
Fe/Fe3C@C nanoparticles encapsulated in N-doped graphene–CNTs framework as an efficient bifunctional oxygen electrocatalyst for robust rechargeable Zn–air batteries†
Qichen
Wang
abc,
Yongpeng
Lei
 *ab,
Zhiyan
Chen
c,
Nan
Wu
d,
Yaobing
Wang
*e,
Bing
Wang
d and
Yingde
Wang
*d
*ab,
Zhiyan
Chen
c,
Nan
Wu
d,
Yaobing
Wang
*e,
Bing
Wang
d and
Yingde
Wang
*d
aSchool of Aeronautics and Astronautics, Science and Technology on High Strength Structural Materials Laboratory, Central South University, Changsha 410083, China. E-mail: lypkd@163.com
bCollege of Basic Education, National University of Defense Technology, Changsha 410073, China
cCollege of Materials Science and Engineering, Central South University of Forestry and Technology, Changsha 410004, China
dScience and Technology on Advanced Ceramic Fibers and Composites Laboratory, National University of Defense Technology, Changsha 410073, China. E-mail: wyd502@163.com
eKey Laboratory of Design and Assembly of Functional Nanostructures, Fujian Provincial Key Laboratory of Nanomaterials, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China. E-mail: wangyb@fjirsm.ac.cn
First published on 10th November 2017
Abstract
3d transition metals or their derivatives encapsulated in nitrogen-doped nanocarbon show promising potential in non-precious metal oxygen electrocatalysts. Herein, we describe the simple construction of a bifunctional oxygen electrocatalyst with a “framework-active sites” structure, namely Fe/Fe3C@C (Fe@C) nanoparticles encapsulated in 3D N-doped graphene and bamboo-like CNTs (Fe@C–NG/NCNTs). The Fe@C structure provides additional electrons on the carbon surface, promoting the oxygen reduction reaction (ORR) on adjacent Fe–Nx active sites. The 3D NG hybrid with a bamboo-like CNTs framework facilitates fast reactant diffusion and rapid electron transfer. The optimized sample displays excellent ORR and oxygen evolution reaction (OER) activity, with a potential difference of only 0.84 V; this places it among the best bifunctional ORR/OER electrocatalysts. Most importantly, Zn–air batteries using Fe@C–NG/NCNTs as the cathode catalyst deliver a peak power density of 101.2 mW cm−2 and a specific capacity of 682.6 mA h g−1 (energy density of 764.5 W h kg−1). After 297 continuous cycle tests (99 h), the rechargeable batteries using Fe@C–NG/NCNTs show a voltage gap increase of only 0.13 V, almost half that of Pt/C + Ir/C (0.22 V) under the same conditions. This work provides new insight into advanced electrocatalysts utilizing the structural features of host nanocarbon materials and guest active species toward energy conversion.
Introduction
Metal–air batteries and fuel cells are regarded as the most promising next-generation energy conversion devices due to their high energy densities and zero carbon emissions.1,2 Unfortunately, the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) suffer from sluggish dynamics; thus, high overpotential is required to drive these reactions.3 Currently, Pt-based catalysts are effective ORR catalysts, and Ru/Ir-based catalysts are generally regarded as the best OER electrocatalysts; however, the limited supply, high cost and inferior durability of these materials impede their large-scale commercialization. In order to alleviate the dependency on precious metals and further enhance electrocatalytic performance, it is of great importance to engineer advanced electrocatalysts with well-defined nanostructures and optimized compositions based on earth-abundant materials.4–6 For ORR, considering the low solubility of oxygen (10−4 mol L−1 at 25 °C) in 6 M KOH electrolyte,7 it is a great challenge to effectively transfer O2 to the active sites. For OER, the timely transfer of evolved O2 molecules along three-dimensional (3D) channels is also a key issue.Among various carbon materials,8,9 sp2-hybridized 1D carbon nanotubes (CNTs) and 2D graphene (G) possess great advantages of high electronic conductivity, excellent chemical stability, etc.2,8 Assembling CNTs and graphene into a 3D interconnected network is promising, considering that 3D structures can prevent aggregation and significantly enhance the stability of electrocatalysts. For example, Zhu et al. reported N-doped G/CNTs hybrids prepared by multi-step chemical vapor deposition (CVD).10 Li and co-workers synthesized CNTs–G complexes by partly exfoliating the outer wall of multi-walled CNTs,11 which requires extremely harsh oxidation conditions followed by annealing in NH3. These synthetic routes are either complicated, afford low yields or use poisonous gases. Hence, the rational design and development of 3D G/CNTs architectures combined with plentiful active sites obtained by simple processes is highly desirable.
Carbon materials functionalized with transition-metal-based inorganic nanoparticles (NPs) (such as metal,12 metal oxides,13 sulfides,14 nitrides,15 and carbides16) display synergistic effects and show significant improvements in electrocatalysts. Recently, our group prepared FeCo@NC core–shell nanospheres supported on graphene as an efficient bifunctional oxygen electrocatalyst.19 We found that the bimetallic FeCo@NC NPs contributed greatly to the ORR and OER. Meanwhile, carbide (e.g. Fe3C)-based composites have received attention owing to their facile preparation, good activity and long-term durability.17,18 The inner metal@C can dramatically promote the ORR activity of the neighboring Fe–Nx.20 Thus, it is rational to design and develop a highly efficient and robust bifunctional ORR/OER electrocatalyst from the point of view of improving the charge/electronics transport. In this respect, simple construction of Fe3C@C on a 3D G/CNTs framework with plentiful Fe–Nx active sites is attractive.
Considering this, we have constructed a bifunctional oxygen electrocatalyst with a “framework-active sites” structure, namely Fe/Fe3C@C NPs encapsulated in 3D N-doped graphene–CNTs (Fe@C–NG/NCNTs). The facile pyrolysis simultaneously realizes a 3D NG/NCNTs conductive network, Fe–Nx active sites, a Fe@C structure and high N doping (6.78 at%), resulting in excellent ORR/OER behavior in alkaline media. The potential difference between ORR and OER is only 0.84 V. Using Fe@C–NG/NCNTs as an air cathode for Zn–air batteries, a peak power density of 101.3 mW cm−2 and a high energy density of 764.5 W h kg−1 at the discharge current density of 10 mA cm−2 were obtained. The constructed batteries can steadily discharge over 40 h with negligible voltage change. Furthermore, after 99 h, the rechargeable batteries using Fe@C–NG/NCNTs show a voltage gap increase of only 0.13 V, almost half that of Pt/C + Ir/C (0.22 V) under the same conditions. Considering its excellent conductivity and structural superiority, the concept of a 3D structure consisting of graphene–CNTs is not only applicable in electrocatalysts but can also be applied in the fields of Li-S batteries, Na-ion batteries, supercapacitors, etc.
Results and discussion
As illustrated in Fig. 1, the 3D framework composed of NG and NCNTs was obtained by a simple pyrolysis strategy. During synthesis, graphitic carbon nitride (g-C3N4) homogeneously appeared on the reduced GO surface (ESI, Fig. S1†) at 550 °C for 4 h due to in situ polymerization of dicyandiamide.21 Then, Fe3+ was reduced to metallic iron by carbothermal reduction at 800 °C; the iron then reacted with free C atoms, leading to the generation of Fe/Fe3C NPs composed of α-Fe and Fe3C phase, as confirmed by X-ray diffraction (XRD) patterns (ESI, Fig. S2†).22 Thus, g-C3N4 plays two roles in providing a C source for the growth of well-defined bamboo-like CNTs catalyzed by the active iron species and an N source that is incorporated into both the CNTs and graphene in Fe@C–NG/NCNTs due to decomposition of g-C3N4 and released CN gases (e.g., C2N2+, C3N2+, C3N3+) above 560 °C, according to the TGA results (ESI, Fig. S3†). Interestingly, the Fe/Fe3C NPs could promote graphitization of the surrounding C atoms;23 thus, the Fe/Fe3C NPs were encapsulated by a graphitic carbon layer and the Fe/Fe3C@C structure was formed.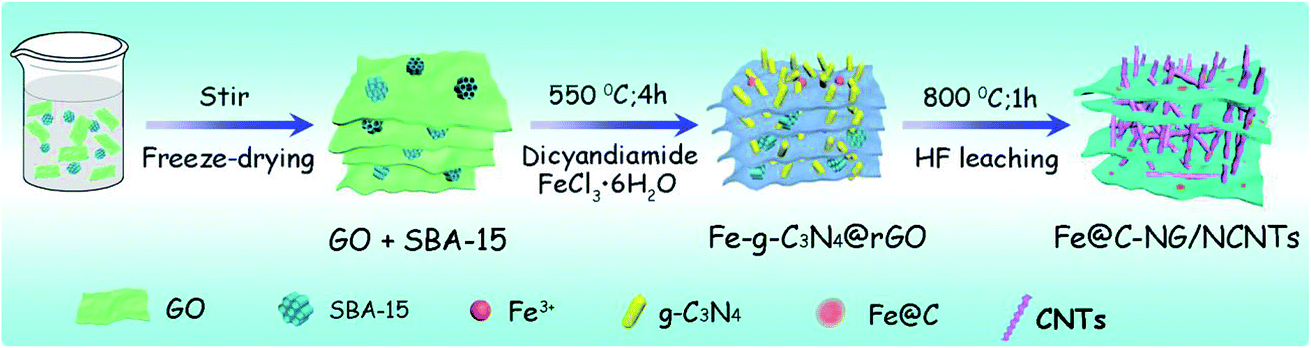 | ||
| Fig. 1 Schematic of the preparation of Fe@C–NG/NCNTs. | ||
Scanning electron microscopy (SEM) images of the resulting Fe@C–NG/NCNTs are shown in Fig. 2a and ESI, Fig. S4.† The well-defined bamboo-like CNTs grown in situ on the graphene surface with stable covalent linkages constructed a porous 3D interconnected carbon skeleton, facilitating fast reactant diffusion and rapid electron transfer. The transmission electron microscope (TEM) images of Fe@C–NG/NCNTs in Fig. 2b and c show that the Fe/Fe3C NPs appeared as black dots on top of or inside the CNTs and were distributed randomly on the graphene. Noteworthily, the bamboo-like CNTs can provide more graphene edges and thus introduce plentiful ORR active sites.24 The Fe/Fe3C NPs were further characterized by high-resolution TEM (HRTEM) measurements. The lattice fringes with spacings of 0.21 and 0.36 nm correspond to crystalline Fe3C and the graphitic carbon layer,25 respectively. Also, the inner Fe/Fe3C NPs are closely wrapped with several carbon layers with a thickness of about 5 nm, as shown in Fig. 2d and e. This geometric confinement of Fe@C (Fig. 2f) not only suppresses dissolution and agglomeration of the inner Fe/Fe3C NPs in harsh conditions, but also enriches the electron density on the carbon surface, thus promoting the surface reaction.26 To confirm the distribution of Fe species in detail, high-angle annular dark-field scanning TEM (HAADF-STEM) was performed (ESI, Fig. S5a–c†). As shown in the images, two types of Fe species exist in the carbon framework; one is atomically dispersed, and the other has a few crystalline phases associated with Fe/Fe3C. Notably, these crystalline Fe/Fe3C phases are surrounded by atomically dispersed Fe atoms. Electron energy loss spectroscopy (EELS) analysis (ESI, Fig. S5d†) taken from the red spot shows the co-existence of Fe and N species. Furthermore, elemental mapping analysis of Fe@C–NG/NCNTs (Fig. 2g) validates the strong Fe signals (with sizes of 10 to 100 nm) and shows that N, O and C elements are uniformly distributed in the 3D carbon matrix.
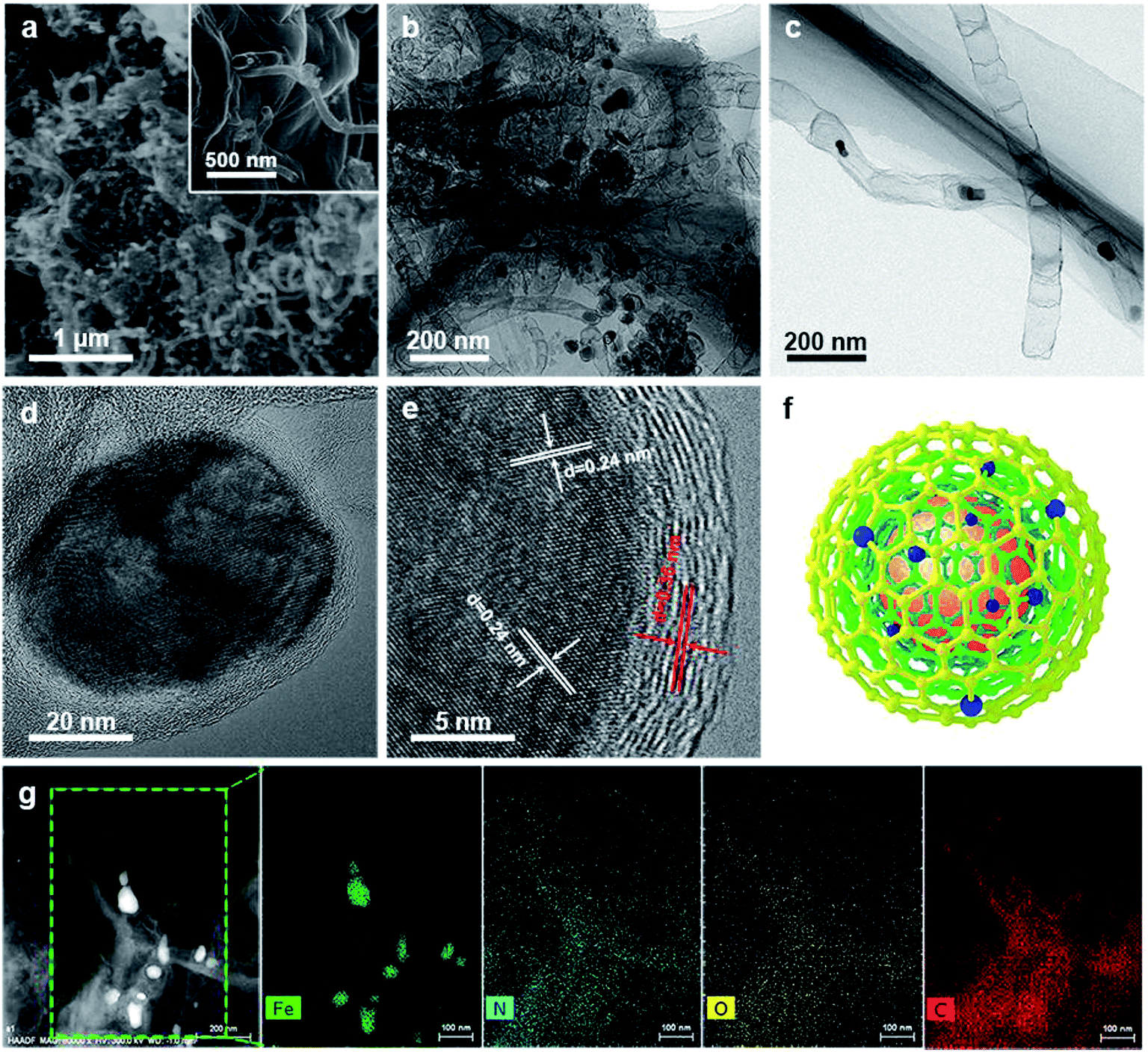 | ||
| Fig. 2 (a) SEM image, (b, c) TEM images of Fe@C–NG/NCNTs and (d, e) HRTEM images of an Fe@C nanoparticle. (f) Schematic of the Fe@C structure. (g) HAADF-STEM image and corresponding elemental mapping images of Fe, N, O and C atoms for Fe@C–NG/NCNTs. | ||
The Raman spectra of Fe@C–NG/NCNTs in Fig. S6 (ESI†) display characteristic D-bands at 1337 cm−1 and G-bands at 1572 cm−1, corresponding to disordered carbon and the E2g vibration of sp2-hybridized graphitic carbon, respectively.27 The ID/IG ratio of 0.7 indicates a higher graphitization degree than those of previously reported Fe/Fe3C catalysts,19,24,28 which can be attributed the co-existence of bamboo-like CNTs and graphene. The BET surface areas and porosities of the as-prepared samples were investigated by N2 adsorption/desorption isotherms (ESI, Fig. S7†). The BET surface area of Fe@C–NG/NCNTs is 117.6 m2 g−1 with meso–macroporous features; this favors mass transport and exposure of active sites. It was noted that SBA-15 played an essential role in preventing restacking of the GO sheets during the thermal treatment processes. As a comparison, the BET surface area of the sample prepared in the absence of SBA-15 was only 17.7 m2 g−1.
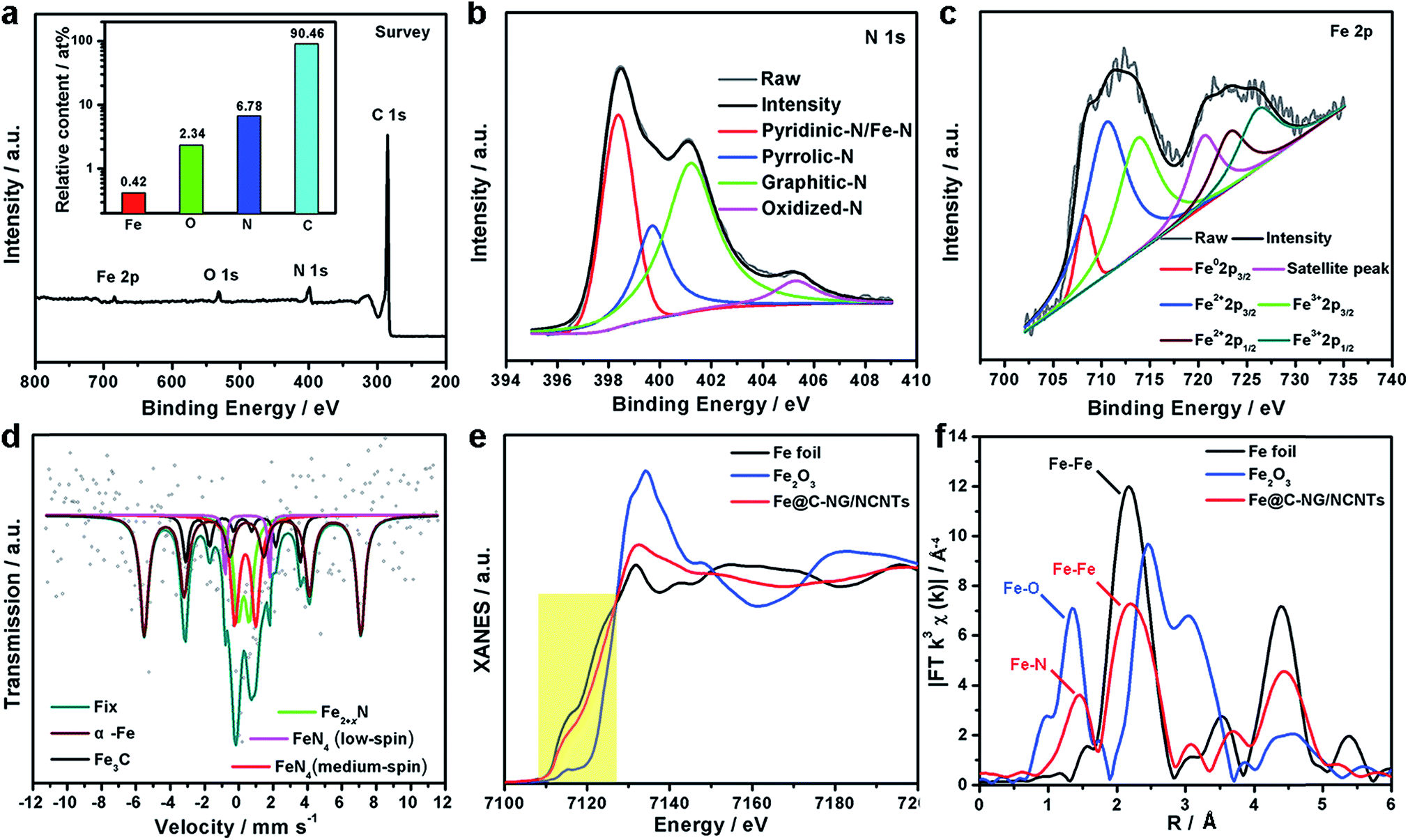 | ||
| Fig. 3 (a) XPS survey spectrum, (b) high-resolution N 1s spectrum, and (c) high-resolution Fe 2p spectrum of the Fe@C–NG/NCNTs sample. (d) Experimental 57Fe Mössbauer transmission spectra measured at 293 K for Fe@C–NG/NCNTs and their fittings with spectral components. (e) Fe K-edge of XANES spectra for Fe foil, Fe2O3 and Fe@C–NG/NCNTs (the yellow area highlights the near-edge absorption energy). (f) EXAFS spectra for the Fe K-edges of Fe foil, Fe2O3 and Fe@C–NG/NCNTs. | ||
XPS was performed to probe the composition and chemical states of the surface elements (Fig. 3a and ESI, Fig. S8†). Fe@C–NG/NCNTs shows the presence of C 1s, N 1s, O 1s and Fe 2p signals in the hybrids; their relative contents are 90.46, 6.78, 2.34 and 0.42 at%, respectively. Specifically, the Fe content may be relatively low because a few Fe/Fe3C NPs were encapsulated by graphitized carbon layers and could not be detected.29 The N 1s curve (Fig. 3b) was fitted as four peaks located at 398.4, 399.7, 401.2 and 405.3 eV, corresponding to pyridinic-N/Fe–N, pyrrolic-N, graphitic-N and oxidized-N, respectively.30 As shown in Fig. 3c, the Fe signal of Fe@C–NG/NCNTs shows two main peaks at 712.1 and 724.2 eV, attributed to the 2p3/2 and 2p1/2 levels, respectively; this implies the presence of Fe(II) and Fe(III).31 Additionally, an Fe0 peak at 706.8 eV was observed for α-Fe phase in the Fe@C structure. This is because pyridinic-N can coordinate with Fe to form an Fe–Nx configuration, which generally results in high ORR activity.32 The average turnover frequency (TOF) of Fe@C–NG/NCNTs on the basis of the Fe–Nx sites was estimated to be as high as 9.97e per site per s. Additionally, the presence of hydrophilic oxygen-containing-groups (e.g. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O–C and C–OH) undoubtedly enhances the three-phase contact between the electrolyte, reactants and electrode (ESI, Fig. S9†).
O, C–O–C and C–OH) undoubtedly enhances the three-phase contact between the electrolyte, reactants and electrode (ESI, Fig. S9†).
To better understand the nature of the Fe–Nx configuration, 57Fe Mössbauer spectroscopy (Fig. 3d) was performed.33 The two sextets shown in the figure in wine and black correspond to α-Fe and Fe3C, respectively, in accordance with the above XRD results. The two doublets in red and magenta are assigned to square-planar Fe(II)N4 coordinations with Fe(II) in low- and medium-spin states, and the green curve is assigned to Fe2+xN.34 These Fe–N species are believed to exist on the surface of Fe@C–NG/NCNTs, as evidenced by XPS. It is known that adsorption of O2 is the rate-limiting step in ORR,35 and theoretical calculations suggest that the adsorption energy of O2 ia −0.79 eV for the Fe–N4 sites. This value is lower than that of the Pt (111) surface (−0.69 eV), indicating the strong binding of O2 to the Fe–N4 active sites.36 Furthermore, we performed X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) studies to study the Fe K-edge of Fe@C–NG/NCNTs. As shown in Fig. 3e, the Fe K-edge XANES curve was different from those of standard Fe foil and Fe2O3, suggesting that the Fe species possess positive charges and that the central Fe atom is coordinated by N or O atoms. The Fe K-edge EXAFS spectra shown in Fig. 3e and S10 (ESI,†) exhibit peaks at 1.50 Å, assigned to Fe–N scattering paths, and the Fe–Fe peak at about 2.20 Å was also distinguished. The EXAFS fitting parameters were presented in Table S1 (ESI†). These results show that atomically dispersed iron atoms and metallic iron both exist in Fe@C–NG/NCNTs, consistent with the HAADF-STEM observations and EELS results.
The electrocatalytic ORR performance of Fe@C–NG/NCNTs was firstly examined by rotating disk electrode (RDE) measurements in 0.1 M KOH electrolyte (ESI, Fig. S11†). Linear sweep voltammogram (LSV) curves demonstrate that the highest ORR activity was obtained at 800 °C. Therefore, 800 °C was chosen as the optimal pyrolysis temperature to prepare comparison samples. Fe@C–NG/NCNTs displayed a prominent ORR cathodic peak at 0.803 V (vs. RHE) (Fig. 4a), which is slightly more positive than that of Pt/C (0.801 V). As shown in Fig. 4b, the onset potential (Eonset) and half-potential (E1/2) were 0.93 V and 0.84 V, respectively, which are comparable to those of Pt/C (0.97 V and 0.85 V, respectively). For comparison, the ORR activity of Fe@C–NG/NCNTs is also better than that of typical bamboo-like CNTs-based electrocatalysts.7,37–39Fig. 4c reveals that the diffusion-limiting current (JL) increased steadily as the rotation rates ranged from 400 to 1600 rpm. The nearly parallel Koutecky–Levich (K–L) plots (inset of Fig. 4c) demonstrate a good linear relationship between J−1 and ω−1/2 from 0.65 to 0.80 V, implying first-order reaction kinetics with respect to O2 concentration.40 The electron transfer numbers (n) were calculated to be 3.90 to 4.00, suggesting a near 4e transfer process with high efficiency. Remarkably, the mass-transfer-corrected Tafel slope of Fe@C–NG/NCNTs (49 mV dec−1) is lower than that of Pt/C (68 mV dec−1) (Fig. 4d), which confirms a more favorable ORR kinetic process.41 The electrochemical impendence spectra (ESI, Fig. S12†) show that the charge transfer resistance (35.2 Ω) of Fe@C–NG/NCNTs is obviously lower than that of Pt/C (42.5 Ω), suggesting a faster charge transfer process. We infer that high conductivity should originate from the 3D structure constructed from graphene and bamboo-like CNTs with a high graphitization degree.
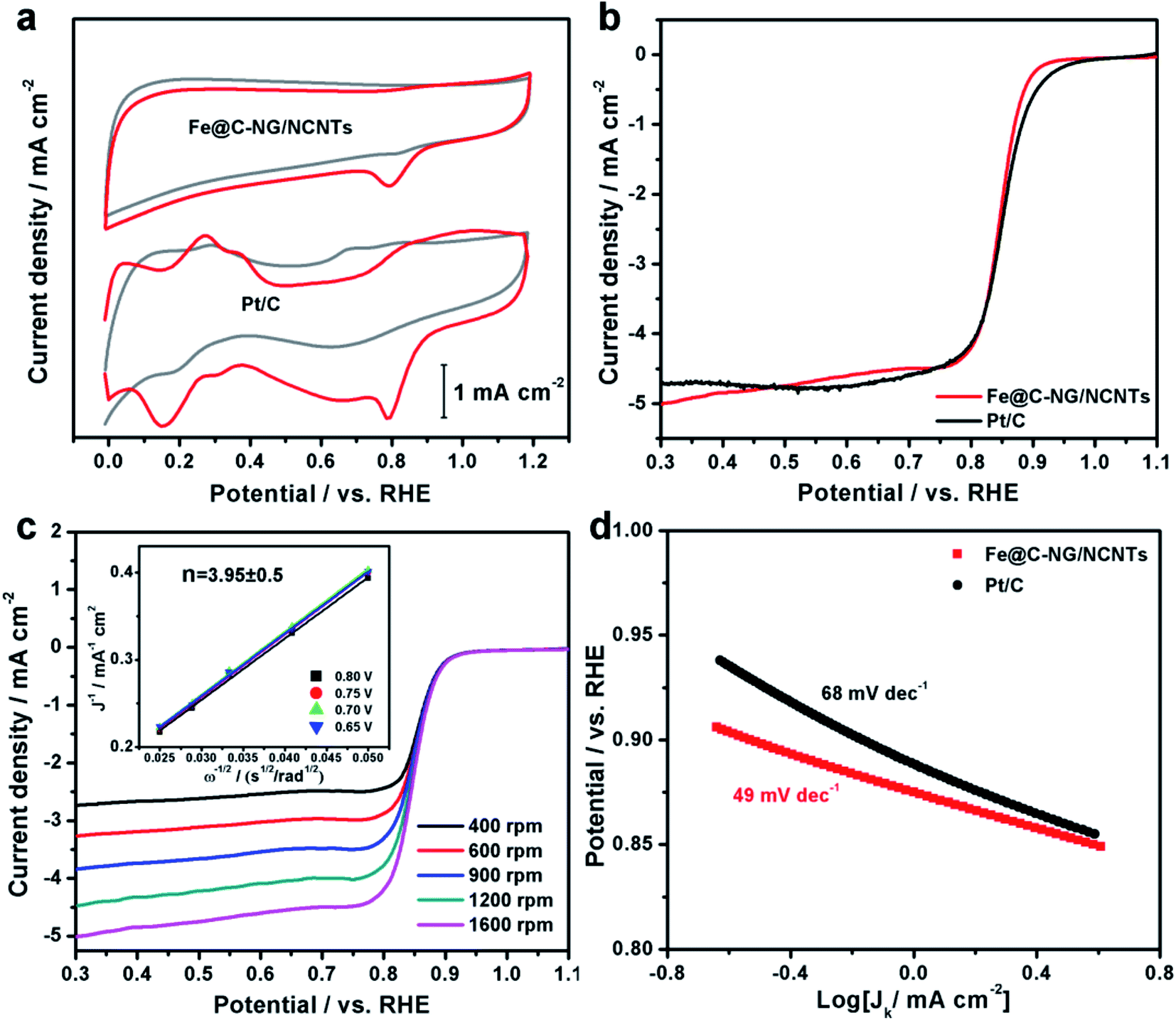 | ||
| Fig. 4 (a) CV curves, (b) LSV curves of Fe@C–NG/NCNTs and Pt/C in 0.1 M KOH. (c) LSV curves of Fe@C–NG/NCNTs at different rotation rates (the inset in Fig. 4c shows the fitted K–L plots) and (d) Tafel plots. | ||
An ideal electrocatalyst for ORR should also display good tolerance to fuel molecules and long-term stability. The current–time (I/t) chronoamperometric responses of the Fe@C–NG/NCNTs and Pt/C catalysts at 0.80 V were determined in O2-saturated 0.1 M KOH at 1600 rpm (ESI, Fig. S13†). After addition of 3 M methanol to the electrolyte, a stable current response for Fe@C–NG/NCNTs was observed, implying good tolerance to methanol, whereas the current of Pt/C displayed a significant shift in the opposite direction. Furthermore, Fe@C–NG/NCNTs exhibited current retention (78.4%) after 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s, which is higher than that of the Pt/C catalyst (59.1%) under the same conditions. This result proves the excellent stability of Fe@C–NG/NCNTs.
000 s, which is higher than that of the Pt/C catalyst (59.1%) under the same conditions. This result proves the excellent stability of Fe@C–NG/NCNTs.
To determine the origin of the superior ORR activity of Fe@C–NG/NCNTs, we performed four control experiments. In Fig. S14 (ESI,†), the metal-free control sample (denoted as MF), whose synthesis procedure was similar to that of Fe@C–NG/NCNTs but without addition of FeCl3, displayed poor ORR activity with regard to Eonset (0.86 V) and JL (2.69 mA cm−2). The Fe@C–NCNTs catalyst was prepared by the same procedure as that used for synthesis of Fe@C–NG/NCNTs, without addition of GO. The ORR activity of Fe@C–NCNTs was slightly inferior to that of Fe@C–NG/NCNTs. Furthermore, as is known, SCN− has a strong affinity to Fe ion in Fe–Nx species; this was used to identify the Fe-based active sites.9,42 After adding a certain amount of SCN− to KOH solution, the obvious negative shifts of E1/2 and JL indicate that the Fe–Nx species are the active sites of Fe@C–NG/NCNTs. On the other hand, we used ball-milling followed by hot acid leaching to destroy and remove the Fe@C structure.43 The obtained product was named Fe-BL. As shown in Fig. S15,† only a few Fe/Fe3C NPs were retained, and the corresponding XRD pattern (ESI, Fig. S2b†) showed a negligible Fe3C peak. In comparison with Fe@C–NG/NCNTs, JL and E1/2 of Fe-BL showed decrements of 1.38 mA cm−2 and 102 mV, respectively. The electrochemically active surface area was estimated by the double layer capacitance (Cdl), which was measured using CV.44 The Cdl of Fe-BL was confirmed to be 9.77 mF cm−2 (ESI, Fig. S16†), which is close to that of Fe@C–NG/NCNTs (10.55 mF cm−2). Hence, the large difference in ORR activity between Fe-BL and Fe@C–NG/NCNTs may be related to the destruction of the Fe@C structure and partial removal of the Fe/Fe3C NPs. Indeed, the co-existence of the Fe@C structure and Fe–Nx active sites in Fe@C–NG/NCNTs catalyst delivered higher ORR activity.
Furthermore, the electrocatalytic OER performance was also characterized by LSV curves measured in 1 M KOH solution. As shown in Fig. 5a, the overpotentials for Fe@C–NG/NCNTs, Pt/C and RuO2/C at the current density of 10 mA cm−2 were 450, 720 and 370 mV, respectively. Meanwhile, the smaller Tafel slope of 163 mV dec−1 for Fe@C–NG/NCNTs (Fig. 5b) indicates an efficient kinetic process. The stability was also tested by continuous cycling CVs from 1.50 to 1.80 V. After 1000 cycles, the LSV curves showed only a slight shift to a higher potential, suggesting good stability (Fig. 5c). ΔE (the potential gap between OER and ORR, ΔE = EOER: j=10 − EORR: 1/2) is generally an important indicator to reflect bifunctional activity towards ORR/OER.19 A lower ΔE value represents better bifunctional activity. The value for Fe@C–NG/NCNTs was estimated to be 0.84 V, as shown in Fig. 5d, which is superior to that of Pt/C catalyst (1.05 V) and also exceeds those of excellent reported bifunctional catalysts (ESI, Table S1†). These results demonstrate that Fe@C–NG/NCNTs is among the most effective bifunctional electrocatalysts for ORR and OER. The origin of the OER activity was also discussed (ESI, Fig. S17†). The MF sample shows poor OER activity. In contrast, there is clear improvement of the OER activity for the Fe-containing samples at 10 mA cm−2. Fe@C–NCNTs displays similar OER activity to Fe@C–NG/NCNTs. After removal of the Fe@C nanoparticles, Fe-BL exhibits a clear OER activity loss with an increase in overpotential of 67 mV at 10 mA cm−2 compared to Fe@C–NG/NCNTs. This implies that the Fe@C nanoparticles have OER activity. It is well known that SCN− ion has a suppressive effect on Fe–Nx sites. After a certain amount of SCN− was added, the OER polarization curve of Fe@C–NG/NCNTs shifted immediately. Therefore, we infer that Fe–Nx, the Fe@C nanoparticles and doped N species contribute to the OER activity, while Fe–Nx may play a major role.
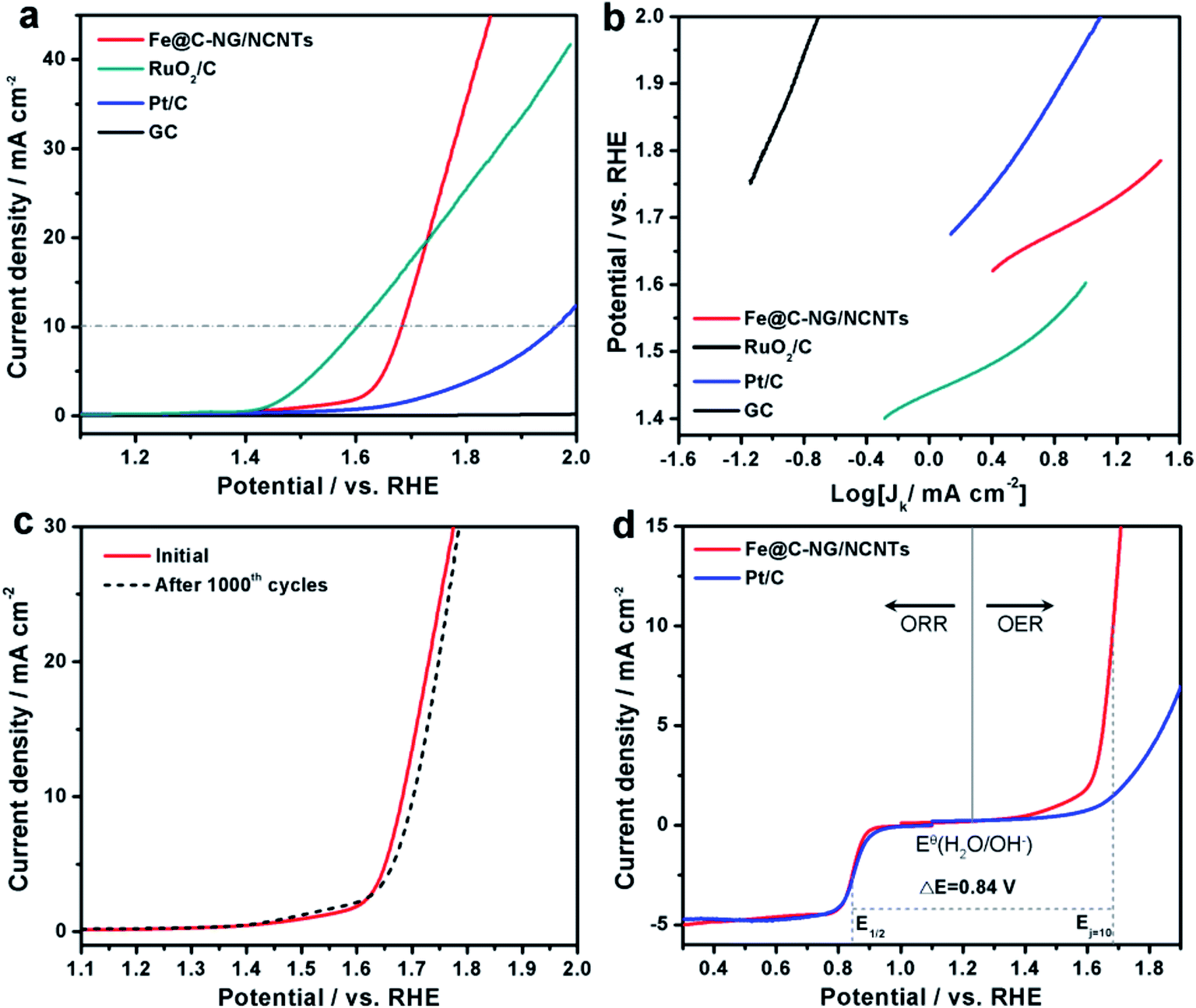 | ||
| Fig. 5 (a) LSV curves of GC, Fe@C–NG/NCNTs, Pt/C and RuO2/C electrode in 1 M KOH. (b) Corresponding Tafel plots. (c) LSV curves for Fe@C–NG/NCNTs before and after 1000 cycles. (d) Overall polarization curves of Fe@C–NG/NCNTs and Pt/C over the entire ORR and OER region. | ||
Furthermore, we constructed primary Zn–air batteries employing Fe@C–NG/NCNTs-loaded carbon paper as an air cathode (1.0 mg cm−2) and Zn foil as the anode measured in 6.0 M KOH electrolyte (ESI, Fig. S18†). A mixed Pt/C + Ir/C (1:1 by weight) sample was also tested as a reference. Typical galvanodynamic charge/discharge polarization curves and the corresponding power densities are shown in Fig. 6a. The open-circuit voltage (OCV) and maximum power density of the Zn–air batteries assembled from Fe@C–NG/NCNTs were 1.37 V and 101.3 mW cm−2, respectively, which are nearly equal to those of the Pt/C + Ir/C air electrode (1.36 V and 101.9 mW cm−2, respectively) under the same conditions. In addition, Fig. 6b exhibits a voltage plateau of 1.12 V at the discharge current density of 10 mA cm−2 over 40 h without an obvious voltage change. Next, the specific capacity of the Fe@C–NG/NCNT-based batteries normalized to the mass of consumed Zn electrode was determined to be 682.6 mA h g−1, corresponding to a high gravimetric energy density of 764.5 W h kg−1. These values are comparable to or even better than other recently reported results (ESI, Table S2†). To further demonstrate the practical application of these batteries, a red light-emitting diode (LED, 3.0 V) was powered by two series-connected Fe@C–NG/NCNTs-based Zn–air batteries.
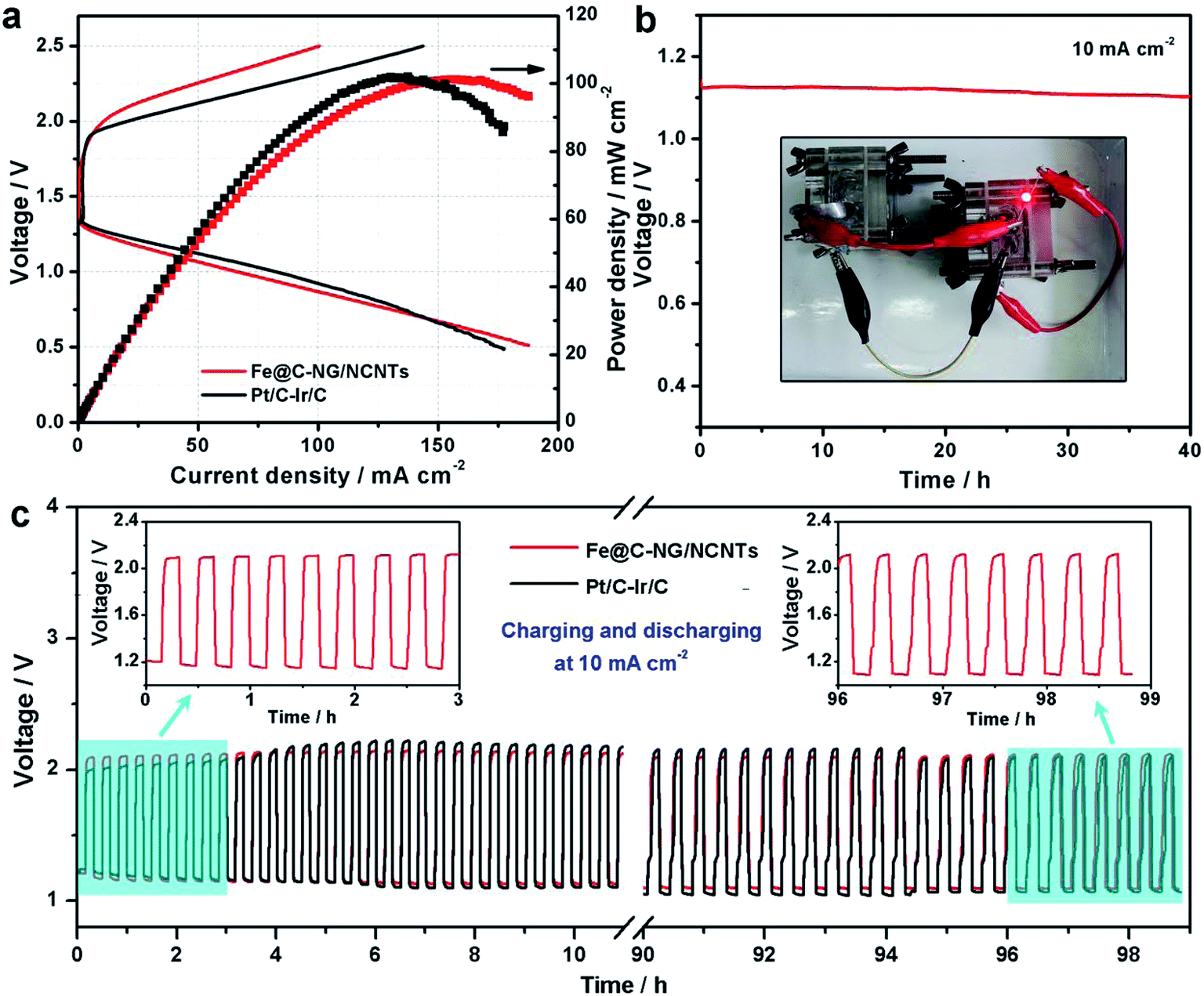 | ||
| Fig. 6 (a) Galvanodynamic charge/discharge polarization curves and power density curves of the Zn–air batteries assembled from the Fe@C–NG/NCNTs and mixed Pt/C + Ir/C (1:1 by weight) air electrodes, (b) galvanostatic discharge curves of Zn–air batteries with Fe@C–NG/NCNTs as the catalyst at 5 and 10 mA cm−2; the inset of (b) shows a photograph of a red LED (∼3.0 V) powered by two liquid Zn–air batteries with Fe@C–NG/NCNTs air–cathodes connected in series. (c) Charging–discharging cycling performance of rechargeable Zn–air batteries assembled with Fe@C–NG/NCNTs and mixed Pt/C + Ir/C electrocatalysts at a constant current density of 10 mA cm−2. | ||
Moreover, a rechargeable Zn–air battery was also assembled with 6 M KOH and 0.2 M Zn acetate as the electrolyte. When cycled at a constant current density of 10 mA cm−2 at 20 min per cycle, the round-trip voltage (the voltage difference between the charging and discharging voltage) was retained very well for more than 99 h (up to 297 cycles). For example, the battery showed a voltage gap of 0.89 V in the 1st cycle (57.42% of the voltaic efficiency). After 297 continuous cycle tests, the battery using a commercial Pt/C + Ir/C electrode showed a voltage gap increase of 0.22 V, almost twice of Fe@C–NG/NCNTs electrode (0.13 V) under the same conditions. Thus, the rechargeable Zn–air batteries using Fe@C–NG/NCNTs as air electrodes display more robust performance than those using Pt/C + Ir/C.
Based on the systematic compositional and structural analysis of Fe@C–NG/NCNT, we propose that three important aspects account for its excellent performance: (1) constructed from N-doped bamboo-like CNTs and graphene sheets, the 3D Fe@C–NG/NCNTs possess robust structural stability, high electronic conductivity and rapid electron/mass transport ability. Also, their high N doping (6.78 at%) favours the adsorption of oxygen species. (2) The active Fe–Nx sites account for the excellent electrochemical performance. (3) The encapsulation structure of Fe@C is assumed to protect the inner Fe/Fe3C NPs, which can continually boost the activity of Fe–Nx sites. Thus, the 3D interconnected NG/bamboo-like CNTs improve the mass transport, and the Fe@C NPs provide additional electrons on the carbon surface, thus promoting O2 reduction on adjacent Fe–Nx active sites.
Conclusion
In summary, Fe/Fe3C@C NPs encapsulated in 3D N-doped graphene–CNTs was fabricated with atomically dispersed Fe–Nx active sites via simple pyrolysis. The Fe–Nx active sites were verified by 57Fe Mössbauer spectroscopy, XANES measurements and SCN− poisoning tests. Also, the Fe@C structure further enhances the catalytic activity of the Fe–Nx sites. As a result, the as-fabricated Fe@C–NG/NCNTs shows E1/2 of 0.84 V and a low Tafel slope of 49 mV dec−1. Also, Fe@C–NG/NCNTs exhibits good OER activity, with a ΔE value of 0.84 V in alkaline media. Furthermore, this catalyst was integrated into rechargeable Zn–air batteries, which showed much higher rechargeability and cyclic durability than batteries containing commercial Pt/C + Ir/C. This work not only provides a novel oxygen electrocatalyst with full utilization of the structural features of the host nanocarbon materials and guest active species, but also promotes the development of advanced noble-metal-free electrocatalysts for use in other energy conversion and storage devices.Experimental section
Chemicals
All the chemical reagents were used as received. Graphite powder (99.95% purity, Aladdin), iron(III) chloride hexahydrate (FeCl3·6H2O), sulfuric acid (H2SO4, 98%), phosphorous acid (H3PO4, 85%), hydrochloric acid (HCl, 40%) potassium permanganate (KMnO4, 99%), hydrogen peroxide (30%), ethanol (99.7%), and dicyandiamide were acquired from Tianjin Guangfu Fine Chemical Reagent Research Institute. SBA-15 (12 nm) was supplied by Nanjing XFNANO Institute. Nafion solution (5 wt%, Dupont D520) and Pt/C (20 wt%, JM) were supplied by Shanghai Hesen Electric Co., Ltd. Polytetrafluoroethylene (PTFE) emulsion (60 wt%, USA) was bought from Dupont. Nitrogen (N2) and oxygen (O2) with a purity of 99.99% were supplied by Hunan Xianggang Co., Ltd. Deionized (DI) water was produced in our lab.Preparation of Fe@C–NG/NCNTs
GO was prepared by an improved Hummers' method from natural flake graphite powder. SBA-15 (0.5 g) was added to the GO suspension (1 mg mL−1) and sonicated for 2 h. This mixed solution was freeze-dried for 48 h. Next, the obtained power (0.2 g) was mixed with FeCl3·6H2O (0.05 g) and dicyandiamide (1.0 g) and transferred to the centre of a tube furnace. After pumping and purging the system with flowing N2 three times, the temperature was ramped at 3 °C min−1 up to 550 °C for 4 h; the reactor was then heated to 800 °C at a rate of 3 °C min−1 with a holding time of 1 h. After that, the furnace was naturally cooled to room temperature under protection of N2. The resulting black powder was then washed with 10% HF for 24 h to remove SBA-15 and unstable Fe species, followed by drying at 80 °C overnight. Thus, Fe@C–NG/NCNTs was obtained.Characterization
The crystal phases were identified by X-ray diffraction (XRD, Bruker AXS D8 ADVANCE) with Cu Kα (λ = 1.5406 Å) radiation at a scanning rate of 2θ = 0.02° per step. X-ray photoelectron spectra (XPS) were recorded on a Thermo Scientific ESCALAB 250Xi machine with an Al Kα source. The morphologies and structures of the samples were characterized using a high-resolution transmission electron microscope (HRTEM, JEM-2100) operated at 200 kV and a scanning electron microscope (SEM, Quanta 450 ESEM, FEI) operated at 10 kV. Raman measurements were performed on a Bruker RAM II instrument with a laser wavelength of 532 nm. The specific surface areas and the pore size distributions of the samples were estimated from nitrogen adsorption isotherms (ASAP 2020 at 77 K, USA) by means of the Brunauer–Emmett–Teller (BET) equation and the Barrett–Joyner–Halenda (BJH) model, respectively. The 57Fe Mösbauer spectra of the prepared samples were recorded on a Topologic 500A spectrometer and a proportional counter at room temperature. The X-ray absorption fine structure spectra (Fe K-edge) were collected at the 1W1B station in the Beijing Synchrotron Radiation Facility (BSRF; the storage rings were operated at 2.5 GeV with a maximum current of 250 mA). The data were collected at room temperature in transmission mode using a N2-filled ionization chamber (Si (111) monochromator for Fe K-edge). All samples were pelletized as disks 13 mm in diameter using graphite powder as a binder (ground thoroughly with a mortar and pestle). The acquired EXAFS data were processed according to the standard procedures using the ATHENA module implemented in the IFEFFIT software package. The EXAFS spectra were obtained by subtracting the post-edge background from the overall absorption and then normalizing with respect to the edge-jump step.Electrochemical measurements
The electrochemical measurements were conducted in a typical three-electrode electrochemical system (CHI 760e, Chenhua, China) at room temperature using Pt wire as the counter electrode and SCE with saturated KCl solution as the reference electrode. All the potentials were calibrated to the reversible hydrogen electrode (RHE) according to the Nernst equation. The working electrode was fabricated as follows: the catalyst ink was prepared by blending 6 mg catalyst with 40 μL Nafion solution dispersed in 1 mL of water-isopropanol solution with a volume ratio of 3:1 by sonicating for at least 60 min to form a homogeneous ink. 5 μL catalyst ink was then drop-casted onto a glassy carbon electrode 4 mm in diameter with 0.24 mg cm−2 loading for all samples. Before ORR testing, N2/O2 was bubbled into the KOH solution for at least 30 min to ensure O2 saturation during the process. The cyclic voltammograms (CVs) were measured in N2/O2-saturated KOH solution. RDE tests were measured in O2-saturated 0.1 M KOH with a scan rate of 50 mV s−1. Also, linear sweep voltammetry (LSV) curve data after iR correction were collected with a rotating speed ranging from 400 to 1600 rpm on RDE with a scan rate of 10 mV s−1. Electrochemical impedance spectroscopy (EIS) measurements were carried out by applying an AC voltage with 5 mV amplitude in a frequency range from 200 kHz to 100 mHz in 0.1 M KOH solution.
In order to estimate the four-electron selectivities of the catalysts, the electron transfer number was calculated based on the Koutecky–Levich (K–L) equation.
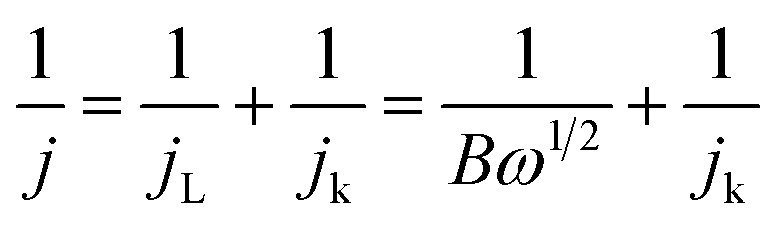
| B = 0.2nFC0(D0)2/3v−1/6 |
| jk = nFkC0 |
485 C mol−1); C0 is the bulk concentration for O2 (1.2 × 10−6 mol cm−3) dissolved in 0.1 M KOH solution; D0 is the diffusivity of O2 (1.9 × 10−5 cm2 s−1); v is the kinematic viscosity of the electrolyte; and k is the electron-transferred rate constant.
The electrocatalytic OER activity was performed in 1 M KOH solution. RDE tests were recorded in the potential window range from −0.20 to 1.00 V with a scan rate of 5 mV s−1. The stability test was performed by continuous cycling of CVs from 1.50 to 1.80 V for 1000 cycles.
Zn–air battery measurements
The Fe@C–NG/NCNTs-based air electrodes used for the Zn–air batteries were composed of a stainless steel mesh (SSM) and a gas diffusion layer (GDL) on the air-facing side as well as a catalyst ink layer (CIL) on the water-facing side. Firstly, the GDL was constructed from carbon black dispersed in ethanol, followed by dropping of a PTFE emulsion with a mass ratio of PTFE emulsion to carbon black of 7:3. After stirring for 1 h and drying at 80 °C overnight to remove excess ethanol, the obtained dough-like paste was rolled to form a film with a thickness of 0.25 mm. Next, the film was rolled onto SSM (mesh 40 × 40, type 304) and then sintered at 360 °C for 1 h. The CIL was prepared by loading the catalyst ink onto the other side of the SSM by drop-casting. The catalyst loading was 1.0 mg cm−2. Electrochemical reactors with a total volume of 28 mL were constructed to test the properties of the primary Zn–air batteries. A polished zinc plate with a thickness of 0.3 mm and 6 M KOH were used as the anode and electrolyte for the Zn–air batteries, respectively. For the rechargeable Zn–air batteries, the electrolyte used was 6 M KOH with 0.2 M Zn acetate to ensure reversible Zn electrochemical reactions at the anode. The galvanostatic discharge (ORR) and discharge–charge (ORR–OER) cycling stability were determined using a LAND testing system. Both the discharge and charge currents and corresponding power densities were normalized to the effective surface areas of the air–cathode electrodes. The specific capacity was calculated according to the equation: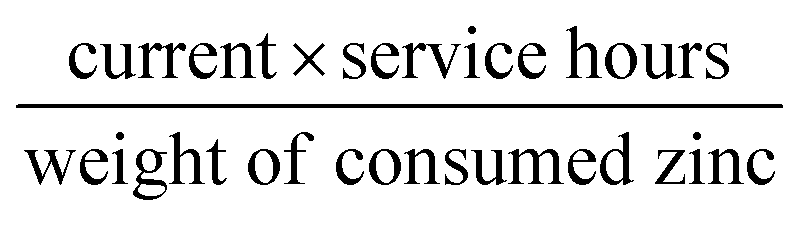
The energy density was calculated according to the equation:
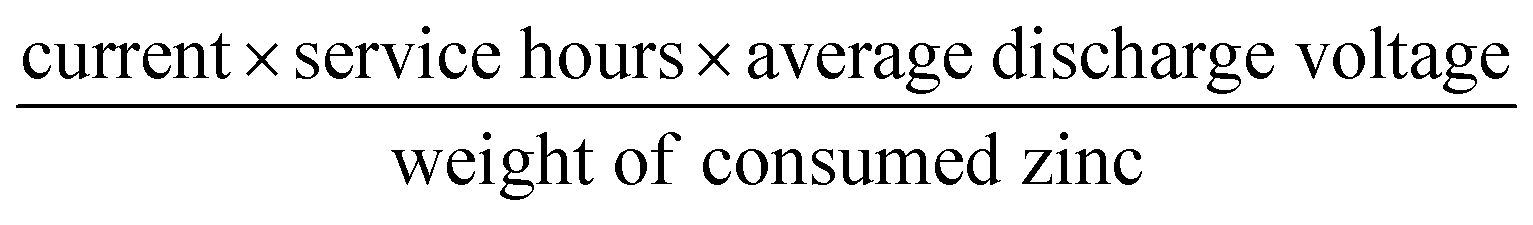
The average turn-over frequency (TOF) data were estimated according to ref. 1 (Angew. Chem., Int. Ed., 2015, 128, 1377) as below:
As the number of N atoms is 16.14 times that of the Fe atoms from the XPS data, we assume that all the Fe atoms on the surface of the catalyst coordinate with nitrogen atoms to form the FeNx/C (x = 2 or 4) active sites, and every active site only contains one Fe atom. Additionally, we assume that the Fe/Fe3C NPs and N atoms that do not coordinate with Fe atoms do not contribute to the ORR activity. The active Fe content (WFe, g Fe/g catalyst) can be calculated from the XPS data:
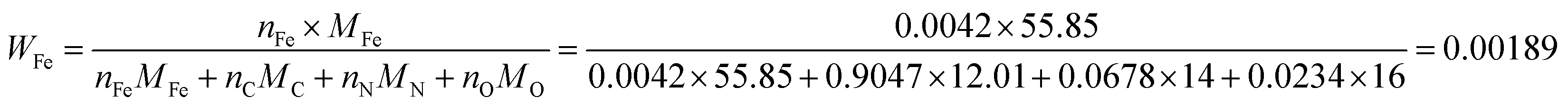
The density of active sites is:
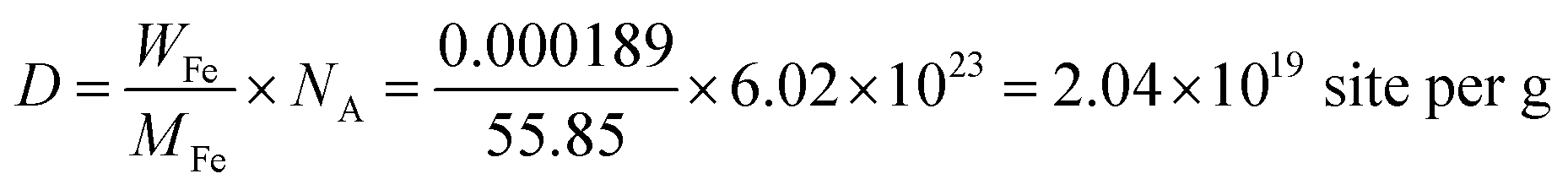
The capacitive current density at 0.8 V is ∼0.2 mA cm−2 from the CV curves in N2 at a scan rate of 50 mV s−1. The LSV curves were recorded at a scan rate of 10 mV s−1. The capacitive current density (icap) from LSV must be lower than 0.2 mA cm−2 due to the lower scan speed. Because icap is much lower than the measured current density (i0.8 V) (4.20 mA cm−2) from the LSV curves (icap ≪ i0.8 V), we assume that the measured current density (i0.8 V) is equal to the faradic current density:
| JF0.8 V = J0.8 V − Jcap ≈ J0.8 V |
Diffuse current density data can be obtained from the LSV curve at 1600 rpm for Fe@C–NG/NCNTs. The kinetic current density is calculated using the K–L equation (mcat; catalyst loading: 0.24 mg cm−2):
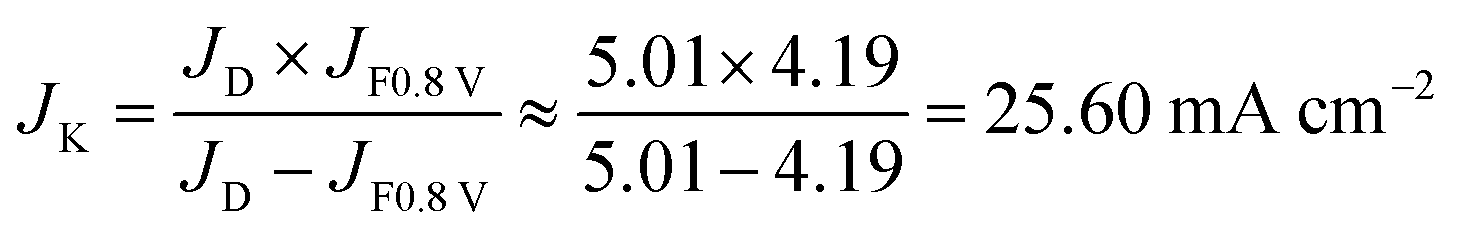
Then, the kinetic current density is normalized to the mass of the catalyst at 0.80 V (vs. RHE):
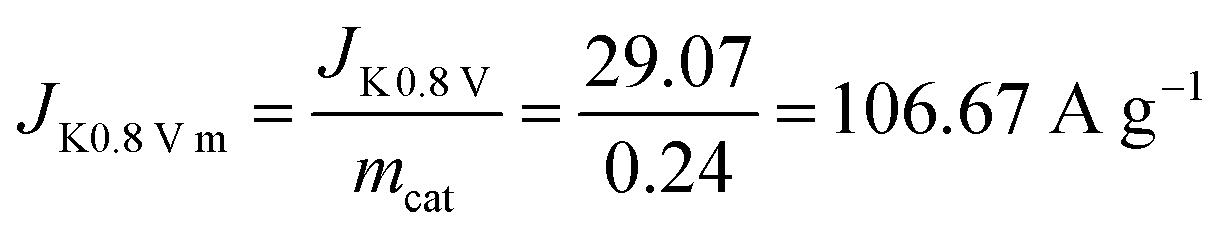
Then the TOF is:
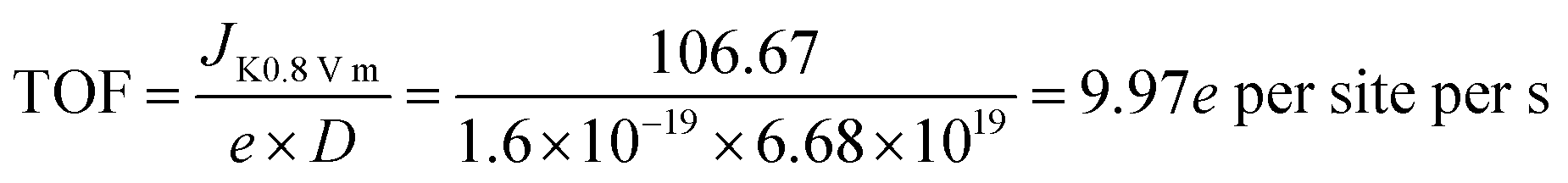
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Yongpeng Lei thanks the Research Project of NUDT (ZK16-03-32). Zhiyan Chen acknowledges support from the Research Foundation of Education Bureau of Hunan Province (16K102). Bing Wang acknowledges support from the National Natural Science Foundation of China (61701514). Yingde Wang is grateful for support from the National Natural Science Foundation of China (51773226). Yongpeng Lei thank the 1W1B station for XAFS measurements in Beijing Synchrotron Radiation Facility (BSRF).References
- H. J. Cui, Z. Zhou and D. Z. Jia, Mater. Horiz., 2017, 4, 7 RSC.
- Z. L. Wang, D. Xu, J. J. Xu and X. B. Zhang, Chem. Soc. Rev., 2014, 43, 7746 RSC.
- (a) W. Xia, A. Mahmood, Z. B. Liang, R. Q. Zou and S. J. Guo, Angew. Chem., Int. Ed., 2015, 55, 2650 CrossRef PubMed; (b) R. Ma, X. Ren, B. Y. Xia, Y. Zhou, C. Sun, Q. Liu, J. Liu and J. Wang, Nano Res., 2016, 9, 808 CrossRef CAS; (c) Y. Lei, Q. Shi, C. Han, B. Wang, N. Wu, H. Wang and Y. Wang, Nano Res., 2016, 9, 2498 CrossRef; (d) J. Lu, W. Zhou, L. Wang, J. Jia, Y. Ke, L. Yang, K. Zhou, X. Liu, Z. Tang, L. Li and S. Chen, ACS Catal., 2016, 6, 1045 CrossRef CAS.
- (a) M. H. Shao, Q. W. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2015, 116, 3594 CrossRef PubMed; (b) N. Bing, O. Chen, X. Xu, J. Zhuang and X. Wang, Adv. Mater., 2017, 29, 1701354 CrossRef PubMed.
- L. Z. Bu, J. B. Ding, S. J. Guo, X. Zhang, D. Su, X. Zhu, J. L. Yao, J. Guo, G. Lu and X. Q. Huang, Adv. Mater., 2015, 27, 7204 CrossRef CAS PubMed.
- J. J. Mao, Y. J. Chen, J. J. Pei, D. S. Wang and Y. D. Li, Chem. Commun., 2016, 52, 5985 RSC.
- J. S. Lee, G. Nam, J. Sun, S. Higashi, H. W. Lee, S. Lee, W. Chen, Y. Cui and J. Cho, Adv. Energy Mater., 2016, 6, 1601052 CrossRef.
- (a) W. H. He, Y. Wang, C. H. Jiang and L. H. Lu, Chem. Soc. Rev., 2016, 45, 2396 RSC; (b) S. Nardecchia, D. Carriazo, M. L. Ferrer, M. C. Gutiérrez and F. D. Monte, Chem. Soc. Rev., 2013, 42, 794 RSC.
- (a) Q. C. Wang, Z. Y. Chen, N. Wu, B. Wang, W. He, Y. P. Lei and Y. D. Wang, ChemElectroChem, 2017, 4, 514 CrossRef CAS; (b) G. Zhang, X. Jin, H. Li, L. Wang, C. Hu and X. Sun, Sci. China Mater., 2016, 59, 337 CAS.
- Y. Zhu, L. Li, C. Zhang, G. Casillas, Z. Sun, Z. Yan, G. Ruan, Z. Peng, A.-R. O. Raji, C. Kittrell, R. H. Hauge and J. M. Tour, Nat. Commun., 2012, 3, 1225 CrossRef PubMed.
- Y. G. Li, W. Zhou, H. L. Wang, L. M. Xie, Y. Y. Liang, F. Wei, J.-C. Idrobo, S. J. Pennycook and H. J. Dai, Nat. Nanotechnol., 2012, 7, 394 CrossRef CAS PubMed.
- J. Deng, P. J. Ren, D. H. Deng and X. H. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100 CrossRef CAS PubMed.
- (a) Z. Q. Liu, H. Cheng, N. Li, T. Y. Ma and Y. Z. Su, Adv. Mater., 2016, 28, 3777 CrossRef CAS PubMed; (b) Y. Cheng, S. Dou, M. Saunders, J. Zhang, J. Pan, S. Wang and S. P. Jiang, J. Mater. Chem. A, 2016, 4, 13881 RSC; (c) Y. Cheng, S. Dou, J. P. Veder, S. Wang, M. Saunders and S. P. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 8121 CrossRef CAS PubMed; (d) Q. Wu, L. Jiang, L. Qi, E. Wang and G. Sun, Int. J. Hydrogen Energy, 2014, 39, 3423 CrossRef CAS; (e) S. Gul, J. W. D. Ng, T. F. Jaramillo and J. Yano, Phys. Chem. Chem. Phys., 2015, 17, 8901 RSC.
- S. Dou, L. Tao, J. Huo, S. Y. Wang and L. M. Dai, Energy Environ. Sci., 2016, 9, 1320 CAS.
- H. Yin, C. Z. Zhang, F. Liu and Y. L. Hou, Adv. Funct. Mater., 2014, 24, 2930 CrossRef CAS.
- (a) Z. Yan, H. Wang, M. Zhang, Z. Jiang, T. Jiang and J. Xie, Electrochim. Acta, 2013, 95, 218 CrossRef CAS; (b) Z. Yan, M. Zhang, J. Xie, H. Wang and W. Wei, J. Power Sources, 2013, 243, 48 CrossRef CAS; (c) Y. Hou, T. Z. Huang, Z. H. Wen, S. Mao, S. M. Cui and J. H. Chen, Adv. Energy Mater., 2014, 4, 1400337 CrossRef.
- H. Huang, X. Feng, C. C. Du, S. Y. Wu and W. B. Song, J. Mater. Chem. A, 2015, 3, 4976 CAS.
- Z. Y. Wu, X. X. Xu, B. C. Hu, H. W. Liang, Y. Lin, L. F. Chen and S. H. Yu, Angew. Chem., Int. Ed., 2015, 54, 8179 CrossRef CAS PubMed.
- (a) N. Wu, Y. P. Lei, Q. C. Wang, B. Wang, C. Han and Y. D. Wang, Nano Res., 2017, 10, 2332–2343 CrossRef CAS; (b) C. Y. Su, H. Cheng, W. Li, Z. Q. Liu, N. Li, Z. Hou, F. Q. Bai, H. X. Zhang and T. Y. Ma, Adv. Energy Mater., 2017, 7, 1602420 CrossRef; (c) Q. Liu, Y. Wang, L. Dai and J. Yao, Adv. Mater., 2016, 28, 3000 CrossRef CAS PubMed.
- W. J. Jiang, L. Gu, L. Li, Y. Zhang, X. Zhang, L. J. Zhang, J. Q. Wang, J. S. Hu, Z. D. Wei and L. J. Wan, J. Am. Chem. Soc., 2016, 138, 3570 CrossRef CAS PubMed.
- M. Q. Wang, W. H. Yang, H. H. Wang, C. Chen, Z. Y. Zhou and S. G. Sun, ACS Catal., 2014, 4, 3928 CrossRef CAS.
- Z. H. Wen, S. Q. Ci, F. Zhang, X. L. Feng, S. M. Cui, S. Mao, S. L. Luo, Z. He and J. H. Chen, Adv. Mater., 2012, 24, 1399 CrossRef CAS PubMed.
- L. Zhang, X. Wang, R. Wang and M. Hong, Chem. Mater., 2015, 27, 7610 CrossRef CAS.
- S. N. Stamatin, I. Hussainova, R. Ivanov and P. E. Colavita, ACS Catal., 2016, 6, 5215 CrossRef CAS.
- W. Y. Yang, X. J. Liu, X. Y. Yue, J. B. Jia and S. J. Guo, J. Am. Chem. Soc., 2015, 137, 1436 CrossRef CAS PubMed.
- Z. S. Wu, S. B. Yang, Y. Sun, K. Parvez, X. L. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082 CrossRef CAS PubMed.
- Q. Shi, Y. D. Wang, Z. M. Wang, Y. P. Lei, B. Wang, N. Wu, C. Han, S. Xie and Y. Z. Gou, Nano Res., 2016, 9, 317 CrossRef CAS.
- J. S. Li, S. L. Li, Y. J. Tang, M. Han, Z. H. Dai, J. C. Bao and Y. Q. Lan, Chem. Commun., 2015, 51, 2710 RSC.
- J. Wei, Y. Liang, Y. X. Hu, B. Kong, G. P. Simon, J. Zhang, S. P. Jiang and H. T. Wang, Angew. Chem., Int. Ed., 2015, 55, 1355 CrossRef PubMed.
- H. L. Fei, J. C. Dong, M. J. A. Jimenez, G. L. Ye, N. D. Kim, E. L. G. Samuel, Z. W. Peng, Z. Zhu, F. Qin, J. M. Bao, M. J. Yacaman, P. M. Ajayan, D. L. Chen and J. M. Tour, Nat. Commun., 2015, 3, 1225 Search PubMed.
- H. B. Zhang, T. Wang, J. J. Wang, H. M. Liu, T. D. Dao, M. Li, G. G. Liu, X. G. Meng, K. Chang, L. Shi, T. Nagao and J. H. Ye, Adv. Mater., 2016, 28, 3703 CrossRef CAS PubMed.
- A. Kong, X. F. Zhu, Z. Han, Y. Y. Yu, Y. B. Zhang, B. Dong and Y. K. Shan, ACS Catal., 2014, 4, 1793 CrossRef CAS.
- L. Z. Gu, L. H. Jiang, X. N. Li, J. T. Jin, J. H. Wang and G. Q. Sun, Chin. J. Catal., 2016, 37, 539 CrossRef CAS.
- A. Zitolo, V. Goellner, V. Armel, M. T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937 CrossRef CAS PubMed.
- S. Kattel and G. F. Wang, J. Phys. Chem. Lett., 2014, 5, 452 CrossRef CAS PubMed.
- X. X. Yan, K. X. Liu, T. Wang, Y. You, J. G. Liu, P. Wang, X. Q. Pan, G. F. Wang, J. Luo and J. Zhu, J. Mater. Chem. A, 2017, 5, 3336 CAS.
- Z. H. Wen, S. Q. Ci, Y. Hou and J. H. Chen, Angew. Chem., Int. Ed., 2014, 53, 6496 CrossRef CAS PubMed.
- J. W. Xiao, C. Chen, J. B. Xi, Y. Y. Xu, F. Xiao, S. Wang and S. H. Yang, Nanoscale, 2015, 7, 7056 CAS.
- Q. Li, H. Y. Pan, D. Higgins, R. G. Cao, G. Q. Zhang, H. F. Lv, K. B. Wu, J. Cho and G. Wu, Small, 2014, 11, 1443 CrossRef PubMed.
- (a) Q. Shi, Y. P. Lei, Y. D. Wang, H. P. Wang, L. H. Jiang, H. L. Yuan, D. Fang, B. Wang, N. Wu and Y. Z. Gou, Curr. Appl. Phys., 2015, 15, 1606 CrossRef; (b) Y. Zhao, K. Kamiya, K. Hashimoto and S. Nakanishi, J. Phys. Chem. C, 2015, 119, 2583 CrossRef CAS.
- G. X. Zhang, H. X. Luo, H. Y. Li, L. Wang, B. Han, H. C. Zhang, Y. J. Li, Z. Chang, Y. Kuang and X. M. Sun, Nano Energy, 2016, 26, 241 CrossRef CAS.
- Y. J. Chen, S. F. Ji, Y. G. Wang, J. C. Dong, W. X. Chen, Z. Li, R. G. Shen, L. R. Zheng, Z. B. Zhuang, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2017, 56, 6937 CrossRef CAS PubMed.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. F. Li, Angew. Chem., Int. Ed., 2014, 53, 3675 CrossRef CAS PubMed.
- K. G. Qu, Y. Zheng, S. Dai and S. Z. Qiao, Nano Energy, 2015, 19, 373 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta08423d |
| This journal is © The Royal Society of Chemistry 2018 |