courant–Friedrichs–Lewy condition:
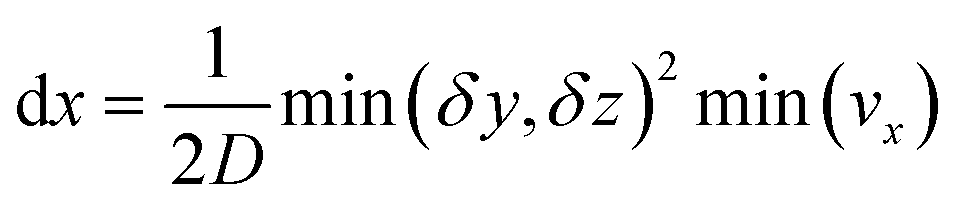 | (7) |
This choice of dx means the step matrix S is independent on D and Q, the flow rate, given by 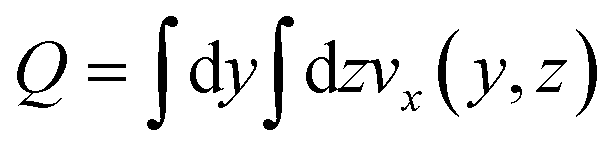 . Defining the dimensionless step size dx′ = dx × D/Q, the number of steps to reach a position L is found to be:
. Defining the dimensionless step size dx′ = dx × D/Q, the number of steps to reach a position L is found to be:
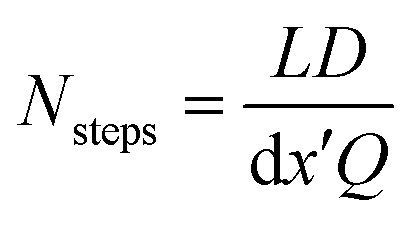 | (8) |
The evolution of an initial concentration distribution c0 can therefore be quickly calculated by repeated matrix squaring. Indeed using Si = S2i, only at most log2(Nsteps) matrix multiplications are necessary. For example 50 is 110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 010 in binary, so c50 = S6·(S5·(S1·c0)). Fig. S6† shows concentration profiles obtained using the above for insulin monomers.
010 in binary, so c50 = S6·(S5·(S1·c0)). Fig. S6† shows concentration profiles obtained using the above for insulin monomers.
3 Results and discussions
3.1 Validation using simple solutions
In order to validate the compatibility of the two device architectures with SRCD, we have proceeded to a range of experiments using model protein systems. BSA monomers and insulin monomers and amyloid fibrils formed from the same proteins40,41 were tested in a range of microfluidic devices under flow and static conditions and compared with the spectra obtained in static mode in dedicated flow cells, using both SRCD and a bench top CD spectropolarimeter Chirascan Plus.In the first set of separation experiments, solutions of insulin monomers (0.2 mg ml−1) or fibres (0.4 mg ml−1) were injected alongside a buffer solution (water) and SRCD spectra were measured in the two measurement chambers, each collecting 50% of the solution, after a diffusion length of 90 mm (Fig. 3a). The high diffusion chamber collects a fraction of the small, high diffusion coefficient molecules, while the low diffusion measurement chambers retains most of the larger molecules, with a lower diffusion coefficient (Fig. 3a and b). The flow rates were 30 and 300 μl h−1 for the insulin and the buffer solutions respectively. The devices were made using the first architecture and the heights of the measurement chambers, corresponding to the path length, ranged typically between 3 and 5 mm. The spectra obtained for the monomers, with a typical high α-helical content (see Fig. 3c), and fibres, with a stronger β-sheet signal (see Fig. S7†), are in good agreement with the results obtained using benchtop CD instrument in static mode and confirm that neither the devices, nor the flow rate, induce any artefact (see Fig. S8† for the spectra and secondary structure of undiluted solutions using a benchtop CD instrument).
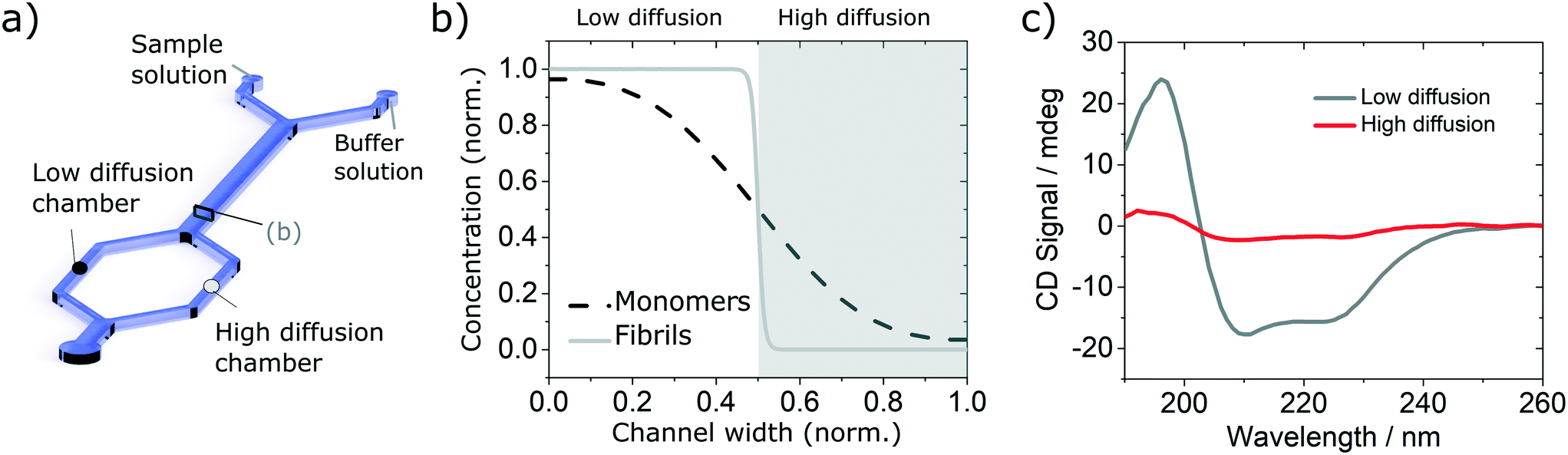 | ||
| Fig. 3 Validation of the diffusion based separation using simple solutions of insulin monomers. (a) Simplified sketch of the device used with indication of the low (black circle) and high (grey circle) diffusion chambers. (b) Diffusion profile and fractions collected at the end of the diffusion channel. (c) SRCD spectra of insulin monomer in each chamber. | ||
In order to verify the efficiency of the diffusion-based separation, we have measured the concentration of each fraction and compared it with the expected theoretical value. The concentration ratios were measured by dividing the amplitude of the CD signal at 208 nm (monomer) and 222 nm (fibre) in each chamber by the corresponding amplitude of the total concentration. These values were compared to the concentration expected due to diffusion, as calculated using the area under the theoretical concentration profile (Fig. 3b). The measured and expected (calculated) fraction in each chamber show excellent agreement as detailed in Table 1.
| Measured | Calculated | |
|---|---|---|
| Amyloid fibrils (222 nm) | 2.3% | 1.1% |
| Monomers (208 nm) | 13.8% | 11.7% |
We also measured the concentration profile of a solution of insulin monomers (2 mg ml−1) across a 2000 μm wide microfluidic channel, using the second architecture with a 50 μm high channel, and flow rates of 30 and 300 μl h−1 for the monomers and buffer solution respectively. The SRCD signal amplitude at 208 nm was measured continuously for 1 minute in 5 positions across the channel width. The normalised average amplitude points (and standard deviation) show a good match with the expected concentration profile as shown in Fig. 4.
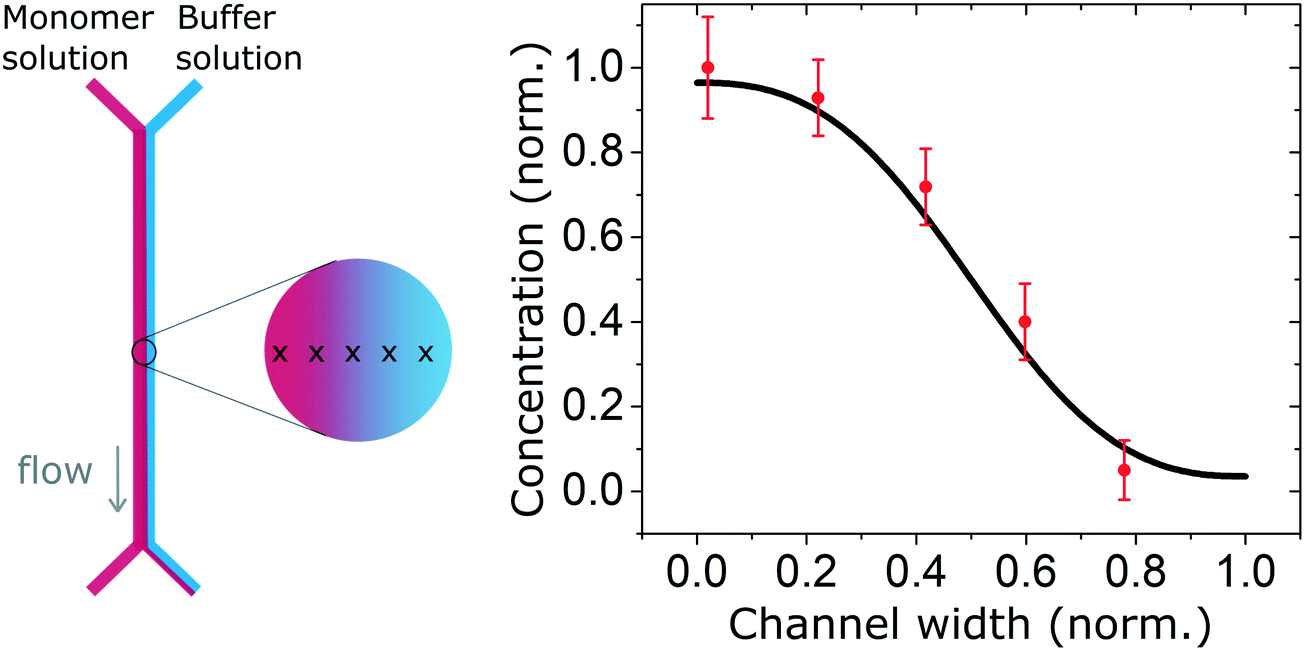 | ||
| Fig. 4 Concentration profile of a solution of insulin monomers (2 mg ml−1) across a 2000 μm wide microfluidic channel with flow rates of 30 and 300 μl h−1 for the monomers and buffer solution respectively. The high energy photon beam was positioned using the motorised stage in 5 locations across the channel and the SRCD signal amplitude, taken at 208 nm, was measured continuously during one minute and averaged. The measured profile shows a good agreement with the expected (simulated) profile. | ||
Finally, we performed a time-dependent dilution experiment, using BSA (0.2 mg ml−1, 60 μl h−1) in a Y-junction channel fabricated with a 3D printed master mould based on the second architecture. The measurement, limited by the time needed to acquire a full spectrum, is performed at the end of the channel, across its entire width, where the original sample gets diluted due to the progressive introduction of the aqueous buffer solution from 0 μl h−1 to 60 μl h−1. The spectra shown in Fig. 5 do not exhibit any distortions, as confirmed after normalisation (Fig. S5.b†). For faster experiments, continuous measurement at a single wavelength can be performed.
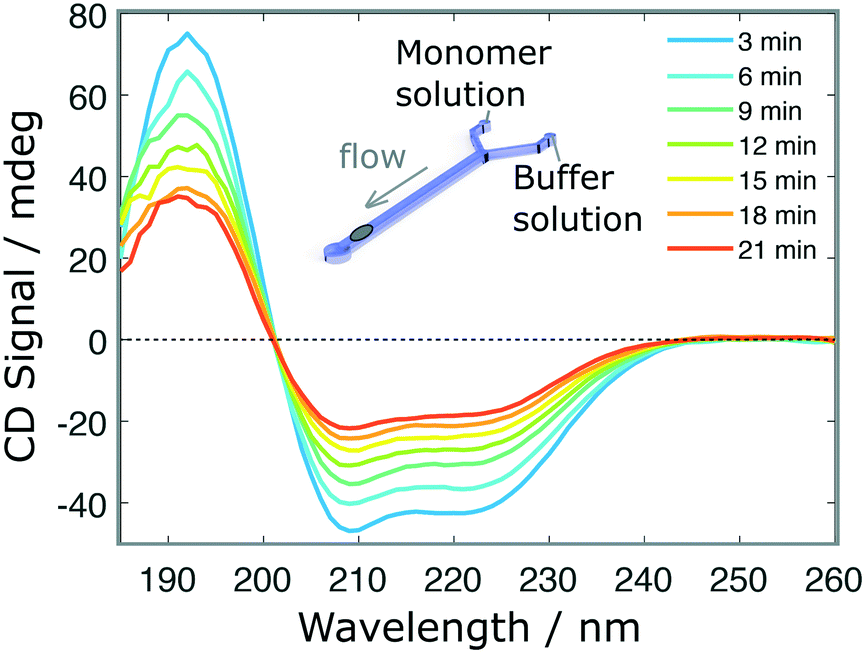 | ||
| Fig. 5 Time-dependent measurements. SRCD spectra of BSA protein (initial concentration 0.3 mg ml−1) in a dilution experiment as a function of time. In this case, the channel is initially loaded with the sample solution and the buffer is introduced progressively at 30 μl h−1 in a Y-junction microfluidic device fabricated by soft-lithography using a 3D printed master mould. | ||
The validation results presented above, obtained using simple protein solutions, constitute a proof that the different microfluidics device architectures proposed are compatible with SRCD.
3.2 Resolution in complex mixtures
Finally, we explore the possibility to resolve complex protein mixtures using the methods and devices detailed earlier. To this effect, we explored solutions comprised of insulin monomers and fibrils, mixed at different concentrations depending on the devices used. The case of a microfluidic device of 25 μm path length used to resolve a 1:1 mixture of monomers (3 mg ml−1) and fibrils (1 mg ml−1) is shown in Fig. 6. By analysing the spectrum of the mixture and calculating the secondary structures, it is not possible to resolve the different fractions that compose it, without an a priori knowledge of the individual fractions. However, if one can isolate a single constituent it then becomes possible to resolve the mixture. The case of a 2 component mixture is straight forward and is demonstrated experimentally in this manuscript (Fig. 6). The case of more complex mixtures is discussed later.
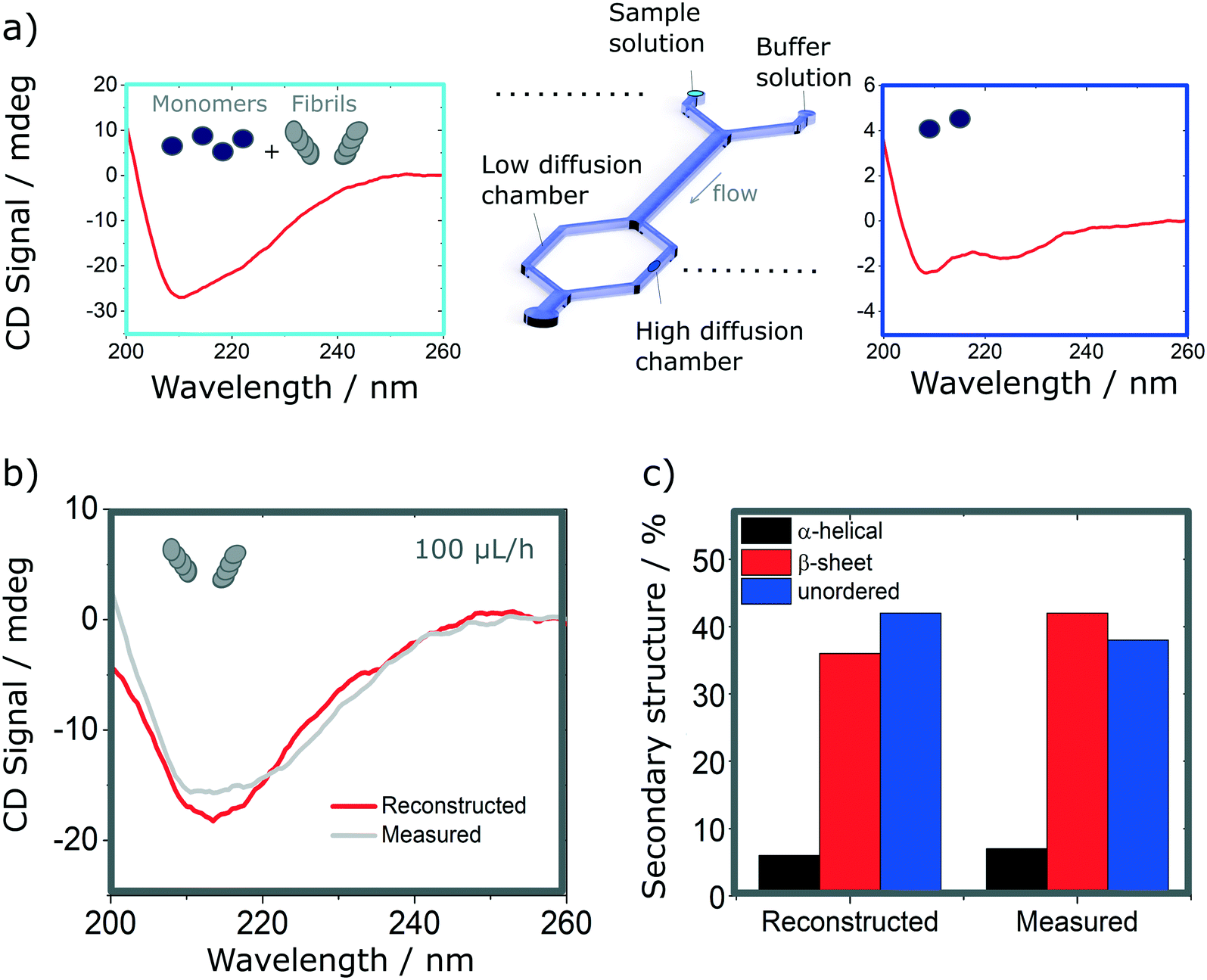 | ||
| Fig. 6 Resolution of mixtures of insulin monomer and fibrils using analytical diffusion-based microfluidics separation and SRCD. It is not possible to resolve the mixture from its SRCD spectrum (a, left). However, by isolating the monomers (a, right), it is possible to reconstruct the fibrils spectrum after subtracting the spectrum of the resolved fraction from the that of the total mixture. (b). The spectrum shows good agreement with the measured spectrum in the same conditions (b) as confirmed by the analysis of their secondary structures (c). | ||
Using the H-filter microfluidic separation device architecture presented herein, we isolate a fraction of monomers from the mixture. Using an 18 cm long diffusion channel, one can direct 22% of monomers and no fibres (0%) in the high-diffusion chamber by collecting one third of the flow (see Fig. 6a, right hand-side spectrum). The amplitude of the spectrum collected is then adjusted by a multiplication factor (in this case, 100/22 = 4.55) to account for the 100% of monomer present in the mixture and subtracted from the spectrum obtained for the mixture. The resulting reconstructed spectrum, shown in Fig. 6b, compares well with the spectrum of a fibril solution (of the same concentration) measured in the same chamber. This observation is confirmed by the analysis of the secondary structure, calculated using BeStSel, a method for the secondary structure determination and fold recognition from protein circular dichroism spectra.42Fig. 6c shows that the α-helical content for the reconstructed and measured spectra are identical (6 and 7% for the reconstructed and measured spectra respectively). The β-sheet and unordered contents, however, show small discrepancies. The β-sheet content is of 36% for the reconstructed and 42% for the measured spectra while the unordered content is of 42 and 38% for the reconstructed and measured spectra respectively. The differences observed can be attributed to small distortions of the spectrum that can arise due to variation of the microfluidics chip position with respect to the photon beam between the measurements, the measurement in dynamic mode and resulting flow fluctuations, as well as the low concentration collected, imposed by the necessity to exclude fibres from the high-diffusion chamber. It is noted also that even though monomers are stable at room temperature and neutral pH, they can start to aggregate with the fibrils, which act as catalysts.43 However, in this case, such conversion is very slow and, therefore, the content of the solutions studied is expected to be stable for the duration of the experiments presented herein (see Fig. S9† for details).
The approach described above can also be adapted to resolve more complex mixtures. Microfluidic diffusion-based separation enables the exclusion of species above a given hydrodynamic radius, in a single separation step. Even though it would theoretically be possible to isolate the smallest molecule in the complex mixture (e.g. a monomer), it has been shown that it is not possible to precisely resolve biomolecules unless their hydrodynamic radii differ by at least a factor three.44 Therefore, in order to increase the separation resolution, one should combine the approach with other high resolution microfluidic separation techniques, such as free-flow electrophoresis that enables the selection of biomolecules based on their electrophoretic mobility.45–47 However, ultimately, the detection of a single constituent from a mixture will be limited by the sensitivity of the measurement technique. Nevertheless, the separation of a heterogeneous mixture into less complex, yet well-resolved, fractions is expected to provide further insights into complex biological phenomena.
4 Conclusions
The study of heterogeneous mixtures of proteins is challenging yet important for a number of practical applications. Current biophysical methods and devices are usually not well-suited to study, in molecular detail, heterogeneous mixtures in their native environment while retaining the temporal information. In this manuscript, we show that the combination of microfluidic free-flow separation and label-free bulk measurement techniques such as synchrotron radiation circular dichroism can overcome such issues. In particular, we demonstrate that a diffusional sizing microfluidic device can be used to isolate into a well-resolved fraction, insulin monomers from a heterogeneous mixture of insulin monomers and fibrils. Using the spectrum from this fraction, it is possible to extract information about the missing fraction and finally resolve the entire heterogeneous mixture.In order to make the results presented in this study available to a broad scientific community, we have developed new fabrication methods to integrate far-UV compatible measurement chambers (confined between quartz windows) into PDMS based microfluidic devices fabricated using conventional soft-lithography approaches. Two device architectures, which enable the measurement of a wide range of concentrations, are presented, characterised and validated using the highly collimated and high photon-flux microbeam light available at Diamond Light Source B23 beamline for SRCD.
The device architecture presented herein can also be used in combination with other sensing modalities requiring far-UV transparency and more generally, the principle can also be adapted to other bulk measurement techniques. In summary, the possibility to precisely separate a heterogeneous mixture into well-resolved, simpler fractions opens up a range of opportunities for the study of complex biological phenomena. In particular, such developments open up interesting perspective to study protein misfolding and aggregation diseases such as Alzheimer's and Parkinson's diseases and may provide time-resolved information about the protein aggregation pathway.
Authors contributions
G. S., T. P. J. K. and J. C. conceived the project. C. B., T. K., D. C., I. C. M., P. K. C., T. J., R. H. and J. C. performed the experiments. I. C. M., Y. Z., Q. A. E. P., T. J. and R. H. provided resources. C. B., Q. A. E. P., R. H., G. S. and J. C. analysed the results. M. D., G. S., T. P. J. K. and J. C. supervised the work. C. B. and J. C. wrote the original manuscript. All authors commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013) through the ERC grant PhysProt (agreement no. 337969, T. P. J. K.). We gratefully acknowledge financial support from the Biotechnology and Biological Sciences Research Council (agreement no. BB/J002119/1), the Wellcome Trust, the Frances and Augustus Newman Foundation, the Lundbeck Foundation (C. B.), the Nanotechnologies Doctoral Training Centre in Cambridge (NanoDTC Cambridge EP/L015978/1, T. K.), the Institutional Links grant, ID 352360246, under the Newton-Katip Celebi Fund partnership (J. C.), the European Union's Horizon 2020 research and innovation programme under grant agreement no. 674979-NANOTRANS (Q. A. E. P.), the MSCA-IF grant 660351-EmuCam (D. C.), and CONACYT and Cambridge Trust (I. C. M.).Notes and references
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS.
- S. Brahms and J. Brahms, J. Mol. Biol., 1980, 138, 149–178 CrossRef CAS.
- W. K. Surewicz, H. H. Mantsch and D. Chapman, Biochemistry, 1993, 32, 389–394 CrossRef CAS.
- J. Seo, W. Hoffmann, S. Warnke, X. Huang, S. Gewinner, W. Schöllkopf, M. T. Bowers, G. von Helden and K. Pagel, Nat. Chem., 2017, 9, 39–44 CAS.
- A. E. Smith, Z. Zhang, G. J. Pielak and C. Li, Curr. Opin. Struct. Biol., 2015, 30, 7–16 CrossRef CAS.
- K. Wuthrich, Science, 1989, 243, 45–50 CrossRef CAS.
- G. Meisl, X. Yang, E. Hellstrand, B. Frohm, J. B. Kirkegaard, S. I. a. Cohen, C. M. Dobson, S. Linse and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9384–9389 CrossRef CAS PubMed.
- M. H. Horrocks, S. F. Lee, S. Gandhi, N. K. Magdalinou, S. W. Chen, M. J. Devine, L. Tosatto, M. Kjaergaard, J. S. Beckwith, H. Zetterberg, M. Iljina, N. Cremades, C. M. Dobson, N. W. Wood and D. Klenerman, ACS Chem. Neurosci., 2016, 7, 399–406 CrossRef CAS PubMed.
- M. Pfammatter, M. Andreasen, G. Meisl, C. G. Taylor, J. Adam- cik, S. Bolisetty, A. Sánchez-Ferrer, D. Klenerman, C. M. Dobson, R. Mezzenga, T. P. J. Knowles, A. Aguzzi and S. Hornemann, Anal. Chem., 2017, 89, 12306–12313 CrossRef CAS PubMed.
- M. R. H. Krebs, E. H. C. Bromley and A. M. Donald, J. Struct. Biol., 2005, 149, 30–37 CrossRef CAS PubMed.
- J. Adamcik, J.-M. Jung, J. Flakowski, P. De Los Rios, G. Dietler and R. Mezzenga, Nat. Nanotechnol., 2010, 5, 423–428 CrossRef CAS PubMed.
- A. F. Oberhauser, P. E. Marszalek, M. Carrion-Vazquez and J. M. Fernandez, Nat. Struct. Mol. Biol., 1999, 6, 1025–1028 CrossRef CAS PubMed.
- A. W. P. Fitzpatrick, B. Falcon, S. He, A. G. Murzin, G. Murshudov, H. J. Garringer, R. A. Crowther, B. Ghetti, M. Goedert and S. H. W. Scheres, Nature, 2017, 547, 185–190 CrossRef CAS.
- M. Bucciantini, E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson and M. Stefani, Nature, 2002, 416, 507–511 CrossRef CAS PubMed.
- F. S. Ruggeri, G. Longo, S. Faggiano, E. Lipiec, a. Pastore and G. Dietler, Nat. Commun., 2015, 6, 7831 CrossRef CAS PubMed.
- D. Pinotsi, C. H. Michel, A. K. Buell, R. F. Laine, P. Mahou, C. M. Dobson, C. F. Kaminski, G. S. Kaminski Schierle and G. S. K. Schierle, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3815–3819 CrossRef CAS PubMed.
- M. H. Horrocks, L. Rajah, P. Jönsson, M. Kjaergaard, M. Vendruscolo, T. P. J. Knowles and D. Klenerman, Anal. Chem., 2013, 85, 6855–6859 CrossRef CAS PubMed.
- F. S. Ruggeri, J. Habchi, A. Cerreta and G. Dietler, Curr. Pharm. Des., 2016, 22, 3950–3970 CrossRef CAS.
- A. T. Petkova, R. D. Leapman, Z. Guo, W.-M. Yau, M. P. Mattson and R. Tycko, Science, 2005, 307, 262–265 CrossRef CAS PubMed.
- D. Pinotsi, A. K. Buell, C. M. Dobson, G. S. Kaminski Schierle and C. F. Kaminski, ChemBioChem, 2013, 14, 846–850 CrossRef CAS PubMed.
- G. W. Somsen, W. Morden and I. D. Wilson, J. Chromatogr. A, 1995, 703, 613–665 CrossRef CAS.
- F. J. Moy, K. Haraki, D. Mobilio, G. Walker, R. Powers, K. Tabei, H. Tong and M. M. Siegel, Anal. Chem., 2001, 73, 571–581 CrossRef CAS.
- H. Liu, S. J. Berger, A. B. Chakraborty, R. S. Plumb and S. A. Cohen, J. Chromatogr., B, 2002, 782, 267–289 CrossRef CAS.
- M. Elizabeth, M. Ahmed and M. Nick, J. Synchrotron Radiat., 2004, 11, 314–318 CrossRef PubMed.
- P. G. Righetti, N. Campostrini, J. Pascali, M. Hamdan and H. Astner, Eur. J. Mass Spectrom., 2004, 10, 335–348 CrossRef CAS PubMed.
- J. F. Carpenter, T. W. Randolph, W. Jiskoot, D. J. A. Crommelin, C. R. Middaugh and G. Winter, J. Pharm. Sci., 2010, 99, 2200–2208 CrossRef CAS PubMed.
- T. Arakawa, D. Ejima, T. Li and J. S. Philo, J. Pharm. Sci., 2010, 99, 1674–1692 CrossRef CAS PubMed.
- J. Charmet, P. Arosio and T. P. J. Knowles, J. Mol. Biol., 2018, 430, 565–580 CrossRef CAS PubMed.
- T. A. Duncombe, A. M. Tentori and A. E. Herr, Nat. Rev. Mol. Cell Biol., 2015, 16, 554–567 CrossRef CAS PubMed.
- T. P. J. Knowles, D. A. White, A. R. Abate, J. J. Agresti, S. I. A. Cohen, R. A. Sperling, E. J. De Genst, C. M. Dobson, D. A. Weitz, E. J. D. Genst, C. M. Dobson and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14746–14751 CrossRef CAS PubMed.
- J. J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J.-C. Baret, M. Marquez, A. M. Klibanov, A. D. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4004–4009 CrossRef CAS PubMed.
- J. P. Brody and P. Yager, Sens. Actuators, A, 1997, 58, 13–18 CrossRef CAS.
- R. Hussain, T. Jávorfi and G. Siligardi, J. Synchrotron Radiat., 2012, 19, 132–135 CrossRef PubMed.
- A. S. Kane, A. Hoffmann, P. Baumgärtel, R. Seckler, G. Reichardt, D. A. Horsley, B. Schuler, O. Bakajin, P. Baumga, R. Seckler, G. Reichardt, D. A. Horsley, B. Schuler and O. Bakajin, Anal. Chem., 2008, 80, 9534–9541 CrossRef CAS PubMed.
- P. K. Challa, T. Kartanas, J. Charmet and T. P. J. Knowles, Biomicrofluidics, 2017, 11, 014113 CrossRef PubMed.
- Y. Xia and G. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS.
- G. Silligardi and R. Hussain, Structural Proteomics, Springer New York, New York, NY, 2015, vol. 1261 Search PubMed.
- M. Spiga and G. L. Morino, Int. Commun. Heat Mass Transfer, 1994, 21, 469–475 CrossRef CAS.
- J. L. Jiménez, E. J. Nettleton, M. Bouchard, C. V. Robinson, C. M. Dobson and H. R. Saibil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9196–9201 CrossRef.
- M. I. Ivanova, S. A. Sievers, M. R. Sawaya, J. S. Wall and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18990–18995 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y.-H. Lee, Y. Goto, M. Refregiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed.
- S. I. a. Cohen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, J. Mol. Biol., 2012, 421, 160–171 CrossRef CAS.
- P. Arosio, T. Muller, L. Rajah, E. V. Yates, F. A. Aprile, Y. Zhang, S. I. A. Cohen, D. A. White, T. W. Herling, E. J. De Genst, S. Linse, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, ACS Nano, 2016, 10, 333–341 CrossRef CAS.
- T. W. Herling, P. Arosio, T. Müller, S. Linse and T. P. J. Knowles, Phys. Chem. Chem. Phys., 2015, 17, 12161–12167 RSC.
- B. R. Fonslow and M. T. Bowser, Anal. Chem., 2006, 78, 8236–8244 CrossRef CAS PubMed.
- M. Shao, X. Xing and C.-C. Liu, Electroanalysis, 1994, 6, 245–249 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc00757h |
| This journal is © The Royal Society of Chemistry 2019 |