
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCooperative activation of X–H (X = H, C, O, N) bonds by a Pt(0)/Ag(I) metal-only Lewis pair†
Nereida
Hidalgo
 ,
Celia
Maya
and
Jesús
Campos
*
,
Celia
Maya
and
Jesús
Campos
*
Instituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universidad de Sevilla and Consejo Superior de Investigaciones Científicas (CSIC), Avenida Américo Vespucio 49, 41092 Sevilla, Spain. E-mail: jesus.campos@iiq.csic.es
First published on 17th May 2019
Abstract
Metal-only Lewis pairs made of Pt(0)/Ag(I) combinations have been previously reported, but their cooperative reactivity remains unexplored. Here we demonstrate that these systems are capable of synergistically activating a wide variety of X–H (X = H, C, O, N) bonds, including those in dihydrogen, alkynes, water and ammonia.
Since the seminal works of Werner at the end of the 19th century, the chemistry of transition metal complexes has been mostly defined by the acidic character of the metal centre, to which basic ligands can bind by means of dative bonds. In contrast, the potential of transition metals to behave as electron donors has remained less explored, despite its fundamental role in oxidative addition reactions or in the coordination of π-acidic ligands.1 More recently, the interest in electron-rich transition metal complexes capable of coordinating σ-acceptor ligands (Z-type ligands) has grown enormously.2 This unusual reverse flux of electrons provides the metal centre with uncommon stereoelectronic properties and reactivity patterns.3 In this context, the use of unsaturated transition metal fragments instead of p-block σ-acceptors has become a successful strategy to access heterobimetallic compounds containing M→M dative bonds.4 A fascinating class among these bimetallic entities are metal-only Lewis pairs (MOLPs),5 in which two metal fragments are held together exclusively through a M–M interaction. Within this field, the use of group 11 metals acting as Lewis acids for the synthesis of MOLPs is well-documented.6 In the case of silver, accessing these bimetallic targets acquires particular relevance in connection to the use of silver species as halide abstractors7 or transmetallation reagents,8 as well as due to the photoluminescent properties of silver-containing heterometallic compounds.9 In addition, TM→Ag dative bonds have served as an experimental gauge to calibrate the basicity of transition metals.10 Moreover, metal–metal interactions involving silver centres are key in catalysis, where the so-called silver effect has been regularly invoked in a variety of transition-metal catalysed transformations.11 Likewise, well-defined silver-based heterobimetallic catalysts can outperform their monometallic parent precursors and provide unusual selectivities.12 On this basis, and following our recent results on bimetallic bond activation using the basic Pt(0) compound [Pt(PtBu3)2] (1) in combination with a sterically crowded electrophilic phosphine gold(I) fragment,13 we decided to investigate the reactivity of Pt(0)/Ag(I) MOLPs. The latter systems find precedent in the recent literature but their cooperative reactivity remains unexplored.14 A prior work demonstrating the cooperative activation of water using (1) and Cu(I) salts, further encouraged us to pursue these research aims.15
The formation of Pt→Ag adducts 2 and 3 proceeds readily in benzene or dichloromethane upon mixing [Pt(PtBu3)2] (1) and the corresponding silver salt in the absence of light (Scheme 1), resulting in the instant coloration of the solution from colourless to bright yellow. A chemical shift towards slightly lower frequencies is recorded by 31P{1H} NMR monitoring, accompanied by a pronounced decrease in the 1JPPt coupling constant (2: δ = 99.6 ppm, 1JPPt = 3298 Hz; 3: δ = 99.2 ppm, 1JPPt = 3244 Hz) with respect to precursor 1 (δ = 100.2 ppm, 1JPPt = 4410 Hz). The diminished 1JPPt values in 2 and 3 are expected considered the reduced s character of P–Pt bonds as a consequence of the new Pt→Ag interaction. These data are similar to the NMR changes observed in the formation of a trimetallic compound of formula [(PCy3)2Pt→Ag+←Pt(PCy3)2] previously described by the group of Braunschweig.14c However, in our case the use of slightly coordinating anions such as triflimide (NTf2−, [N(SO2CF3)2]−) or triflate (OTf−, [OSO2CF3]−) seems to prevent the formation of the trimetallic compound in favour of the bimetallic MOLPs 2 and 3. This was demonstrated by the reaction of 1 with 0.5 equivalents of silver salts AgNTf2 or AgOTf, which results in the formation of equimolar mixtures of unreacted 1 and compounds 2 and 3, respectively. Interestingly, the use of silver reagents containing less coordinating counteranions (i.e. BF4− and PF6−) also led to Pt/Ag MOLPs, as inferred from 31P{1H} NMR studies, but those displayed limited stability. More precisely, they evolved towards the cyclometalation of one of the tert-butyl fragments of a PtBu3 ligand to yield compounds [PtII(κ2P,C-PtBu2CMe2CH2)(PtBu3)]+ (δ(31P{1H}) = 55.9, 23.2 ppm (d, 2JPP = 319 Hz)) and [PtII(PtBu3)2H]+ (δ(31P{1H}) = 86.3 ppm, 1JPPt = 2723 Hz; δ(1H) = −33.0 ppm (1JHPt = 2224 Hz)).15 The same reactivity was noticed upon dissolving compounds 1 or 2 in tetrahydrofuran. An identical transformation was latterly reported using a ferrocenium salt as the one-electron oxidant,16 a role presumably played by the silver salt in the compounds reported herein.
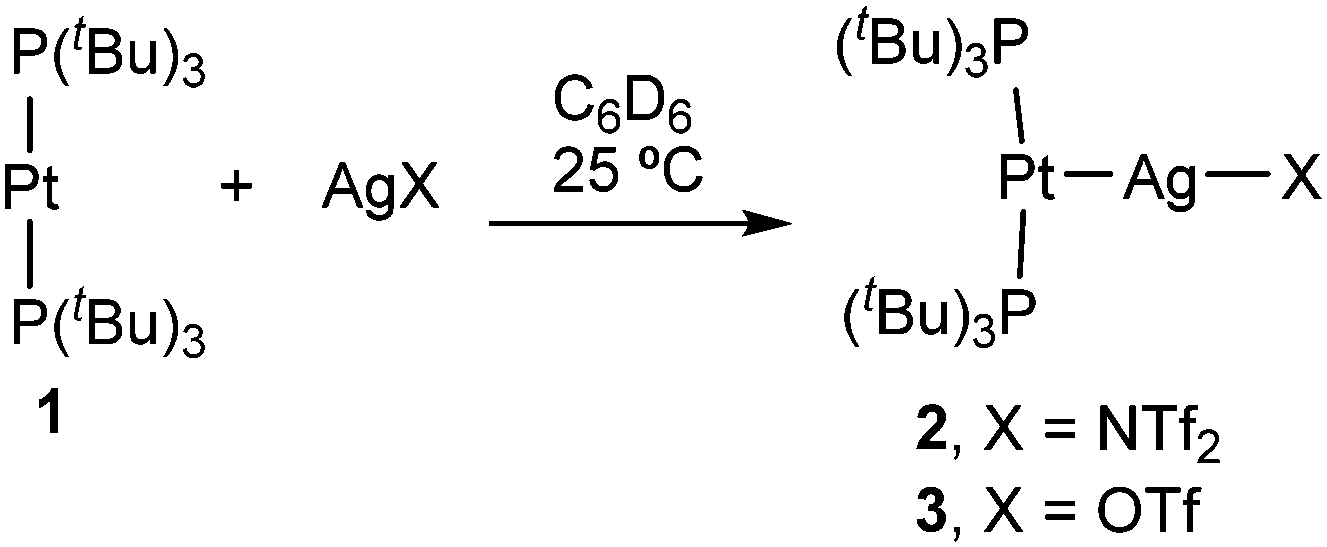 | ||
| Scheme 1 Synthesis of Pt(0)/Ag(I) metal-only adducts (NTf2− = [N(SO2CF3)2]−; OTf− = [OSO2CF3]−). | ||
The molecular structures of compounds 2 and 3 were authenticated by single-crystal X-ray diffraction studies, confirming the proposed bimetallic formulation of the metal–metal core. While compound 2 crystallized as a monomer in the solid state (Fig. 1), the structure of 3 reveals a dimeric configuration with two triflate anions as bridging fragments (Fig. S1, ESI†). Nevertheless, diffusion NMR experiments ruled out a dimeric formulation as the main species in solution. The structures are otherwise comparable to previous Pt(0) Lewis adducts,13,14c,17 exhibiting slightly distorted T-shaped configurations around the metal centre (P–Pt–P = 167.07(5) (2) and 174.69(4)° (3)). The Pt–P bond distances (2.30–2.32 Å) are elongated in comparison to precursor 1 (2.25 Å).18 This may be attributed to the release of electrons from the platinum centre towards the silver atom, in analogy to the Pt–P bond lengthening observed upon one-electron oxidation of precursor 1, whose origin is still a matter of debate.19
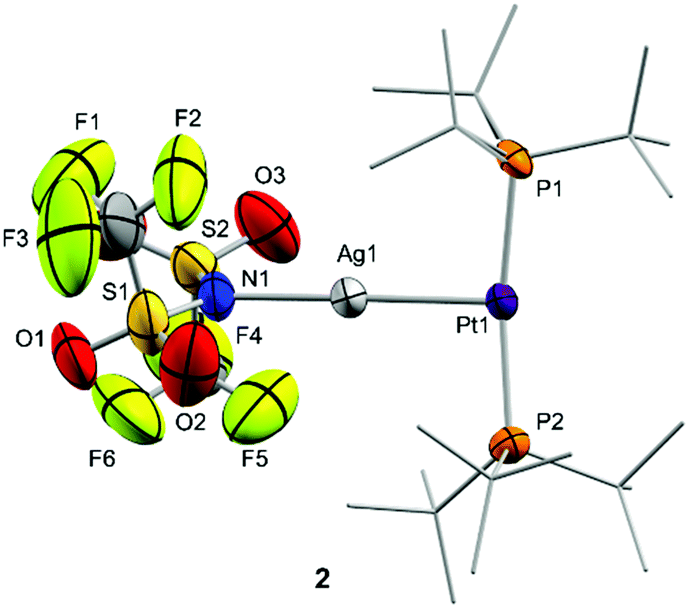 | ||
| Fig. 1 ORTEP diagram of compound 2; for the sake of clarity hydrogen atoms are excluded and some substituents have been represented in wireframe format, while thermal ellipsoids are set at 50% probability. | ||
Given the superior stability of compound 2 over 3, we chose the former to investigate its reactivity and compare the outcomes with its independent components, that is, compounds 1 and AgNTf2. We examined the reactivity of 2 towards the activation of hydrogen, alkynes and polar X–H bonds (X = O, N), including those in water and ammonia. Compound 2 cleanly evolves in the presence of H2 under mild conditions (1 atm, −20 °C) to yield the heterobimetallic Pt(II)/Ag(I) dihydride 4 that contains a terminal and a bridging hydride (Scheme 2). Related Pt/Ag heterobimetallic hydrides have been previously prepared by mixing platinum(II) dihydrides with silver salts.20 Nevertheless, it is important to remark that neither precursor 1 nor AgNTf2 exhibit any reactivity towards dihydrogen even under harsher conditions (4 atm, 60 °C). This demonstrates that both metals are required for the cleavage of the H–H bond to take place. The course of the reaction can be monitored by 31P{1H} NMR spectroscopy, following the appearance of a new resonance at 94.6 ppm (1JPPt = 2660 Hz) due to the heterobimetallic dihydride 4. This compound exhibits fluxional behavior in solution, as evinced by a single and distinctive low-frequency 1H NMR resonance (−4.89 ppm) at 25 °C due to the two hydride ligands. This signal appears as an apparent double quartet due to coupling to 107,109Ag (1JAgH = 120 Hz) and 31P (2JHP = 10 Hz), and it is flanked by 197Pt satellites (1JHPt = 778 Hz). The dynamic process is quenched at temperatures below −50 °C, at which the initial signal is split into two resonances at −3.98 (1JAgH = 182, 1JHPt = 570 Hz) and −5.21 (1JAgH = 44, 1JHPt = 984 Hz) ppm due to the bridging and terminal hydrides, respectively (Fig. S4, ESI†). The barrier for the exchange was determined by standard line-shape analysis (ΔG‡298 = 13.2 kcal mol−1). It could result from either fast tilting of the square-planar Pt(H)2(PtBu3)2 fragment or rapid dissociation/coordination of the silver salt. In fact, the lability of the Pt→Ag bond is attested by the reaction of 1 with 0.5 equivalents of AgNT2 under H2 atmosphere, which forms an equimolar mixture of compound 4 and unreacted 1 after 5 hours and eventually evolves to a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the former and Pt(H)2(PtBu3)2, along with variable amounts of [PtII(PtBu3)2H]+ as a recurrent side-product. Having in mind that precursor 1 does not react with H2 under our experimental conditions, the formation of Pt(H)2(PtBu3)2 may imply transfer of silver from hydride 4 to unreacted 1 forming compound 2, which could subsequently be hydrogenated by the synergistic action of silver.
:1 mixture of the former and Pt(H)2(PtBu3)2, along with variable amounts of [PtII(PtBu3)2H]+ as a recurrent side-product. Having in mind that precursor 1 does not react with H2 under our experimental conditions, the formation of Pt(H)2(PtBu3)2 may imply transfer of silver from hydride 4 to unreacted 1 forming compound 2, which could subsequently be hydrogenated by the synergistic action of silver.
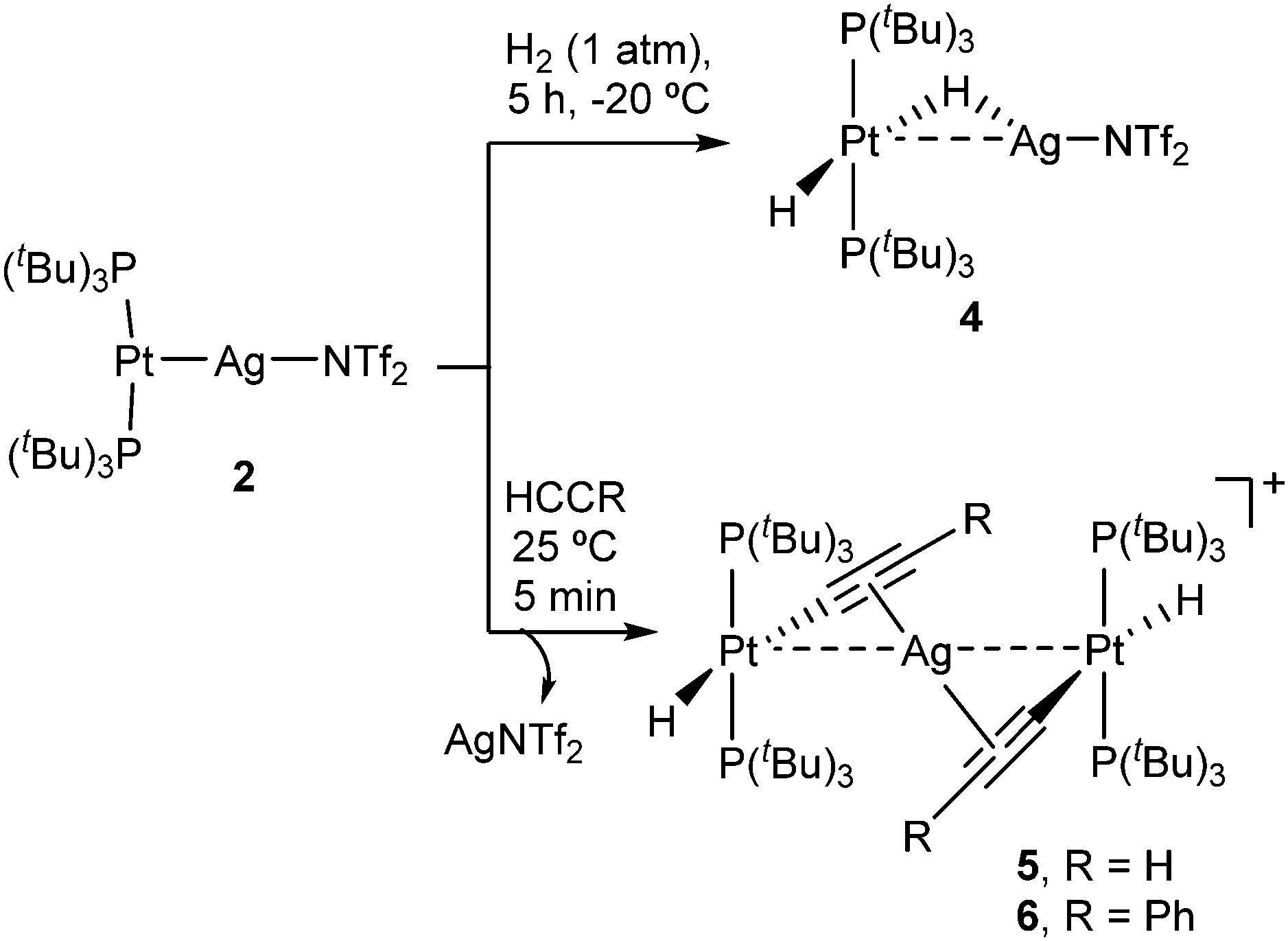 | ||
| Scheme 2 Cooperative activation of H2 and alkynes mediated by 2. | ||
We next investigated the reactivity of 2 towards acetylene and phenylacetylene (Scheme 2). Compound 1 acts as a catalyst for the polymerization of C2H2, provoking the rapid precipitation of a dark purple solid upon exposure to the gas. The formation of the polymer is, however, visibly inhibited in the presence of the silver salt, in which case the trinuclear [(PtBu3)2(H)Pt(μ-CCH)Ag(μ-CCH)Pt(H)(PtBu3)2] (5) is obtained in almost quantitative spectroscopic yield. Analogous reactivity is derived from the addition of phenylacetylene, resulting in the formation of the heterobimetallic dibridged bisacetylide 6. Similarly to our studies with H2, neither precursor 1 nor the silver salt exhibit any reactivity towards phenylacetylene as monometallic species. Compounds 5 and 6 are related to other Pt/Ag bridging acetylides that have been prepared by the addition of silver salts to pre-formed platinum(II) acetylides,21 an approach that contrasts to the bimetallic cooperative alkyne activation described herein. The new heterobimetallic compounds are characterized by 31P{1H} NMR resonances at 83.4 (5) and 82.8 (6) ppm flanked by 195Pt satellites (1JPPt = 2782 and 2759 Hz, respectively), shifted to lower frequencies by ca. 16 ppm with respect to their precursor 2. These compounds are thermally unstable in solution, particularly complex 5. However, we managed to grow single-crystals of the latter species. X-ray diffraction studies revealed the trimetallic formulation of the dibridged σ,π bisacetylide (Fig. 2). The Pt⋯Ag distance is markedly elongated (3.634(9) Å) compared to the metallic Lewis pair 2 (2.658(1) Å), indicating that the dative Pt→Ag bond is no longer present. The σ,π-bridged acetylide fragments are characterized by short bond distances with the silver nuclei (dAg-centroid 2.216(6) Å), along with relatively lengthened Pt–C bond distances (dPt–C 2.107(12) Å) compared to other compounds of formula [Pt(H)(CCR)(PR3)2] (ca. 1.9–2.0 Å).22 The formulation of compounds 5 and 6 as trimetallic bisacetylides was corroborated in solution by the reactions of equimolar mixtures of 1 and 2 with the two investigated alkynes, leading to compounds 5 and 6, respectively, accompanied by complete consumption of the two platinum precursors, that is, using only one equivalent of silver per two platinum nuclei.
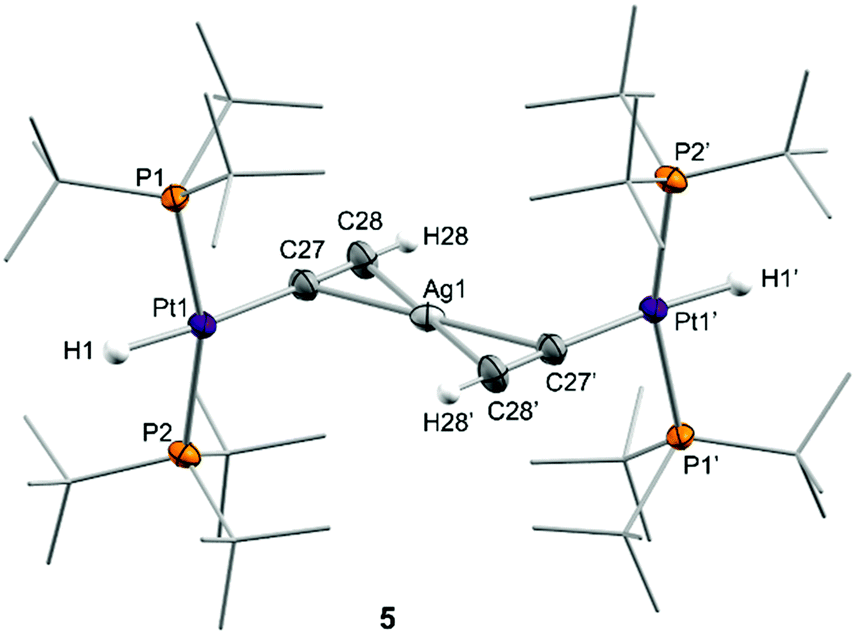 | ||
| Fig. 2 ORTEP diagram of compound 5; for the sake of clarity most hydrogen atoms and the triflimide counteranion have been excluded and some substituents have been represented in wireframe format, while thermal ellipsoids are set at 50% probability. | ||
We additionally tested the reactivity of 2 towards the activation of polar X–H (X = O, N) bonds. Before discussing these results, it is pertinent to highlight that none of the reagents tested for X–H bond cleavage exhibited any reactivity towards precursor 1. However, addition of methanol (5 equiv.) to a C6D6 solution of 2 provides formation of the previously reported22 cationic T-shaped platinum hydride 7 (ca. 80%) after around one hour at 25 °C (Scheme 3). This compound is characterized by a distinctive low-frequency 1H NMR resonance at −33.0 ppm (1JPPt = 2224 Hz). Its molecular formulation was further confirmed by X-ray diffraction studies (Fig. S2, ESI†). More interestingly, analogous reactivity arises from the addition of water or ammonia, yielding the corresponding cationic Pt-hydride complexes further stabilized by coordination of a water (8a) or ammonia (8b) ligand, respectively. While the activation of water (50 equiv.) in benzene solution takes up to 12 hours to reach ca. 90% completion, likely due to water immiscibility, using wet acetonitrile yields [PtH(CH3CN)(PtBu3)2]+ (8c) instantly. The reaction with ammonia (1 bar) requires heating at 60 °C to form compound 8b in ca. 60% yield after 16 hours.
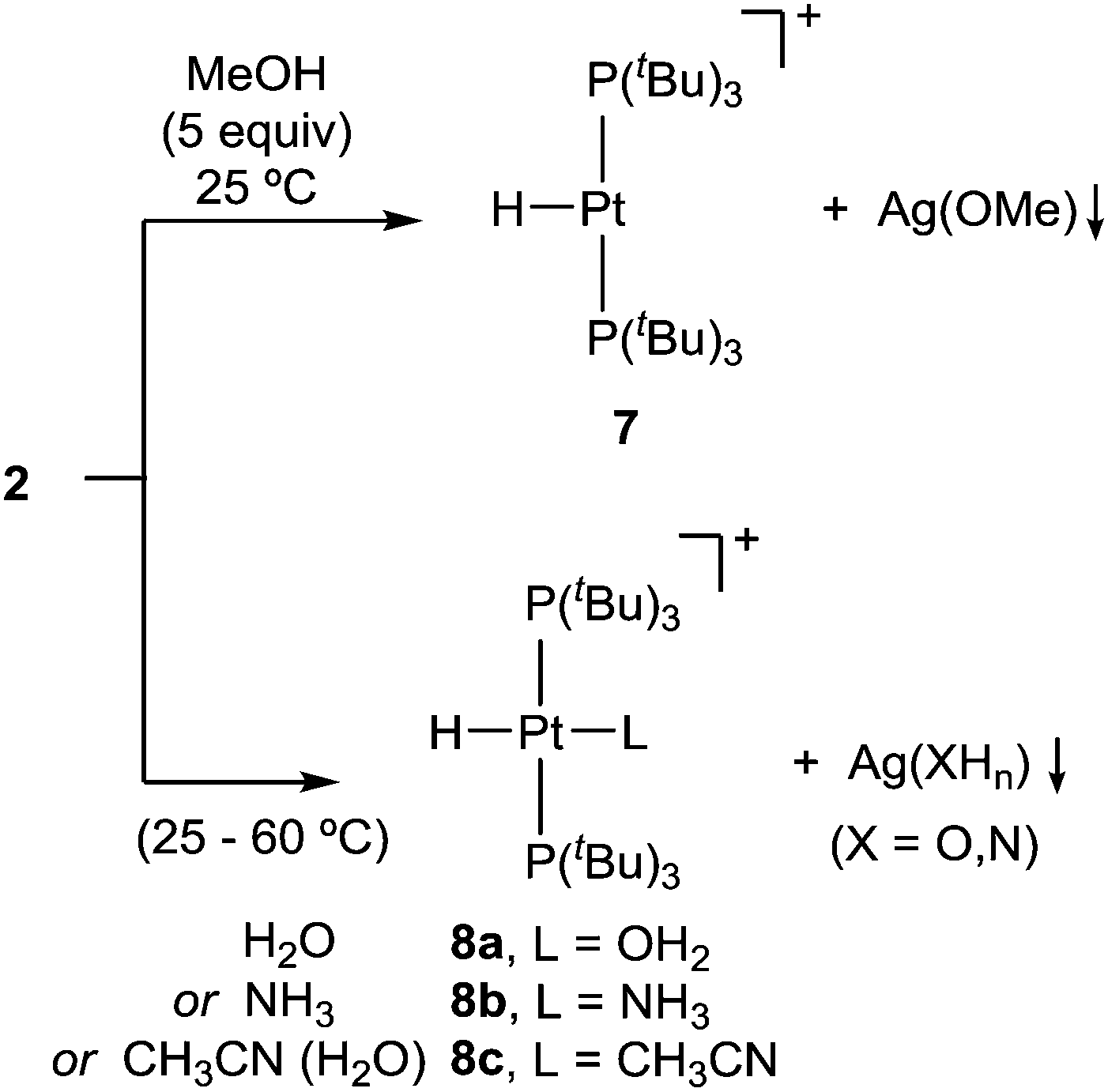 | ||
| Scheme 3 Reactivity of compound 2 towards polar X–H (X = O, N) bonds. | ||
The activation of X–H bonds in methanol, water and ammonia is accompanied by the appearance of greyish precipitates that we attribute to the corresponding silver alkoxide, hydroxide or amide salts, respectively. The formation of compounds 8a–c was unequivocally confirmed by 1H and 31P{1H} NMR spectroscopy.23 The solid structure of the aquo/hydride complex 8a was validated by X-ray diffraction studies (Fig. S3, ESI†).
As stated in the introduction, the cooperative activation of water using a bimetallic Pt(0)/Cu(I) pair has been recently described.15 However, the cleavage of N–H bonds in ammonia remains a challenge in transition metal chemistry, where formation of Werner-type adducts is typically preferred.24 Thus, compound 2 adds to the limited list of transition metal systems capable of activating ammonia under mild conditions by virtue of the synergistic cooperation between the two metals.
In summary, the reactivity of the Pt(0)/Ag(I) MOLP 2 markedly differs from that of its independent metal components. Thus, while [Pt(PtBu3)2] (1) does not react with H2, phenylacetylene, methanol, water or ammonia, the presence of a silver salt readily facilitates the activation of these molecules by X–H (X = H, C, O, N) bond cleavage. These results demonstrate the usefulness of MOLP systems for small molecule activation and, as an immediate consequence, for catalytic applications, a research avenue that we are currently pursuing in our group.
This work has been supported by the European Research Council (ERC Starting Grant, CoopCat, Project 756575). We are grateful to M. Roselló for valuable discussions. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, John Wiley & Sons, New Jersey, 6th edn, 2014 Search PubMed.
- (a) A. Amgouneab and D. Bourissou, Chem. Commun., 2011, 47, 859 RSC; (b) G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2016, 45, 1065 RSC; (c) M. Sircoglou, S. Bontemps, M. Mercy, N. Saffon, M. Takahashi, G. Bouhadir, L. Maron and D. Bourissou, Angew. Chem., Int. Ed., 2007, 46, 8583 CrossRef CAS PubMed.
- (a) D. You, H. Yang, S. Sen and F. P. Gabbaï, J. Am. Chem. Soc., 2018, 140, 9644 CrossRef CAS PubMed; (b) F. Inagaki, C. Matsumoto, Y. Okada, N. Maruyama and C. Mukai, Angew. Chem., Int. Ed., 2015, 54, 818 CrossRef CAS PubMed.
- (a) M. R. Ringenberg, Chem. – Eur. J., 2019, 25, 2396 CrossRef CAS PubMed; (b) M. Devillard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Back, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917 CrossRef CAS PubMed; (c) R. J. Eisenhart, L. J. Clouston and C. C. Lu, Acc. Chem. Res., 2015, 48, 2885 CrossRef CAS PubMed; (d) J. P. Krogman, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2011, 133, 14582 CrossRef CAS PubMed; (e) T.-P. Lin, C. R. Wade, L. M. Pérez and F. P. Gabbaï, Angew. Chem., Int. Ed., 2010, 49, 6357 CrossRef CAS PubMed.
- J. Bauer, H. Braunschweig and R. D. Dewhurst, Chem. Rev., 2012, 112, 4329 CrossRef CAS PubMed.
- (a) P. J. Malinowsky and I. Krossing, Angew. Chem., Int. Ed., 2014, 53, 13460 CrossRef PubMed; (b) D. E. Janzen, L. F. Mehne, D. G. VanDerveer and G. J. Grant, Inorg. Chem., 2005, 44, 8182 CrossRef CAS PubMed; (c) Z. Xie, T. Jelínek, R. Bau and C. A. Reed, J. Am. Chem. Soc., 1994, 116, 1907 CrossRef CAS; (d) T. Yamaguchi, F. Yamazaki and T. Ito, J. Am. Chem. Soc., 2001, 123, 743 CrossRef CAS PubMed; (e) G. Wang, Y. S. Ceylan, T. R. Cundari and H. V. R. Dias, J. Am. Chem. Soc., 2017, 139, 14292 CrossRef CAS PubMed; (f) S. Takemoto, T. Tsujimoto and H. Matsuzaka, Organometallics, 2018, 37, 1591 CrossRef CAS.
- (a) G. Weber, F. Rominger and B. F. Straub, Eur. J. Inorg. Chem., 2012, 2863 CrossRef; (b) G. Sipos, P. Gao, D. Foster, B. W. Skelton, A. N. Sobolev and R. Dorta, Organometallics, 2017, 36, 801 CrossRef CAS; (c) K. Sasakura, K. Okamoto and K. Ohe, Organometallics, 2018, 37, 2319 CrossRef CAS.
- (a) M. Baya, Ú. Belío, D. Campillo, I. Fernández, S. Fuertes and A. Martín, Chem. – Eur. J., 2018, 24, 13879 CrossRef CAS PubMed; (b) I. Meana, P. Espinet and A. C. Albéniz, Organometallics, 2014, 33, 1 CrossRef CAS; (c) M. Asay, B. Donnadieu, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 4796 CrossRef CAS PubMed.
- (a) J. Moussa, L. M. Chamoreau, M. P. Gullo, A. Degli Esposti, A. Barbieri and H. Amouri, Dalton Trans., 2016, 45, 2906 RSC; (b) L. R. Falvello, J. Forniés, E. Lalinde, B. Menjón, M. A. García-Monforte, M. T. Moreno and M. Tomás, Chem. Commun., 2007, 3838 RSC; (c) K. M.-C. Wong, C.-K. Hui, K.-L. Yu and V. W.-W. Yam, Coord. Chem. Rev., 2002, 229, 123 CrossRef CAS.
- H. Braunschweig, C. Brunecker, R. D. Dewhurst, C. Schneider and B. Wennemann, Chem. – Eur. J., 2015, 21, 19195 CrossRef CAS PubMed.
- (a) A. Homs, I. Escofet and A. M. Echavarren, Org. Lett., 2013, 15, 5782 CrossRef CAS PubMed; (b) D. Weber and M. R. Gagné, Org. Lett., 2009, 11, 4962 CrossRef CAS PubMed; (c) C. Chen, C. Hou, Y. Wang, T. S. A. Hor and Z. Weng, Org. Lett., 2014, 16, 524 CrossRef CAS PubMed.
- M. K. Karunananda and N. P. Mankad, J. Am. Chem. Soc., 2015, 137, 14598 CrossRef CAS PubMed.
- J. Campos, J. Am. Chem. Soc., 2017, 139, 2944 CrossRef CAS PubMed.
- (a) B. R. Barnett, C. E. Moore, P. Chandrasekaran, S. Sproules, A. L. Rheingold, S. DeBeerde and J. S. Figueroa, Chem. Sci., 2015, 6, 7169 RSC; (b) B. R. Barnett and J. S. Figueroa, Chem. Commun., 2016, 52, 13829 RSC; (c) H. Braunschweig, R. D. Dewhurst, F. Hupp and C. Schneider, Chem. Commun., 2014, 50, 15685 RSC.
- S. Jamali, S. Abedanzadeh, N. K. Khaledi, H. Samouei, Z. Hendi, S. Zacchini, R. Kiaa and H. R. Shahsavari, Dalton Trans., 2016, 45, 17644 RSC.
- T. Troadec, S.-Y. Tan, C. J. Wedge, J. P. Rourke, P. R. Unwin and A. B. Chaplin, Angew. Chem., Int. Ed., 2016, 55, 3754 CrossRef CAS PubMed.
- (a) N. Hidalgo, S. Bajo, J. J. Moreno, C. Navarro-Gilabert, B. Q. Mercado and J. Campos, submitted; (b) J. Bauer, H. Braunschweig, A. Damme and K. Radacki, Angew. Chem., Int. Ed., 2012, 51, 10030 CrossRef CAS PubMed.
- S. Otsuka, T. Yoshida, M. Matsumoto and K. Nakatsu, J. Am. Chem. Soc., 1976, 98, 5850 CrossRef CAS.
- M. C. MacInnis, J. C. DeMott, E. M. Zolnhofer, J. Zhou, K. Meyer, R. P. Hughes and O. V. Ozerov, Chem, 2016, 1, 902 CAS.
- (a) A. Alhinati, F. Demartin, L. M. Venanzi and M. K. Wolfer, Angew. Chem., Int. Ed. Engl., 1988, 27, 563 CrossRef; (b) A. Albinati, H. Lehner, L. M. Venanzi and M. Wolfer, Inorg. Chem., 1987, 26, 3933 CrossRef CAS.
- (a) Q.-H. Wei, L.-J. Han, Y. Jiang, X.-X. Lin, Y.-N. Duan and G.-N. Chen, Inorg. Chem., 2012, 51, 11117 CrossRef CAS PubMed; (b) Z. Dai, A. J. Metta-Magaña and J. E. Nuñez, Inorg. Chem., 2014, 53, 7188 CrossRef CAS PubMed; (c) S. Yamazaki, A. J. Deeming, D. M. Speel, D. E. Hibbs, M. B. Hursthousec and K. M. A. Malik, Chem. Commun., 1997, 177 RSC.
- (a) A. Furalani, S. Licoccia, M. V. Russo, A. C. Villa and C. Guastini, J. Chem. Soc., Dalton Trans., 1982, 2449 RSC; (b) J. R. Berenguer, M. Bernechea and E. Lalinde, Organometallics, 2007, 26, 1161 CrossRef CAS; (c) I. Ara, J. R. Berenguer, E. Eguizabal, J. Forniés, J. Gómez, E. Lalinde and J. M. Saez-Rocher, Organometallics, 2000, 19, 4385 CrossRef CAS.
- R. G. Goel and R. C. Srivasta, Can. J. Chem., 1983, 61, 1352 CrossRef CAS.
- Selected examples of ammonia activation by transition metal complexes: (a) J. Zhao, A. S. Goldman and J. F. Hartwig, Science, 2005, 307, 1080 CrossRef CAS PubMed; (b) C. M. Fafard, D. Adhikari, B. M. Foxman, J. Mindiola and O. V. Ozerov, J. Am. Chem. Soc., 2007, 129, 10318 CrossRef CAS PubMed; (c) M. G. Scheibel, J. Abbenseth, M. Kinauer, F. W. Heinemann, C. Würtele, B. de Bruin and S. Schneider, Inorg. Chem., 2015, 54, 9290 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of new compounds, X-ray diffraction details and NMR spectra. CCDC 1909266–1909270. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc03008e |
| This journal is © The Royal Society of Chemistry 2019 |