
Low fouling strategies for electrochemical biosensors targeting disease biomarkers
Nianzu
Liu
a,
Zhenying
Xu
a,
Aoife
Morrin
b and
Xiliang
Luo
 *a
*a
aKey Laboratory of Optic-electric Sensing and Analytical Chemistry for Life Science, MOE, Shandong Key Laboratory of Biochemical Analysis, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, China. E-mail: xiliangluo@qust.edu.cn
bSchool of Chemical Sciences, National Centre for Sensor Research, INSIGHT Centre for Data Analytics, Dublin City University, Dublin 9, Ireland
First published on 7th January 2019
Abstract
Over the past few years, different biomarkers related to various disease states have been identified, and the assay of these disease biomarkers, which can reveal the health status and stages of diseases, has received increasing attention. A broad range of analytical techniques have been applied to the detection of these biomarkers, with electrochemical analysis becoming one of the most promising approaches due to high sensitivity, accuracy, wide measurement range and low-cost and portable instrumentation requirements. However, one of the biggest challenges in this field relates to the operation of electrochemical sensors in biological media, as biofouling deleteriously affects the functioning of the electrochemical sensing interface when exposed to biological fluid. In this mini review, recent trends in the development of low fouling electrochemical biosensors based on different antifouling materials are summarized and discussed. Also, an outlook on the opportunities and challenges with regard to future development of low fouling electrochemical biosensors capable of assaying biomarkers in clinical samples is presented.
 Nianzu Liu | Miss Nianzu Liu received her B.S. degree in Marine Science from Qingdao University of Science and Technology in 2016. She is currently a PhD candidate majoring in Applied Chemistry in the College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, under the supervision of Prof. Xiliang Luo. Her scientific research interests focus on biochemical analysis, nanobiosensing and electrochemistry. |
 Zhenying Xu | Miss Zhenying Xu received her B.S. degree in New Energy Materials and Engineering from Qingdao University of Science and Technology in 2018. She is currently a postgraduate student majoring in Analytical Chemistry in Qingdao University of Science and Technology, under the supervision of Prof. Xiliang Luo. Her scientific research interests focus on biochemical analysis and electrochemistry. |
 Aoife Morrin | Dr Aoife Morrin obtained her PhD in Electroanalytical Chemistry in 2004 at Dublin City University (DCU) under Prof. Malcolm Smyth. Following her PhD she gained post-doctoral experience at the National Centre for Sensor Research at DCU. She took up an academic position in the School of Chemical Sciences at DCU as a lecturer in Analytical Science in 2008. She has an active research group where a lot of her focus has been on responsive materials for chemical and biochemical sensing applications. She is funded by Science Foundation Ireland for a work programme on wearables and epidermal sensing as a means to collect health diagnostic information. |
 Xiliang Luo | Prof. Xiliang Luo received his PhD degree from Nanjing University in 2005. He then worked as a postdoctoral fellow in Dublin City University, Arizona State University and the University of Pittsburgh successively. In 2011, he became a research assistant professor in the Department of Bioengineering, University of Pittsburgh, and a senior Marie Curie Fellow in the Department of Chemistry, University of Oxford. He then joined the Qingdao University of Science and Technology at the end of 2011 as a Taishan Scholar professor. His scientific interests are focused on biochemical analysis, electrochemistry and conducting polymers, and he has published more than 100 papers in peer reviewed journals. |
Introduction
In the past decade, the number of verified disease biomarkers has increased dramatically.1 The ability to rapidly, sensitively and accurately determine disease biomarkers in a quantitative way is crucial in disease monitoring and clinical diagnosis. Therefore, much importance has been attached to the development of new technologies for early disease diagnosis based on these biomarkers,5 using optical techniques including surface plasmon resonance (SPR),6–8 surface enhanced Raman spectroscopy (SERS),9–11 fluorescence,12–14 and phosphorescence as well as electrochemical techniques including voltammetry, impedance spectroscopy, etc. Electrochemistry has been demonstrated to be capable of biomarker detection with high sensitivity in a format that is convenient, simple, and low-cost.18,19 Because electrochemical formats operate using recognition elements typically confined to an electrode surface that interfaces directly with the solution of interest, analysis of real clinical sample solutions (such as cell lysate, plasma, blood serum, saliva or urine) becomes challenging not only due to selectivity issues on account of the complexity of the matrix but also due to biofouling arising from the nonspecific protein adsorption and undesired cell adhesion.Biofouling of electrode interfaces arising from these adsorption and adhesion processes induces temporal changes, which is a ubiquitous and problematic phenomenon. Biofouling hampers the selectivity of electrochemical sensors and deteriorates signal-to-noise (S/N) ratios over time. Critically, this can lead to false-positive or false-negative responses when it comes to disease marker diagnostics.21,22 A versatile strategy for alleviating this problem is to integrate functional materials with the electrode surface to enhance surface hydrophilicity by inducing a thin hydration layer on the electrode surface. This strategy has been successful in that it has the capability to make surfaces highly nonfouling.24,25 At present, much effort is focused on the development of efficient antifouling materials and grafting methods for their assembly on electrodes. Indeed, as viable innovations in this area are being generated, the door for the commercial translation of sensors for ambitious applications in the clinical domain is opened. For instance, the U.S. Food and Drug Administration (FDA) has just this year approved an implantable glucose sensor (based on fluorescence) which can be left in situ for up to 90 days, demonstrating the recent advances in biocompatible, antifouling coatings.
Materials with admirable antifouling properties include polyethylene glycol (PEG),26–28 zwitterionic materials,15,20,29 biomimetic materials8 and synthetic peptides.32–34 These have been integrated into electrochemical biosensors as biofoulant protection coatings, and convincing results are being reported widely in the literature. This review will provide a high-level overview of these recent advances. This research is driven by the need for reliable and stable sensing interfaces that can be used for highly sensitive and selective biomarker detection in real clinical samples. Key strategies used in the design of these antifouling coatings for electrochemical bio-interfacial structures are discussed in the following sections.
Biosensor fouling
Nonspecific biomolecule or microbial adsorption is a persistent and pervasive menace for interfaces exposed to biological fluids. On in vivo devices such as implants or catheters, protein adsorption may promote bacterial colonization and infection and cause adverse immunological responses. Furthermore, the function of in vitro devices that are in contact with protein-rich biological materials, such as biosensors and microarrays, may be either partially or completely impaired. This review focuses on strategies for overcoming adhesion, that is, to minimize or even avoid irreversible adsorption at the surface.39 It is important to understand the interactions between proteins and biological interfaces and the basic mechanism behind biofouling formation, and the theoretical and practical aspects of nonspecific protein adhesion on the surface. This understanding is critical in the rational design of antifouling surfaces for biosensing.The biofouling process is triggered once the electrode interface is exposed to the analytical sample of interest upon formation of an electrical double layer which mediates the adsorption and conformation of biomolecules which reach the surface by mass transfer. This is known as the transport phase, which triggers the adhesion phase.41 Typically, the nonspecific adsorption process is determined by thermodynamics as the entropy gain and enthalpy loss in the system, resulting in an adsorption energy preference.43 A conditioning layer consisting of lipids and glycoproteins is then formed within seconds to minutes at the surface, providing a suitable environment for microbial colonization, further protein attachment, and biofilm formation.44 The foulants adhere to the surface through numerous interactions, including hydrophobicity, charge attraction and surface nucleation.45
Antifouling interfaces resist biofilm formation by making the interaction of the interface with its biological environment reversible. To do this, the adhesion of bacteria, proteins or other molecules to the surface must be minimized. Most antifouling materials have a low water interface energy making the settling of the foulant energetically unfavorable. Furthermore, for high molecular weights of PEG, steric repulsion can be used to explain the mechanism for protein resistance. Furthermore, the attachment of biomolecules to antifouling surfaces requires surface dehydration which for PEG, for example, is thermodynamically unfavourable because it leads to constraint of the PEG polymer chains that previously possessed high conformational entropy. In the presence of PEG, low adhesion enthalpy changes are obtained while entropy is lost, resulting in unfavorable change in free energy leading to prevention of biofouling.
Engineering an antifouling surface requires surface chemistries that allow the binding of water molecules to the surface, compared to other materials (Fig. 1 shows the basic mechanisms of fouling and antifouling). This implies that antifouling surfaces must be very hydrophilic (or even superhydrophilic, i.e. contact angle < 10 degrees). An antifouling effect, achieved with the formation of this surface-confined aqueous layer, reduces the magnitude of the formed electric double layer (Fig. 2). The induced hydrophilicity is capable of reducing non-polar interactions between functional groups and these surfaces. Thus the electrical neutrality of antifouling materials can help reduce electrostatic interactions with charged foulants.46,47 Moreover, these antifouling materials may prevent adsorption by steric hindrance.43,48 In addition, parameters such as surface roughness, Lewis acidity and stiffness also play roles in resisting fouling.49
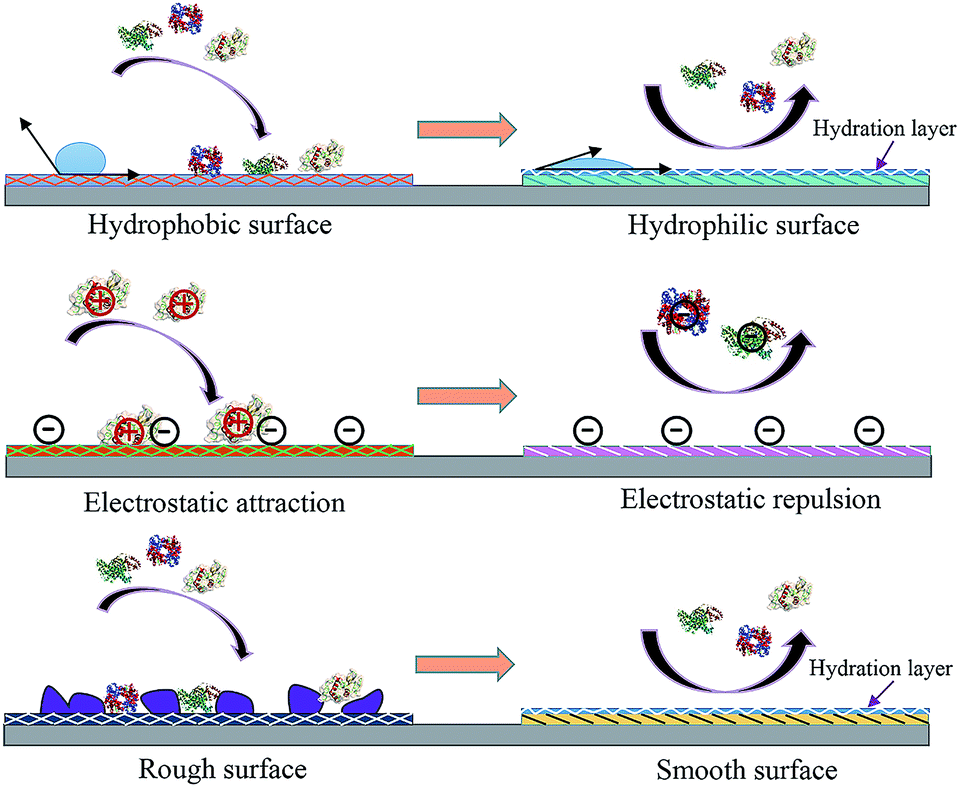 | ||
| Fig. 1 Antifouling materials’ surface properties and their correlation with interface fouling and antifouling. | ||
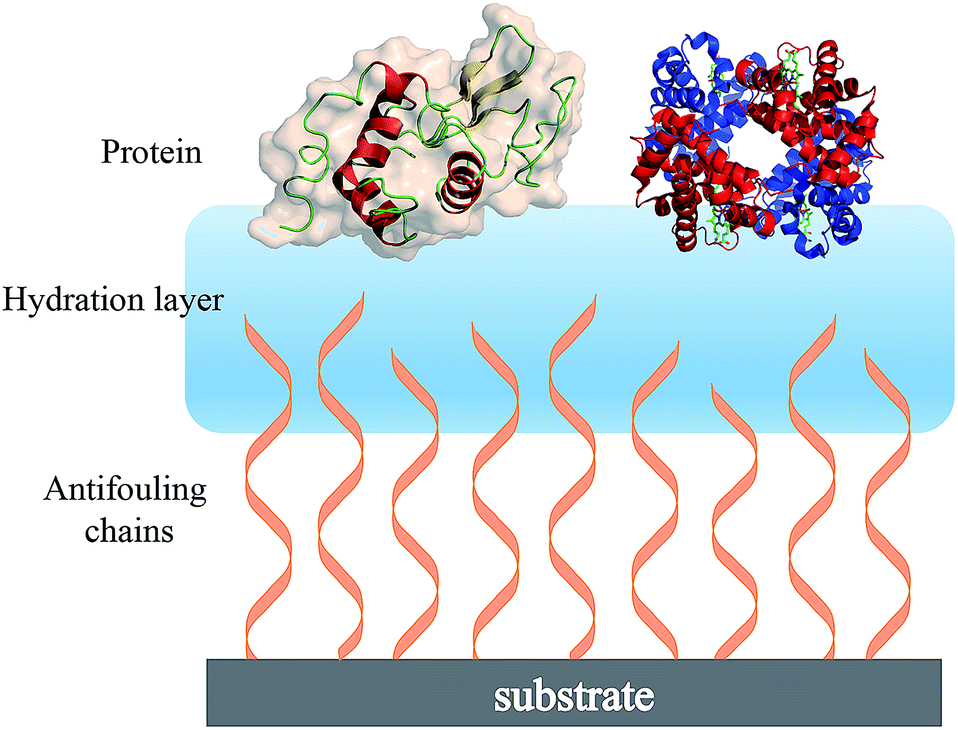 | ||
| Fig. 2 Schematic showing the hydration layer on a solid substrate surface having terminally attached antifouling chains. | ||
There are a large number of polymer-based materials that can prevent or slow down biofilm formation by minimising inter-molecular interactions with the surrounding environment. Three functional properties of these materials guide the design of new nonfouling materials: electrical neutrality, hydrophilicity, and ability to act as a hydrogen bond acceptor rather than a hydrogen bond donor.50
Many materials have been well documented for their antifouling abilities, for example PEG.26–28 The extraordinary antifouling ability of PEG is attributed to its extensive hydration, large excluded volume, steric hindrance, notable conformational flexibility and high chain mobility.51–53 However, the polyether structure and terminal hydroxyl groups of PEG are susceptible to oxidative and thermal degradation, which significantly limits its utility in long-term applications.54–56 Zwitterion-based materials as antifouling alternatives have been brought forward because of robust hydration properties via ionic solvation, long-term durability, environmental stability and facile functionalization.54,57,58 Indeed, the dipole arrangement of water molecules in the hydration layer formed via electrostatic interactions with the charged zwitterion is closer to that of free water than the orientation of water molecules in the hydration layer formed via hydrogen bonds with PEG.59 However, the facile and reproducible synthesis of zwitterion-derived films remains challenging.60 Certain peptides also possess remarkable antifouling properties, benefiting from their notable hydrophilicity and charge neutrality, and have been effective as a component in the design of electrochemical biosensor interfaces.
Based on the characteristic properties of antifouling materials, surface modification efforts have focused on several aspects of antifouling materials. These include inducing super-hydrophilicity into these surface layers, neutralizing electrostatic surface charge, surface roughness reduction, and utilizing steric repulsion effects which have all contributed to the in-depth understanding of the respective materials and their specific antifouling mechanisms.61,62 In relation to sensing interfaces, achieving nonfouling is challenging as the grafting density required for efficient nonfouling compromises the loading of molecular recognition probes that can be immobilized on the sensing surface. Aspects such as these are examined in the following sections, according to their relevance to different functions of the antifouling materials and the electrochemical biosensor strategy deployed.
Antifouling electrochemical biosensors constructed with different materials
Antifouling materials can be classed according to chemical composition, and include PEG-based materials, zwitterionic polymers, and peptides. Peptides can further be divided into hydrophilic peptides, zwitterionic peptides and amphipathic peptides. Hydrophilicity and charge neutrality are commonly considered as key to the antifouling performance of a material. When hydrophilic materials are exposed to aqueous solutions, water molecules accumulate on the surface to form a hydration layer, which can inhibit the proteins and other contaminants from interacting with the surface.63 Zwitterionic polymers can resist charged biomolecules, including proteins, from adsorbing on the interface due to its electrical neutrality. In the following sections, three categories of antifouling materials used in electrochemical biosensors are introduced, namely PEG-based polymers, zwitterionic compounds, and peptides. Other materials that are beginning to show promise in the literature are also discussed below (see also Table 1).| Component construct | Modification strategies | Sensing target | Detection method | Sensing performance | Working media | Ref. |
|---|---|---|---|---|---|---|
| Mixed interface of PEG and ATP | Michael addition reaction | ATP | EIS | 0.1–1000 pM | Human plasma and cancer cell extracts | 4 |
| LOD: 0.1 pM | ||||||
| PEG/AuNPs/PANI | Covalent bonding | AFP | DPV | 0.01–1000 pg mL−1 | Human serum | 16 |
| LOD: 0.007 pg mL−1 | ||||||
| AuNPs/PEDOT/PEG | Electrodeposition | AFP | EIS | 0.001–10 fg mL−1 | 10% human serum | 17 |
| LOD: 0.0003 fg mL−1 | ||||||
| PEGylated PANI nanofibers | Covalent bonding | BRCA1 | EIS | 0.01–1000 pM | Human serum | 23 |
| LOD: 0.0038 pM | ||||||
| PEG/AuNPs/GCE composite | Covalent bonding | BRCA1 | EIS | 50.0 fM to 1.0 nM | Serum | 30 |
| LOD: 1.72 fM | ||||||
| GCE/PEDOT–citrate/Ni2+/peptide/ssDNA | Covalent bonding | BRCA1 | DPV | 1.0 × 10−16 to 1.0 × 10−10 M | 5% human plasma | 37 |
| LOD: 0.03 fM | ||||||
| MCH/Apt–Pep/AuNPs/GE | Self-assembled | ATP | EIS | 1.1 pM to 5 nM | 1% human whole blood | 38 |
| LOD: 0.1 pM | ||||||
| AFP–aptamer/peptide/Au | Self-assembled | AFP | DPV | 0.01–100.0 pg mL−1 | Real human serum | 40 |
| LOD: 3.1 fg mL−1 | ||||||
| Peptides/PANI/GCE | Covalent bonding | IgG | DPV | 1.0–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ng mL−11 000 ng mL−11 |
Real patient serum | 42 |
| LOD: 0.26 ng mL− |
| Component construct | Modification strategies | Sensing target | Detection method | Sensing behaviour | Working media | Ref. |
|---|---|---|---|---|---|---|
| Capture probe/MCH/peptide/Au | Self-assembled | BRCA1 | EIS | 1 fM to 10 pM | 10% human serum | 2 |
| LOD: 0.3 fM | ||||||
| Peptide/aptamer/porous Au/GCE | Self-assembled | IgG | DPV | 0.1–10 pg mL−1 | 5% serum | 3 |
| LOD: 42 fg mL−1 | ||||||
| PSBEDOT–GOx | Electropolymerization | Glucose | i–t | 0.1–0.5 mM | 100% human blood plasma | 15 |
| Pt–PANI–GOx–pSBMA sensor | Electrochemically mediated atom transfer radical polymerization (eATRP) | Glucose | i–t | 1.1–5 mM | Undiluted bovine serum | 20 |
| LOD: 0.05 mM | ||||||
| EDOT-co-EDOTPC/GCE | Electropolymerization | C-Reactive protein | EIS | 10–160 nM | 3.3 M KCl solution | 31 |
| LOD: 37 nM | ||||||
| MB-rep/probe/Au/HA/Popd/CoO/GCE | Electrodeposition | microRNA-24 | SWV | 0.1 pM to 0.1 μM | Human serum | 35 |
| LOD: 33.3 fM | ||||||
| Fe3O4@Au@PEG@HA NPs | Covalent bonding | Brucellosis antibody | DPV | 10−15 to 10−10 g mL−1 | 100% serum | 36 |
| LOD: 0.36 fg mL−1 |
PEG-based materials
PEG has been studied extensively as an antifouling material and has been widely used to successfully resist protein adsorption.64,65 There are several environmental factors that influence the antifouling performance of PEG, such as temperature, pH, and ionic strength.66 For example, a temperature change from 25 °C to 37 °C induced the conformational change of PEG chains from a protein-repulsive state to a protein-attractive state, which led to increased fibrinogen adsorption onto grafted PEG over this temperature range demonstrating the importance of temperature control.67 On account of high hydrophilicity, PEG-based materials have been successfully used in low-fouling electrode interfaces in electrochemical assays.68 For example, Wang et al. developed an ultrasensitive impedimetric antifouling biosensor for adenosine triphosphate (ATP) based on a self-polymerized polydopamine (PDA)-modified electrode surface, which was further functionalized via self-assembly of PEG and ATP aptamers.4 This aptasensor was used to detect ATP in human blood plasma with high precision (%RSD 2.71–4.18), and in cancer cell extracts the limit of detection (LOD) was calculated to be 0.29 (±0.02) mM (n = 3), showing excellent promise for sensitive determination of ATP in complex media. The antifouling properties of the aptasensor were investigated (Fig. 3) whereby it was shown that the use of PEG in the interface design dramatically reduces the interfacial resistance to charge transfer after exposure to human plasma. This demonstrates that the PEG-containing self-assembled layers can resist protein adsorption, which is attributed to enhanced hydrophilicity.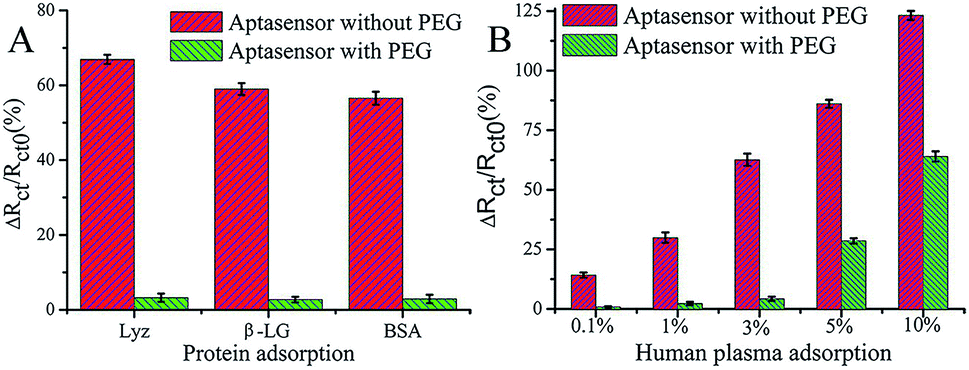 | ||
| Fig. 3 Charge-transfer resistance changes of the aptasensor with and without PEG after incubation in different proteins (A) and human plasma (B). The aptasensor with and without PEG corresponds to 6-mercaptohexanol (MCH)/aptamer–PEG/PDA/glassy carbon electrode (GCE) and MCH/aptamer/PDA/GCE, respectively. Adapted with permission from ref. 4. Copyright (2017) American Chemical Society. | ||
Another example of using PEG to resist protein attachment was reported by Hui et al.16 They reported a mixed self-assembled sensing interface to detect alpha-fetoprotein (AFP) via electrochemically deposited polyaniline (PANI) nanowires, which were functionalized with PEG polymer chains and gold nanoparticles (AuNPs) (Fig. 4). The signal response range of the resulting biosensor was 0.01 pg mL−1 to 1.0 ng mL−1 with an LOD of 0.007 pg mL−1. They assessed the signal response of the antifouling biosensor in pepsinogen (PG), human serum albumin (HSA), trypsinogen (Try), γ-globulin from bovine blood (GLO) and single-stranded DNA to detect the selectivity of the biosensor toward its target AFP. The result showed that there was a very small and negligible signal response to PG, HSA, Try, GLO and DNA compared with target AFP, which demonstrated great selectivity of this biosensor. They also showed that after incubation in human serum, the analytical signal was 77.1% of the original signal, indicating the antifouling properties of PEG-based polymers. The same year, another novel ultrasensitive and low fouling electrochemical biosensor was reported to detect AFP through electrochemical polymerization of poly(3,4-ethylenedioxythiophene) (PEDOT) and a PEG derivative, 4-arm PEG terminated with thiol groups.17 The LOD was 0.0003 fg mL−1 (S/N = 3), and it was capable of assaying target AFP in 10% v/v human serum samples.
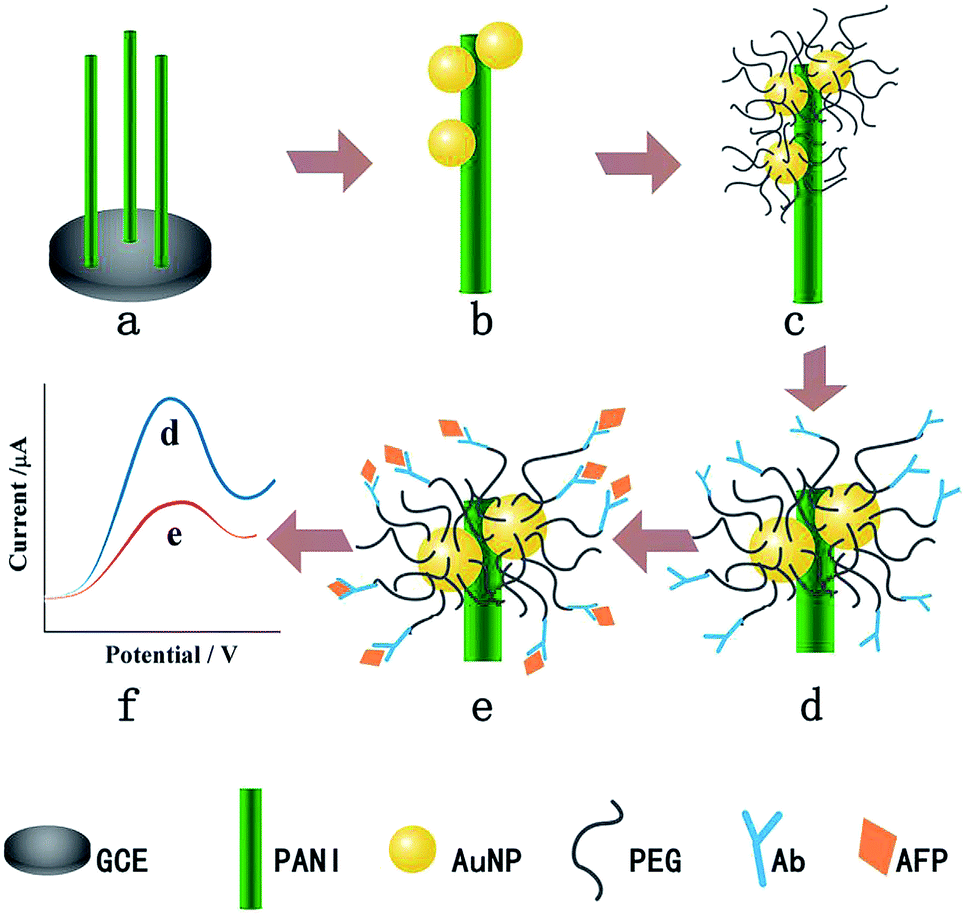 | ||
| Fig. 4 The fabrication process of the AFP immunosensor. (a) PANI nanowires deposited on the GCE, (b) AuNP electrodeposition, (c) PEG modification, (d) AFP antibody immobilization, (e) AFP target capturing and (f) DPV current signal recording. Adapted with permission from ref. 16. Copyright (2016) Elsevier. | ||
Hui et al. synthesized novel highly biocompatible composite nanofibers to detect breast cancer susceptibility gene (BRCA1) through the grafting of PEG polymer chains onto PANI nanofibers.23 The differential pulse voltammetry (DPV) peak current was used to measure the BRCA1 in human serum samples, which showed a linear range across 0.01 pM to 1 nM with an LOD of approx. 0.0038 pM. The same group also reported a label-free and low fouling electrochemical impedance-based biosensor based on a cross-linked amine-terminated PEG layer which was used as the antifouling layer and as the base layer for subsequent immobilization of gold nanoparticles and BRCA1 complementary single-stranded 19-mer oligonucleotides. This complementary strand was used to capture and hybridise with a specific target BRCA1 sequence.30 The linear range was from 50.0 fM to 1.0 nM, and the LOD was calculated to be 1.72 fM. The antifouling capability of the biosensor was assessed in different solutions (immobilized probe S1, target probe S2, one-base mismatch S3, three-base mismatch S4 and non-complementary S5 DNA sequences, and bovine serum albumin BSA). High selectivity towards the target probe S2 was demonstrated and attributed to the PEG layer. Such examples demonstrate that PEG-based materials exhibit good antifouling properties, and have potential for application in electrochemical biosensors as it can be integrated into bio-interfaces in a range of ways without compromising on the sensitivity of the sensor. However, due to the oxidative effects that PEG is susceptible to, it is not suitable for long-term deployment, i.e. as antifouling coatings in implantable sensors for long term deployment.
Zwitterionic polymers
Zwitterionic polymers have found increasing application as next generation biomaterials because zwitterionic materials have been demonstrated to effectively resist nonspecific adsorption of protein in complex media. Zwitterionic surfaces can form a hydration layer with both hydrogen bonds and ionic solvation, thus indicating that zwitterionic polymers could have greater antifouling properties than PEG.69 Zwitterionic antifouling materials can be divided into three types, phosphorylcholines (pPC),70–72 poly(sulfobetaine) (pSB)73,74 and poly(carboxybetaine) (pCB).75 In addition to antifouling properties, zwitterionic polymers have several other advantages, including good stability and low cost.Wu et al. fabricated a zwitterionic poly(sulfobetaine-3,4-ethylenedioxythiophene) (PSBEDOT)–glucose oxidase (GOx) sensor using a single-step electro-polymerization method (Fig. 5).15 The sensor based on this PSBEDOT–GOx surface displayed high linearity (R2 = 0.9874) over a low glucose concentration range from 0.1 to 0.5 mM. They compared the antifouling properties of PSBEDOT–GOx with PEDOT–GOx immobilized films and showed that the zwitterion-based film displayed significantly greater protein resistance to both fibrinogen and 100% blood plasma. Another enzyme-based biosensor based on antifouling zwitterionic polymer films was developed using sulfobetaine methacrylate monomers which were polymerized on a coarse enzyme-adsorbed electrode surfaces by electrochemically mediated atom transfer radical polymerization (eATRP).20 In this study, Hu et al. used enzyme-linked immunosorbent assay (ELISA) assays to show its antifouling performance where non-pSBMA-coated samples adsorbed much more protein than coated samples calculated by the relative optical density values versus bare Pt surface.
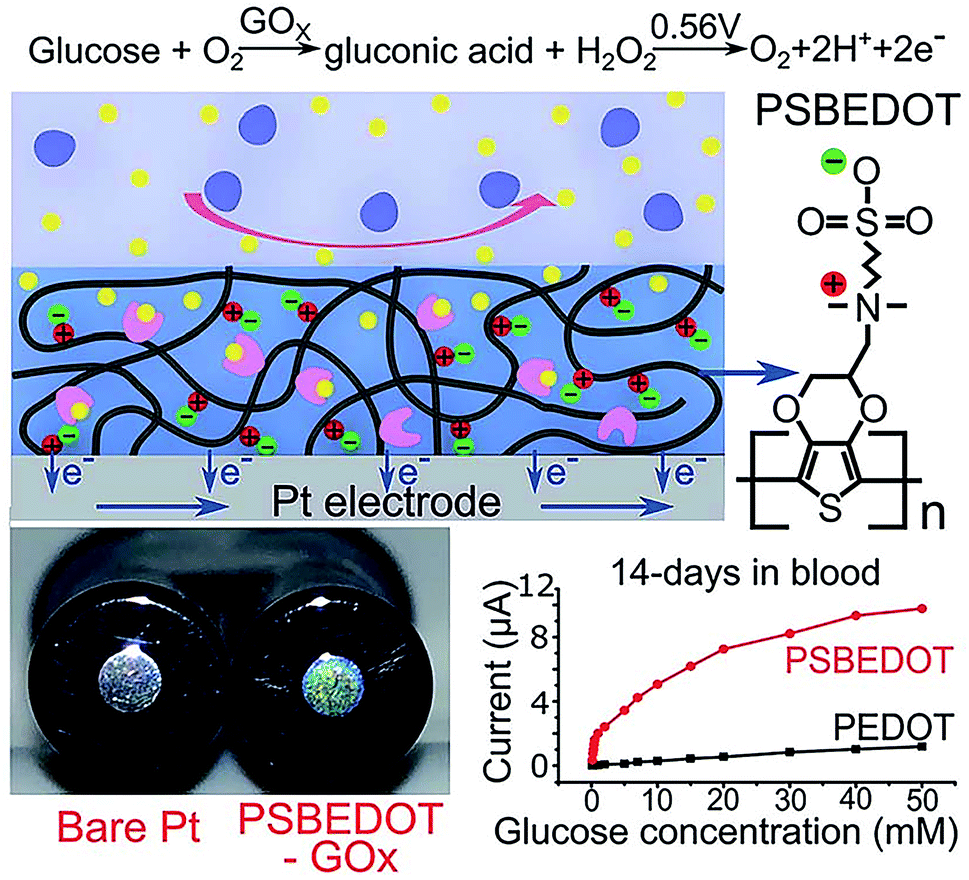 | ||
| Fig. 5 Zwitterionic poly(sulfobetaine-3,4-ethyl-enedioxythiophene) (PSBEDOT) glucose biosensor. Adapted with permission from ref. 15. Copyright (2018) The Royal Society of Chemistry. | ||
A 3,4-ethylenedioxythiophene (EDOT) derivative bearing a zwitterionic phosphorylcholine group was investigated for the detection of the acute-phase biomarker human C-reactive protein (CRP) via interaction of CRP with the immobilized phosphorylcholine group.31 This phosphorylcholine group was also expected to resist nonspecific adsorption of protein. This biosensor achieved a detection range of 10–160 nM with an LOD of 37 nM. Compared with the above sensors, it possesses high sensitivity and selectivity and importantly, it covers the dynamically changing CRP levels in circulation during the acute phase. While it remains to be seen how it performs in clinical samples, it represents an interesting approach whereby the same functional group tethered to the conducting polymer chain serves both as the recognition unit and as the basis for its proposed antifouling properties. Future work on such materials will include improving the ionic and electronic conductivities of zwitterion-based polymers so as to maximize their signal transduction properties as well as their antifouling properties.
Peptides
Peptides are another class of promising candidates as antifouling materials. Peptides have an overall neutral charge and have great hydrophilicity because of their amino acid composition, which exists as zwitterionic residues. The deprotonated carboxyl groups (–COO−) and protonated amine groups (–NH3+) easily form common hydrophilic interactions which can help to reduce protein adsorption. In addition, due to charge neutrality, the adsorption of charged proteins is not favourable on peptide functionalized surfaces. These peptides have been used in biosensor design to achieve antifouling or low fouling surfaces. For example, a highly sensitive and selective biosensor based on a zwitterionic peptide (EKEKEKE) was recently reported for BRCA1 determination. The working electrode interface comprised a citrate-doped PEDOT layer functionalized with a zwitterionic peptide via nickel cation coordination. This film was further functionalized with a DNA probe to construct an electrochemical biosensor for BRCA1 (Fig. 6).37 A good linear range was obtained from 1.0 × 10−16 to 1.0 × 10−10 M, and the LOD was as low as 0.03 fM (S/N = 3), which was an order of magnitude lower than previous work (approx. 1.72 fM).76 In order to test the antifouling properties of the aptasensor, the DPV response changes of the biosensor in HSA solution and human plasma were recorded. The electrode modified with the peptide showed significantly smaller DPV response changes compared with the electrode without the peptide, indicating the promising antifouling ability of these zwitterionic peptides.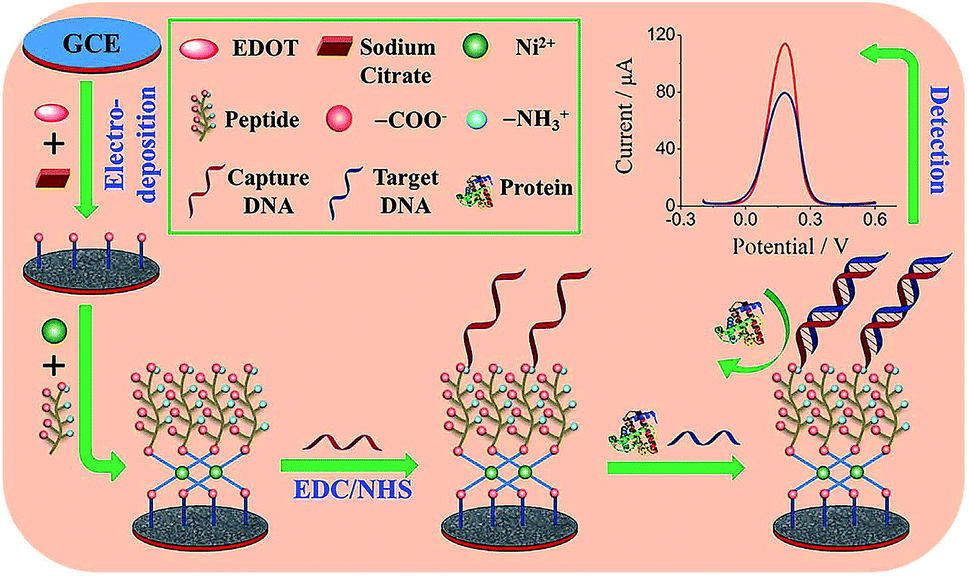 | ||
| Fig. 6 Schematic illustration of the fabrication process of the DNA sensor. Adapted with permission from ref. 37. Copyright (2017) Elsevier. | ||
Wang et al. suggested another antifouling strategy based on peptides where they built a sensing interface based on AuNPs which included an excellent antifouling peptide material (EKEKEKE-PPPPC) along with an ATP aptamer in order to detect ATP.38 In order to evaluate the antifouling properties of the biosensor, they recorded the EIS response changes of the aptasensor after soaking in protein solution. When the electrode modified with the peptide was compared to one without the peptide, the signal changes were significantly smaller for the electrode modified with the peptide (Fig. 7A). As shown in Fig. 7B, the antifouling abilities of the biosensor are demonstrated further in human plasma and whole blood, also with very positive results. Another electrochemical DNA biosensor for BRCA1 detection based on the same antifouling peptide (EKEKEKE-PPPPC) has been since reported, but this time in the form of a self-assembled monolayer (SAM). 19-mer BRCA1 related sequence-specific oligonucleotides were immobilised on top of this peptide-based SAM (Fig. 8).2 This sensitive and selective impedance-based assay was characterized as having a linear range of 1.0 fM to 10.0 pM and an LOD of 0.3 fM. No significant analytical response changes were observed after immersion into either single protein solutions or diluted serum.
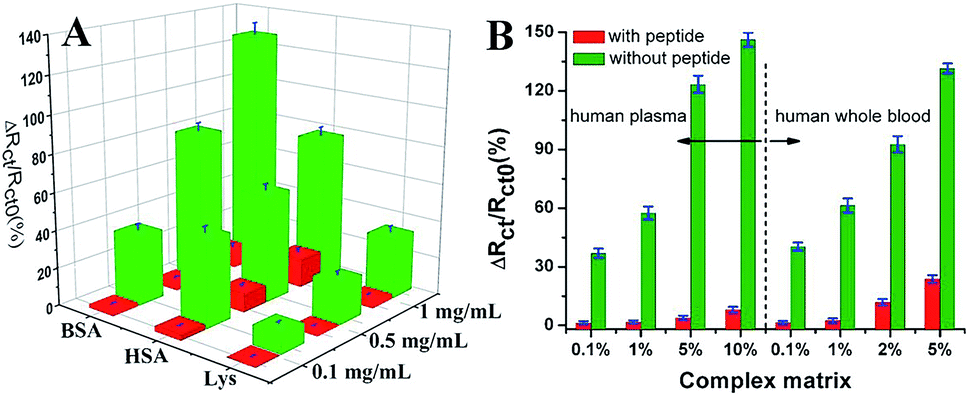 | ||
| Fig. 7 (A) Charge-transfer resistance changes of the aptasensor with (red cuboids) and without (green cuboids) peptide after incubation in different protein solutions. (B) Charge-transfer resistance changes of the aptasensor with (red column) and without (green column) peptide after incubation in a complex matrix. Adapted with permission from ref. 38. Copyright (2018) Elsevier. | ||
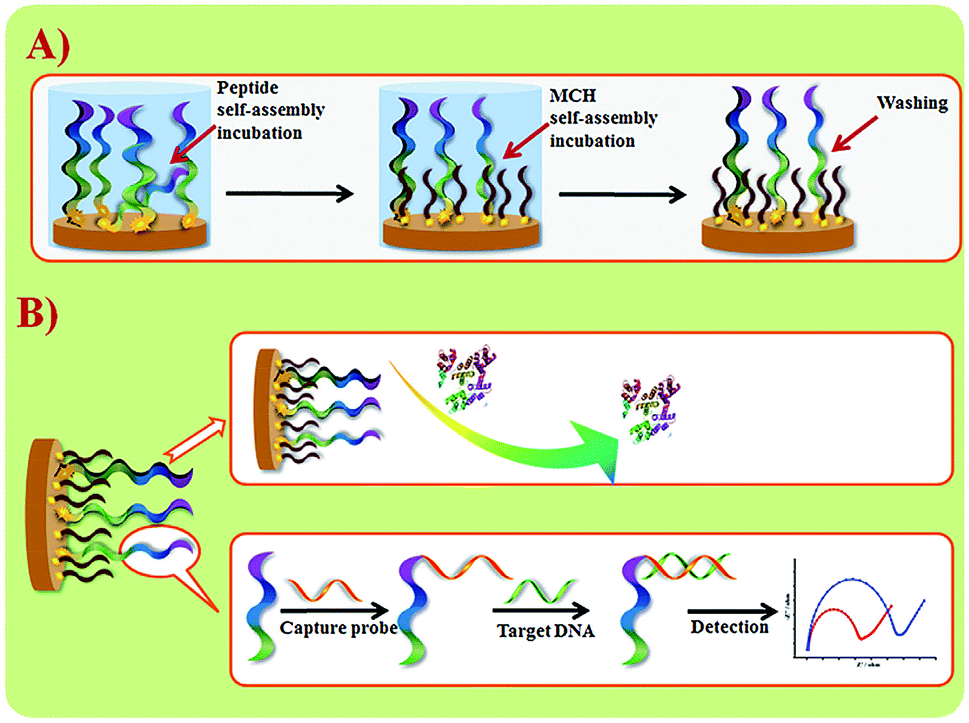 | ||
| Fig. 8 (A) Schematic illustration of peptide SAM formation on a Au electrode surface. (B) DNA biosensor fabrication and sensing procedures. Adapted with permission from ref. 2. Copyright (2017) Elsevier. | ||
Another antifouling interface based on mixed self-assembled aptamers and a newly designed multifunctional zwitterionic peptide (EESKSESKSGGGGC) was designed for AFP detection.40 The peptide showed excellent antifouling performance in 5 mg mL−1 HSA and 1–2% serum. Such a low fouling capability is ascribed to the prominent hydrophilicity and remarkable charge neutrality of the newly designed zwitterionic peptide. Under the optimized sensing conditions, the electrochemical aptasensor shows a good linear range of 10.0 fg mL−1 to 100.0 pg mL−1 and an LOD of 3.1 fg mL−1. Another interesting approach used a multi-functional peptide where the anchoring, recognition and antifouling functions were integrated into a single peptide. This peptide was modified onto PANI nanowire arrays.42 This offered an elegant design based on a single sequence that demonstrated excellent function in neat serum and real clinical samples (Fig. 9). This biosensor showed good analytical performance with a linear range of 1 ng mL−1 to 10 μg mL−1 with an LOD of 0.26 ng mL−1.
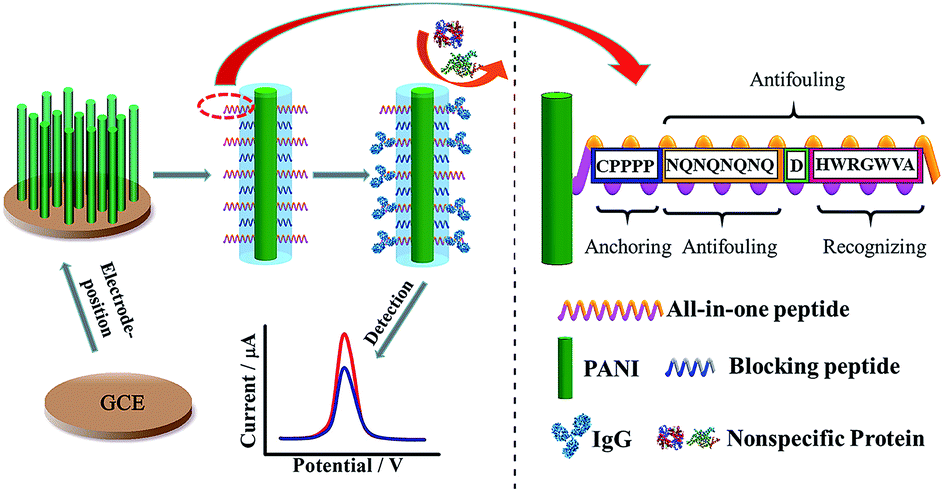 | ||
| Fig. 9 Schematic illustration of the preparation of a PANI supported peptide-based amperometric biosensor. Adapted with permission from ref. 42. Copyright (2018) American Chemical Society. | ||
Peptides have several advantages over other biofouling materials in that they can be engineered to be multifunctional and are biocompatible. However, their synthesis is tedious and their cost is high, prompting research into other materials that can exhibit equivalent antifouling properties.
Other antifouling materials
There are other antifouling materials such as polysaccharide based protein-resistant surfaces, hyperbranched polymers, polyoxazolines, and poly(hydroxy functional acrylates). Many of them have yet to be demonstrated as tools in the development of low fouling electrochemical biosensors.Hyaluronic acid (HA) is an anionic, nonsulfated glycoaminoglycan that has recently shown promise as an antifoulant in electrochemical sensing materials. HA materials include disaccharide units, which have amide and carboxyl groups providing the donors and acceptors for hydrogen bonds, resulting in the hydration strength of these acids against biofouling. Wang et al. have used HA in an electrochemical microRNA biosensor which was prepared via chemical grafting of a Methylene Blue labeled reporter (MB-Rep) duplex onto a nanostructured surface of electrodeposited cobalt oxide and poly(o-phenylenediamine), followed by the attachment of HA and gold nanoclusters.35 Square-wave voltammetric (SWV) responses of the antifouling HA modified interface in the presence of the target resulted in a linear range of 10−13 to 10−6 M with an LOD of 33.3 fM. They used impedance spectroscopy to investigate the response of the HA-based biosensor to the target after soaking in 0%, 1%, 5%, 10%, 20%, 50% and 100% human serum (v/v) for 30 min. An electrode modified with HA had no significant changes in the Rct for all the studied serum concentrations, indicating excellent antifouling ability of the modified electrode. Another interesting electrochemical biosensor designed for the detection of brucellosis antibody was reported, which was based on a functionalized magnetic nanoparticle (Fe3O4@Au@PEG@HA NP).36 The current response of the immunosensor was assessed in BP26, VirB5, BSA and brucellosis antibody. The immunosensor showed a high signal response to brucellosis antibody and almost no response to nonspecific proteins, which demonstrated the excellent selectivity of the sensor. This biosensor showed impressive antibody responses in 100% serum from 10−15 to 10−11 g mL−1, with an LOD of 0.36 fg mL−1.
Conclusions and perspectives
Significant progress has been made in the determination of a range of protein disease markers in complex clinical matrices conveniently, quickly, sensitively and accurately on account of innovations around overcoming biofouling. Several antifouling materials have been developed, including PEG and a range of PEG substitutes to combat protein-based biofouling. Although tremendous effort has been invested in the development of antifouling electrochemical biosensors, the field is now only beginning to translate these innovations into commercial opportunities. Further research is required to achieve robust approaches to fabricate surfaces that can resist protein adsorption from complex biological media. We have reviewed some of the strategies reported for designing effective antifouling electrochemical biosensors. Strategies being deployed are positively impacting biofouling issues, but some of them are linked with other shortcomings related to stability, toxicity, challenging synthesis and manufacturing methods, and reduced sensor sensitivity. There is much research opportunity in this field based for example on investigating new monomers of peptide or zwitterionic polymers based on novel structures. All in all, there is a promising future for high quality antifouling materials, and the development of new materials will be an active research area as it relates to electrochemical biosensors for both analysis of clinical media and in vivo deployment. In addition to the development of new antifouling polymer chains, there is also scope for new surface grafting/anchoring strategies. Continued application of existing as well as new anchoring strategies combined with new or existing antifouling chemical and biochemical architectures will continue.Once the development of reasonably mature disease biomarker electrochemical detection strategies is combined with suitable and effective antifouling advances, the commercial potential of biosensors for disease biomarkers could be realized within a reasonably short timeframe. Antifouling materials are some of the key enablers for the translation of biosensors from the research laboratory into true clinical applications.
Conflicts of interest
There are no conflicts to declare.Abbreviation
| PEG | Polyethylene glycol |
| ATP | Adenosine triphosphate |
| EIS | Electrochemical impedance spectroscopy |
| LOD | Limit of detection |
| AuNPs | Gold nanoparticles |
| PANI | Polyaniline |
| AFP | Alpha-fetoprotein |
| DPV | Differential pulse voltammetry |
| PEDOT | Poly(3,4-ethylenedioxythiophene) |
| BRCA1 | Breast cancer susceptibility gene |
| GCE | Glassy carbon electrode |
| MCH | 6-Mercaptohexanol |
| GE | Gold electrode |
| IgG | Immunoglobulin G |
| PSBEDOT | Poly(sulfobetaine-3,4-ethylenedioxythiophene) |
| GOx | Glucose oxidase |
| i–t | i–t curve |
| pSBMA | Poly(sulfobetaine methacrylate) |
| EDOT | 3,4-Ethylenedioxythiophene |
| EDOTPC | EDOT derivative bearing phosphorylcholine |
| MB | Methylene blue |
| HA | Hyaluronic acid |
| Popd | Poly(o-phenylenediamine) |
| CoO | Cobalt oxide (CoO) |
| SWV | Square-wave voltammetry |
Acknowledgements
The authors acknowledge the support of the National Natural Science Foundation of China (21422504, 21675093), the Taishan Scholar Program of Shandong Province of China (ts20110829) and the financial support from Science Foundation Ireland (SFI) 13/CDA/2155.Notes and references
- X. Luo and J. J. Davis, Chem. Soc. Rev., 2013, 42, 5944–5962 RSC.
- M. Cui, Y. Wang, H. P. Wang, Y. M. Wu and X. L. Luo, Sens. Actuators, B, 2017, 244, 742–749 CrossRef CAS.
- Y. Wang, M. Cui, M. Jiao and X. Luo, Anal. Bioanal. Chem., 2018, 410, 5871–5878 CrossRef CAS PubMed.
- G. X. Wang, Q. J. Xu, L. Liu, X. L. Su, J. H. Lin, G. Y. Xu and X. L. Luo, ACS Appl. Mater. Interfaces, 2017, 9, 31153–31160 CrossRef CAS PubMed.
- N. Liu, Z. Liu, H. Han and Z. Ma, J. Mater. Chem. B, 2014, 2, 3292–3298 RSC.
- H. Lisalova, E. Brynda, M. Houska, I. Visova, K. Mrkvova, X. C. Song, E. Gedeonova, F. Surman, T. Riedel, O. Pop-Georgievski and J. Homola, Anal. Chem., 2017, 89, 3524–3531 CrossRef CAS PubMed.
- C. Guo, F. Su, Y. Song, B. Hu, M. Wang, L. He, D. Peng and Z. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 41188–41199 CrossRef CAS PubMed.
- H. J. Ye, Y. Q. Xia, Z. Q. Liu, R. L. Huang, R. X. Su, W. Qi, L. B. Wang and Z. M. He, J. Mater. Chem. B, 2016, 4, 4084–4091 RSC.
- F. Sun, H. C. Hung, A. Sinclair, P. Zhang, T. Bai, D. D. Galvan, P. Jain, B. W. Li, S. Y. Jiang and Q. M. Yu, Nat. Commun., 2016, 7, 13437 CrossRef CAS PubMed.
- K. Sivashanmugan, P. C. Liu, K. W. Tsai, Y. N. Chou, C. H. Lin, Y. Chang and T. C. Wen, Nanoscale, 2017, 9, 2865–2874 RSC.
- F. Sun, J. R. Ella-Menye, D. D. Galvan, T. Bai, H. C. Hung, Y. N. Chou, P. Zhang, S. Y. Jiang and Q. M. Yu, ACS Nano, 2015, 9, 2668–2676 CrossRef CAS PubMed.
- S. Xu, L. Jiang, Y. Nie, J. Wang, H. Li, Y. Liu, W. Wang, G. Xu and X. Luo, ACS Appl. Mater. Interfaces, 2018, 10, 26851–26858 CrossRef CAS PubMed.
- S. Xu, Y. Nie, L. Jiang, J. Wang, G. Xu, W. Wang and X. Luo, Anal. Chem., 2018, 90, 4039–4045 CrossRef CAS PubMed.
- S. Xu, Z. Su, Z. Zhang, Y. Nie, J. Wang, G. Ge and X. Luo, J. Mater. Chem. B, 2017, 5, 8748–8753 RSC.
- H. Y. Wu, C. J. Lee, H. F. Wang, Y. Hu, M. Young, Y. Han, F. J. Xu, H. B. Cong and G. Cheng, Chem. Sci., 2018, 9, 2540–2546 RSC.
- N. Hui, X. T. Sun, Z. L. Song, S. Y. Niu and X. L. Luo, Biosens. Bioelectron., 2016, 86, 143–149 CrossRef CAS PubMed.
- M. Cui, Z. Song, Y. Wu, B. Guo, X. Fan and X. Luo, Biosens. Bioelectron., 2016, 79, 736–741 CrossRef CAS PubMed.
- M. Martín, P. Salazar, R. Álvarez, A. Palmero, C. López-Santos, J. L. González-Mora and A. R. González-Elipe, Sens. Actuators, B, 2017, 240, 37–45 CrossRef.
- M. Labib, E. H. Sargent and S. O. Kelley, Chem. Rev., 2016, 116, 9001 CrossRef CAS PubMed.
- Y. Hu, B. Liang, L. Fang, G. Ma, G. Yang, Q. Zhu, S. Chen and X. Ye, Langmuir, 2016, 32, 11763–11770 CrossRef CAS PubMed.
- E. van Andel, I. de Bus, E. J. Tijhaar, M. M. J. Smulders, H. F. J. Savelkoul and H. Zuilhof, ACS Appl. Mater. Interfaces, 2017, 9, 38211–38221 CrossRef CAS PubMed.
- T. R. Walsh and M. R. Knecht, Chem. Rev., 2017, 117, 12641–12704 CrossRef CAS PubMed.
- N. Hui, X. Sun, S. Niu and X. Luo, ACS Appl. Mater. Interfaces, 2017, 9, 2914–2923 CrossRef CAS PubMed.
- Z. L. Zhi, Y. J. Su, Y. W. Xi, L. Tian, M. Xu, Q. Q. Wang, S. Padidan, P. Li and W. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 10383–10397 CrossRef CAS PubMed.
- S. R. Meyers and M. W. Grinstaff, Chem. Rev., 2012, 112, 1615–1632 CrossRef CAS PubMed.
- D. Z. Yao, W. Q. Zhao, L. M. Zhang and Y. Tian, Analyst, 2017, 142, 4215–4220 RSC.
- T. J. Plegue, K. M. Kovach, A. J. Thompson and J. A. Potkay, Langmuir, 2018, 34, 492–502 CrossRef CAS PubMed.
- P. Jolly, A. Miodek, D. K. Yang, L. C. Chen, M. D. Lloyd and P. Estrela, ACS Sens., 2016, 1, 1308–1314 CrossRef CAS.
- J. G. Wu, S. C. Wei, Y. Chen, J. H. Chen and S. C. Luo, Langmuir, 2018, 34, 943–951 CrossRef CAS PubMed.
- W. T. Wang, X. J. Fan, S. H. Xu, J. J. Davis and X. L. Luo, Biosens. Bioelectron., 2015, 71, 51–56 CrossRef CAS PubMed.
- T. Goda, M. Toya, A. Matsumoto and Y. Miyahara, ACS Appl. Mater. Interfaces, 2015, 7, 27440–27448 CrossRef CAS PubMed.
- H. Ye, L. Wang, R. Huang, R. Su, B. Liu, W. Qi and Z. He, ACS Appl. Mater. Interfaces, 2015, 7, 22448–22457 CrossRef CAS PubMed.
- T. Date, T. Sawada and T. Serizawa, Soft Matter, 2013, 9, 3469 RSC.
- X. Yu, J. Xiao and F. Dang, Langmuir, 2015, 31, 5891–5898 CrossRef CAS PubMed.
- W. Wang, S. Jayachandran, M. R. Li, S. H. Xu and X. L. Luo, Microchim. Acta, 2018, 185, 156 CrossRef PubMed.
- S. Lv, J. Sheng, S. Zhao, M. Liu and L. Chen, Biosens. Bioelectron., 2018, 117, 138–144 CrossRef CAS PubMed.
- G. X. Wang, R. Han, X. L. Su, Y. N. Li, G. Y. Xu and X. L. Luo, Biosens. Bioelectron., 2017, 92, 396–401 CrossRef CAS PubMed.
- G. Wang, X. Su, Q. Xu, G. Xu, J. Lin and X. Luo, Biosens. Bioelectron., 2018, 101, 129–134 CrossRef CAS PubMed.
- C. J. Wilson, R. E. Clegg, D. I. Leavesley and M. J. Pearcy, Tissue Eng., 2005, 11, 1–18 CrossRef CAS PubMed.
- M. Cui, Y. Wang, M. Jiao, S. Jayachandran, Y. Wu, X. Fan and X. Luo, ACS Sens., 2017, 2, 490–494 CrossRef CAS PubMed.
- J. Jass, S. Surman and J. T. Walker, Medical Biofilms: Detection, Prev. Control, 2003, pp. 1–28 Search PubMed.
- N. Z. Liu, N. Hui, J. J. Davis and X. L. Luo, ACS Sens., 2018, 3, 1210–1216 CrossRef CAS PubMed.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- M. S. Selim, M. A. Shenashen, S. A. El-Safty, S. A. Higazy, M. M. Selim, H. Isago and A. Elmarakbi, Prog. Mater Sci., 2017, 87, 1–32 CrossRef CAS.
- T. Mérian and J. M. Goddard, J. Agric. Food Chem., 2012, 60, 2943–2957 CrossRef PubMed.
- D. Rana and T. Matsuura, Chem. Rev., 2010, 110, 2448–2471 CrossRef CAS PubMed.
- V. Kochkodan, D. J. Johnson and N. Hilal, Adv. Colloid Interface Sci., 2014, 206, 116–140 CrossRef CAS PubMed.
- S. Jeon and J. Andrade, J. Colloid Interface Sci., 1991, 142, 159–166 CrossRef CAS.
- J. A. Lichter, K. J. Van Vliet and M. F. Rubner, Macromolecules, 2009, 42, 8573–8586 CrossRef CAS.
- E. Ostuni, R. G. Chapman, R. E. Holmlin, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 5605–5620 CrossRef CAS.
- V. A. Liu, W. E. Jastromb and S. N. Bhatia, J. Biomed. Mater. Res., 2002, 60, 126–134 CrossRef CAS PubMed.
- M. Amiji and K. Park, J. Biomater. Sci., Polym. Ed., 1993, 4, 217–234 CrossRef CAS PubMed.
- I. Szleifer, Biophys. J., 1997, 72, 595–612 CrossRef CAS PubMed.
- A. Hucknall, S. Rangarajan and A. Chilkoti, Adv. Mater., 2009, 21, 2441–2446 CrossRef CAS.
- Y. Arima, M. Toda and H. Iwata, Biomaterials, 2008, 29, 551–560 CrossRef CAS PubMed.
- R. Webster, E. Didier, P. Harris, N. Siegel, J. Stadler, L. Tilbury and D. Smith, Drug Metab. Dispos., 2007, 35, 9–16 CrossRef CAS PubMed.
- C. Gao, G. Li, H. Xue, W. Yang, F. Zhang and S. Jiang, Biomaterials, 2010, 31, 1486–1492 CrossRef CAS PubMed.
- Q. Shao and S. Jiang, Adv. Mater., 2015, 27, 15–26 CrossRef CAS PubMed.
- P. Singha, J. Locklin and H. Handa, Acta Biomater., 2017, 50, 20–40 CrossRef CAS PubMed.
- K. Ishihara, H. Nomura, T. Mihara, K. Kurita, Y. Iwasaki and N. Nakabayashi, J. Biomed. Mater. Res., 1998, 39, 323–330 CrossRef CAS PubMed.
- S. Lowe, N. M. O'Brien-Simpson and L. A. Connal, Polym. Chem., 2015, 6, 198–212 RSC.
- G. Gunkel, M. Weinhart, T. Becherer, R. Haag and W. T. Huck, Biomacromolecules, 2011, 12, 4169–4172 CrossRef CAS PubMed.
- B. Liu, X. Liu, S. Shi, R. Huang, R. Su, W. Qi and Z. He, Acta Biomater., 2016, 40, 100–118 CrossRef CAS PubMed.
- S. Roy, J. H. Soh and J. Y. Ying, Biosens. Bioelectron., 2016, 75, 238–246 CrossRef CAS PubMed.
- R. Chhasatia, M. J. Sweetman, F. J. Harding, M. Waibel, T. Kay, H. Thomas, T. Loudovaris and N. H. Voelcker, Biosens. Bioelectron., 2017, 91, 515–522 CrossRef CAS PubMed.
- L. Liu, W. Li and Q. Liu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2014, 6, 599–614 CAS.
- S. R. S. N. V. Efremova and D. E. Leckband, Langmuir, 2001, 17, 7628–7636 CrossRef.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed.
- W. Wang, Y. Lu, J. B. Xie, H. Zhu and Z. Q. Cao, Chem. Commun., 2016, 52, 4671–4674 RSC.
- T. Goda, M. Tabata, M. Sanjoh, M. Uchimura, Y. Iwasaki and Y. Miyahara, Chem. Commun., 2013, 49, 8683–8685 RSC.
- S. Jiang and Z. Cao, Adv. Mater., 2010, 22, 920–932 CrossRef CAS PubMed.
- C. Rodriguez Emmenegger, E. Brynda, T. Riedel, Z. Sedlakova, M. Houska and A. B. Alles, Langmuir, 2009, 25, 6328–6333 CrossRef CAS PubMed.
- J. Wang, Biosens. Bioelectron., 2006, 21, 1887–1892 CrossRef CAS PubMed.
- Y. Chang, Y.-J. Shih, C.-J. Lai, H.-H. Kung and S. Jiang, Adv. Funct. Mater., 2013, 23, 1100–1110 CrossRef CAS.
- J. E. Krause, N. D. Brault, Y. T. Li, H. Xue, Y. B. Zhou and S. Y. Jiang, Macromolecules, 2011, 44, 9213–9220 CrossRef CAS.
- W. Wang, X. Fan, S. Xu, J. J. Davis and X. Luo, Biosens. Bioelectron., 2015, 71, 51–56 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |