
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Critical review of the molecular design progress in non-fullerene electron acceptors towards commercially viable organic solar cells†
Andrew
Wadsworth
 *a,
Maximilian
Moser
a,
Adam
Marks
a,
Mark S.
Little
a,
Nicola
Gasparini
bc,
Christoph J.
Brabec
bd,
Derya
Baran
c and
Iain
McCulloch
ac
*a,
Maximilian
Moser
a,
Adam
Marks
a,
Mark S.
Little
a,
Nicola
Gasparini
bc,
Christoph J.
Brabec
bd,
Derya
Baran
c and
Iain
McCulloch
ac
aDepartment of Chemistry and Centre for Plastic Electronics, Imperial College London, London, SW7 2AZ, UK. E-mail: andrew.wadsworth11@imperial.ac.uk
bInstitute of Materials for Electronics and Energy Technology (I-MEET), Friedrich-Alexander-University Erlangen-Nuremberg, Martensstraße 7, 91058 Erlangen, Germany
cPhysical Sciences and Engineering Division, KAUST Solar Center (KSC), King Abdullah University of Science and Technology (KAUST), KSC Thuwal 23955-6900, Saudi Arabia
dBavarian Center for Applied Energy Research (ZAE Bayern), Haberstrasse 2a, 91058 Erlangen, Germany
First published on 26th April 2018
Abstract
Fullerenes have formed an integral part of high performance organic solar cells over the last 20 years, however their inherent limitations in terms of synthetic flexibility, cost and stability have acted as a motivation to develop replacements; the so-called non-fullerene electron acceptors. A rapid evolution of such materials has taken place over the last few years, yielding a number of promising candidates that can exceed the device performance of fullerenes and provide opportunities to improve upon the stability and processability of organic solar cells. In this review we explore the structure–property relationships of a library of non-fullerene acceptors, highlighting the important chemical modifications that have led to progress in the field and provide an outlook for future innovations in electron acceptors for use in organic photovoltaics.
1. Introduction
Fullerene-based acceptors, such as phenyl-C60-butyric acid methyl ester (PC60BM), its C70 analogue (PC70BM) and indene-C60 bisadduct (ICBA), have long been the dominant electron accepting materials used in bulk heterojunction solar cells; with promising results being obtained when these acceptors are used in combination with low-bandgap electron donating polymers. Despite their success, however, many problems and limitations still persist in organic solar cells that cannot be addressed without replacing this aging class of acceptors. The emergence of alternatives to fullerene-based electron acceptors has revitalized the field of organic photovoltaics (OPVs) somewhat over the past few years.Fullerenes possess a number of advantageous properties, allowing them to produce highly efficient solar cells and their initial success in the field of organic photovoltaics. Many of the properties that have allowed fullerene acceptors to excel are derived from the 3D-conjugated cage structure inherent to these molecules. For example, the lowest unoccupied molecular orbitals (LUMOs) of the fullerene acceptors are delocalized across the entire 3D surface of the C60 or C70 cages, allowing efficient and isotropic electron transport.1 This delocalisation of the molecular orbitals across the 3D fullerene cages also provides the acceptors with the ability to undergo weak π–π interactions, such that small scale aggregation of the fullerene acceptors can occur forming nanoscale pure and mixed domains in the bulk heterojunction.2 The formation of domains on the lengthscale of the exciton diffusion length (5–15 nm for organic semiconductor blends) is necessary for efficient exciton splitting and free charge generation in active layer blends.3,4
However, the same 3D cage structures are responsible for some of the most significant drawbacks of fullerene acceptors. The highly symmetric nature of the wavefunctions render the optical transitions forbidden, impeding the ability of the fullerenes to absorb photons in the UV-visible region of the solar spectrum, thereby limiting the contribution of the acceptor towards the photogenerated current of the solar cells and condemning them to rely mainly on p-type (Channel-I) excitation. PC70BM was designed to overcome this issue; the lower symmetry of the C70 cages leads to a greater number of allowed optical transitions within the molecule, enhancing the ability of the acceptor to harvest photons. It must be noted that this is still dramatically lower in intensity than the absorption of the donor polymer in the UV-visible region of the solar spectrum, and thus Channel-I excitation is still mainly predominant.5 The delocalisation of the LUMO across the 3D cages also presents an issue for the fullerene acceptors, whereby it is difficult to chemically modify the LUMO by the inclusion of additional functional groups on the C60 cage. There have been some successful attempts to shift the LUMO level of the acceptors by the addition of functional groups, such as methano- and diphenyl methano-adducts, or the inclusion of amines or fluorine atoms on the phenyl unit of the adduct, however only small shifts (<0.2 eV) have been reported, with poorer synthetic yields.6–9 The inability to alter the frontier molecular orbitals (FMOs) results in poor tunability of the absorption spectrum of these acceptors, and hence limits the photocurrent that can be produced in the bulk heterojunction. Additionally, the open circuit voltage (VOC) achieved in organic solar cells has been shown to display a dependence on the difference in energy between the highest occupied molecular orbital (HOMO) of the donor and the LUMO of the acceptor. Therefore, the ability to tune the LUMO of the acceptor is critical to maximizing the VOC that an organic solar cell can achieve, and this is not straightforward when using fullerene based acceptors. The strong tendency of fullerenes to aggregate can cause long-term morphological stability issues in fullerene-containing solar cells. Whilst the aggregation of the acceptors can be favourable up to a point, aiding in the formation of the correct morphology in the bulk heterojunction in the short-term, this aggregation continues after the active layer has been cast; leading to microscale aggregates forming over time in the blend.10 These large aggregates that form over time are far larger than the exciton diffusion length leading to significant exciton relaxation and recombination of free charge carriers in the blends. In an operational solar cell, fullerene acceptors have also been shown to migrate to the device's anode over time, this eventually leads to delamination of the device, rendering it inoperational.11 Additionally, the relatively poor solubility of fullerenes, a result of their strong tendency to aggregate, can also be problematic in the short-term. Without the use of high-boiling additives such as 1,8-diiodooctane (DIO) and 1-chloronaphthalene (CN), fullerene acceptors tend to form microscale domains and aggregates. However the use of these halogenated additives has been shown to be detrimental to the long-term photostability of active layers.12 Therefore, eliminating the need for these additives is necessary to produce photo- and morphologically stable organic solar cells.
Despite the rather significant shortcomings of the fullerene based acceptors, they have remained prevalent in the field of organic solar cells owing to their favourable electron accepting and transport properties, which have been difficult to replicate, and practically, their ready availability from a range of chemical suppliers. In lieu of replacing the fullerene acceptors, there has been a focus on improving solar cell performance through the rational design of the donor polymers and strategic device engineering over the past several years.13 As stated above, the relatively weak absorption of the fullerene acceptors in the UV-visible region limits most fullerene containing solar cells to Channel-I excitation, where the donor polymer is largely responsible for exciton generation, therefore low-bandgap donor polymers with broad absorption were developed in order to improve the photocurrent that could be achieved, since they are able to absorb light of longer wavelengths. Push–pull copolymers are able to achieve low bandgaps by making use of molecular orbital hybridization of electron rich and electron poor units in the conjugated polymer backbone, which effectively reduces the bandgap. Polymers such as PTB7-Th and PffBT4T-2OD make use of push–pull hybridization to achieve long wavelength light absorption, in addition to high hole mobility and favourable aggregation properties, to produce solar cells that were able to achieve power conversion efficiencies (PCEs) exceeding 10 and 11% respectively, when used in combination with fullerene acceptors.14,15 Another approach to improve the photon harvesting capabilities of the active layer is by employing a ternary system, whereby a third organic semiconductor is added to the active layer. The inclusion of a second donor polymer, which predominantly absorbs in a different region of the spectrum to the other donor polymer, allows a greater fraction of photons to be absorbed, and so the photocurrent can be improved.16,17 The use of ternary systems can also provide a means of improving the VOC in fullerene-containing solar cells. If the additional polymer component has a deeper lying HOMO the VOC can be raised in comparison to the corresponding binary device.18,19 Unfortunately, ternary solar cells that contain two polymer components tend to be quite difficult to fabricate with optimal phase separation; this is a result of the unfavourable mixing of polymers due to a lack of entropic driving force. To address the poor morphological stability of fullerene-containing active layers, crosslinking has been employed to create a more robust microstructure within the active layer. Examples where the donor polymer and the fullerene acceptor have been crosslinked have both been shown successfully;20–22 a key conclusion from these reports is that it is preferable to crosslink at a site that does not perturb the conjugated system (i.e. on the side chains of the materials).23 Whilst crosslinking in the active layer has been shown to improve the morphological stability of blends, it often results in decreased PCE, an increased risk of electrode delamination and requires synthetically complex and expensive derivatives of donor polymers or fullerene acceptors.23 Overcoming the need for high-boiling halogenated additives for effective fullerene containing bulk heterojunctions has also been addressed in a recently reported system.24 The devices were fabricated from an entirely non-halogenated processing conditions, while still achieving a high PCE (11.7%). By replacing the high-boiling additive (DIO) with a non-halogenated equivalent, 2-phenyl naphthalene (PN), the active layer photostability should be improved.
Although the aforementioned approaches have overcome many of the issues presented by fullerenes, they bring new problems of their own into focus. A more elegant approach to address the drawbacks of fullerene acceptors is to replace them with strategically designed electron accepting materials. Non-fullerene acceptors (NFAs), which have been specifically designed to match the electron accepting and transport properties possessed by fullerenes, and also to overcome the poor optical properties and long term morphological instability associated with fullerene acceptors, provide an attractive alternative to the use of fullerenes and employing the other strategies mentioned above. Most NFAs, similar to donor polymers, make use of push–pull hybridisation, allowing them to absorb strongly in the visible and near IR region of the solar spectrum. As such, these acceptors are able to absorb a greater fraction of photons and consequently form excitons to be split into free charge carriers; this is n-type (Channel-II) excitation. If both the donor and the acceptor are able to absorb photons in different regions of the spectrum, the total fraction of excitons being utilized is increased and the photocurrent can be maximized. Chemical modification of these structures allow a greater degree of control over the FMOs of the acceptors, leading to a wider range of possible donor polymers to be used, and the ability to achieve a much higher VOC in devices. Another common feature of NFAs is the use of steric hindrance or the inclusion of solubilising alkyl chains in order to gain some control over their aggregation properties; rendering them easier to process in common organic solvents than their fullerene counterparts. Beyond these common features, NFAs employ a wide range of novel approaches in an attempt to improve upon the standard set by fullerene acceptors. This has yielded a diverse range of exciting new materials that have already begun to push the field of organic photovoltaics to new heights (Fig. 1).
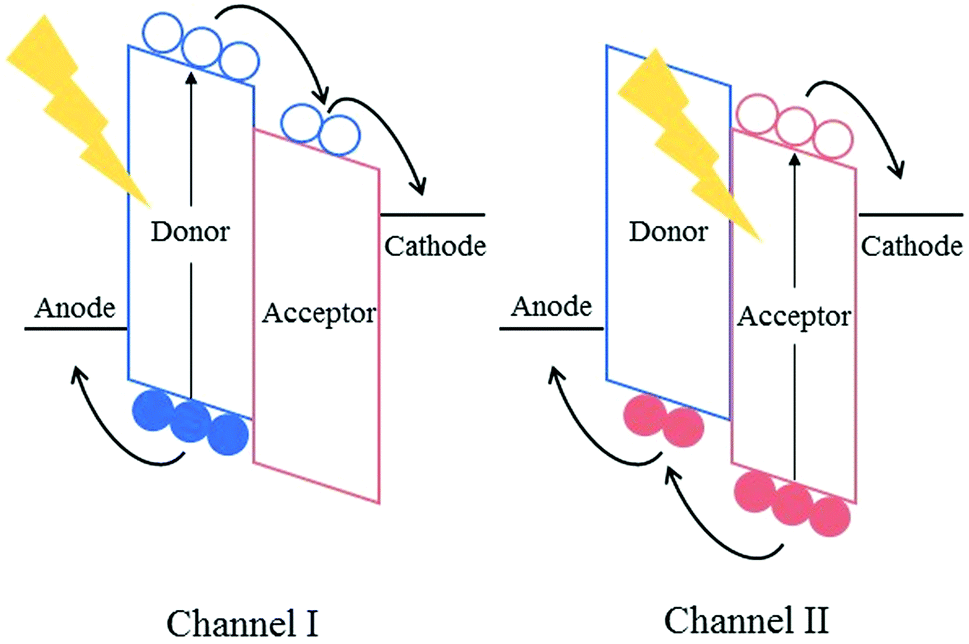 | ||
| Fig. 1 Channel I and II excitation in organic solar cells. | ||
Care must be taken when comparing these acceptors across the reported literature, with a number of factors affecting the optoelectronic properties of the acceptors and the J–V characteristics of OPV devices. For example, particular caution must be used when drawing comparison between the reported energy levels of the acceptors, since there is no universal procedure for measuring them. Photo-electron spectroscopy in air (PESA) and cyclic voltammetry (CV) are both commonly employed to measure the ionization potentials (IP) and electron affinity (EA) of an organic semiconductor. However, the variety of conditions and reference values used in CV measurements can lead to quite different measured values for the same material, particularly the values used to define the vacuum level, and comparisons must therefore be made with care. In terms of J–V characteristics, there are factors that must be accounted for when comparing the performance of OPV devices, even if the same donor polymer is being used: (i) the device architecture can play a huge role in the performance of devices – vertical phase separation in the active layer can lead to electron donor or acceptor rich layers in the blend and depending on whether one extracts the electrons from the top of the device (conventional) or the bottom (inverted), the electrons may have to travel through a donor or acceptor rich region before being extracted, affecting charge carrier mobilities and recombination rates in the bulk heterojunction,25,26 (ii) the electron and hole transport layers (ETL and HTL respectively), can also have a significant influence on device performance – the choice of these layers can affect the ease of extraction of free charges at the contacts of the device, along with recombination and resistive losses (Fig. 2).27–29
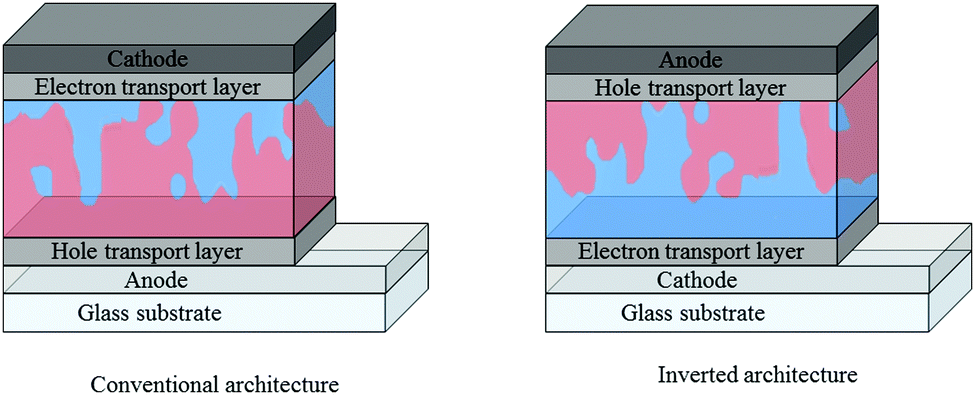 | ||
| Fig. 2 Conventional and inverted architectures employed in bulk heterojunction organic solar cells, where the electron donor is denoted by red regions and the electron acceptor by blue regions. | ||
In this review, we document the great strides that have been made by non-fullerene acceptors over the past few years. We discuss the main classes of NFAs and relate their molecular and device properties to the key structural characteristics of the materials. By highlighting these strategic design principles, we aim to provide a foundation for future innovation in the field and move closer towards the end goal of commercially viable large scale organic photovoltaics.
Early frontrunners in the field of non-fullerene acceptors also included subphthalocyanines (SubPCs), subnaphthalocyanines (SubNCs) and truxenones (Fig. 3). SubPCs are a subcategory of phthalocyanines, consisting of three fully conjugated diiminoisoindole moieties affording an aromatic macrocyclic structure surrounding a central boron atom. Their initial success in OPVs was closely tied to their favourable energy levels affording high VOC, strong absorption coefficients, in excess of 3.5 × 105 cm−1, and excellent thermal and chemical stabilities.30–32 Subnaphthalocyanines (SubNCs), the higher homologue of SubPCs have also been developed and share their same advantageous properties. The highest PCE of binary OPV blends employing either SubPCs or SubNCs as electron acceptor was reported in 2015 and yielded 6.86%. Since then, additional research efforts into this class of NFAs were unable to eclipse this benchmark. Another drawback of SubPCs and SubNCs is their more energy and capital expensive vacuum-processing, thus also contributing to their decline. Similar to SubPCs, truxenones were another type of rotationally symmetrical vacuum-deposited small molecule NFAs, characterised by their easily tuneable molecular curvature and in turn their optoelectronic and morphological properties. Truxenones’ inherently low crystallisation tendency often resulted in poor charge transport properties thus severely limiting the FF of devices. Consequently, the PCEs of donor:truxenone systems were never able to exceed 3%.33–35
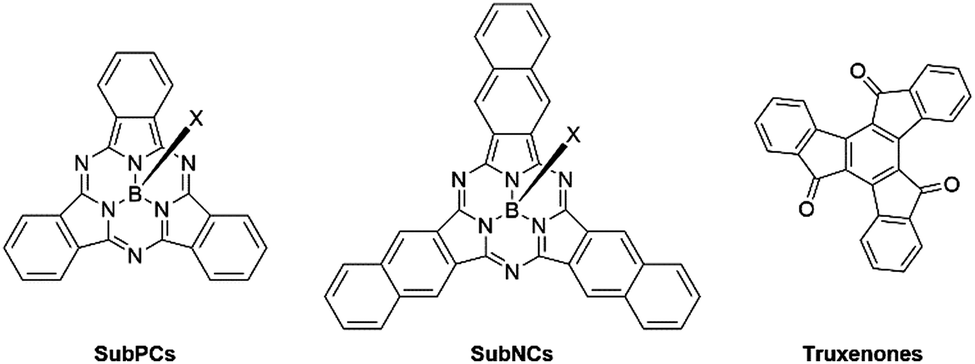 | ||
| Fig. 3 General structure of SubPC, SubNC and truxenone NFAs. | ||
2. Acceptor–donor–acceptor calamitic small molecules
Despite being a relatively new class of electron acceptors, acceptor–donor–acceptor (A–D–A) calamitic-type small molecules appear to be among the most promising replacements for fullerenes to have been reported, with PCEs now exceeding 11% being achieved regularly. Their classification as A–D–A type acceptors is derived from their generic structure of an electron rich donor central core flanked on either side by electron deficient acceptor units. They have been designed in a modular fashion, hence tuning the FMOs and absorption spectra can be easily and readily achieved by substituting one electron donating (or electron withdrawing) unit with another. The LUMO of these molecules is mostly located on the electron withdrawing (acceptor) units on the periphery of the molecule, and the HOMO is mainly located on the electron rich (donor) core. As such, any structural changes on the periphery have a much greater effect on the LUMO than the HOMO, and alterations to the donor core have a greater impact on the HOMO of the acceptor, allowing independent control over both the HOMO and LUMO levels. Another advantage of these A–D–A small molecules, in comparison to the extended rigid fused ring acceptors, is the relative ease with which they can be synthesized. Also, like all small molecules, these A–D–A type acceptors do not suffer from the batch-to-batch variations in molecular weight, polydispersity and purity that is regularly seen in polymers.Fluorene, carbazole, indaceno[1,2-b:5,6-b′]dithiophene (IDT), indacenodithieno[3,2-b]thiophene (IDTT), and their derivatives, are the most commonly used donor units in the core of these molecules, and established dye based moieties, such as diketopyrrolopyrrole (DPP), indandione and rhodanine derivatives, are most commonly used on the electron withdrawing periphery of the molecules. The main differences between many of these acceptors lies within the π-conjugated spacer unit (if one is included at all) between the donor unit in the core and acceptor units, allowing further tuning of the HOMO, LUMO and bandgap. Chemical modifications to the dye based end groups are also commonly employed to easily tune the optoelectronic and structural properties of these acceptors.
2.1. Fluorene and carbazole based acceptors
The fluorene moiety was among the first to be used as the donor unit in A–D–A type NFAs, owing to its simple synthesis and ready availability, in addition to the facile inclusion of solubilizing chains allowing a degree of control over structural properties (Fig. 4). One of the first examples of such an NFA was reported in 2014, where alkylated carbazole and fluorene cores were flanked by thiophenes and 3-ethylrhodanine end groups to create Cz-RH and Flu-RH respectively (Table 1).36 These acceptors possessed high lying LUMOs (approx. −3.5 eV), to aid in maximizing the VOC, which was achieved with the use of an electron donating thiophene spacer unit. Another important feature of the thiophene spacer was to ensure that the acceptor was planar. Where phenyl–phenyl links are used, the molecular backbone tends to twist to avoid the steric clash of ortho-hydrogens. However, in phenyl–thienyl links, this steric strain is much less as the ortho-hydrogens are much further from one another, hence the molecular backbone no longer has to twist to avoid steric strain. When used in combination with P3HT, Cz-RH and Flu-RH were able to achieve PCEs of 2.56% and 3.08% respectively, where the high VOC (1.03 V for both acceptors) played a large role in the success of these NFAs. The main limitation of the solar cell performance was modest JSC, which can be partly attributed to the fact that the NFAs absorb in the same region of the solar spectrum as P3HT (Eg = 2.10 eV), limiting the fractions of photons that can be harvested in this system. This design concept was then developed further with the acceptor, F(DPP)2B2.37 Again, this acceptor utilized an alkylated fluorene as the electron donating core, and contained thiophene spacer groups, but was flanked by alkylated diketopyrrolopyrrole (DPP) moieties on the periphery of the molecule. Strongly electron withdrawing DPP units were employed to narrow the bandgap of the acceptor (Eg = 1.82 eV), such that complimentary absorption could be achieved. Using P3HT as the donor material, solar cells were able to achieve an exceptionally high VOC of 1.18 V, owing to the high lying LUMO (−3.4 eV), however the JSC and fill factor (FF) could not be improved upon, relative to Flu-RH and Cz-RH containing devices. This design was also utilized as a basis to develop FBR.38 Again, it contained the alkylated fluorene donor as the core, but was this time flanked by electron withdrawing benzothiadiazole (BT) and 3-ethylrhodanine units on the periphery. Whilst the increased electron withdrawing character on the molecule should theoretically serve to narrow the bandgap, this is mitigated by the phenyl–phenyl link between the fluorene and BT units. Geometry optimization calculations, using Density Functional Theory (DFT), estimated a ∼35° twist in the backbone (using B3LYP/6-31G* level) to avoid the steric clash of ortho-hydrogen atoms, as discussed above, and thus yielding a wide bandgap of 2.14 eV. The inclusion of the strongly electron withdrawing BT spacer served to lower the LUMO level of the acceptor to −3.6 eV, which would thereby lead to a decrease in the VOC relative to the aforementioned acceptors. Solar cells using P3HT as the donor were able to achieve a VOC of 0.82 V, but much improved JSC and FF (7.95 mA cm−2 and 0.63 respectively), which can be attributed to the use of an inverted device architecture rather than a conventional architecture, in addition to the improved electron accepting abilities of this molecule. Overall this led to an impressive PCE of 4.1%. However, it was found that the device was limited by two major factors: (i) the very similar absorption spectra of FBR and P3HT, limiting the photocurrent that could be achieved, (ii) the amorphous nature of the acceptor, owing to its twisted structure, which led to it becoming molecularly mixed with the P3HT. As a result of the molecular mixing, the acceptor was unable to aggregate to form the nanoscale domains and percolating networks needed for efficient exciton separation and extraction of free charges in the blend, and thus large nongeminate recombinative losses were observed in the system. Good morphological stability under extended thermal annealing was demonstrated, where the acceptor did not form large aggregates, which is often seen in fullerene containing devices. When FBR was later combined with the low bandgap donor polymer PffBT4T-2DT, it was able to achieve a PCE of 8.00%.39 The improved PCE when FBR was used in combination with the low bandgap polymer can be traced back to a great improvement in both the VOC and JSC in devices (1.13 V and 11.7 mA cm−2 respectively). The increase in VOC is a result of the lower lying HOMO level of PffBT4T-2DT, in comparison to P3HT, and the increase in photocurrent can be attributed to the complimentary absorption of the donor and acceptor leading to greater spectral coverage by the active layer blend. DICTF is another NFA that has built upon the structure of Flu-RH, but instead of replacing the thiophene spacer, the rhodanine end group had been replaced with a modified indandione derivative, 2-(2,3-dihydro-3-oxo-1H-inden-1-ylidene) propanedinitrile, often referred to as a dicyanovinylindanone (DCI) group.40 The logic behind this strategy was to keep the molecule synthetically simple and inexpensive, by retaining the fluorene donor unit in the core, whilst attempting to lower the bandgap, which is usually relatively wide (>2.0 eV) in fluorene based acceptors. This strategy was effective in decreasing the bandgap to 1.82 eV, among the lowest with fluorene core units, by pushing the LUMO deeper. From grazing incidence wide-angle X-ray scattering (GIWAXS) measurements, it was shown that DICTF displayed a preference to π-stack in a face-on orientation. When combined with PTB7-Th, a low bandgap polymer that also tends to display face-on π-stacking, the resultant OPV devices were able to achieve a substantially larger JSC than any other fluorene based NFAs (16.6 mA cm−2). Reasonably high and balanced charge carrier mobilities were observed in the blend (μe = 1.93 × 10−4 cm2 V−1 s−1 and μh = 3.82 × 10−4 cm2 V−1 s−1), and along with much stronger absorption by this medium bandgap acceptor, are likely to be the cause of this particularly high photocurrent. Because DICTF has a relatively deep LUMO, a modest VOC of 0.86 V was achieved, however the impressive JSC resulted in a PCE of 7.93%. This acceptor was later improved upon by using a fluorene-based core that includes extended conjugation by fusing the thiophene spacers to the fluorene, affording FDICTF.41 In fusing these units together, greater planarization and thus a greater degree of conjugation along the molecular backbone was possible; this led to a narrowing of the bandgap to 1.62 eV and the extinction coefficient was almost three times as large as that of DICTF. This increased conjugation also led to a change in the energy of the FMOs; with the LUMO being raised slightly (∼0.1 eV), and the HOMO being raised more significantly (∼0.3 eV). Due to the red-shift in absorption spectrum, relative to DICTF, the donor polymer that was chosen to blend with FDICTF was the medium bandgap PBDB-T, in order to ensure complimentary absorption profiles between the donor and acceptor. The OPV devices using this blend are among the highest that use fluorene-based acceptor molecules, attaining a PCE of 10.06%. Excellent photon harvesting and good matching of the energy levels afforded a satisfactory VOC and JSC to be achieved, 0.95 V and 16.09 mA cm−2 respectively. An impressive FF of 0.67 was achieved; with a greater degree of phase separation in the active layer. The stronger tendency to aggregate possessed by the more planar FDICTF is likely to have facilitated the formation of phase separated donor and acceptor domains, whereas no clear domains were seen in DICTF blends. Fusing adjacent conjugated units together has proven effective in lowering the bandgap of the acceptor and increasing its tendency to aggregate and phase separate from the donor polymer, resulting in improved photovoltaic performance in blends. However the added synthetic complexity associated with this strategy could be problematic in the potential scale-up of such an acceptor.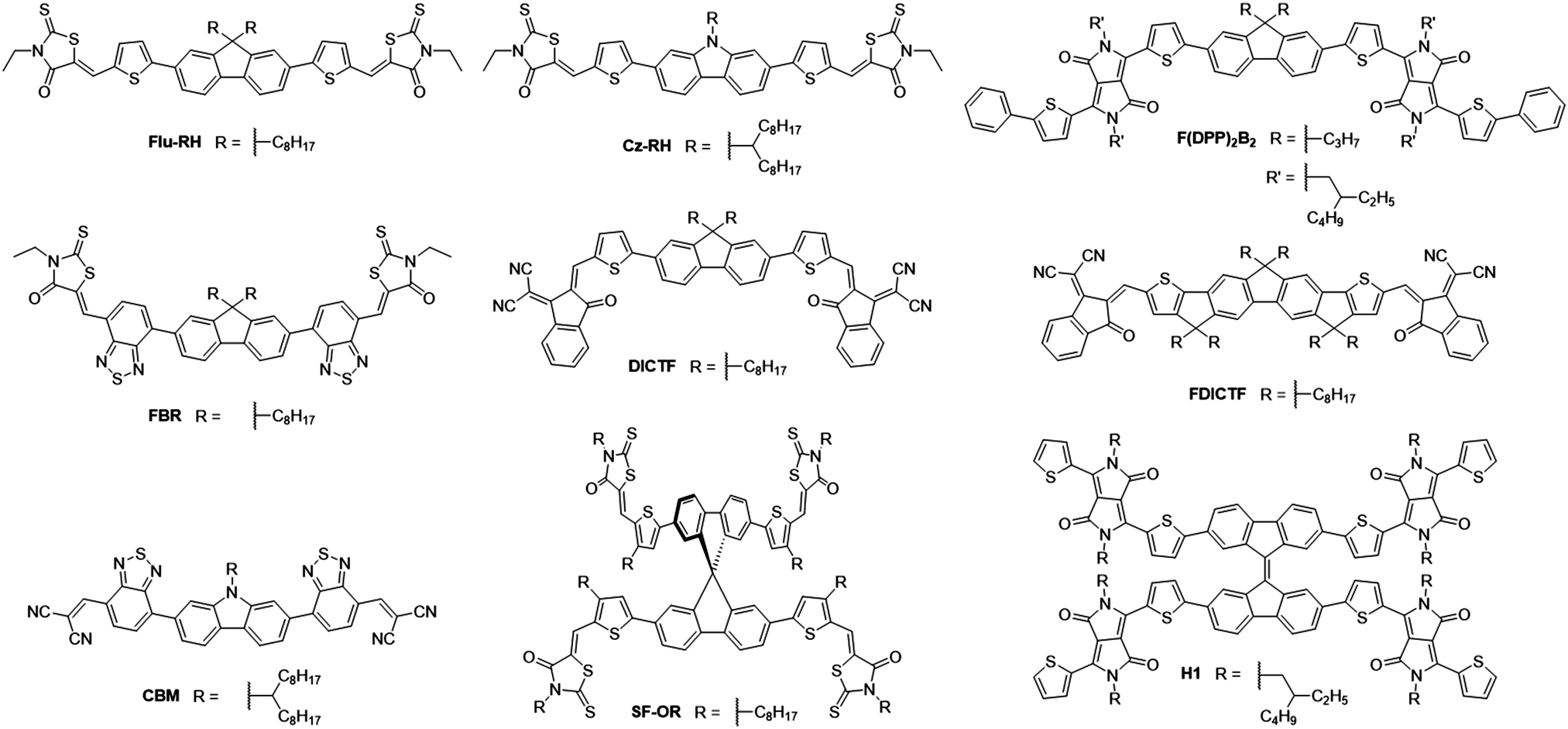 | ||
| Fig. 4 Fluorene and carbazole based small molecule A–D–A type acceptors. | ||
The basic design principles of fluorene containing A–D–A molecules was expanded by synthesizing a series of NFAs using electron rich central units (fluorene, carbazole and cyclopentadithiophene) flanked by benzothiadiazole units. However, the 3-ethylrhodanine groups were replaced by dicyanovinyl (DCV) moieties on the periphery; a strategy that has previously been used in the design of small molecule donor materials.42 The reasoning for the use of DCV end groups was to promote planarity, through favourable π–π interactions, with the aim of improving the charge carrier mobility of the acceptors. CBM, the carbazole containing acceptor, was able to achieve the best performance of the series with a PCE of 5.3% with the low bandgap polymer PTB7-Th. Though the photocurrent was reasonably high, the devices suffered from a lower VOC, especially when considering the low lying HOMO of PTB7-Th, due to the deeper LUMOs achieved in the acceptors when the DCV units are included. Additionally, these devices suffered from low FF (0.53), which also limits their performance; this is likely to be a result of an unfavourable morphology in the blends.
SF-OR, a spirobifluorene derivative of Flu-RH made use of a 3D structure and inherent twisting to improve upon the performance and properties of the early A–D–A type acceptors.43 The twisted 3D structure was adopted to suppress the crystallinity and aggregation of the acceptor, acting as a method to ensure domains are on the order of the nanoscale (13.3 and 9.6 nm for donor and acceptor domains respectively). The optoelectronic properties of this acceptor are very similar to those of Flu-RH, with comparable FMOs and bandgap, this is because the 90° twist in the spirobifluorene unit (predicted by geometry optimization based on DFT at B3LYP/6-31G* level) acts to break the conjugation, forming what is essentially two Flu-RH molecules attached at the fluorene bridge. As such, SF-OR devices, employing P3HT as the donor polymer, exhibited a similar VOC (0.97 V) to the Flu-RH devices. However, the JSC and FF showed significant improvement (7.5 mA cm−2 and 0.65 respectively). The 3D structure of SF-OR was shown to inhibit micrometer-scale aggregation in the blend, leading to an interpenetrating donor:acceptor network on the correct length-scale. H1 is another acceptor that makes use of a bifluorene type donor moiety at its core, this time a bifluorenylidene.44 The bifluorenylidene moiety was attached to four thiophene-flanked diketopyrrolopyrrole (DPP) units, affording an ‘H shaped’ NFA. The planarity of each DPP–fluorene–DPP section of the acceptor can be attributed to the phenyl–thienyl links between the bifluorenylidene and DPP units, and in combination with the strongly electron-withdrawing character of the DPP units, H1 was able to achieve a reasonably narrow bandgap of 1.67 eV and a low lying LUMO. The highly twisted double bond (calculated to be 40° using DFT at B3LYP/6-311G(d,p)) that links the fluorene units together acts to suppress the excessive aggregation, often seen in highly planar acceptors, and also improve the NFA's ability to accept electrons. Upon accepting an electron, a radical anion forms, which can be stabilized effectively. The anion is stabilized by one of the fluorene units, and the radical is stabilized by the other. When blended with P3HT, H1 was able to achieve an impressive VOC of 1.17 V, however could only exhibit a modest JSC and FF (7.74 mA cm−2 and 0.60). This led to an overall PCE of 5.42%, which is among the best efficiencies achieved with P3HT as the donor polymer. Considering the low bandgap of H1, one would expect that the complimentary absorption of the donor polymer and NFA would lead to a high photocurrent in devices. However, this was not achieved in devices and is the parameter that limited the PCE. The low photocurrent is likely to be a result of a large amount of non-geminate recombination in the blend, caused by an intimately mixed donor:acceptor morphology; a result of the twisted nature of the acceptor.
| Acceptor | Optical Eg (eV) | HOMO (eV) | LUMO (eV) | V OC (V) | J SC (mA cm−2) | FF | Electron mobilitya (cm2 V−1 s−1) | Hole mobilityb (cm2 V−1 s−1) | PCEc (%) | Donor | Additive | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by space charge limited current (SCLC) measurements using electron only devices. b Determined by space charge limited current (SCLC) measurements using hole only devices. c Average PCE values are shown in parentheses. | ||||||||||||
| Cz-RH | 2.05 | −3.50 | −5.53 | 1.03 | 4.69 | 0.53 | — | — | 2.56 (—) | P3HT | — | 36 |
| Flu-RH | 2.10 | −3.53 | −5.58 | 1.03 | 5.70 | 0.52 | — | — | 3.08 (—) | P3HT | — | 36 |
| F(DPP)2B2 | 1.82 | −3.39 | −5.21 | 1.18 | 5.35 | 0.50 | 2.80 × 10−4 | 4.30 × 10−5 | 3.17 (—) | P3HT | — | 37 |
| FBR | 2.14 | −3.57 | −5.70 | 0.82 | 7.95 | 0.63 | 2.60 × 10−5 | — | 4.11 (—) | P3HT | — | 38 |
| FBR | 2.14 | −3.57 | −5.70 | 1.13 | 11.70 | 0.63 | 3.80 × 10−4 | — | 8.00 (7.80) | PffBT4T-2DT | — | 39 |
| DICTF | 1.82 | −3.79 | −5.67 | 0.86 | 16.61 | 0.56 | 1.93 × 10−4 | 3.82 × 10−4 | 7.93 (7.63) | PTB7-Th | — | 40 |
| FDICTF | 1.63 | −3.71 | −5.43 | 0.95 | 16.09 | 0.67 | 2.40 × 10−5 | 3.37 × 10−5 | 10.06 (9.81) | PBDB-T | — | 41 |
| CBM | 2.02 | −4.13 | −6.05 | 0.88 | 10.60 | 0.53 | 1.90 × 10−6 | 1.00 × 10−4 | 5.30 (5.00) | PTB7-Th | 2.0% DIO | 42 |
| SF-OR | 2.15 | −3.25 | −5.50 | 0.97 | 7.50 | 0.65 | 6.71 × 10−6 | 8.49 × 10−5 | 4.70 (4.46) | P3HT | — | 43 |
| H1 | 1.67 | −3.84 | −5.51 | 1.17 | 7.74 | 0.60 | 2.40 × 10−3 | — | 5.40 (—) | P3HT | — | 44 |
2.2. Indacenodithiophene and indacenodithienothiophene based acceptors
Despite the early success of fluorene and carbazole based A–D–A type NFAs, it became apparent that more strongly absorbing, narrow bandgap, acceptors were desirable, particularly for complementary absorption when using scalable medium bandgap donor polymers such as P3HT. Many of the fluorene based acceptors also suffered from sub-optimal morphologies as a result of intimate mixing in the active layer blends, limiting the FF and photocurrent that could be achieved. With this is mind, more electron donating and planar units were identified to be used in the core of A–D–A type molecules to create narrow bandgap NFAs with enhanced self-aggregation properties. IDT and its derivatives emerged as strong candidates and have subsequently been used extensively in A–D–A type acceptors, owing to the strong electron donating and planar structures that these units possess, the relatively straightforward synthesis and good stability of these units, compared to benzodithiophene (BDT) and other typical donor moieties employed in push–pull copolymers (Fig. 5).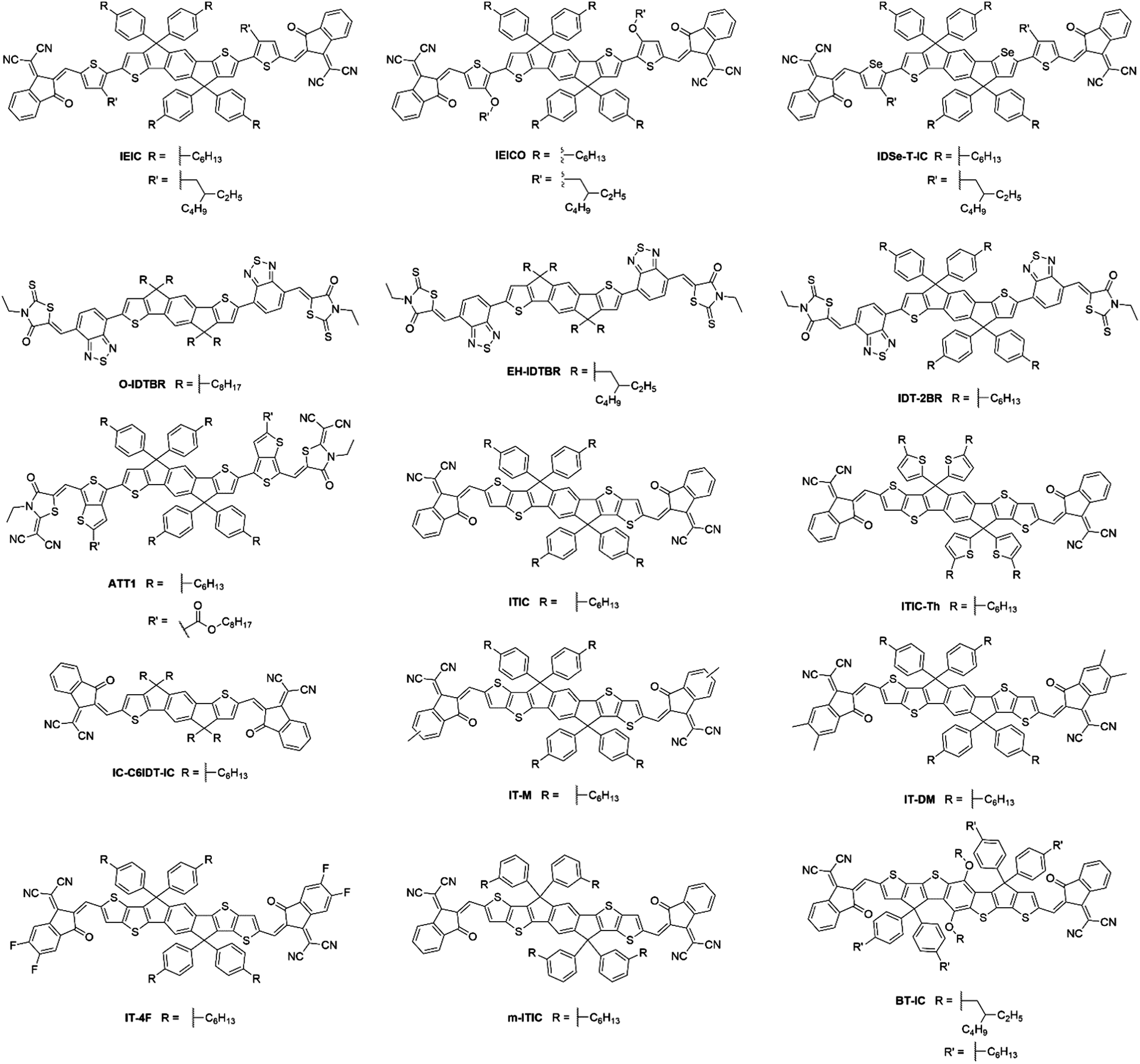 | ||
| Fig. 5 IDT and IDTT based small molecule A–D–A type acceptors. | ||
IEIC, an acceptor containing the IDT core flanked by thiophene spacer units and DCI end groups, was among the first A–D–A acceptors to incorporate the IDT unit at its core, in 2014.45 This molecule can be considered as analogous to DICTF with the difference being the replacement of the fluorene unit at the core with IDT and using phenylhexyl solubilizing chains. The stronger electron donating character, and added planarity associated with thiophene–thiophene links, led to IEIC being able to achieve a bandgap of 1.57 eV. By reducing the twisting in the molecule, the effective conjugation was increased, thereby narrowing the bandgap. Also, the greater overlap of HOMO and LUMO spatial distribution improves the oscillator strength of the acceptor, increasing the absorption coefficient of IEIC, compared to the values previously seen in many of the fluorene containing A–D–A type acceptors. Devices were fabricated using PTB7-Th as the donor material, despite the similar absorption profiles of the donor and acceptor. A result of this poor spectral coverage was a modest JSC of 13.55 mA cm−2. A relatively poor FF of 0.48 was achieved in these devices, likely to be a result of imbalanced hole and electron mobilities in the blend (μe = 1.0 × 10−4 cm2 V−1 s−1 and μh = 4.5 × 10−4 cm2 V−1 s−1), and domains of around 30 nm; somewhat larger than the exciton diffusion length in organic materials (Table 2). This can be attributed to the highly planar nature of the acceptor, inducing excessive crystallisation. A VOC of 0.97 V was reached in these cells, which, when considering that the difference between the HOMO of the donor and the LUMO of the acceptor is only ∼1.4 eV, is evidence of surprisingly low losses in this system. Despite the low FF, the devices were able to achieve a PCE of 6.3%, the highest at the time for small molecule acceptor devices. Devices using IEIC were later improved upon when PffT2-FTAZ-2DT was instead chosen as the donor polymer.46 This polymer possessed a medium bandgap, allowing the active layer to achieve greater spectral coverage. Additionally, this polymer was able to form a more favourable morphology with the IEIC (∼20 nm domains) and more balanced charge transport properties, leading to a much improved FF of 0.62. Overall, by changing the polymer to obtain a more favourable morphology and charge transport properties, an increase in PCE to 7.30% was achieved. This highlights the importance of pairing the acceptor with a polymer that is able to form favourable morphologies, as well as the correct energetics and complimentary absorption profiles. A small structural change to the structure of IEIC was designed where alkoxy, rather than alkyl, solubilizing groups on the thiophene spacers were employed resulting in IEICO.47 Using DFT calculations (geometry optimization based on DFT at B3LYP/6-31G(d,p) level), it was found that whilst the inclusion of alkoxy chains had no effect on the HOMO and LUMO distributions, the stronger electron donating nature of alkoxy chains acted to raise the HOMO level by ∼0.2 eV, thereby narrowing the bandgap of the acceptor to 1.34 eV. IEICO was then blended with PBDTTT-E-T to produce solar cell devices. The narrower bandgap of the acceptor led to improved spectral coverage and a broader EQE was reported, this improved photon harvesting was reflected by an improved JSC of 17.7 mA cm−2. A more preferable active layer morphology was also observed in comparison to their IEIC reference device, and diminished bimolecular recombination was observed, overall leading to the achievement of an 8.40% PCE. Another analogue of IEIC was reported in which the sulfur atoms in the IDT unit had been replaced by selenium atoms, producing IDSe-T-IC.48 This acceptor possessed similar energy levels to IEIC; with a small amount of narrowing of the bandgap (1.52 eV). This is typical upon substituting thiophene for selenophene type moieties as the larger chalcogen atoms reduce the chalcogenophene's aromaticity, thereby increasing the quinoidal character of the unit and narrowing the bandgap. Possessing a slightly narrower bandgap and being blended with the donor polymer J51, which has a complimentary absorption to IDSe-T-IC, devices using this blend were able to achieve a JSC of 15.2 mA cm−2, and with similar FMOs it was able to achieve a VOC of 0.91 V in devices. The morphology of the blend contained a fibrous, interpenetrating structure, which is important for efficient charge transport and reducing recombination in the blend. The presence of this morphological feature is likely to be the reason for the improved FF observed relative to IEIC and IEICO devices. As a result, IDSe-T-IC was able to exhibit an impressive performance of 8.58% PCE. Whilst they can lead to improved performance, selenophene containing acceptors are unlikely to be suitable candidates for commercialization due to toxicity and environmental concerns associated with them.
| Acceptor | Optical Eg (eV) | HOMO (eV) | LUMO (eV) | V OC (V) | J SC (mA cm−2) | FF | Electron mobilitya (cm2 V−1 s−1) | Hole mobilityb (cm2 V−1 s−1) | PCEc (%) | Donor | Additive | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by space charge limited current (SCLC) measurements using electron only devices. b Determined by space charge limited current (SCLC) measurements using hole only devices. c Average PCE values are shown in parentheses. | ||||||||||||
| IEIC | 1.57 | −3.82 | −5.42 | 0.97 | 13.55 | 0.48 | 1.00 × 10−4 | 4.50 × 10−4 | 6.31 (6.08) | PTB7-Th | — | 45 |
| IEIC | 1.57 | −3.82 | −5.42 | 1.00 | 12.70 | 0.62 | 2.10 × 10−4 | 1.90 × 10−4 | 7.30 (7.20) | PffT2-FTAZ-2DT | — | 46 |
| IEICO | 1.34 | −3.95 | −5.32 | 0.82 | 17.70 | 0.58 | 4.60 × 10−4 | 1.50 × 10−3 | 8.40 (8.30) | PBDTTT-E-T | — | 47 |
| IDSe-T-IC | 1.52 | −3.79 | −5.45 | 0.91 | 15.20 | 0.62 | 7.72 × 10−5 | 8.25 × 10−5 | 8.58 (8.21) | J51 | — | 48 |
| O-IDTBR | 1.63 | −3.88 | −5.51 | 0.73 | 14.10 | 0.62 | 4.70 × 10−6 | — | 6.38 (6.30) | P3HT | — | 49 |
| O-IDTBR | 1.63 | −3.88 | −5.51 | 0.83 | 14.70 | 0.65 | 3.40 × 10−6 | — | 7.80 (—) | P3HT | — | 50 |
| EH-IDTBR | 1.68 | −3.90 | −5.58 | 0.76 | 12.10 | 0.62 | 6.10 × 10−6 | 6.80 × 10−4 | 6.05 (—) | P3HT | — | 49 |
| EH-IDTBR | 1.68 | −3.90 | −5.58 | 1.02 | 17.20 | 0.63 | — | — | 11.09 (10.70) | PffBT4T-2DT | — | 51 |
| IDT-2BR | 1.68 | −3.69 | −5.52 | 0.84 | 8.91 | 0.68 | 2.00 × 10−4 | 2.60 × 10−4 | 5.12 (5.04) | P3HT | 3.0% CN | 52 |
| IDT-2BR | 1.68 | −3.69 | −5.52 | 1.02 | 13.90 | 0.60 | 1.70 × 10−4 | 6.70 × 10−4 | 7.70 (7.60) | PTB7-Th | — | 53 |
| IDT-2BR1 | 1.61 | −3.67 | −5.37 | 0.95 | 15.20 | 0.60 | 3.90 × 10−4 | 1.50 × 10−4 | 8.70 (8.60) | PTB7-Th | 3.0% CN | 54 |
| ATT-1 | 1.54 | −3.63 | −5.50 | 0.87 | 16.48 | 0.70 | 2.40 × 10−4 | 5.13 × 10−4 | 10.07 (9.89) | PTB7-Th | 1.0% DIO | 55 |
| ITIC | 1.59 | −3.78 | −5.51 | 0.81 | 14.21 | 0.59 | 1.10 × 10−4 | 4.30 × 10−5 | 6.80 (6.58) | PTB7-Th | — | 56 |
| ITIC | 1.59 | −3.78 | −5.51 | 0.95 | 17.87 | 0.67 | 1.00 × 10−3 | 1.00 × 10−3 | 11.34 (11.03) | PBQ-4F | 5.0% IPA | 57 |
| ITIC | 1.59 | −3.78 | −5.51 | 0.90 | 16.81 | 0.74 | — | — | 11.21 (10.68) | PBDB-T | 0.5% DIO | 64 |
| ITIC-Th | 1.60 | −3.93 | −5.66 | 0.88 | 16.24 | 0.67 | 6.10 × 10−4 | 3.00 × 10−4 | 9.60 (9.30) | PDBT-T1 | 1.0% CN | 58 |
| ITIC-Th | 1.60 | −3.93 | −5.66 | 0.93 | 17.60 | 0.69 | — | — | 10.88 (10.50) | PTFB-O | — | 60 |
| IC-C6IDT-IC | 1.62 | −3.91 | −5.69 | 0.89 | 15.05 | 0.65 | 2.90 × 10−4 | 5.10 × 10−5 | 8.71 (8.57) | PBDB-T | — | 61 |
| IT-M | 1.60 | −3.98 | −5.58 | 0.94 | 17.44 | 0.74 | 1.10 × 10−4 | 3.33 × 10−4 | 12.05 (11.48) | PBDB-T | 1.0% DIO | 62 |
| IT-DM | 1.63 | −3.93 | −5.56 | 0.97 | 16.48 | 0.71 | 4.70 × 10−5 | 2.29 × 10−4 | 11.29 (10.79) | PBDB-T | 1.0% DIO | 62 |
| IT-4F | 1.51 | −4.14 | −5.66 | 0.88 | 20.88 | 0.71 | 4.32 × 10−4 | 3.25 × 10−4 | 13.10 (—) | PBDBT-SF | 0.5% DIO | 64 |
| m-ITIC | 1.58 | −3.82 | −5.52 | 0.91 | 18.31 | 0.71 | 1.30 × 10−4 | 1.54 × 10−4 | 11.77 (11.49) | J61 | — | 66 |
| BT-IC | 1.43 | −3.85 | −5.43 | 0.90 | 17.75 | 0.66 | 7.60 × 10−4 | 3.53 × 10−4 | 10.46 (10.28) | J71 | — | 67 |
O-IDTBR and EH-IDTBR, also made use of the IDT core; replacing the fluorene donor unit in the analogous acceptor, FBR.49 The use of a more electron donating core, still flanked by BT and 3-ethylrhodanine units, resulted in a much narrower bandgap (1.63 eV). However, another important feature contributing to the reduced bandgap is the planarization that occurs when using IDT. The thienyl–phenyl link between the IDT and BT units is much less sterically strained and the single crystal X-ray structure exhibited no twist in O-IDTBR's backbone, compared to the DFT calculated 34° twist in FBR. Again, in reducing the twisting in the molecule, the effective conjugation was increased, producing a narrowing of the bandgap, and the greater oscillator strength of the acceptor increased the absorption coefficient in O-IDTBR and EH-IDTBR compared to FBR. Additionally, the planarity of these acceptors also allows greater self-aggregation, which was necessary to avoid the molecular mixing that limited the performance of FBR devices. When O-IDTBR devices were fabricated with P3HT as the donor polymer, they were able to improve considerably upon the performance of FBR. Whilst a small drop in VOC was observed (0.73 V), which can be attributed to the deeper lying LUMO, a vast improvement was seen in JSC, reaching 14.4 mA cm−2. This resulted in a PCE of 6.38%, which is amongst the highest for P3HT solar cells. The photocurrent was able to almost double due to the complimentary absorption of the donor and acceptor in the blend, and the formation of nanoscale acceptor domains, reduced recombination in the blend. Another notable feature of this system was the excellent oxidative stability in comparison to a number of fullerene devices, using either P3HT, PTB7-Th or PffBT4T-2DT as the donor polymer. After 1200 h in air, the low bandgap PTB7-Th:PCBM and PffBT4T-2DT:PCBM devices had dropped to less than 1% of their initial PCE, the P3HT:PCBM cell had dropped to about 10% of its initial PCE but the P3HT:O-IDTBR device retained over 73% of its initial PCE. O-IDTBR has also been used successfully in ternary solar cells, with P3HT as the polymer donor and O-IDFBR as a second electron acceptor.50O-IDFBR can be considered as analogous to O-IDTBR, differing in that it uses an indeno[1,2-b]fluorene moiety as its electron rich core. This acceptor possesses similar phenyl–phenyl links as had been previously seen in FBR, affording a medium bandgap. In this system the O-IDFBR is used to improve upon the performance of O-IDTBR:P3HT binary solar cells. The inclusion of the second NFA afforded: (i) a greater VOC (0.87 V), due to its higher lying LUMO; (ii) slightly improved JSC (14.70 mA cm−2) due to the improved spectral coverage given by using a medium and narrow bandgap acceptor; (iii) an improvement in FF (0.65), as a result of the a more favourable energy cascade in the ternary blend, and thus lower recombinative losses. Improvements in each of the VOC, JSC and FF led to a significant improvement in PCE, achieving a maximum of 7.80%, the highest reported efficiency for a single junction device with P3HT. The ability to produce reasonably high efficiency devices with P3HT as the donor represents an important benchmark for commercial OPV; P3HT is likely to be one of very few polymer donors that can be produced on the industrial scale currently. As such, developing high efficiency and stable devices using P3HT should be an area of focus to move closer to commercially viable OPV technologies. EH-IDTBR is a branched chain analogue of O-IDTBR, and was able to achieve a similar performance when used with P3HT.49 Interestingly, a recent study reported that when used in combination with the low bandgap polymer PffBT4T-2DT, OPV devices were able to achieve 11.1% PCE using a non-chlorinated processing solvent, mesitylene, and without the use of additives.51 The development of non-chlorinated device processing is important, as many chlorinated solvents are banned from use in industrial printing due to their inherent toxicity both to humans and the environment. Thus, for OPV to be viable on a large scale, alternative processing systems must be explored. By using EH-IDTBR with a low bandgap polymer, possessing a deeper lying HOMO than P3HT, the devices were able to achieve a VOC of 1.02 V, and despite the similar absorption profile of the donor and acceptor, a JSC of 17.2 mA cm−2. The high photocurrent is likely to be due to excellent harvesting of photons in the 500–700 nm region of the spectrum and a favourable active layer morphology. Additionally, the devices processed from mesitylene were in fact able to exceed the efficiency achieved by chlorobenzene (CB) processed devices, the traditional solvent of choice in OPV processing, and presented better reproducibility, shelf-life and operating stability than those processed from CB.
IDT-2BR is analogous to the IDTBR acceptors however it contains phenylhexyl solubilizing chains on the IDT core.52 The only optoelectronic change that the addition of the phenyl units in the solubilizing chains had caused was a slight raising of the HOMO and LUMO levels by ∼0.1 eV. As such, when incorporated into devices with P3HT, a small improvement in VOC to 0.84 V was observed, and an improvement in FF to 0.68 was also reported. However a drastically reduced JSC of just 8.91 mA cm−2 was attained by the IDT-2BR devices, which led to an overall PCE of 5.12%. The fact that these devices were fabricated in a conventional (ITO/PEDOT:PSS/active layer/Ca/Al) architecture whereas the O-IDTBR devices were fabricated using an inverted (ITO/ZnO/active layer/MoO3/Ag) architecture may account for the vast difference in JSC seen between these two analogous acceptors, however the high surface roughness (15.7 nm) and a sub-optimal morphology may also have caused the drop in photocurrent in the IDT-2BR devices. IDT-2BR has also been used with PTB7-Th in OPV devices; achieving a VOC of 1.02 V and a JSC of 13.4 mA cm−2, this blend was able to produce a PCE of 8.3%.53 The JSC was relatively modest for a low bandgap acceptor used in combination with a high performance polymer, which is likely to be a result of a sub-optimal morphology leading to significant recombination in the blend. However, the non-crystalline nature of the acceptors did lead to excellent thermal and morphological stability exhibited by the blends, something that is considered to be a major issue in fullerene containing devices. A more recent study, which also made use of PTB7-Th as the donor, compared IDT-2BR to IDT-2BR1, an analogous acceptor with n-hexyl solubilizing chains rather than phenylhexyl chains.54 It was found that IDT-2BR1 performed significantly better in devices, attaining a maximum PCE of 8.7%, in comparison to 8.3% when IDT-2BR was used. Again, the disparity between the two devices was mainly manifested in a much lower photocurrent when using IDT-2BR as the acceptor, it is suggested that this is a result of the n-hexyl chains of IDT-2BR1, which allowed stronger 3D intermolecular interactions with the donor material and greater electron mobilities (μe = 3.9 × 10−4 cm2 V−1 s−1 and μh = 1.5 × 10−4 cm2 V−1 s−1 for the IDT-2BR1 blend cf. μe = 1.7 × 10−4 cm2 V−1 s−1 and μh = 6.7 × 10−4 cm2 V−1 s−1 for the IDT-2BR blend). Therefore, despite a slightly more simple synthetic route available for IDT-2BR and other IDT based acceptors that make use of arylalkyl rather than alkyl side chains, this study seems to suggest that the inclusion of the phenyl units on the solubilizing chains lead to inferior intermolecular interactions, a less favourable morphology and thus poorer performance in devices. The widespread use of the IDT moiety in A–D–A type acceptors means that any distinction in performance between alkyl and aryl–alkyl IDT should be further investigated; allowing focus to shift to the more suitable IDT analogue (Fig. 6).
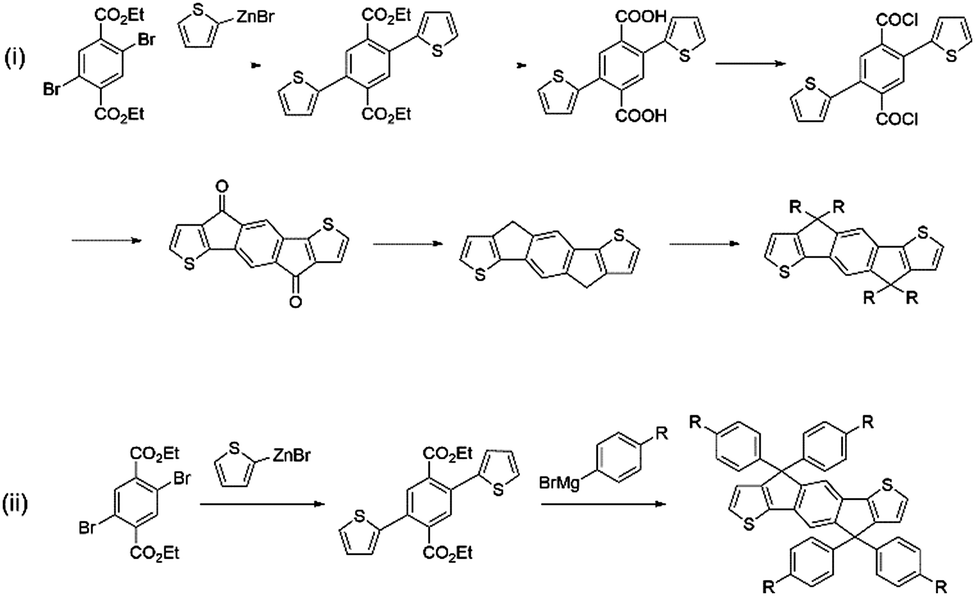 | ||
| Fig. 6 Synthetic procedures for the preparation of IDT with (i) aliphatic alkyl chains and (ii) aromatic alkyl chains at the carbon bridgehead positions. | ||
The design of IDT-2BR was further extended by substituting the electron withdrawing BT spacer unit with thieno[3,4-b]thiophene (TT) and dicyanovinyl moieties were included on the rhodanine units, in the acceptor ATT-1.55 This acceptor exhibited a reduced bandgap (1.54 eV) due to the inclusion of the strongly electron withdrawing dicyanovinyl rhodanine and a small increase in planarity, arising from the inclusion of the TT unit. When OPV devices were made, using PTB7-Th as the donor, an improved PCE of 10.07% was reported, relative to the 8.30% with IDT-2BR. The narrowing of the bandgap of ATT-1 arose from a lower lying LUMO, and therefore a decrease in VOC to 0.87 V, however the narrower bandgap and stronger extinction coefficient of this acceptor led to improved photon harvesting, when compared to IDT-2BR, and thus a substantial increase in JSC to 16.48 mA cm−2. It was necessary to use DIO in order to achieve this high efficiency, by enhancing the acceptor crystallinity and reducing the tendency for the donor and acceptor to mix. Though this led to a substantially improved performance, systems that require the use of additives such as DIO are not particularly attractive from an industrial viewpoint, as a result of the photostability problems that they have been noted to cause.
ITIC can be considered as a further development of the acceptor IEIC, this acceptor made use of IDTT as the core, rather than IDT, and did not include a π-conjugated spacer, with the electron deficient DCI units on the periphery.56 The extension of the electron donating core and removal of the π-conjugated spacer in ITIC produced a very similar bandgap (1.59 eV) to that exhibited by IEIC (Fig. 7). However, the HOMO and LUMO values were shifted upwards by ∼0.1 eV. When paired with PTB7-Th, the OPV devices exhibited a VOC of 0.81 V; this is slightly lower than seen for IEIC, which may be a result of increased energetic losses in this system. The devices showed a similarly high JSC of 14.22 mA cm−2, but the most marked improvement was in FF (0.59) due to more closely balanced hole and electron mobilities in the blend (μe = 1.1 × 10−4 cm2 V−1 s−1 and μh = 4.3 × 10−5 cm2 V−1 s−1 for the ITIC blend cf. μe = 1.0 × 10−4 cm2 V−1 s−1 and μh = 4.5 × 10−4 cm2 V−1 s−1 for the IEIC blend), thereby reducing recombination. This resulted in a maximum PCE of 6.8% being achieved. There have since been several improvements made using this acceptor with medium and wide bandgap polymer donors. However, the most notable of these is the recent report where ITIC was paired with a wide bandgap polymer PBQ-4F to produce devices that were able to achieve 11.34% PCE.57 An improvement in JSC was observed arising from the improved photon harvesting that is possible with the complimentary absorption of the polymer and acceptor. A vastly improved FF (0.67) can be considered as a result of much higher hole and electron mobilities, that were also more closely balanced (μe = 1.0 × 10−3 cm2 V−1 s−1 and μh = 1.0 × 10−3 cm2 V−1 s−1), leading to reduced recombinative losses in the blend. Strong π–π interactions between the donor polymer, likely a result of PBQ-4F's planar structure, improved the charge transport properties of the blend. Importantly, the active layers of the high efficiency devices reported here are also processed from a relatively benign non-chlorinated solvent system, tetrahydrofuran (THF) with 5% isopropanol as an additive. As highlighted above for the PffBT4T-2DT:EH-IDTBR solar cells that were also processed from non-chlorinated solvents, this is an important step towards realising commercially viable OPV. ITIC-Th is an analogue of ITIC in which the phenyl units of the solubilizing sidechains have been replaced by thienyl groups.58 Relative to its phenyl containing counterpart, ITIC-Th possesses slightly deeper lying HOMO and LUMO levels; a result of the electron withdrawing σ-inductive effect from the thienyl moieties. Additionally, the inclusion of the thienyl–alkyl chains led to enhanced intermolecular interactions, and therefore an improved electron mobility for ITIC-Th (μe = 6.1 × 10−4 cm2 V−1 s−1 for the ITIC-Th blend cf. μe = 2.6 × 10−4 cm2 V−1 s−1 for the ITIC blend). In devices with PDBT-T1, this acceptor was able to reach 9.6% PCE, with a high JSC of 16.24 mA cm−2 resulting from the complimentary absorption and preferred morphology in the bulk heterojunction. The outstanding feature of this acceptor is a high FF of 0.67 being achieved, mainly as a product of the high and balanced charge carrier mobilities exhibited by this blend. However, it must be noted that to achieve the optimal morphology, 1-chloronaphthalene (CN), a chlorinated high boiling point additive was needed. Whilst this can improve the performance in OPV devices, if they persist in the active layer they can significantly lower the morphological stability of the blends over time, leading to microscale phase separation.59 Building upon this promising result, ITIC-Th was combined with a less crystalline, medium bandgap donor polymer, PTFB-O.60 This allowed the formations of much smaller domains in the bulk heterojunction (∼30 nm) and high PL quenching was observed, suggesting that the excitons can be split into free charges more efficiently and non-geminate recombinative losses can be minimized. This led to the impressive photocurrent and fill factor (17.6 mA cm−2 and 0.69) and ultimately a PCE of 10.88% without the use of any additives.
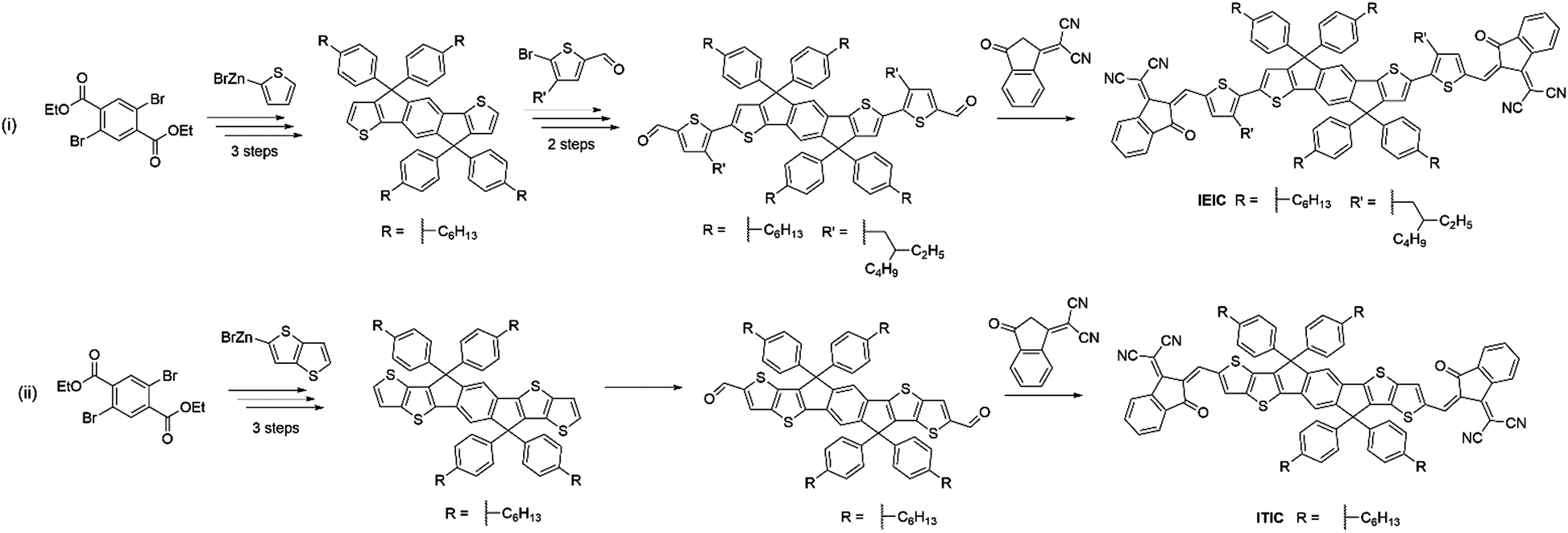 | ||
| Fig. 7 Comparison of the synthetic routes required for (i) IDT based IEIC and (ii) IDTT based ITIC. | ||
IC-C6IDT-IC makes use of an alkylated IDT core, in this case with n-hexyl chains rather than the phenyl or thienyl alkyl chains. Again, use is made of the DCI end group, without the use of a π-conjugated spacer in the molecule.61 This afforded a bandgap of 1.62 eV despite its structural simplicity, relative to the aforementioned IDT and IDTT based acceptors, and possessed HOMO and LUMO levels similar to the IDTBR acceptors. IC-C6IDT-IC was paired with a medium bandgap donor polymer in devices (PDBT-T1) and was among the first examples of A–D–A type NFA solar cells to reach a PCE of over 8%, in this case reaching 8.71%. Through the complimentary absorption profiles of the donor and acceptor, along with the strong absorption coefficient, this blend was able to reach photocurrents of 15.05 mA cm−2. Preferential face-on π–π ordering and appropriate length scale phase separation was suggested to lead to relatively good charge carrier mobilities in the blend (μe = 2.9 × 10−4 cm2 V−1 s−1 and μh = 5.1 × 10−5 cm2 V−1 s−1), further contributing to the high JSC and FF (0.67) exhibited by this blend.
A further development in the design of ITIC based acceptors by introducing either one or two methyl groups onto the phenyl ring of the DCI unit, to create IT-M and IT-DM.62 The aim of these modifications was to raise the LUMO slightly, in order to improve the VOC, without causing additional morphological disruption, hence very short methyl units were used. The inclusion of the weakly electron donating methyl groups had the desired effect, with IT-M possessing a LUMO which was 0.04 eV higher than their value measured for ITIC, and IT-DM possessing a LUMO that was 0.09 eV higher. The acceptors were paired with the donor polymer PBDB-T, which had already shown reasonable success with ITIC; achieving 11.2%.63IT-M was able to achieve an improved VOC of 0.94 V (relative to the 0.90 V achieved in PBDB-T:ITIC devices) as a result of the higher lying LUMO, but also had an superior JSC which was ∼1 mA cm−2 higher (17.44 mA cm−2) than the ITIC reference device. The result of which was a PCE of 12.05%, among the highest currently reported fullerene-free single-junction OPVs. IT-DM was able to further improve on the VOC to 0.97 V, however the JSC and FF dropped slightly, leading to a still impressive 11.29% PCE. The poorer JSC and FF in devices containing IT-DM were attributed to a slight reduction in domain purity, which led to a small increase in exciton dissociation efficiency and asymmetric charge transport properties. A similar strategy has since been employed in the design of IT-4F.64 In this case the phenyl units of the DCI end group each contain 2 fluorine atoms, rather than the methyl groups used in IT-M and IT-DM. The inclusion of the fluorine atoms was aimed to narrow the bandgap further, and improve both intra- and intermolecular interactions through the non-covalent F⋯H and S⋯F interactions that can often be observed in fluorinated molecules.65 As expected the fluorination of the DCI end group led to a lowering of both the HOMO and LUMO levels due to the strong electron withdrawing nature of the fluorine atoms. Stronger π–π interactions led to a broadened and red shifted absorption spectrum, along with an enhanced extinction coefficient due to enhanced intramolecular charge transfer, in IT-4F. The improved intermolecular interactions also led to a slight gain in the electron mobility of the acceptor (μe = 4.32 × 10−4 cm2 V−1 s−1 for the IT-4F blend cf. 3.13 × 10−4 cm2 V−1 s−1 for the ITIC blend). As a result of the lowered FMOs of IT-4F, PBDB-T was no longer the optimal candidate to be used in bulk heterojunctions as the offset between the HOMO of the donor and the LUMO of the acceptor would not have been able to produce a respectable VOC in devices. Instead a modified version of the polymer (PBDB-T-SF) was developed, which was fluorinated on the thienyl units of the side-chains. This again led to a lowering of the FMOs and similar improvements in the optical and charge carrier transport properties. OPV devices utilizing IT-4F and PBDB-T-SF were able to achieve an exceptional 13.1% PCE, the highest reported in single-junction polymer solar cells to date. Despite having a lower VOC than devices using most ITIC based acceptors (0.88 V), a vast improvement in JSC to 20.88 mA cm−2 was apparent as a result of the enhanced photon harvesting made possible by the more strongly absorbing components. It must be recognized that the chemical modification of the polymer also contributed to the improved performance, highlighting the benefit of tuning the donor polymer to better suit the acceptor. These devices were also shown to display excellent storage stability under N2, still achieving 11.99% after 1700 h. m-ITIC, another derivative of ITIC has also been reported in which the position of the solubilizing alkyl substituent has been moved from the para- to the meta-position on the phenyl rings.66 The design strategy behind this side-chain isomerization was to tune the intermolecular self-assembly of the acceptor without altering the optoelectronic properties of the acceptor substantially. The FMOs remained very similar to those of ITIC, and no obvious changes in the optical properties were reported. The isomerization of the side-chain did however have a significant impact on the crystallinity of the acceptor, whereby the para-alkyl–phenyl version (ITIC) had poorer self-organization than the meta-alkyl–phenyl m-ITIC; this was apparent from better defined scattering peaks in GIWAXS and a longer crystalline correlation length (CCL). Additionally, m-ITIC adopted a predominantly face-on crystalline orientation in these devices, compared to the co-existence of both edge-on and face-on crystallites in ITIC devices. The more crystalline nature and preferential face-on orientation of crystallites in m-ITIC gave rise to a greater electron mobility (μe = 1.30 × 10−4 cm2 V−1 s−1 for the m-ITIC blend cf. μe = 1.05 × 10−4 cm2 V−1 s−1 for the ITIC blend). The acceptor was blended with a medium bandgap donor polymer J61, with reference devices using ITIC also fabricated. The virtually identical FMOs of the two acceptors led to a very similar VOC in both cases (0.91 V), however the improved electron mobility of m-ITIC led to a more balanced charge transport in the blend, resulting in reduced recombination in the bulk heterojunction. This was reflected in a large increase in FF for the m-ITIC devices relative to the reference ITIC devices; 0.71 and 0.66 respectively. This led to an overall improvement in PCE from 10.57% to 10.77% upon modifying the acceptor to contain the meta-phenyl–alkyl chains. Therefore, in addition to the consideration of alkyl vs. aryl–alkyl chains on the acceptor, isomerization of the aryl–alkyl chains can also play a role in fine tuning the structural and morphological properties of NFAs.
BT-IC, an extremely low bandgap acceptor, comprised of a BDT core unit, which was fused with cyclopentadithiophene units on either side to produce a fused seven ring system, flanked by the electron deficient DCI end groups.67 This acceptor included electron donating alkoxy chains on the BDT part of the fused ring core to raise the HOMO, and narrow the bandgap further without lowering the LUMO, thus avoiding the possibility of a lower VOC. The added electron-donating nature of this core unit allowed a remarkably low bandgap of 1.43 eV to be achieved, and still retained an extinction coefficient that was comparable to ITIC. This allowed greater spectral coverage when blended with J71, a medium bandgap donor polymer, resulting in a respectable JSC of 17.75 mA cm−2. The strategy of including alkoxy chains to raise the HOMO and narrow the bandgap, without impacting the LUMO, also proved to be successful with a VOC of 0.90 V being achieved. Overall this resulted in a PCE of 10.46%, however the increased synthetic complexity relative to ITIC is not offset by any outstanding improvements in performance.
There are several key points that can be taken away from the evolution of the A–D–A type acceptors discussed above. The early fluorene based acceptors showed considerable promise, but ultimately the push–pull character and conjugation afforded in many of these materials was not sufficient to harvest many of the high-energy photons effectively. Additionally, the inherent twisting in a number of these acceptors, caused by the steric clash of ortho-hydrogen atoms in the phenyl–phenyl linkages, led to poorly aggregating materials that mixed too finely with the donor polymer in active layer blends. The IDT based acceptors that evolved from the above issues have led to significant progress. Increased conjugation and push–pull character has allowed narrow bandgaps to be accessed in NFAs, and therefore improved photon absorption. While, the extended planar structures achieved a greater tendency for aggregation of the acceptors in blends, often leading to favourable lengthscale percolating networks of donor and acceptor materials in the blends. The relative aggregation tendency of the acceptor, necessary for optimal device performance, depends heavily on the crystallinity of the donor polymer used. However, a general rule is apparent that if the acceptor does not exhibit a strong tendency to aggregate and phase separate it will molecularly mix with the polymer, resulting in large recombinative losses and reduced electron mobility. Alternatively, acceptors with a high aggregation tendency will often form domains that are far larger than the exciton diffusion length, leading to a larger fraction of excitons relaxing before reaching the interface and are therefore wasted. Hence, a balance between these two extremes must be reached in order to achieve donor and acceptor domains that are on the same lengthscale as the exciton diffusion length, leading to an optimal blend morphology. As the successful A–D–A structures have become more apparent, diligent work to tune the solubilizing chains, located both on the core and the end groups, has led to a fine-tuning of the aggregation properties of the acceptors and consequently improved bulk-heterojunction morphologies and device performance. Though this has allowed incremental improvements in PCE, care must be taken not to compromise the synthetic complexity of these acceptors in the pursuit of higher efficiencies such that they are not viable to produce on an industrial scale. Further to this, the inclusion of a handful of studies reporting OPV devices fabricated from non-chlorinated solvents whilst using A–D–A type NFAs highlights another advantage this class of acceptors holds, in addition to the highest efficiencies currently reported. With >11% PCE now achieved on a routine basis, it may allow a shift of focus to improve the stability of materials in devices and lowering the costs associated with the preparation of these acceptors. Another important consideration is to design NFAs that are compatible with scalable donor polymers. Though the most exceptional efficiencies have been achieved with low bandgap polymers, P3HT remains to be the only truly scalable donor to date. Therefore, further optimization of the A–D–A type acceptors should have some focus, at least, on improving upon the best P3HT:NFA systems that have been reported.
3. Perylene diimide based acceptors
Perylene-3,4:9,10-tetracarboxylic acid diimides (PDIs) are a class of π-conjugated molecules that over the past three decades have found extensive applications as high-performance organic semiconductors, and have found relative success in the field of organic photovoltaics. Whilst their electron-withdrawing character arises from their dicarboxylic acid imide groups at the 3,4- and 9,10-peri-positions, their polycyclic aromatic skeleton acts as electron-donating unit. The optoelectronic properties of this class of NFAs can further be influenced through the inclusion of alkyl, aryl or heteroaryl substituents at their core (1,2,5,6,7,8,11,12) positions. Since the imide nitrogen is not conjugated to the aromatic system, functional group substitution at these positions does not tend to affect the FMOs and is instead used to tune the self-assembly properties of these acceptors, thus allowing for a partially independent modulation of the optoelectronic and morphological properties. Compared to A–D–A type acceptors, the vast aromatic system of PDIs is highly beneficial to their charge carrier mobilities with electron mobilities over 1 cm2 V−1 s−1 having been reported.68 Moreover, their good electron accepting properties and excellent thermal, chemical and photochemical stability add to their attractiveness.69,70 The main drawback of PDIs as NFAs is their extended π-scaffold often leading to micrometer-sized aggregates in blends, which in turn leads to insufficiently large donor–acceptor interfaces for efficient exciton splitting. Consequently, over the past 30 years numerous molecular engineering strategies have been dedicated towards striking a balance between the formation of donor-PDI domains that are sufficiently small to allow for efficient charge separation, yet large enough to ensure percolating networks for high charge carrier mobilities.71 Three main approaches have been developed towards optimising PDI's optoelectronic and morphological properties in OPV blends, these rely on chemical modifications at either the imide, bay (1,6,7,12) or ortho (2,5,8,11) positions of the PDI core (Fig. 8).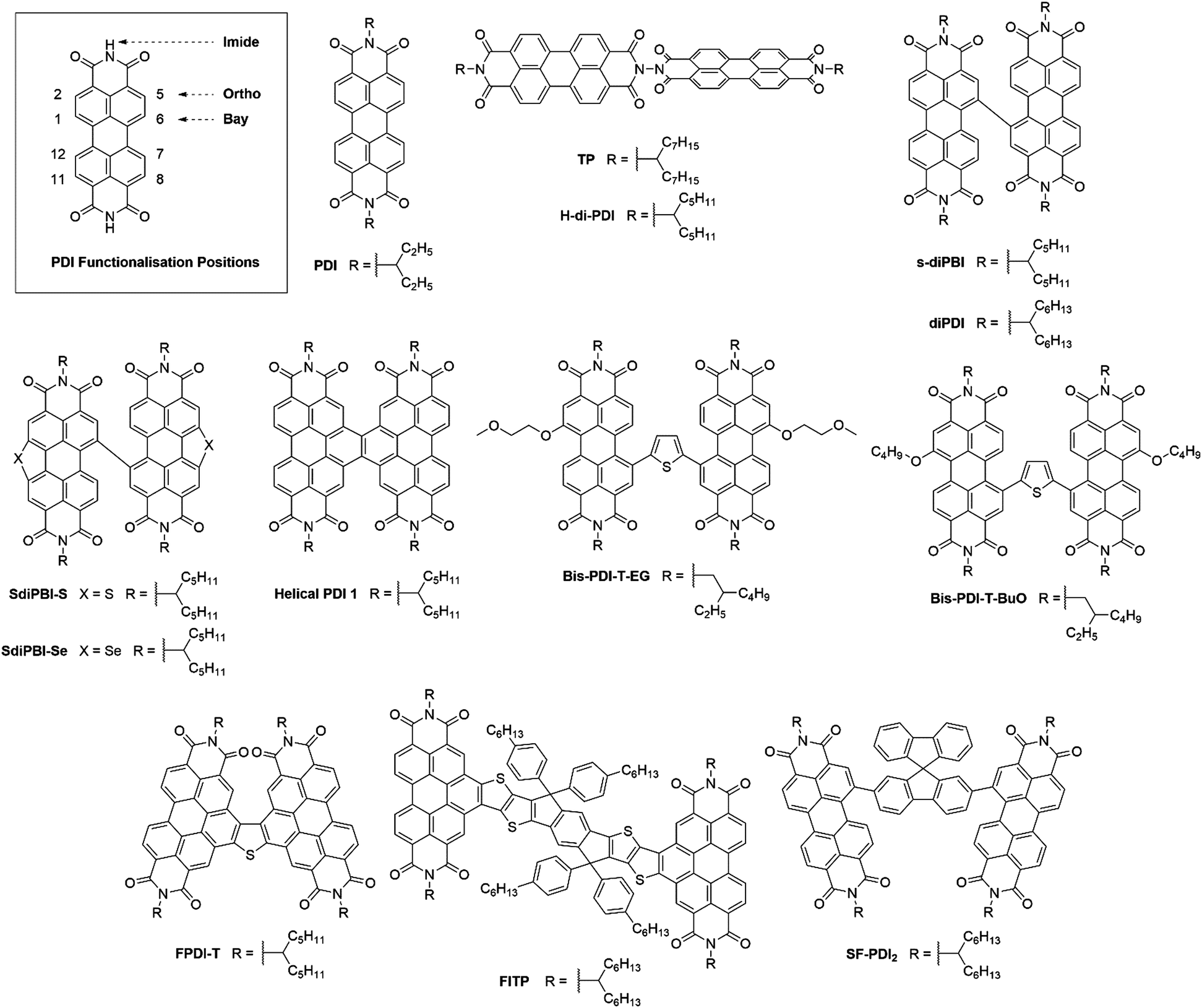 | ||
| Fig. 8 Monomeric and dimeric PDI based small molecule acceptors. | ||
3.1. PDI monomers
The oldest and simplest strategy to control the aggregation tendency of PDIs involves the inclusion of solubilising side chains. A well-known and highly studied example thereof is PDI, bearing pendant 1-ethyl-propyl chains at either imide nitrogen atom. Although initial investigations of PDI as a NFA only yielded devices with PCEs <1%, successful fabrication of devices with a PCE of 3.0% was achieved, when blending PDI with the low bandgap small molecule donor p-DTS(FBTTh2)2 (Table 3).72,73 The increase in the photocurrent (7.4 mA cm−2) was attributed to the complementary absorption of the donor and the acceptor leading to greater spectral coverage compared to the previously reported active layer blends. Further incrementation of the performance of p-DTS(FBTTh2)2:PDI based devices was achieved by optimising the donor:acceptor ratio from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1.3:1. Due to p-DTS(FBTTh2)2's greater molar extinction coefficient compared to PDI, increasing the donor percentage in the blend allowed for 10% more intense light absorption compared to the 1:1 reference blend.74 Additionally, transmission electron microscopy (TEM) revealed smaller phase domains, around 20 nm, in the 1.3:1 blends that should allow for more efficient exciton splitting. Photoluminescence quenching measurements confirmed the more suitable blend morphology as the fluorescence from the donor and the acceptor were minimised at the higher donor concentration in the blend. Better charge generation by tuning of the optical and morphological properties of the blend thus rationalise the 35% improved photocurrent in devices with a maximum PCE of 5.13%.
:1 to 1.3:1. Due to p-DTS(FBTTh2)2's greater molar extinction coefficient compared to PDI, increasing the donor percentage in the blend allowed for 10% more intense light absorption compared to the 1:1 reference blend.74 Additionally, transmission electron microscopy (TEM) revealed smaller phase domains, around 20 nm, in the 1.3:1 blends that should allow for more efficient exciton splitting. Photoluminescence quenching measurements confirmed the more suitable blend morphology as the fluorescence from the donor and the acceptor were minimised at the higher donor concentration in the blend. Better charge generation by tuning of the optical and morphological properties of the blend thus rationalise the 35% improved photocurrent in devices with a maximum PCE of 5.13%.
| Acceptor | Optical Eg (eV) | HOMO (eV) | LUMO (eV) | V OC (V) | J SC (mA cm−2) | FF | Electron mobilitya (cm2 V−1 s−1) | Hole mobilityb (cm2 V−1 s−1) | PCEc (%) | Donor | Additive | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by space charge limited current (SCLC) measurements using electron only devices. b Determined by space charge limited current (SCLC) measurements using hole only devices. c Average PCE values are shown in parentheses. | ||||||||||||
| PDI | — | — | — | 0.78 | 7.4 | 0.52 | 1.70 × 10−4 | 7.40 × 10−5 | 3.00 (—) | p-DTS(FBTTh2)2 | 0.4% DIO | 73 |
| PDI | — | −5.87 | −3.82 | 0.8 | 10.1 | 0.64 | 4.50 × 10−3 | 1.61 | 5.13 (5.07) | p-DTS(FBTTh2)2 | 0.4% DIO | 74 |
| TP | — | — | — | 0.77 | 9.0 | 0.46 | 1.47 × 10−4 | 2.74 × 10−4 | 3.20 (—) | PBDTTT-C-T | — | 75 |
| H-di-PDI | 2.09 | −5.85 | −3.74 | 0.79 | 13.1 | 0.60 | 4.30 × 10−4 | 2.30 × 10−2 | 6.41 (6.19) | PTB7-Th | 1.0% DIO and 2.0% CN | 76 |
| s-diPBI | 2.10 | −5.94 | −3.84 | 0.73 | 10.6 | 0.47 | — | — | 3.63 (—) | PBDTTT-C-T | 1.5% DIO and 1.5% CN | 77 |
| s-diPBI | 2.08 | −5.95 | −3.87 | 0.87 | 8.3 | 0.61 | — | 1.00 × 10−3 | 4.39 (—) | PBDB-T | 1.5% DIO and 1.5% CN | 78 |
| s-diPBI | 2.09 | −6.13 | −4.04 | 0.80 | 12.0 | 0.59 | 3.32 × 10−5 | 4.36 × 10−2 | 5.90 (5.73) | PTB7-Th | 1.0% DIO and 2.0% CN | 79 |
| SdiPBI-S | 2.20 | −6.05 | −3.85 | 0.90 | 12.0 | 0.66 | 2.80 × 10−3 | 1.20 × 10−3 | 7.16 (6.90) | PDBT-T1 | 0.75% DIO | 80 |
| SdiPBI-Se | 2.22 | −6.09 | −3.87 | 0.91 | 12.8 | 0.70 | 4.80 × 10−3 | 3.60 × 10−3 | 8.47 (8.23) | PDBT-T1 | 0.25% DIO | 81 |
| Helical PDI 1 | — | — | — | 0.80 | 13.5 | 0.55 | 3.40 × 10−4 | 2.90 × 10−4 | 6.05 (5.94) | PTB7-Th | 1.0% DIO and 1.0% CN | 82 |
| Bis-PDI-T-EG | 1.81 | −5.65 | −3.84 | 0.85 | 8.9 | 0.54 | 1.00 × 10−3 | 3.00 × 10−3 | 4.03 (3.91) | PBDTTT-C-T | 5.0% DIO | 83 |
| Bis-PDI-T-EG | 1.81 | −5.65 | −3.84 | 0.84 | 12.8 | 0.56 | 6.06 × 10−3 | 1.03 × 10−2 | 6.10 (6.00) | PBDTTT-C-T | 1.5% DIO | 84 |
| Bis-PDI-T-EG | 1.80 | −5.64 | −3.84 | 0.89 | 13.2 | 0.59 | 1.87 × 10−4 | 1.63 × 10−4 | 7.24 (6.94) | PDBT-T1 | 3.0% DIO | 85 |
| Bis-PDI-T-BuO | 1.79 | −5.65 | −3.86 | 0.89 | 12.3 | 0.58 | 2.30 × 10−5 | 4.10 × 10−4 | 6.36 (6.18) | PDBT-T1 | 2.0% DIO | 86 |
| FPDI-T | 2.22 | −5.98 | −3.77 | 0.93 | 12.0 | 0.58 | 1.63 × 10−4 | 5.92 × 10−2 | 6.72 (6.48) | PTB7-Th | 2.0% CN | 87 |
| FITP | 1.73 | −5.28 | −3.75 | 0.99 | 13.2 | 0.56 | 3.66 × 10−4 | 5.60 × 10−4 | 7.33 (—) | PTB7-Th | 2.0% CN | 88 |
| SF-PDI2 | 2.07 | −5.90 | −3.83 | 0.98 | 10.7 | 0.57 | 1.80 × 10−4 | 2.30 × 10−3 | 6.30 (6.00) | PffBT4T-2DT | — | 89 |
| SF-PDI2 | 2.37 | −5.99 | −3.62 | 1.11 | 13.3 | 0.64 | — | — | 9.50 (—) | P3TEA | 2.5% ODT | 90 |
3.2. PDI dimers
An alternative strategy to disrupt PDIs’ cofacial stacking involves the use of dimeric PDI acceptors with twisted conformations. TP was conveniently synthesised in a four-step route with an overall yield of 34%, whereby the two PDI units are connected by a hydrazine linkage and rotated orthogonally to each other.75 The use of a highly twisted 3D structure was intended to inhibit the excessive aggregation often observed in PDI containing blends, leading to a percolating donor:acceptor network on the correct length scale, rather than the unfavourable formation of micrometer sized domains observed in active layers where aggregation has not been suppressed. When used in combination with the low bandgap polymer donor PBDTTT-C-T, this acceptor was able to achieve a PCE of 3.20%. The good photovoltaic performance was attributed to the optimum domain sizes around 10 nm in the active layer allowing for efficient charge generation and transport. TP's design concept was then developed further with the acceptor H-di-PDI. Again, this acceptor utilised a hydrazine linked PDI dimer, but employed pentyl–hexyl rather than heptyl–octyl solubilising chains.76 Shortened side-chains allowed for tighter molecular packing, such that higher charge carrier mobilities could be achieved (μe = 4.3 × 10−4 cm2 V−1 s−1 and μh = 2.3 × 10−2 cm2 V−1 s−1 for the H-di-PDI blend cf. μe = 1.47 × 10−4 cm2 V−1 s−1 and μh = 2.74 × 10−4 cm2 V−1 s−1 for the TP blend). Using PTB7-Th as the donor material, devices were able to achieve a much improved PCE of 6.41%. Whilst PTB7-Th's lower bandgap allowed for greater spectral coverage hence photocurrents, the higher charge carrier mobilities ensured a 30% larger fill factor (Fig. 9).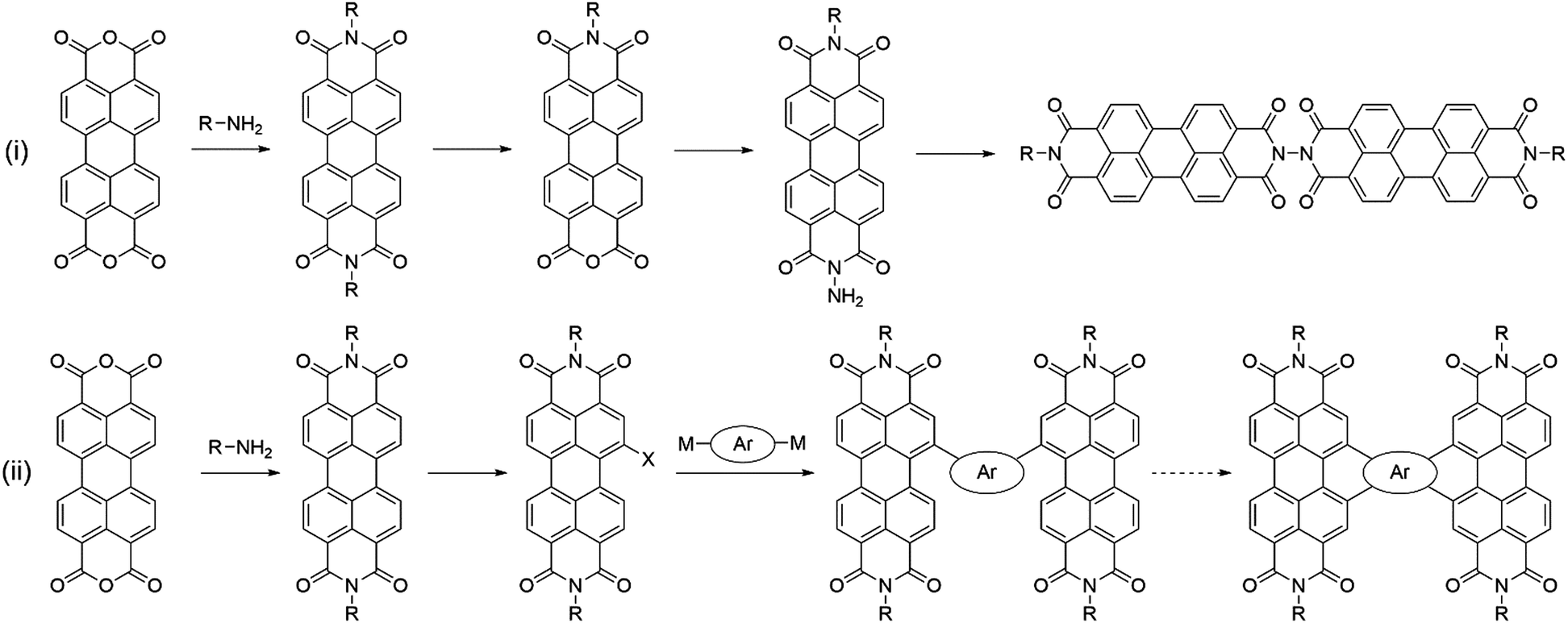 | ||
| Fig. 9 Comparison of the general synthetic routes required for (i) imide linked and (ii) bay linked PDI dimers. | ||
Twisted conformations in PDI dimers can also arise via linkage of the two monomers at their bay positions. Amongst the simplest and highest performing bay-linked PDI dimers is s-diPBI, in which the two PDI units are covalently bound through a C–C single bond at their 1-position. The 70° dihedral angle (estimated by geometry optimization using DFT with B3LYP/6-31G(d) basis sets) across the two PDI planes gives rise to the three-dimensionality of the NFA and is designed to improve the photovoltaic performance by reducing the acceptor's inherent aggregation tendency. Another important feature of the large twist angle is the resulting break in conjugation thus conferring the acceptor a relatively high lying LUMO (approx. −3.8 eV) to aid in maximising the VOC. The first devices employing s-diPBI as NFA were fabricated by spin-coating the active layer from a PBDTTT-C-T:s-diPBI blend, affording cells with a PCE of 3.63%.77 It was found that despite the acceptors high-lying LUMO, the performance was limited predominantly by poorly matched energy levels between the polymer donor and the acceptor thereby limiting the VOC of the cells. Based on this observation, PDBTTT-C-T was replaced with PBDTBDD, whose deeper HOMO yielded an almost 20% larger VOC thus boosting performance to 4.39%.78 Further enhancement of the efficiency of s-diPBI based solar cells was achieved by switching from a conventional to an inverted device architecture.79 In the inverted device, more intense light absorption due to reduced thin-film interference arising from more carefully managed refractive index differences led to better optical distribution. Furthermore, deposition of a PC61BM-SAM on the ZnO ETL led to reduced charge recombination at the active layer-ZnO interface, as suggested by the increased shunt resistance and decreased reverse saturation current density. Both of these factors were responsible for an almost 50% incremental increase in the JSC compared to the conventional reference, which in turn also explained the improved PCE of 5.90%. Based on s-diPBI's molecular scaffold, SdiPBI-S was developed in a 5-step synthesis.80 Again, the core of the NFA consisted of two bay-linked PDI monomers, yet this time the external bay positions were annulated with sulfur atoms. Heteroannulation in the bay regions was achieved by Stille coupling of SdiPBI-S's tetrachloro precursor with bis(tributyltin)sulfide and was employed to induce a more twisted conformation in the acceptor, thereby raising its LUMO energy and consequentially also the VOC in devices. Moreover, a more pronounced 3D character should also inhibit microscale aggregation in the active layer leading to more suitably tuned phase domains. In conventional architecture devices PDBT-T1:SdiPBI-S blends reached a noteworthy PCE of 7.16%. Albeit using different donors and device architectures, the increased performance of SdiPBI-S compared to s-diPBI was attributed to the increased torsional angle between the two PDI planes in SdiPBI-S, which reduces the conjugation between the two PDI monomers. Consequently, the acceptor's LUMO was raised, thereby contributing to the increased VOC in devices. Another ramification of SdiPBI-S's higher lying LUMO was a hypsochromically shifted absorption band leading to greater donor:acceptor spectral complementarity and ultimately a higher JSC, reaching 11.98 mA cm−2. Inspired by the successes of this molecular engineering strategy and in the pursuit of higher PCEs, the selenophene analogue of SdiPBI-S, SdiPBI-Se was synthesised.81 It was envisaged that selenium's more diffuse and polarisable electron cloud would improve orbital overlap and intermolecular interactions thus increase the charge carrier mobility in the acceptor. GIWAXS and space-charge limited current measurements confirmed the above hypothesis by revealing shorter lamellar stacking distances and more equilibrated electron and hole mobilities in the PDBT-T1:SdiPBI-Se blend compared to the SdiPBI-S reference (μe = 4.8 × 10−3 cm2 V−1 s−1 and μh = 3.6 × 10−3 cm2 V−1 s−1 for the SdiPBI-Se blend cf. μe = 2.8 × 10−3 cm2 V−1 s−1 and μh = 1.2 × 10−3 cm2 V−1 s−1 for the SdiPBI-S blend). This also rationalises the then unprecedented FF of 0.70 and impressive PCE of 8.42% obtained in champion devices.
More complex bay-linked PDI structures with vinyl or aromatic bridging moieties have also shown significant potential as substitutes for fullerenes in OPVs. The general synthesis of such bay-substituted PDI dimers follows a three-step route involving imidisation of the commercially available perylene-3,4,9,10-tetracarboxylic dianhydride, followed by mono-bromination at the 1-position using elemental bromine and finally a palladium catalysed cross-coupling reaction to join the three fragments. Amongst the structurally simplest derivatives of this class is Helical PDI 1, whose PDI subunits are linked at their bay positions via a two-carbon bridge giving rise to a fully conjugated aromatic system.82 The aim of this modification was to generate a highly delocalised π-system to confer intense photon absorption. The steric clashes between the C–H bonds at the internal ortho positions on the other hand give rise to the helical 3D structure of the NFA. In a blend with PTB7-Th, a maximum PCE of 6.05% was recorded. The high efficiency of Helical PDI 1's was rationalised by femtosecond transient absorption spectroscopy (TAS), which indicated exciton photogeneration in both the donor and the acceptor domains. It was speculated that device performance could potentially be further incremented by minimising the number and extent of recombination mechanisms in the cells. Three dimensionality in PDIs can also arise through the use of heteroaromatic π-bridges. A thienyl linked PDI dimer, Bis-PDI-T-EG was reported where the use of a more electron donating bridging unit in the acceptor was intended to generate a push–pull structure similar to the one in calamitic shaped small molecule NFAs, which in turn should enhance both the optical transition intensity and width.83 Cells were spin-coated from a PBDTTT-C-T:Bis-PDI-T-EG blend and gave a maximum PCE of 4.03%. The narrowed bandgap of the acceptor led to ameliorated spectral coverage and a broader EQE was reported, this improved photon harvesting was reflected by a JSC of 8.86 mA cm−2. On the other hand, atomic force microscopy (AFM) showed that a potentially limiting factor in the photocurrent, and therefore photovoltaic performance, was the unsuitably large phase domains in the active layer. In fact, in a later publication further raising of the efficiency of the Bis-PDI-T-EG acceptor was possible by fine tuning the film-forming kinetics of the active layer.84 TEM revealed that judicious regulation of the solvent additive content and the solvent vapour annealing process resulted in larger fibril sizes in the active layer, thereby favouring the charge carrier mobilities and fill factor (μe = 6.06 × 10−3 cm2 V−1 s−1 and μh = 1.03 × 10−2 cm2 V−1 s−1 for the fibrillar Bis-PDI-T-EG blend cf. μe = 1.0 × 10−3 cm2 V−1 s−1 and μh = 3.0 × 10−4 cm2 V−1 s−1 for the initial Bis-PDI-T-EG blend). Furthermore, light-power-dependent J–V curves also indicated a decrease in both monomolecular and bimolecular recombination losses in the optimised blend, thus explaining the almost 50% larger JSC and PCE of 6.08%. Over the course of a year, the OPV performance of this model donor–acceptor system was improved once again, by replacing the PBDTTT-C-T donor with the lower band gap polymer PBDT-TS1, thereby favouring photon absorption across a broader wavelength range and consequently improving the JSC.85 Incorporation of the molecular PDINO species in the Ca electron transporting layer on the other hand aided electron mobility, factor (μe = 1.87 × 10−3 cm2 V−1 s−1 for the Bis-PDI-T-EG blend using a PDINO ETL cf. μe = 1.16 × 10−3 cm2 V−1 s−1 for the regular Bis-PDI-T-EG blend) and hence the FF, which also contributes to the higher PCE of 7.24% of the devices. Concomitantly to the optimisation of Bis-PDI-T-EG based devices, a boost in OPV PCE was attempted through side-chain engineering of the methoxy-capped ethylene glycol units.86 Substitution by 4-butylalkoxy moieties afforded Bis-PDI-T-BuO, whose side chains were envisaged to adjust the intermolecular interactions with the donor by adopting an antiperiplanar rather than gauche conformation. Moreover, the reduced flexibility of the n-butoxyl chains should also lead to slightly increased π–π stacking distances and therefore more suitably tuned phase domains. When blended with the same polymer donor (PBDT-TS1) as in the best performing Bis-PDI-T-EG device a PCE of 6.36% was obtained. The decreased performance was suggested to be a result of the excessively large aggregation of the acceptor in the active layer, leading to poor exciton dissociation therefore accounting for the reduced photocurrent. FPDI-T is analogous to the Bis-PDI-T-EG and Bis-PDI-T-BuO acceptors, however it contains a fully fused thiophene linker and different solubilising chains.87 Ring fusion between the aromatic linker and the two PDI monomers was suggested to be an effective way to rigidify the PDI acceptor, thereby favouring its morphology in blends.75 DFT calculations confirmed the success of this strategy by demonstrating a significantly decreased dihedral angle in FPDI-T, thereby planarising the molecule and aiding π–π stacking in OPV blends. GIWAXS reinforced this hypothesis as FPDI-T possessed a more intense and narrow out-of-plane π–π stacking peak. A result of the tighter molecular packing was an improved charge carrier transport, which was reflected in the excellent hole and electron mobilities (μe = 1.63 × 10−4 cm2 V−1 s−1 and μh = 5.92 × 10−2 cm2 V−1 s−1 for the FPDI-T blend). Consequently this is reflected in the high PCE of 6.72% obtained for PTB7-Th:FPDI-T based devices. Another high-performance PDI with a fully fused nonacyclic IDTT core, FITP, was presented.88 The selection of the IDTT core was based on its extended conjugation length and strong electron-donating nature. UV-vis spectroscopy highlighted the advantages of FITP's more extended aromatic backbone by showing improved photon absorption at longer wavelengths compared to FPDI-T. The 10% higher JSC in PTB7-Th:FITP devices is further proof of the maximised charge generation and responsible for the improved PCE of 7.33%. An alternative aromatic bridging moiety to have shown considerable promise in bay-linked PDI dimers is 9,9′-spirobifluorene. 9,9′-Spirobifluorene was selected as a bridge in SF-PDI2 because of its helical shape and high lying LUMO, which should benefit SF-PDI2's morphological and optoelectronic properties respectively.89 AFM confirmed the favourable morphology of SF-PDI2 in PffBT4T:SF-PDI2 blends revealing domain sizes around 20–30 nm. The high photoluminescence quenching efficiency of 93% suggests that well-sized donor–acceptor domains extend also beyond the surface of the film. The judiciously tuned LUMO energy of SF-PDI2 by inclusion of 9,9′-spirobifluorene was reflected in the large VOC of 0.98 V obtained in the optimised solar cells with a PCE of 6.30%. Further device optimisation was performed by pairing SF-PDI2 with the low bandgap polymer P3TEA to achieve an impressive PCE of 9.50% with an at the time record VOC of 1.11 V.90 The large VOC can be rationalised through the low voltage loss of only 0.61 V in the devices, the origin of which stems from the lack of any sub-bandgap charge transfer state absorption and minimised non-radiative recombination mechanisms. Perhaps an even more striking aspect of the photovoltaic performance data is that despite an apparently almost negligible energy offset of 0.05 eV between P3TEA and SF-PDI2's LUMOs, charge generation and separation remained efficient as demonstrated by the high JSC. Another advantageous property of SF-PDI2 over other PDI-based NFAs is its ease of synthesis, as SF-PDI2 was obtained in three steps from the commercially available and inexpensive perylene-3,4,9,10-tetracarboxylic dianhydride. Moreover, the use of a Suzuki rather than Stille cross-coupling reaction between bis(pinacolato)spirobifluorene and the bay position monobrominated perylene diimide precursor also avoids the use of any highly toxic organotin reagents, thus further adding to SF-PDI2 industrial applicability.
3.3. PDI trimers
To mimic fullerene's spherical shape, hence its favourable isotropic charge transport, three-dimensional PDI trimers have been developed (Fig. 10). This approach was designed to also benefit the morphological properties of the donor–acceptor blend, as the constituent PDI monomers of the NFA are rotated in different directions, thereby reducing PDIs’ inherent aggregation tendency. One of the earliest PDI trimers was S(TPA-PDI).91 This acceptor utilises a triphenylamine core joined to three PDI arms leading to an NFA with a star-shaped structure. Moreover, the use of an electron rich central unit and electron deficient end units induces a significant push–pull character in the molecule intended to favour photon absorption across a broad range of wavelengths. When blended with the low bandgap polymer donor PBDTTT-C-T, good photovoltaic performance with a PCE of 3.32% was reported (Table 4). The main limitation of the solar cell performance was the poor FF of 0.34, which can be predominantly attributed to the poorly balanced and moderate hole and electron mobilities (μe = 2.32 × 10−5 cm2 V−1 s−1 and μh = 7.17 × 10−4 cm2 V−1 s−1 for the S(TPA-PDI) blend). Based on S(TPA-PDI)'s molecular design, B(PDI)3 was developed in which a phenyl moiety was selected as a replacement for S(TPA-PDI)'s triarylamine core.92 Although substitution of the sp3 hybridised nitrogen central unit by the more planar sp2 hybridised phenyl group no longer afforded a star-shaped structure, a 51.4° dihedral angle (from DFT calculations using B3LYP/6-31G basis sets) between the phenyl and PDI planes ensured retention of a twisted molecular geometry, thereby suppressing PDI's strong aggregation tendency. Grazing incidence X-ray diffraction indicated crystal sizes of around 5 nm in the active layer, which in turn benefited JSC. Short π–π stacking distances around 1.5 nm for both the PTB7-Th donor and the S(TPA-PDI) acceptor were also found, consequently allowing for the formation of good charge-transport networks, which is highlighted in the almost 50% improved FF of PTB7-Th:B(PDI)3 devices compared to S(TPA-PDI) based cells. Devices spin-casted from a PTB7-Th:B(PDI)3 blend ultimately yielded a PCE of 5.65%. In an attempt to further exploit the structurally favourable properties of the benzene core, two novel C3 symmetric NFAs, TPH and TPH-Se, were designed.93 In comparison to B(PDI)3, these acceptors featured a fully-annulated aromatic core, which was achieved through a Pd-catalysed Suzuki cross-coupling followed by subsequent photocyclization. The aim of this modification was to generate a highly delocalised π-system to confer intense photon absorption as well as favourable charge carrier mobilities. Solar cells for TPH and TPH-Se were fabricated from PDBT-T1:NFA blends, yielding impressive performances with a PCE of 8.28% and 9.28% respectively. The notable photovoltaic performance of both devices was attributed to the stronger and broader optical absorption of both acceptors compared to S(TPA-PDI) and B(PDI)3 and the ideally-sized domains in both donor–acceptor combinations (14.70 nm and 14.20 nm for TPH and TPH-Se respectively), as indicated by resonant soft X-ray scattering (R-SoXS). It was speculated that the tighter molecular packing in TPH-Se arising from increased Se–Se interactions in neighbouring molecules accounts for the slightly higher and more balanced electron and hole mobilities in PDBT-T1:TPH-Se compared to PDBT-T1:TPH thus also the superior FF and photovoltaic performance (μe = 2.2 × 10−3 cm2 V−1 s−1 and μh = 1.7 × 10−3 cm2 V−1 s−1 for the TPH-Se blend cf. μe = 1.5 × 10−3 cm2 V−1 s−1 and μh = 1.0 × 10−3 cm2 V−1 s−1 for the TPH blend) (Fig. 11). | ||
| Fig. 10 Trimeric and tetrameric PDI based small molecule acceptors. | ||
| Acceptor | Optical Eg (eV) | HOMO (eV) | LUMO (eV) | V OC (V) | J SC (mA cm−2) | FF | Electron mobilitya (cm2 V−1 s−1) | Hole mobilityb (cm2 V−1 s−1) | PCEc (%) | Donor | Additive | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by space charge limited current (SCLC) measurements using electron only devices. b Determined by space charge limited current (SCLC) measurements using hole only devices. c Average PCE values are shown in parentheses. | ||||||||||||
| S(TPA-PDI) | 1.76 | −5.40 | −3.70 | 0.88 | 11.3 | 0.34 | 2.32 × 10−5 | 7.17 × 10−4 | 3.32 (3.22) | PBDTTT-C-T | 5.0% DIO | 91 |
| B(PDI)3 | 2.14 | −6.00 | −3.86 | 0.83 | 13.1 | 0.52 | 4.20 × 10−5 | 1.75 × 10−4 | 5.65 (—) | PTB7-Th | 3.0% CN | 92 |
| TPH | 2.19 | −6.02 | −3.83 | 1.00 | 12.3 | 0.64 | 1.50 × 10−3 | 1.00 × 10−3 | 8.28 (8.15) | PDBT-T1 | 0.25% DIO | 93 |
| TPH-Se | 2.17 | −5.97 | −3.80 | 1.00 | 13.0 | 0.72 | 2.20 × 10−3 | 1.70 × 10−3 | 9.28 (8.98) | PDBT-T1 | 0.75% DIO | 94 |
| Ta-PDI | 2.05 | −6.03 | −3.81 | 0.78 | 17.1 | 0.69 | 2.70 × 10−4 | 3.60 × 10−4 | 9.15 (8.91) | PTB7-Th | — | 95 |
| H-tri-PDI | 2.09 | −6.01 | −3.93 | 0.73 | 16.5 | 0.60 | 1.40 × 10−5 | 1.20 × 10−4 | 7.25 (—) | PDBT-TS1 | 7.0% DPE | 95 |
| hPDI3 | 2.37 | −6.23 | −3.86 | 0.81 | 14.5 | 0.67 | 1.50 × 10−4 | 1.00 × 10−4 | 7.90 (7.70) | PTB7-Th | 1.0% DIO | 96 |
| TPE-PDI4 | 2.05 | −5.77 | −3.72 | 0.91 | 11.7 | 0.52 | 1.00 × 10−3 | — | 5.53 (5.44) | PBDTT-S-TT | — | 97 |
| TPPz-PDI4 | 2.10 | −5.86 | −3.76 | 0.99 | 12.5 | 0.56 | 2.30 × 10−3 | — | 7.10 (6.90) | PffBT-T3(1,2)-2 | — | 98 |
| TPB | 1.82 | −5.71 | −3.89 | 0.79 | 17.9 | 0.58 | 6.00 × 10−6 | 1.08 × 10−5 | 8.47 (8.11) | PTB7-Th | 8.0% DPE | 99 |
 | ||
| Fig. 11 Synthetic routes employed for TPH and TPH-Se involving photocyclization to achieve a fully-annulated aromatic core. | ||
Whilst each of the previously discussed trimeric PDI NFAs employ an electron-donating central building block, the first PDI derivative containing an electron-deficient core, Ta-PDI, was recently reported.94 Due to the electron-poor nature of the triazine π-bridge, an inversion of the typical arrangement of the halide and organometallic functionalities on the PDI and aromatic linker coupling partners used in the Pd-catalysed cross-coupling reaction of Ta-PDI was needed, thus setting a synthetic precedent for future PDI oligomers with electron-poor cores. Inverted architecture OPV devices were spin-casted from a PTB7-Th:Ta-PDI blend to yield an outstanding photovoltaic performance with PCE of 9.15%. Although using a different donor, Ta-PDI based devices yielded a significantly higher JSC of 17.1 mA cm−2 than each of the previously discussed trimeric PDI NFAs, which can be related to its higher EQE of almost 80% across a broad spectral range. Furthermore, the impressive JSC can be attributed to efficient exciton dissociation as indicated by charge dissociation probability P(E,T) measurements, yielding a value of 98%. The VOC of PTB7-Th:Ta-PDI devices on the other hand was significantly below those of devices fabricated from the other trimeric acceptors, which can be attributed to the electron-withdrawing nature of the triazine core downshifting the LUMO of the acceptor (Fig. 12).
 | ||
| Fig. 12 Synthetic route employed for Ta-PDI including inversion of the halide and organometallic coupling functionalities on the PDI and aromatic core. | ||
Following previous work on imine coupled PDI monomers, the trimeric N-linked PDI non-fullerene acceptor, H-tri-PDI was designed.75,95 Whilst it was envisaged that the inclusion of an additional PDI unit in the acceptor would enhance photon absorption, thereby benefiting JSC in the devices, retention of the 90° dihedral angle between the PDI planes (estimated using DFT calculations with the B3LYP/6-31G* basis sets) should continue to disrupt the acceptor's aggregation tendency and favour the blend morphology. The success of this design strategy is reflected in the R-SoXS data of PBDT-TS1:T-tri-PDI blends, which indicates characteristic mode length scales in the order of 15 nm. These findings also account for the remarkable JSC of 16.5 mA cm−2 in the champion PBDT-TS1:H-tri-PDI devices with a PCE of 7.25%. Another example of improved photovoltaic performance upon inclusion of an additional repeat unit in a PDI dimer is Helical PDI 1 and its trimeric analogue, hPDI3.82,96 Similar to Helical PDI 1, hPDI3 was synthesised by fusing three PDI units together at their bay positions with ethylene bridging units. Whilst both Helical PDI 1 and hPDI3 display nonplanar structures due to the steric congestion at their ortho positions, DFT calculations demonstrated that hPDI3 can exist as two isoenergetic conformers by inversion of its helicity at either two-carbon junction. Because of these conformational dynamics and the 3D molecular structure, PTB7-Th:hPDI3 blends exhibit highly-favourable intercalating donor–acceptor networks of approx. 20 nm in size. In combination with PTB7-Th and hPDI3's excellent spectral complementarity this led to an excellent JSC of 14.5 mA cm−2 which contributes to the devices’ high PCE of 7.90%.
3.4. PDI tetramers
The structurally most complex and most recently developed class of PDI based NFAs are PDI tetramers. Amongst the early examples of these molecules was TPE-PDI4.97 Because of steric clashes the four phenyl rings in TPE-PDI4's tetraphenylethylene core are twisted by approx. 55° relative to the plane of the central double bond, thereby conferring TPE-PDI4 a four-wing propeller shaped molecular structure. This highly twisted conformation ensures good solvent processability in common organic solvents, as well as the formation of smooth thin films with a root-mean square roughness of 0.207 nm as confirmed by X-ray diffraction measurements. AFM measurements support the favourable active layer morphology, as PTB7-Th:TPE-PDI4 blends demonstrate average surface features between 20–30 nm in size. The 55° torsional angle in TPE-PDI4's core (estimated from DFT calculations using B3LYP/6-31G* basis sets) also ensures minimal conjugation between the four PDI units in the acceptor, which in turn leads to a high lying LUMO thus accounting for the remarkable VOC of 0.91 V in the OPV devices. Ultimately, champion devices yielded a PCE of 5.53%. In a recent study, TPE-PDI4's central tetraphenylethylene moiety was replaced by a tetraphenylpyrazine unit to afford TPPz-PDI4.98 It was hypothesised that TPPz-PDI4's larger core should reduce steric clashes between the four phenyl units, therefore reduce the extent of molecular twisting and thereby favouring charge carrier mobilities. The success of this design strategy is reflected in DFT calculations (using B3LYP/6-31G* basis sets), which revealed a 40° dihedral angle between TPPz-PDI4's pyrazine and phenyl units (cf. 55° dihedral angle between TPE-PDI4's ethylene and phenyl groups). Space-charge-limited current electron mobility measurements also highlighted the positive outcome of the molecular engineering strategy as TPPz-PDI4 had an electron mobility more than double of TPE-PDI4 (μe = 2.3 × 10−3 cm2 V−1 s−1 for the TPPz-PDI4 blend cf. μe = 1.0 × 10−3 cm2 V−1 s−1 for the TPE-PDI4 blend). Devices based on PffBT-T3(1,2)-2:TPPz-PDI4 thus afforded improved photovoltaic performance compared to PTB7-Th:TPE-PDI4, with a FF of 0.56 and PCE of 7.10%. In contrast to the minimal conjugation between PDI units in TPE-PDI4, a benzodithiophene-thienyl (BDT-Th) molecular backbone was utilised in TPB, whereby the two PDI caps flanking the BDT core possessed dihedral angles of 50.2° and 58.9° respectively, hence a twist angle of 9° between the PDI groups, estimated from DFT calculations using B3LYP/6-31G(d).97,99 This led to almost parallel equatorial PDI moieties and therefore a partially conjugated PDI–BDT–PDI backbone, which was envisaged to favour exciton splitting. Specifically, during charge transfer from the donor (PTB7-Th) to a PDI unit in the acceptor the transmitted electron can be delocalised to the opposite PDI unit thereby reducing the electron–hole binding energy. This hypothesis was supported by photoluminescence data, which showed almost completely quenched PTB7-Th and TPB luminescence, thus suggesting efficient charge separation. Alongside suitable donor–acceptor domain sizes and intense absorption across the entire visible spectrum, this rationalises the excellent photovoltaic performance of PTB7-Th:TPB blends with a PCE of 8.47%.Overall, a range of molecular engineering strategies have been developed over the past decade to optimise both the optoelectronic and morphological properties of PDI based acceptors in OPVs, leading to PCEs as high as 9.50%. Whilst initial design concepts relied predominantly on the inclusion of electron-donating moieties between two or more PDI units, in recent years examples of oligomeric PDI acceptors with electron neutral or electron withdrawing π-bridges have also demonstrated considerable promise as NFAs in OPVs. The industrial scalability of PDI NFAs is helped by the commercial availability of the perylene tetracarboxylic dianhydride precursor, thus reducing the synthesis of some PDI NFAs to just three steps. Reports of PDI based devices processed from non-chlorinated solvents have also been rather scarce, thus requiring further attention from the OPV community to allow PDI based solar cells to become industrially viable.
4. Acceptor polymers
The first OPV device employing a polymer acceptor was developed in 1995 and utilised poly(p-phenylenevinylene) derivatives as both the donor and the acceptor.100 Although no power conversion efficiencies were reported, these findings illustrated that all-polymer solar cells not only possess suitable interfaces for charge separation but also percolating networks for charge transport. Numerous classes of polymeric NFAs have since been developed, the most promising include polymeric naphthalene diimide (PNDI) acceptors, polymeric PDI (PPDI) acceptors and terpolymer acceptors (Fig. 13). The general structure of polymer NFAs relies on alternating electron-donating and electron-accepting moieties. Similar to their small molecule counterparts, the energy levels of polymeric NFAs can also be adjusted through chemical modification of both the donor and acceptor units. The inherent polydispersity of polymer NFAs, typically resulting in a distribution of molecular weights, often complicates analysis of the performance of devices and impedes the ability to reliably produce identical batches of the acceptors. Furthermore, the morphology of all-polymer bulk heterojunctions is often difficult to optimize as a result of the unfavourable mixing of two polymeric components. For this reason, progress in polymer acceptors currently lags that of small molecules. However, polymers exhibit excellent flexibility, toughness, processability and continually improving performance that encourages further development and holds promise for potential commercialisation. | ||
| Fig. 13 NDI and PDI based polymeric acceptors. | ||
4.1. Polymeric naphthalene diimide acceptors
The development of the first PNDI NFA, P(NDI2OD-T2), was in 2009, where the low-lying LUMO of −3.9 eV accounted for its excellent stability under ambient conditions.101 The synthesis of P(NDI2OD-T2) required bromination of the commercially available 1,4,5,8-naphthalenetetracarboxylic dianhydride at the 2 and 6 positions, followed by imidisation using 2-octyldodecan-1-amine and lastly polymerisation with 5,5′-bis(trimethylstannyl)-2,2′-bithiophene. Although organic field effect transistor (OFET) data was reported, no OPV devices were fabricated. Two years later, two independent studies reported OPV data for P3HT:P(NDI2OD-T2) blends yielding PCEs of 0.21% and 0.17% respectively (Table 5).102,103 Although in both cases high electron mobilities and broad spectral coverages were reported, scanning transmission X-ray (STXM) microscopy revealed excessively large phase domains between 200–1000 nm in size, which accounted for the poor exciton dissociation and low photocurrent. Subsequent investigations were directed towards reducing P(NDI2OD-T2)s’ aggregation tendency in blends.104 Scanning near-field optical microscopy and AFM demonstrated that by employing solvents with large and polarisable aromatic cores, P(NDI2OD-T2)'s phase domains in the active layer can be significantly decreased consequently leading to an improved JSC of 3.77 mA cm−2 and a PCE of 1.40%. The next OPV performance improvement from P(NDI2OD-T2) based devices was achieved by blending P(NDI2OD-T2) with a low bandgap polymer, PTQ1.105 Better donor–acceptor spectral complementarity and photoluminescence quenching efficiencies of 96% for PTQ1 and 77% for P(NDI2OD-T2) ensured good charge generation thus accounting for the more than doubled JSC and PCE of 4.10%. Further OPV performance optimisation was conducted in 2016 to afford a then highest reported efficiency for OPVs utilising polymeric NFAs.106 The design strategy also relied on donor-polymer substitution, however rather than opting for a lower bandgap polymer, it was hypothesised that a medium bandgap donor should allow for better spectral complementarity with the low bandgap P(NDI2OD-T2) acceptor. In the search for the ideal donor, a fluorine substituted backbone was chosen as a critical attribute to lower the polymer's HOMO, which in turn should afford a larger VOC. With these design criteria in mind, the bifluorinated-benzodithiophene-alt-benzotriazole copolymer, J51 was selected. Utilising a previously reported device configuration, solar cells were fabricated from J51:P(NDI2OD-T2) with a PCE of 8.27%.106 Although both of the initial design strategies proved to be successful, leading to maximised JSC and VOC, an unexpectedly large FF of 0.70 also significantly contributed to the high OPV performance. The excellent FF was ascribed to the high and well-balanced charge carrier mobilities (μe = 2.16 × 10−4 cm2 V−1 s−1 and μh = 2.50 × 10−4 cm2 V−1 s−1 for the J51:P(NDI2OD-T2) blend). Very recently, an unprecedented efficiency of 10.1% for all-polymer solar cells by blending a novel wide-bandgap donor, PTzBI-Si, with P(NDI2OD-T2) was reported.107 Although the resulting devices showed slightly higher VOC, JSC and FF compared to previously reported cells, a more remarkable aspect of these findings was the fact that a record PCE was obtained by spin-coating devices from a non-chlorinated solvent, 2-methyl-tetrafuran, whereby the outstanding solution processability of the new polymer donor was linked to its siloxane capped alkyl chains.| Acceptor | Optical Eg (eV) | HOMO (eV) | LUMO (eV) | V OC (V) | J SC (mA cm−2) | FF | Electron mobilitya (cm2 V−1 s−1) | Hole mobilityb (cm2 V−1 s−1) | PCEc (%) | Donor | Additive | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by space charge limited current (SCLC) measurements using electron only devices. b Determined by space charge limited current (SCLC) measurements using hole only devices. c Average PCE values are shown in parentheses. | ||||||||||||
| P(NDI2OD-T2) | 1.45 | −5.45 | −4.00 | 0.52 | 1.41 | 0.29 | — | — | 0.21 (—) | P3HT | — | 102 |
| P(NDI2OD-T2) | 1.45 | −5.45 | −4.00 | 0.46 | 0.80 | 0.46 | — | — | 0.17 (—) | P3HT | — | 103 |
| P(NDI2OD-T2) | 1.45 | −5.80 | −4.35 | 0.56 | 3.77 | 0.65 | — | — | 1.40 (—) | P3HT | — | 104 |
| P(NDI2OD-T2) | 1.60 | −5.90 | −4.30 | 0.84 | 8.85 | 0.55 | 2.70 × 10−3 | 1.20 × 10−5 | 4.10 (4.00) | PTQ1 | — | 105 |
| P(NDI2OD-T2) | 1.48 | −5.77 | −3.84 | 0.83 | 14.18 | 0.70 | 2.16 × 10−4 | 2.50 × 10−4 | 8.27 (8.10) | J51 | 1.0% DIO | 106 |
| P(NDI2OD-T2) | 1.46 | −5.81 | −3.84 | 0.87 | 15.57 | 0.73 | 2.88 × 10−4 | 1.87 × 10−3 | 10.10 (9.90) | PTz-BI-Si | — | 107 |
| P(NDI2HD-T2) | 1.47 | — | — | 0.82 | 13.97 | 0.53 | 6.23 × 10−5 | 9.79 × 10−5 | 6.11 (6.03) | PTB7-Th | — | 108 |
| P(NDI2TOD-T2) | 1.43 | −5.36 | −3.93 | 0.77 | 11.40 | 0.54 | 2.20 × 10−5 | 6.10 × 10−5 | 4.75 (—) | PTB7-Th | 3.0% DPE | 109 |
| P(NDI2DT-FT2) | — | — | — | 0.81 | 13.53 | 0.62 | 4.90 × 10−4 | 5.50 × 10−4 | 6.71 (6.58) | PTB7-Th | — | 110 |
| P(NDI2HD-T) | 1.85 | −5.64 | −3.79 | 1.06 | 11.22 | 0.56 | 1.55 × 10−5 | 2.84 × 10−5 | 6.64 (6.60) | PBDTTTPD | 1.0% DIO | 111 |
| PNDIS-HD | 1.65 | −5.65 | −4.00 | 0.76 | 7.78 | 0.55 | 1.00 × 10−3 | 2.00 × 10−4 | 3.26 (3.16) | PSEHTT | — | 112 |
| PNDIS-HD | 1.76 | −6.00 | −3.84 | 0.81 | 18.80 | 0.51 | 7.25 × 10−3 | 3.11 × 10−4 | 7.73 (7.21) | PTB7-Th | — | 113 |
| P(IDT-NDI) | 1.51 | −5.75 | −3.84 | 0.93 | 9.55 | 0.60 | 3.06 × 10−5 | 6.58 × 10−5 | 5.33 (5.19) | J51 | — | 114 |
| P(TP) | 1.80 | −5.72 | −3.8 | 0.98 | 9.97 | 0.51 | 1.25 × 10−4 | 8.00 × 10−5 | 5.00 (4.88) | Pil-2T-PS-10 | 2.0% MN | 115 |
| PPDIODT | 1.74 | −5.90 | −3.96 | 0.76 | 15.72 | 0.55 | 1.71 × 10−3 | 5.75 × 10−4 | 6.58 (6.50) | PBDT-TS1 | — | 116 |
| PDI-V | 1.74 | −5.77 | −4.03 | 0.74 | 15.80 | 0.63 | 4.20 × 10−4 | 1.30 × 10−3 | 7.57 (7.30) | PTB7-Th | — | 117 |
| NDP-V | 1.91 | −5.94 | −4.03 | 0.74 | 17.07 | 0.67 | 3.00 × 10−4 | 1.00 × 10−3 | 8.59 (8.48) | PTB7-Th | — | 118 |
| PFPDI-2T | 1.70 | −5.82 | −4.12 | 0.73 | 13.47 | 0.65 | 3.84 × 10−5 | 2.67 × 10−4 | 6.39 (6.31) | PTB7-Th | 3.0% CN | 119 |
| PFPDI-2FT | 1.79 | −5.94 | −4.15 | 0.67 | 13.31 | 0.60 | 3.32 × 10−5 | 2.32 × 10−4 | 5.35 (5.26) | PTB7-Th | 3.0% CN | 119 |
| PNDI-T10 | 1.55 | −6.36 | −4.05 | 0.89 | 12.30 | 0.63 | 2.70 × 10−5 | 7.80 × 10−5 | 6.90 (6.60) | PBDTTS-FTAZ | — | 120 |
| PNDI-T10 | 1.55 | −6.36 | −4.05 | 0.83 | 12.90 | 0.71 | 6.00 × 10−4 | 1.00 × 10−3 | 7.60 (7.40) | PTB7-Th | — | 121 |
| 30PDI | 1.77 | −5.95 | −3.89 | 0.79 | 18.55 | 0.45 | 1.00 × 10−3 | 2.60 × 10−3 | 6.29 (6.17) | PBDTTT-C-T | 3.0% DIO | 122 |
Having demonstrated the potential of PNDI NFAs and successfully optimised initial devices based on P(NDI2OD-T2), the focus of the OPV community shifted to molecular design strategies to ameliorate the performance of PNDI NFAs. Early approaches sought to improve the crystalline behaviour and molecular orientation of P(NDI2OD-T2) by varying the length of its pendant alkyl chains. In a series of PNDI NFAs, the 2-hexyldecyl substituted P(NDI2HD-T2) afforded the highest photovoltaic performance with a PCE of 6.11% when blended with PTB7-Th.108 Notably, the PTB7-Th:P(NDI2HD-T2) combination had a superior efficiency compared to the PTB7-Th:P(NDI2OD-T2) reference, which was ascribed to P(NDI2HD-T2)'s higher and more balanced hole and electron mobilities as well as a more-intermixed and finer blend morphology (μe = 6.18 × 10−5 cm2 V−1 s−1 and μh = 9.79 × 10−5 cm2 V−1 s−1 for the P(NDI2HD-T2) blend cf. μe = 1.31 × 10−5 cm2 V−1 s−1 and μh = 5.71 × 10−5 cm2 V−1 s−1 for the P(NDI2OD-T2) blend). These findings thus suggest that tuning the side chains of PNDIs is a simple, yet effective strategy to enhance their OPV performance. Based on this observation, P(NDI2TOD-T2) was developed, in which the introduction of alkyl-thiophene pendant groups onto the NDI–bithiophene backbone was intended to promote intermolecular interactions.109 The success of this strategy is highlighted by the slightly broader absorption spectra in thin films compared to the P(NDI2OD-T2) reference, as well as the reduced packing distance in blends with PTB7-Th, indicating stronger intermolecular interactions. Although the PTB7-Th:P(NDI2TOD-T2) cells yielded a PCE of 4.75% and marginally outperformed the P(NDI2OD-T2) reference, they were unable to reach the state of the art PTB7-Th:P(NDI2HD-T2) devices. Another layer of complexity to earlier work was added by the introduction of additional fluorine substituents on the polymer backbone, resulting in P(NDI2DT-FT2) acceptor.110 The presence of electron-withdrawing substituents was not only intended to affect the FMOs, but also induce higher molecular organisation to promote higher charge-carrier mobilities. A maximum PCE of 6.71% was reported for the PBDTT-F-T:P(NDI2DT-FT2) blend, outperforming the PBDTT-F-T:P(NDI2OD-T2) reference by more than 25%. AFM and GIWAXS suggested that this was due to the preferential formation of fibrillar nanostructures and face-on stacking in the former blend, both favouring charge carrier transport. These findings corroborate well with the enhanced FF and JSC in the PBDTT-F-T:P(NDI2DT-FT2) devices.
Substitution of the bithiophene monomer by alternative aromatic moieties to modulate the electron density on the polymer and improve device efficiency have also been explored. Following this design principle the NDI–thiophene copolymer P(NDI2HD-T) was developed.111 The selection of a thiophene donor unit was based on its tendency to disrupt NDI's strong aggregation tendency, a result of the large dihedral angles between the thiophene and PDI planes, whilst retaining similar electron donating properties to bithiophene. Cells fabricated from blends of PBDTTTPD:P(NDI2HD-T) afforded a noteworthy PCE of 6.64%. R-SoXS measurements and AFM indicated small and well-intermixed phase domains thus highlighting the success of the molecular engineering strategy employed. To further boost performance, P(NDI2HD-T)'s selenium analogue, PNDIS-HD was synthesised.112 It was hypothesised that the incorporation of a selenium atom would improve orbital overlap between the heteroatom and the aromatic system, thus enhance the charge carrier mobilities. Moreover, Se–Se interactions were also envisaged to increase the crystallinity of the polymer, thereby improve phase separation in blends. Although initial devices only yielded <4% efficiencies, subsequent device optimisation by tuning the donor polymer and the rate of polymer/polymer self-organisation afforded devices with a higher PCE of 7.70%.113 A structurally more complex PNDI, P(NDI-IDT), was designed and consists of alternating NDI and IDT units.114 The IDT core was selected due to its rigidity and coplanarity, which was expected to favour charge carrier mobilities. Moreover, the presence of four hexylphenyl solubilising groups on the IDT backbone was intended to give rise to excellent solution processability and suppress the polymer's aggregation tendency in the solid state. Devices were fabricated by spin-coating J51:P(IDT-NDI) blends onto ITO substrates affording a PCE of 5.33%. Considering that the difference between the HOMO of the donor and the LUMO of the acceptor is only ∼1.6 eV, a relatively high VOC of 0.93 V was obtained. In combination with well-sized phase domains in the active blend, featuring a root mean square roughness of 2.09 nm, this explains the satisfactory photovoltaic performance.
4.2. Polymeric perylene diimide acceptors
P(TP) is similar to P(NDI2HD-T) in that it also employs a thiophene co-monomer, however rather than an NDI acceptor subunit, it includes PDI instead.115 Substitution of NDI by the sterically more hindered PDI resulted in an increased dihedral angle of 60° (estimated from DFT calculations using B3LYP/6-31G*) across the thiophene–PDI bond, which in turn led to a more twisted and less crystalline structure. The introduction of dove-tailed 1-hexylheptyl chains at the imide positions of P(TP) was designed to further hinder the self-aggregation tendency of the PDI units, thus giving rise to excellent solution processability. Devices were prepared by spin-coating PiI-2T-PS10:P(TP) solutions in toluene onto ITO substrates and a PCE of 5.0% was achieved. The use of a non-chlorinated processing solvent during device fabrication compensates for the only moderate performance, as this would potentially allow for the industrial upscaling of solar cells utilizing this NFA. A similar PPDI to P(TP), namely PPDIODT, was reported in which the 1-hexylheptyl pendant chains were replaced by more extended 2-octyldodecyl chains to further boost solubility in environmentally benign solvents.116 Champion devices based on PPDIODT were cast from anisole solution and outperformed P(TP) devices by more than 20%. The smaller phase domains, ∼25 nm in the active layer of PPDIODT cells, compared to ∼50 nm in P(TP) cells led to significantly improved exciton splitting and subsequently improved short circuit current. It was suggested that one of the key factors limiting the efficiencies in the previously mentioned PPDI-based devices was the excessive backbone twist in the PPDIs’ backbones thus resulting in poor polymer crystallinity and low electron mobilities. With this design guideline in mind, the thiophene co-monomer was replaced by a vinylene linker to afford PDI-V.117 DFT calculations, using B3LYP/6-31G* basis sets, showed a significantly decreased dihedral angle of only ∼5° and GIWAXS confirmed the structural regularity of the polymer backbone. The success of this design strategy was ultimately highlighted by the more than 30% higher fill factor in PTB7-Th:PDI-V devices, yielding an improved PCE of 7.11%.In a recent elaboration, half of PDI-V's vinylene units were covalently fused to the bay region of the adjacent PDI cores thus affording a naphthodiperylenetetraimide-vinylene based polymer, NDP-V (Fig. 14).118 The resulting larger aromatic repeat unit and fewer twistable C–C bonds were intended to further boost the acceptor's crystallinity and lead to a more favourable blend morphology. Space charge limited current measurements revealed 50% higher charge carrier mobilities for PTB7-Th:NDP-V blends compared to the PTB7-Th:PDI-V reference (μe = 3.0 × 10−4 cm2 V−1 s−1 and μh = 1.0 × 10−3 cm2 V−1 s−1 for the NDP-V blend cf. μe = 2.0 × 10−4 cm2 V−1 s−1 and μh = 7.6 × 10−4 cm2 V−1 s−1 for the PDI-V blend), thereby partially explaining the improved FF in NDP-V based cells. It was speculated that a slightly higher root-mean-square roughness in PTB7-Th:NDP-V further aided charge collection, hence the FF. Ultimately, PTB7-Th:NDP-V devices achieved an impressive PCE of 8.59%. Another outstanding finding in this work was the retention of PCEs in excess of 8% whilst employing four different solubilising alkyl chains, suggesting that the material morphology is almost entirely dictated by the polymer backbone. Despite these successes, one of the drawbacks of NDP-V is its lengthier and more challenging synthesis compared to structurally more simple polymeric NFAs, such as P(NDI2OD-T2), thus potentially posing an issue for its industrial scale-up. Inspired by the naphthodiperylenetetraimide core, two further acceptors combining this unit once with bithiophene and once with difluorobithiophene were designed to afford PFPDI-2T and PFPDI-2FT respectively.117,119 Bithiophene was chosen as a suitable donor unit because of its electron-rich nature, which in turn should lead to a narrower bandgap and improved photon absorption. The two fluorine atoms on difluorobithiophene on the other hand were envisaged to aid exciton splitting by increasing the HOMO–HOMO offset between the donor and the acceptor whilst also favouring greater backbone planarity through S⋯F dipolar interactions. When combining PFPDI-2T and PFPDI-2FT with the low bandgap polymer PTB7-Th maximum PCEs of 6.39% and 5.35% were obtained respectively, meaning that neither acceptor was able to outperform the previously reported similar NFAs.117,118 The reduced JSC of ∼13 mA cm−2 in both sets of devices was the primary cause for the lower efficiency and was attributed to the larger root-mean-square roughness >1.00 nm leading to excessively large domain sizes in the active layer resulting in increased charge recombination.
 | ||
| Fig. 14 Synthetic routes employed for NDP-V cyclization to achieve an annulated aromatic core. | ||
4.3. Terpolymer acceptors
A key factor limiting the performance of polymeric acceptors in organic photovoltaics is their low fill factor arising from their unbalanced charge carrier mobilities and suboptimal blend morphologies. As discussed previously, significant efforts have been devoted to the molecular engineering of the polymer backbone and side chains, as well as judicious tuning of device processing conditions to overcome these limitations. A more recent strategy to tackle these issues is based on the copolymerisation of multiple existing building blocks, which should allow for a more predictable tuning of the chemical and physical properties. By varying the monomer ratio during the synthesis, careful control over the frontier molecular energy levels, charge transport and film morphology is expected. One of the frontrunning polymers employing this strategy in the context of polymeric NFAs was PNDI-T10, a random ternary polymer comprised of an NDI acceptor unit and a 1:9 ratio of thiophene and bithiophene donor moieties.120 The inclusion of thiophene in the polymer backbone was intended to increase its flexibility by reduction of the chain regularity and the increased torsional angle around the NDI–thiophene linkage. In combination with the high bandgap polymer donor PBDTT-FTAZ, an impressive PCE of 6.9% was achieved. Most notably, the performance of PNDI-T10 was superior to devices of both binary reference polymers PNDI-T and P(NDI2OD-T2), therefore highlighting the success of this molecular engineering approach. Whilst space charge limited current measurements showed a more balanced hole and electron mobility in PNDI-T10 devices thus explaining its higher fill factor (μe = 2.7 × 10−5 cm2 V−1 s−1 and μh = 7.8 × 10−5 cm2 V−1 s−1 for the PNDI-T10 blend cf. μe = 6.8 × 10−6 cm2 V−1 s−1 and μh = 4.2 × 10−5 cm2 V−1 s−1 for the PNDI-T blend and μe = 1.1 × 10−5 cm2 V−1 s−1 and μh = 8.9 × 10−5 cm2 V−1 s−1 for the P(NDI2OD-T2) blend), AFM indicated more carefully tuned phase domains in the active layer leading to improved charge generation. Additional device optimisation was performed and involved replacing PBDTT-FTAZ with the narrow bandgap polymer PTB7-Th.121 Interestingly, although polymer substitution deteriorated the spectral complementarity of the active layer, JSC remained largely unaffected. External quantum efficiency measurements suggested that this is because of the much-improved photon-to-electron conversion at longer wavelengths, primarily due to PTB7-Th's lower bandgap compared to PBDTTs-FTAZ. The improved performance could therefore be attributed to the more than 15% higher FF of 0.71 in the PTB7-Th:PNDI-T10 devices, which stems from the more than 10-fold greater and more balanced charge carrier mobilities (μe = 6.0 × 10−4 cm2 V−1 s−1 and μh = 1.0 × 10−3 cm2 V−1 s−1 for the PTB7-Th:PNDI-T10 blend cf. μe = 2.7 × 10−5 cm2 V−1 s−1 and μh = 7.8 × 10−5 cm2 V−1 s−1 for the PBDTTs-FTAZ:PNDI-T10 blend).
Rather than using two different electron-rich monomers and one electron-poor monomer, a series of polymers from a single donor monomer, selenophene, and two different acceptor monomers, NDI and PDI, was synthesised.122 The aim of this molecular engineering strategy was to attain an optimum blend morphology by varying the ratio of the more crystalline NDI-selenophene and more amorphous PDI-selenophene repeat units. When blended with PBDTTT-C-T it was found that the polymer containing 30% PDI-selenophene repeat units, 30PDI, gave the highest efficiency with a maximum PCE of 6.29%. A particularly impressive feature of the devices was their JSC of 18.55 mA cm−2, so-far the highest reported JSC for all-polymer solar cells. This property was attributed to the average crystalline domain sizes of 5.11 nm that closely match the typical exciton diffusion length ∼5 nm in OPVs. The main factor still limiting device performance was the poor FF of 0.45 arising from severe recombination losses, partially due to the insufficient phase segregation, resulting in the absence of fibrillar donor:acceptor networks that are known to facilitate charge separation and transport.
In summary, most of the research efforts in polymeric NFAs have been dedicated towards optimising the PCE of P(NDI2OD-T2) based OPVs due to P(NDI2OD-T2)'s favourably tuned energy levels, high charge carrier mobilities and facile synthesis. Despite systematically investigating the effects of the solubilising groups and the electron rich co-monomer in this model system, the so-far highest reported PCE for all-polymer solar cells has been reported for the reference P(NDI2OD-T2) system, thus indicating that increases in structural complexity are not always a necessity nor a guarantee for higher PCEs in OPVs. To overcome the limitations posed by PNDIs’ highly crystalline nature, two alternative polymeric NFA classes have been developed. In PPDIs the increased steric bulk of the PDI moiety is used to reduce the backbone planarity by favouring larger dihedral angles around the linkages connecting the two monomers. Morphology control in terpolymers on the other hand is achieved by the variation of the nature and stoichiometry of the chosen repeat units. Overall, polymeric NFAs have not been as successful as their small molecule counterparts, mainly due to their suboptimal blend morphology. Their inherent polydispersity and batch-to-batch variations in molecular weight further complicate device optimisation thus directly impacting the PCE and reproducibility of the devices. If polymer acceptors are to become an industrial reality, these issues must first be tackled in order to exploit their remaining advantageous properties such as their excellent compatibility with industrial printing techniques.
5. Industrial considerations for NFAs
5.1. Synthetic complexity
The most highly cited metric in OPV research papers is the PCE of a given donor:acceptor combination. Whilst its importance in determining the industrial success of OPV materials should not be underestimated, additional factors such as the long-term stability and synthetic complexity of the materials employed are also critical. Previous publications have already been directed towards the cost analysis of OPV technologies, yielding an acceptable cost for commercially viable OPV modules of around 10 € g−1.123,124 Although the cost of fabricating a photovoltaic module entails different contributions, the primary driver arises from the material costs, in particular from the active layer materials.125 The cost of the donor and acceptor employed is in turn primarily dependent on their synthetic accessibility, which broadly speaking is related to the number of synthetic steps (NSS) required.126 Taking the acceptor's core as starting point, Tables S1–S5 (ESI†) detail the commercially available starting materials and NSS required for the various NFAs reported herein. One limitation of this approach is that acceptors featuring highly complex π-bridges and end groups will be favoured, as the additional synthetic complexity poised by these will not be considered. For a more detailed and complete account of the cost and synthetic evaluation of photovoltaic materials the reader is directed elsewhere.123,125From the synthetic complexity data it follows that NFAs employing either fluorene, carbazole, PDI or NDI as their core appear to be more suited for industrial scale-up due to their relatively low NSS. Conversely, IDT and IDTT based acceptors appear to have a synthetic disadvantage due to the increased NSS (5–10) required to afford the target molecule. The primary cause for this drawback is the multistep synthesis of the as of yet commercial availability of IDT/IDTT cores; the issue of synthetic complexity is further compounded in IDT/IDTT based NFAs bearing pendant alkyl rather than aryl chains where three additional chemical transformations are required. Investment in scale-up and reverse engineering of the alkyl IDT may be offset by its prevalent use in charge transport polymers. The higher NSS of IDT/IDTT based NFAs is also largely offset by their superior OPV performance compared to other acceptors, thus effectively leading to a trade-off between synthetic complexity and photovoltaic performance. It is interesting to note that if the starting material for PDI based acceptors was to be synthesised from simpler chemical building blocks as shown in Fig. 15, the NSS required for PDI based acceptors would in theory be very similar compared to the IDT/IDTT based ones. This highlights the importance of developing commercially available intermediates to reduce the NSS and cost of producing electron acceptors.
 | ||
| Fig. 15 Synthetic route employed for the synthesis of the starting material for PDI based acceptors. | ||
5.2. Industrial printing techniques
Another key issue facing the commercialization of OPV, is the ability to produce devices with industrially scalable printing technologies. The need for non-halogenated solvent processing has been discussed above, however the technique used to deposit the active layer must also be considered. Spin-coating is an energy and material intensive printing technique that does not translate well to large scale production,127 however it often produces the highest performance in small area devices for research purposes, and as such, it is used extensively in the device fabrication reported throughout OPV literature. For spin cast active layers the donor:acceptor ratio, solution concentration and solvent choice are optimized such that the aggregation of the donor and acceptor, which occurs as the solvent evaporates, lead to the formation nanoscale interpenetrating domains that are ideal in bulk heterojunctions. The issues presented by spin-coating include: (i) the large amount of active layer material ejected during the spinning of the substrate and (ii) the ability to only cast the active layer of one substrate at a time, rather than in a roll-to-roll (R2R) system.128,129 Also spin coating is such a rapid kinetic process that the thermodynamically favoured phase separation processes are suppressed, leading to significant post-deposition morphology changes. Technologies such as slot-die coating and blade coating provide scalable alternatives to spin coating, and both of these technologies lend themselves well to large area R2R casting, allowing for a high throughput and more economically viable production process. The drying kinetics and aggregation of active layer solutions when blade or slot-die coating are rather different to those of spin coating, and thus require careful optimization of processing conditions, which are likely to differ from those used when spin coating.130,131Table 6 details a number of notable devices that have been produced using slot-die and blade coating to process the active layer. In general, lower PCEs are observed for R2R printed devices at present, however as more research efforts are directed towards tackling the problems of maintaining optimal morphology when using alternative printing techniques this gap is likely to close. In the three cases employing blade coating cited in Table 6, OPVs employing a similar donor:acceptor combination to their spin-coated correspondent yielded almost identical PCEs, highlighting the potential of this particular deposition method for large area organic solar cells. In the case of the FTAZ:IT-M and PBTA-TF:IT-M blends the use of relatively low boiling point solvents contributed towards the high PCEs observed. This is because the longer drying times associated with blade coating compared to spin coating can be circumvented by the use of volatile processing solvents, thus enabling fast solvent evaporation and preventing large scale aggregation in the active layer of blade coated cells. The use of spray coating and push coating have also been reported in the fabrication of fullerene based devices.132,133 The ability to produce devices that are able to achieve >3% PCE using both of these techniques suggest that they may also show promise as suitable alternatives in the production of large area devices.| Acceptor | V OC (V) | J SC (mA cm−2) | FF | PCE (%) | Donor | Coating technique | Ref. |
|---|---|---|---|---|---|---|---|
| IEIC | 0.94 | 6.89 | 0.35 | 2.05 | PTB7-Th | Slot-die | 134 |
| PNDIT | 0.87 | 8.51 | 0.50 | 3.71 | PII2T-PS | Slot-die | 135 |
| O-IDTBR | 0.72 | 12.55 | 0.67 | 6.05 | P3HT | Blade | 136 |
| IT-M | 0.95 | 16.80 | 0.66 | 10.60 | FTAZ | Blade | 137 |
| IT-M | 0.95 | 18.14 | 0.66 | 11.40 | PBTA-TF | Blade | 138 |
| PC61BM | 0.87 | 10.76 | 0.42 | 3.60 | TQ1 | Spray | 139 |
| PC61BM | 0.59 | 8.46 | 0.67 | 3.34 | P3HT | Push | 140 |
6. Outlook & conclusions
To conclude, a plethora of design strategies have been utilized in the pursuit of developing a suitable replacement for the fullerene acceptors used in OPV, and as a result of much of the exciting work discussed in this review an explosion in the field of NFAs has occurred within the last 5 years. The dominance that NFAs have established in recent times can be illustrated by the number of NFA-based devices that now exceed the device performance achieved in the analogous fullerene-containing OSCs (see Table S6 and associated text in the ESI†).Small molecule acceptors currently hold a significant advantage over their polymeric counterparts. The lack of entropic driving force for the polymeric acceptors to mix with a polymer donor has led to several reports of suboptimal morphologies, and thus limited PCEs in devices, although there are a small number of examples where promising efficiencies have been achieved in all-polymer solar cells. The batch-to-batch variations in molecular weight, PDI and regioregularity, currently observed in polymeric materials synthesized in sub gram quantities, leaves them at a disadvantage to small molecules, where batches will always be virtually identical. This inability to produce identical polymer batches on a regular basis provides an obvious impediment to the commercialization of polymeric NFAs. PDI based small molecule NFAs have made telling strides from the early reported acceptors, and by employing π-conjugated bridges and twisted 3D structures it has been possible to control the optoelectronic properties to maximize the VOC in devices, and suppress the microscale aggregation that plagued early materials, resulting in vastly improved blend morphologies. Many of the PDI acceptors are relatively simple from a synthetic viewpoint, and are able to attain PCEs of >8.5% on a regular basis, yielding them exciting candidates to replace fullerenes. However, almost all of these acceptors require high-boiling halogenated additives in order to achieve the desired blend morphology; not only is it unlikely for these additives to be permitted in the printing industry, but they have also been shown to often cause morphological and photo-instability in OPV devices. Thus, developing PDIs with improved solubility and further suppression of aggregation should be of high priority to avoid the need of such additives, rendering them more feasible to use in OPV devices. Of all the classes of NFAs discussed in this work, A–D–A type acceptors appear to be the most attractive by some margin. The IDT and IDTT acceptors are now able to consistently achieve exceptional efficiencies of over 10% due to their high lying LUMOs, narrow bandgaps and controlled aggregation. Though PDIs may have a slight advantage in terms of synthetic simplicity, the A–D–A type acceptors can achieve higher PCEs, without the need for additives in several cases, and a number of highly stable devices have been reported. Additionally, the modular fashion in which the A–D–A acceptors are produced provides a number of opportunities to further tune these acceptors to maximize their performance. A large proportion of the recent success of OPV can be attributed to the quick and strategic evolution of non-fullerene acceptors, and they are likely to play a vital role in the future of organic solar cells as further improvements in their design are realized.
A number of the best performing NFAs have been summarized in Table 7, below. These acceptors are able to achieve amongst the highest efficiencies in their respective classes, whilst often possessing other advantageous features such as: (i) greater synthetic simplicity (SF-PDI2, Ta-PDI and P(NDI2OD-T2)), (ii) the use of non-chlorinated solvent processing (FDICTF, O-IDTBR, EH-IDTBR, Ta-PDI and P(NDI2OD-T2)), (iii) a greater degree of flexibility in the donor polymers they can be paired with (EH-IDTBR, ITIC, ITIC-Th and P(NDI2OD-T2)) and (iv) the ability to process high efficiency devices with industrially viable deposition methods (O-IDTBR and IT-M). Though these acceptors are likely to form the basis for a considerable fraction of further development in the field of NFAs, the wide variety of design strategies and chemical moieties present in each of these acceptors indicates the large number of viable approaches to push the boundaries of NFA performance.
| Acceptor | PCE (%) | Donor | NSS | Additive |
|---|---|---|---|---|
| a Fabricated by spin-coating. b Fabricated by blade-coating. | ||||
| FDICTF | 10.06 | PBDB-T | 8 | — |
| O-IDTBR , | 6.38 | P3HT | 10 | — |
| EH-IDTBR | 11.09 | PffBT4T-2DT | 10 | — |
| ITIC | 11.34 | PBQ-4F | 5 | 5.0% IPA |
| ITIC-Th | 10.88 | PTFB-O | 5 | — |
| IT-M , | 12.05 | PBDB-T | 5 | 1.0% DIO |
| IT-4F | 13.10 | PBDBT-SF | 5 | 0.5% DIO |
| SF-PDI2 | 9.50 | P3TEA | 3 | 2.5% ODT |
| Ta-PDI | 9.15 | PTB7-Th | 4 | — |
| P(NDI2OD-T2) | 10.10 | PTz-BI-Si | 3 | — |
As a general note on the further development of NFAs, care must be taken that the pursuit of an ever-greater PCE does not become the sole point of focus. Through rational design there has been an enhancement in the performance of NFAs over time, however this is often accompanied by a substantial increase in the synthetic complexity of the acceptors or the need for unfavorable solvent systems in processing devices. Whilst this is often necessary to drive the development in this field, it should not outweigh the aims to produce cheap, scalable and highly stable devices, since the ultimate goal remains to be commercially viable OPV. As we approach 14–15% PCE with organic solar cells, the bottle neck in producing large scale OPV will become factors such as the cost and availability of the materials, compatibility with industrial printing processes and stability, rather than insufficient PCE to compete with rival technologies. Though there is some work currently being carried out on these problems, at present there appears to be less value placed on them in academia than there is on chasing a record PCE, which could relegate the field of OPV to the realms of academic curiosity rather than an achievable renewable energy technology should this imbalance in the research persist.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from KAUST and BASF, as well as EPSRC Project EP/G037515/1, EP/M005143/1, EC FP7 Project SC2 (610115), and EC H2020 Project SOLEDLIGHT (643791) for their financial support.References
- Y. He and Y. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- X. Yang, J. Loos, S. Veenstra, W. J. H. Verhees, M. M. Wienk, J. M. Kroon, M. Michels and R. A. J. Janssen, Nano Lett., 2005, 5, 579–583 CrossRef CAS PubMed.
- J. D. A. Lin, O. V. Mikhnenko, J. Chen, Z. Masri, A. Ruseckas, A. Mikhailovsky, R. P. Raab, J. Liu, P. W. M. Blom, M. A. Loi, C. J. Garcia-Cervera, I. W. Samuel and T. Q. Nguyen, Mater. Horiz., 2014, 1, 280–285 RSC.
- Y. Tamai, H. Ohkita, H. Benten and S. Ito, J. Phys. Chem. Lett., 2015, 6, 3417–3428 CrossRef CAS PubMed.
- M. M. Wienk, J. M. Kroon, W. J. H. Verhees, J. Knol, J. C. Hummelen, P. A. van Hal and R. A. J. Janssen, Angew. Chem., Int. Ed., 2003, 42, 3371–3375 CrossRef CAS PubMed.
- F. B. Kooistra, J. Knol, F. Kastenberg, L. M. Popescu, W. J. H. Verhees, J. M. Kroon and J. C. Hummelen, Org. Lett., 2007, 9, 551–554 CrossRef CAS PubMed.
- M. Lenes, G. J. A. H. Wetzelaer, F. B. Kooistra, S. C. Veenstra, J. C. Hummelen and P. W. M. Blom, Adv. Mater., 2008, 20, 2116–2119 CrossRef CAS.
- C. Z. Li, S. C. Chien, H. L. Yip, C. C. Chueh, F. C. Chen, Y. Matsuo, E. Nakamura and A. K. Y. Jen, Chem. Commun., 2011, 47, 10082–10084 RSC.
- Y. He, H. Y. Chen, J. Hou and Y. Li, J. Am. Chem. Soc., 2010, 132, 1377–1382 CrossRef CAS PubMed.
- M. Campoy-Quiles, T. Ferenczi, T. Agostinelli, P. G. Etchegoin, Y. Kim, T. D. Anthopoulos, P. N. Stavrinou, D. D. C. Bradley and J. Nelson, Nat. Mater., 2008, 7, 158–164 CrossRef CAS PubMed.
- S. R. Dupont, M. Oliver, F. C. Krebs and R. H. Dauskardt, Sol. Energy Mater. Sol. Cells, 2012, 97, 171–175 CrossRef CAS.
- B. J. T. de Villers, K. A. O’Hara, D. P. Ostrowski, P. H. Biddle, S. E. Shaheen, M. L. Chabinyc, D. C. Olson and N. Kopidakis, Chem. Mater., 2016, 28, 876–884 CrossRef.
- W. Chen and Q. Zhang, J. Mater. Chem. C, 2017, 5, 1275–1302 RSC.
- Z. He, B. Xiao, F. Liu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Kiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- T. Ameri, J. Min, N. Li, F. Machui, D. Baran, M. Forster, K. J. Schottler, D. Dolfen, U. Scherf and C. J. Brabec, Adv. Energy Mater., 2012, 2, 1198–1202 CrossRef CAS.
- L. Lu, T. Xu, W. Chen, E. S. Landry and L. Yu, Nat. Photonics, 2014, 8, 716–722 CrossRef CAS.
- P. P. Khlyabich, B. Burkhart and B. C. Thompson, J. Am. Chem. Soc., 2011, 133, 14534–14537 CrossRef CAS PubMed.
- J. Zhang, Y. Zhang, J. Fang, K. Lu, Z. Wang, W. Ma and Z. Wei, J. Am. Chem. Soc., 2015, 137, 8176–8183 CrossRef CAS PubMed.
- B. J. Kim, Y. Miyamoto, B. Ma and J. M. J. Fréchet, Adv. Funct. Mater., 2009, 19, 2273–2281 CrossRef CAS.
- J. E. Carlé, B. Andreasen, T. Tromholt, M. V. Madsen, K. Norman, M. Jørgensen and F. C. Krebs, J. Mater. Chem., 2012, 22, 24417–24423 RSC.
- J. W. Rumer, R. S. Ashraf, N. D. Eisenmenger, Z. Huang, I. Meager, C. B. Nielsen, B. C. Schroeder, M. L. Chabinyc and I. McCulloch, Adv. Energy Mater., 2015, 5, 1401426 CrossRef.
- J. W. Rumer and I. McCulloch, Mater. Today, 2015, 8, 425–435 CrossRef.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- Z. Xu, L. M. Chen, G. Yang, C. H. Huang, J. Hou, Y. Wu, G. Li, C. S. Hsu and Y. Yang, Adv. Funct. Mater., 2009, 19, 1227–1234 CrossRef CAS.
- Y. Yan, Z. Liu and T. Wang, Adv. Mater., 2017, 29, 1601674 CrossRef PubMed.
- J. R. Tumbleston, D. H. Ko, E. T. Samulski and R. Lopez, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 205325 CrossRef.
- T. J. K. Brenner, Y. Vaynzof, Z. Li, D. Kabra, R. H. Friend and C. R. McNeill, J. Phys. D: Appl. Phys., 2012, 45, 415101 CrossRef.
- I. Etxebarria, A. Guerrero, J. Alberto, G. Garcia-Belmonte, E. Palomares and R. Pacios, Org. Electron., 2014, 15, 2756–2762 CrossRef CAS.
- K. Cnops, B. P. Rand, D. Cheyns, B. Verreet, M. A. Empl and P. Heremans, Nat. Commun., 2014, 5, 3406 CrossRef PubMed.
- K. Cnops, G. Zango, J. Genoe, P. Heremans, M. V. Martinez-Diaz, T. Torres and D. Cheyns, J. Am. Chem. Soc., 2015, 137, 8991–8997 CrossRef CAS PubMed.
- B. Ebenhoch, N. B. A. Prasetya, V. M. Rotello, G. Cooke and I. D. W. Samuel, J. Mater. Chem. A, 2015, 3, 7345–7352 RSC.
- C. B. Nielsen, E. Voroshazi, S. Holliday, K. Cnops, B. P. Rand and I. McCulloch, J. Mater. Chem. A, 2013, 1, 73 RSC.
- C. B. Nielsen, E. Voroshazi, S. Holliday, K. Cnops, D. Cheyns and I. McCulloch, J. Mater. Chem. A, 2014, 2, 12348–12354 RSC.
- G. Zhang, V. Lami, F. Rominger, Y. Vaynzof and M. Mastalerz, Angew. Chem., Int. Ed., 2016, 55, 3977–3981 CrossRef CAS PubMed.
- Y. Kim, C. E. Song, S.-J. Moon and E. Lim, Chem. Commun., 2014, 50, 8235–8238 RSC.
- H. Shi, W. Fu, M. Shi, J. Ling and H. Chen, J. Mater. Chem. A, 2015, 3, 1902–1905 RSC.
- S. Holliday, R. S. Ashraf, C. B. Nielsen, M. Kirkus, J. A. Röhr, C.-H. Tan, E. Collado-Fregoso, A.-C. Knall, J. R. Durrant, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 898–904 CrossRef CAS PubMed.
- D. Baran, T. Kirchartz, S. Wheeler, S. Dimitrov, M. Abdelsamie, J. Gorman, R. S. Ashraf, S. Holliday, A. Wadsworth, N. Gasparini, P. Kaienburg, H. Yan, A. Amassian, C. J. Brabec, J. R. Durrant and I. McCulloch, Energy Environ. Sci., 2016, 9, 3783–3793 RSC.
- M. Li, Y. Liu, W. Ni, F. Liu, H. Feng, Y. Zhang, T. Liu, H. Zhang, X. Wan, B. Kan, Q. Zhang, T. P. Russell and Y. Chen, J. Mater. Chem. A, 2016, 4, 10409–10413 RSC.
- N. Qiu, H. Zhang, X. Wan, C. Li, X. Ke, H. Feng, B. Kan, H. Zhang, Q. Zhang, Y. Lu and Y. Chen, Adv. Mater., 2017, 29, 1604964 CrossRef PubMed.
- K. Wang, Y. Firdaus, M. Babics, F. Cruciani, Q. Saleem, A. El Labban, M. A. Alamoudi, T. Marszalek, W. Pisula, F. Laquai and P. M. Beaujuge, Chem. Mater., 2016, 28, 2200–2208 CrossRef CAS.
- N. Qiu, X. Yang, H. Zhang, X. Wan, C. Li, F. Liu, H. Zhang, T. P. Russell and Y. Chen, Chem. Mater., 2016, 28, 6770–6778 CrossRef CAS.
- A. Gupta, A. Rananaware, P. S. Rao, D. D. La, A. Bilic, W. Xiang, J. Li, R. A. Evans, S. V. Bhosale and S. V. Bhosale, Mater. Chem. Front., 2017, 1, 1600–1606 RSC.
- Y. Lin, Z.-G. Zhang, H. Bai, J. Wang, Y. Yao, Y. Li, D. Zhu and X. Zhan, Energy Environ. Sci., 2015, 8, 610–616 RSC.
- H. Lin, S. Chen, Z. Li, J. Y. L. Lai, G. Yang, T. McAfee, K. Jiang, Y. Li, Y. Liu, H. Hu, J. Zhao, W. Ma, H. Ade and H. Yan, Adv. Mater., 2015, 27, 7299–7304 CrossRef CAS PubMed.
- H. Yao, Y. Chen, Y. Qin, R. Yu, Y. Cui, B. Yang, S. Li, K. Zhang and J. Hou, Adv. Mater., 2016, 28, 8283–8287 CrossRef CAS PubMed.
- Y. Li, L. Zhong, F.-P. Wu, Y. Yuan, H.-J. Bin, Z.-Q. Jiang, Z. Zhang, Z.-G. Zhang, Y. Li and L.-S. Liao, Energy Environ. Sci., 2016, 9, 3429–3435 RSC.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C.-H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed.
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Röhr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. M. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant and I. McCulloch, Nat. Mater., 2017, 16, 363–369 CrossRef CAS PubMed.
- A. Wadsworth, R. S. Ashraf, M. Abdelsamie, S. Pont, M. Little, M. Moser, Z. Hamid, M. Neophytou, W. Zhang, A. Amassian, J. R. Durrant, D. Baran and I. McCulloch, ACS Energy Lett., 2017, 2, 1494–1500 CrossRef CAS.
- Y. Wu, H. Bai, Z. Wang, P. Cheng, S. Zhu, Y. Wang, W. Ma and X. Zhan, Energy Environ. Sci., 2015, 8, 3215–3221 RSC.
- Q.-Y. Li, J. Xiao, L.-M. Tang, H.-C. Wang, Z. Chen, Z. Yang, H.-L. Yip and Y.-X. Xu, Org. Electron., 2017, 44, 217–224 CrossRef CAS.
- B. Jia, Y. Wu, F. Zhao, C. Yan, S. Zhu, P. Cheng, J. Mai, T. K. Lau, X. Lu, C. J. Su, C. Wang and X. Zhan, Sci. China: Chem., 2017, 60, 257–263 CrossRef CAS.
- F. Liu, Z. Zhou, C. Zhang, T. Vergote, H. Fan, F. Liu and X. Zhu, J. Am. Chem. Soc., 2016, 138, 15523–15526 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- Z. Zheng, O. M. Awartani, B. Gautam, D. Liu, Y. Qin, W. Li, A. Bataller, K. Gundogdu, H. Ade and J. Hou, Adv. Mater., 2017, 29, 1604241 CrossRef PubMed.
- Y. Lin, F. Zhao, Q. He, L. Huo, Y. Wu, T. C. Parker, W. Ma, Y. Sun, C. Wang, D. Zhu, A. J. Heeger, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2016, 138, 4955–4961 CrossRef CAS PubMed.
- L. Chang, H. W. A. Lademann, J. B. Bonekamp, M. Meerholz and A. J. Moulé, Adv. Funct. Mater., 2011, 21, 1779–1787 CrossRef CAS.
- Z. Li, K. Jiang, G. Yang, J. Y. L. Lai, T. Ma, J. Zhao, W. Ma and H. Yan, Nat. Commun., 2016, 7, 13094 CrossRef CAS PubMed.
- Y. Lin, Q. He, F. Zhao, L. Huo, J. Mai, X. Lu, C.-J. Su, T. Li, J. Wang, J. Zhu, Y. Sun, C. Wang and X. Zhan, J. Am. Chem. Soc., 2016, 138, 2973–2976 CrossRef CAS PubMed.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS PubMed.
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganäs, F. Gao and J. Hou, Adv. Mater., 2016, 28, 4734–4739 CrossRef CAS PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- K. Reichenbacher, H. I. Suss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22–30 RSC.
- Y. Yang, Z.-G. Zhang, H. Bin, S. Chen, L. Gao, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 15011–15018 CrossRef CAS PubMed.
- Y. Li, L. Zhong, B. Gautam, H.-J. Bin, J.-D. Lin, F.-P. Wu, Z. Zhang, Z.-Q. Jiang, Z.-G. Zhang, K. Gundogdu, Y. Li and L.-S. Liao, Energy Environ. Sci., 2017, 10, 1610–1620 RSC.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- C. B. Nielsen, S. Holliday, H.-Y. Chen, S. J. Cryer and I. McCulloch, Acc. Chem. Res., 2015, 48, 2803–2812 CrossRef CAS PubMed.
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- W. Zhang, J. Smith, S. E. Watkins, R. Gysel, M. McGehee, A. Salleo, J. Kirkpatrick, S. Ashraf, T. Anthopoulos, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2010, 132, 11437–11439 CrossRef CAS PubMed.
- C. L. Zhan and A. D. Q. Li, Curr. Org. Chem., 2011, 15, 1314–1339 CrossRef CAS.
- A. Sharenko, C. M. Proctor, T. S. van der Poll, Z. B. Henson, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2013, 25, 4403–4407 CrossRef CAS PubMed.
- Y. X. Chen, X. Zhang, C. L. Zhan and J. N. Yao, Phys. Status Solidi A, 2015, 212, 1961–1968 CrossRef CAS.
- R. Shivanna, S. Shoaee, S. Dimitrov, S. K. Kandappa, S. Rajaram, J. R. Durrant and K. S. Narayan, Energy Environ. Sci., 2014, 7, 435–441 RSC.
- C.-H. Wu, C.-C. Chueh, Y.-Y. Xi, H.-L. Zhong, G.-P. Gao, Z.-H. Wang, L. D. Pozzo, T.-C. Wen and A. K.-Y. Jen, Adv. Funct. Mater., 2015, 25, 5326–5332 CrossRef CAS.
- W. Jiang, L. Ye, X. Li, X. Ciao, F. Tan, W. Zhao, J. Hou and Z. Wang, Chem. Commun., 2014, 50, 1024–1026 RSC.
- L. Ye, W. Jiang, W. Zhao, S. Zhang, D. Qian, Z. Wang and J. Hou, Small, 2014, 10, 4658–4663 CrossRef CAS PubMed.
- Y. Zang, C.-Z. Li, C.-C. Chueh, S. T. Williams, W. Jiang, Z.-H. Wang, J.-S. Yu and A. K.-Y. Jen, Adv. Mater., 2014, 26, 5708–5714 CrossRef CAS PubMed.
- D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156–11162 CrossRef CAS PubMed.
- D. Meng, D. Sun, C. Zhong, T. Liu, B. Fan, L. Huo, Y. Li, W. Jiang, H. Choi, T. Kim, J. Y. Kim, Y. Sun, Z. Wang and A. J. Heeger, J. Am. Chem. Soc., 2016, 138, 375–380 CrossRef CAS PubMed.
- Y. Zhong, M. T. Trinh, R. Chen, W. Wang, P. P. Khlyabich, B. Kumar, Q. Xu, C.-Y. Nam, M. Y. Sfeir, C. Black, M. L. Steigerwald, Y.-L. Loo, S. Xiao, F. Ng, X. Y. Zhu and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 15215–15221 CrossRef CAS PubMed.
- X. Zhang, Z. Lu, L. Ye, C. Zhan, J. Hou, S. Zhang, B. Jiang, Y. Zhao, J. Huang, S. Zhang, Y. Liu, Q. Shi, Y. Liu and J. Yao, Adv. Mater., 2013, 25, 5791–5797 CrossRef CAS PubMed.
- X. Zhang, C. Zhan and J. Yao, Chem. Mater., 2015, 27, 166–173 CrossRef CAS.
- X. Zhang, W. Li, J. Yao and C. Zhan, ACS Appl. Mater. Interfaces, 2016, 8, 15415–15421 CrossRef CAS PubMed.
- B. Jiang, X. Zhang, Y. Zheng, G. Yu, J. Yao and C. Zhan, RSC Adv., 2016, 6, 43715–43718 RSC.
- H. Zhong, C.-H. Wu, C.-Z. Li, J. Carpenter, C.-C. Chueh, J.-Y. Chen, H. Ade and A. K.-Y. Jen, Adv. Mater., 2016, 28, 951–958 CrossRef CAS PubMed.
- S. Li, W. Liu, C.-Z. Li, T.-K. Lau, X. Lu, M. Shi and H. Chen, J. Mater. Chem. A, 2016, 4, 14983–14987 RSC.
- J. Zhao, Y. Li, H. Lin, Y. Liu, K. Jiang, C. Mu, T. Ma, J. Y. Lin Lai, H. Hu, D. Yu and H. Yan, Energy Environ. Sci., 2015, 8, 520–525 RSC.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef CAS.
- Y. Lin, Y. Wang, J. Wang, J. Hou, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2014, 26, 5137–5142 CrossRef CAS PubMed.
- S. Li, W. Liu, C.-Z. Li, F. Liu, Y. Zhang, M. Shi, H. Chen and T. P. Russell, J. Mater. Chem. A, 2016, 4, 10659–10665 RSC.
- D. Meng, H. Fu, C. Xiao, X. Meng, T. Winands, W. Ma, W. Wei, B. Fan, L. Huo, N. L. Doltsinis, Y. Li, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2016, 138, 10184–10190 CrossRef CAS PubMed.
- Y. Duan, X. Xu, H. Yan, W. Wu, Z. Li and Q. Peng, Adv. Mater., 2017, 29, 1605115 CrossRef PubMed.
- N. Liang, K. Sun, Z. Zheng, H. Yao, G. Gao, X. Meng, Z. Wang, W. Ma and J. Hou, Adv. Energy Mater., 2016, 6, 1600060 CrossRef.
- Y. Zhong, M. T. Trinh, R. Chen, G. E. Purdum, P. P. Khlyabich, M. Sezen, S. Oh, H. Zhu, B. Fowler, B. Zhang, W. Wang, C.-Y. Nam, M. Y. Sfeir, C. T. Black, M. L. Steigerwald, Y.-L. Loo, F. Ng, X. Y. Zhu and C. Nuckolls, Nat. Commun., 2015, 6, 8242 CrossRef CAS PubMed.
- Y. Liu, C. Mu, K. Jiang, J. Zhao, Y. Li, L. Zhang, Z. Li, J. Y. L. Lai, H. Hu, T. Ma, R. Hu, D. Yu, X. Huang, B. Z. Tang and H. Yan, Adv. Mater., 2015, 27, 1015–1020 CrossRef CAS PubMed.
- H. Lin, S. Chen, H. Hu, L. Zhang, T. Ma, J. Y. L. Lai, Z. Li, A. Qin, X. Huang, B. Tang and H. Yan, Adv. Mater., 2016, 28, 8546–8551 CrossRef CAS PubMed.
- Q. Wu, D. Zhao, A. M. Schneider, W. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7248–7251 CrossRef CAS PubMed.
- J. J. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498–500 CrossRef CAS.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS PubMed.
- J. R. Moore, S. Albert-Seifried, A. Rao, S. Massip, B. Watts, D. J. Morgan, R. H. Friend, C. R. McNeill and H. Sirringhaus, Adv. Energy Mater., 2011, 1, 230–240 CrossRef CAS.
- T. W. Holcombe, J. E. Norton, J. Rivnay, C. H. Woo, L. Goris, C. Piliego, G. Griffini, A. Sellinger, J.-L. Brédas, A. Salleo and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 12106–12114 CrossRef CAS PubMed.
- M. Schubert, D. Dolfen, J. Frisch, S. Roland, R. Steyrleuthner, B. Stiller, Z. Chen, U. Scherf, N. Koch, A. Facchetti and D. Neher, Adv. Energy Mater., 2012, 2, 369–380 CrossRef CAS.
- D. Mori, H. Benten, I. Okada, H. Ohkita and S. Ito, Adv. Energy Mater., 2014, 4, 1301006 CrossRef.
- L. Gao, Z.-G. Zhang, L. Xue, J. Min, J. Zhang, Z. Wei and Y. Li, Adv. Mater., 2016, 28, 1884–1890 CrossRef CAS PubMed.
- B. Fan, L. Ying, P. Zhu, F. Pan, F. Liu, J. Chen, F. Huang and Y. Cao, Adv. Mater., 2017, 29, 1703906 CrossRef PubMed.
- W. Lee, C. Lee, H. Yu, D.-J. Kim, C. Wang, H. Y. Woo, J. H. Oh and B. J. Kim, Adv. Funct. Mater., 2016, 26, 1543–1553 CrossRef CAS.
- X. Wu, Y. Tang, Y. Wang, X. Liu, C. Liu, X. Zhang, Y. Yang, X. Gao, F. Chen, X. Guo and Z.-K. Chen, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3679–3689 CrossRef CAS.
- J. W. Jung, J. W. Jo, C.-C. Chueh, F. Liu, W. H. Jo, T. P. Russell and A. K.-Y. Jen, Adv. Mater., 2015, 27, 3310–3317 CrossRef CAS PubMed.
- T. Kim, J. H. Kim, T. E. Kang, C. Lee, H. Kang, M. Shin, C. Wang, B. Ma, U. Jeong, T. S. Kim and B. J. Kim, Nat. Commun., 2015, 6, 8547 CrossRef CAS PubMed.
- T. Earmme, Y.-J. Hwang, N. M. Murari, S. Subramaniyan and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14960–14963 CrossRef CAS PubMed.
- Y.-J. Hwang, B. A. E. Courtright, A. S. Ferreira, S. H. Tolbert and S. A. Jenehke, Adv. Mater., 2015, 27, 4578–4584 CrossRef CAS PubMed.
- L. Xue, Y. Yang, Z.-G. Zhang, X. Dong, L. Gao, H. Bin, J. Zhang, Y.-X. Yang and Y. Li, J. Mater. Chem. A, 2016, 4, 5810–5816 RSC.
- Y. Zhou, K. L. Gu, X. Gu, T. Kurosawa, H. Yan, Y. Guo, G. I. Koleilat, D. Zhao, M. F. Toney and Z. Bao, Chem. Mater., 2016, 28, 5037–5042 CrossRef CAS.
- S. Li, H. Zhang, W. Zhao, L. Ye, H. Yao, B. Yang, S. Zhang and J. Hou, Adv. Energy Mater., 2016, 6, 1–9 CAS.
- Y. Guo, Y. Li, O. Awartani, J. Zhao, H. Han, H. Ade, D. Zhao and H. Yan, Adv. Mater., 2016, 28, 8483–8489 CrossRef CAS PubMed.
- Y. Guo, Y. Li, O. Awartani, H. Han, J. Zhao, H. Ade, H. Yan and D. Zhao, Adv. Mater., 2017, 29, 1700309 CrossRef PubMed.
- M. Liu, J. Yang, C. Lang, Y. Zhang, E. Zhou, Z. Liu, F. Guo and L. Zhao, Macromolecules, 2017, 50, 7559–7566 CrossRef CAS.
- Z. Li, W. Zhang, X. Xu, Z. Genene, D. Di Carlo Rasi, W. Mammo, A. Yartsev, M. R. Andersson, R. A. J. Janssen and E. Wang, Adv. Energy Mater., 2017, 7, 1602722 CrossRef.
- Z. Li, X. Xu, W. Zhang, X. Meng, W. Ma, A. Yartsev, O. Inganäs, M. R. Andersson, R. A. J. Janssen and E. Wang, J. Am. Chem. Soc., 2016, 138, 10935–10944 CrossRef CAS PubMed.
- Y. J. Hwang, T. Earmme, B. A. E. Courtright, F. N. Eberle and S. A. Jenekhe, J. Am. Chem. Soc., 2015, 137, 4424–4434 CrossRef CAS PubMed.
- F. Machui, M. Hösel, N. Li, G. D. Spyropoulos, T. Ameri, R. R. Sondergaard, M. Jorgensen, A. Scheel, D. Gaiser, K. Kreul, D. Lenssen, M. Legros, N. Lemaitre, M. Vilkman, M. Välimäki, S. Nordman, C. J. Brabec and F. C. Krebs, Energy Environ. Sci., 2014, 7, 2792–2802 RSC.
- L. Lucera, P. Kubis, F. W. Fecher, C. Bronnbauer, M. Turbiez, K. Forberich, T. Ameri, H.-J. Egelhaaf and C. J. Brabec, Energy Technol., 2015, 3, 373–384 CrossRef.
- J. Min, Y. N. Luponosov, C. Cui, B. Kan, H. Chen, X. Wan, Y. Chen, S. A. Ponomarenko, Y. Li and C. J. Brabec, Adv. Energy Mater., 2017, 7, 1700465 CrossRef.
- E. Bundgaard, F. Livi, O. Hagemann, J. E. Carlé, M. Helgesen, I. M. Heckler, N. K. Zawacka, D. Angmo, T. T. Larsen-Olsen, G. A. dos Reitos Benatto, B. Roth, M. V. Madsen, M. R. Andersson, M. Jorgensen, R. R. Sondergaard and F. C. Krebs, Adv. Energy Mater., 2015, 5, 1402186 CrossRef.
- A. Teichler, J. Perelaer and U. S. Schubert, J. Mater. Chem. C, 2013, 1, 1910–1925 RSC.
- R. R. Sondergaard, M. Hösel and F. C. Krebs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 16–34 CrossRef CAS.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2009, 93, 394–412 CrossRef CAS.
- R. Mens, P. Adriaensens, L. Lutsen, A. Swinnen, S. Bertho, B. Ruttens, J. D’Haen, J. Manca, T. Cleij, D. Vanderzande and J. Gelan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 138–145 CrossRef CAS.
- B. Schmidt-Hansberg, M. F. G. Klein, K. Peters, F. Buss, J. Pfeifer, S. Wahlheim, A. Colsmann, U. Lemmer, P. Scharfer and W. Schabel, J. Appl. Phys., 2009, 106, 124501 CrossRef.
- T. Wang, N. W. Scarratt, H. Yi, A. D. F. Dunbar, A. J. Pearson, D. C. Watters, T. S. Glen, A. C. Brook, J. Kinglsey, A. R. Buckley, M. W. A. Skoda, A. M. Donald, R. A. L. Jones, A. Iraqi and D. G. Lidzey, Adv. Energy Mater., 2013, 3, 505–512 CrossRef CAS.
- G. Susanna, L. Salamandra, T. M. Brown, A. Di Carlo, F. Brunetti and A. Reale, Sol. Energy Mater. Sol. Cells, 2011, 95, 1775–1778 CrossRef CAS.
- K. Liu, T. T. Larsen-Olsen, Y. Lin, M. Beliatis, E. Bundgaard, M. Jørgensen, F. C. Krebs and X. Zhan, J. Mater. Chem. A, 2016, 4, 1044–1051 RSC.
- X. Gu, Y. Zhou, K. Gu, T. Kurosawa, Y. Guo, Y. Li, H. Lin, B. C. Schroeder, H. Yan, F. Molina-Lopez, C. J. Tassone, C. Wang, S. C. B. Mannsfeld, H. Yan, D. Zhou, M. F. Toney and Z. Bao, Adv. Energy Mater., 2017, 7, 1602742 CrossRef.
- N. Gasparini, M. Salvador, S. Strohm, T. Heumueller, I. Levchuk, A. Wadsworth, J. H. Bannock, J. C. de Mello, H. Egelhaaf, D. Baran, I. McCulloch and C. J. Brabec, Adv. Energy Mater., 2017, 7, 1700770 CrossRef.
- L. Ye, Y. Xiong, Q. Zhang, S. Li, C. Wang, Z. Jiang, J. Hou, W. You and H. Ade, Adv. Mater., 2018, 30, 1705485 CrossRef PubMed.
- W. Zhao, S. Zhang, Y. Zhang, S. Li, X. Liu, C. He, Z. Zheng and J. Hou, Adv. Mater., 2018, 30, 1704837 CrossRef PubMed.
- C. Cai, Y. Zhang, R. Song, Z. Peng, L. Xia, M. Wu, K. Xiong, B. Wang, Y. Lin, X. Xu, Q. Liang, H. Wu, E. Wang and L. Hou, Sol. Energy Mater. Sol. Cells, 2017, 161, 52–61 CrossRef CAS.
- V. Vohra, W. Mróz, S. Inaba, W. Porzio, U. Giovanella and F. Galeotti, ACS Appl. Mater. Interfaces, 2017, 9, 25434–25444 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cs00892a |
| This journal is © The Royal Society of Chemistry 2019 |