
Catalyst-controlled synthesis of 4-amino-isoquinolin-1(2H)-one and oxazole derivatives†
Ben
Niu
a,
Ruixing
Liu
b,
Yin
Wei
 *b and
Min
Shi
*acb
*b and
Min
Shi
*acb
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, Meilong Road No. 130, Shanghai, 200237, China
bState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, University of Chinese Academy of Sciences, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai, 200032, China. E-mail: weiyin@sioc.ac.cn; mshi@mail.sioc.ac.cn
cState Key Laboratory and Institute of Elemento-organic Chemistry, Nankai University, Tianjin 300071, P. R. China
First published on 8th March 2018
Abstract
A facile synthetic method to access 4-amino-isoquinolin-1(2H)-one and oxazole derivatives was disclosed in this paper. The reaction of N-(pivaloyloxy)-amides with ynamides produced 4-amino-isoquinolin-1(2H)-ones in good yields in the presence of Cp*Rh(III) catalyst through a C–H bond functionalization. Using Sc(OTf)3 as the catalyst, oxazole derivatives were obtained in good yields from the same reaction. This protocol features good functional group tolerance and excellent regioselectivity.
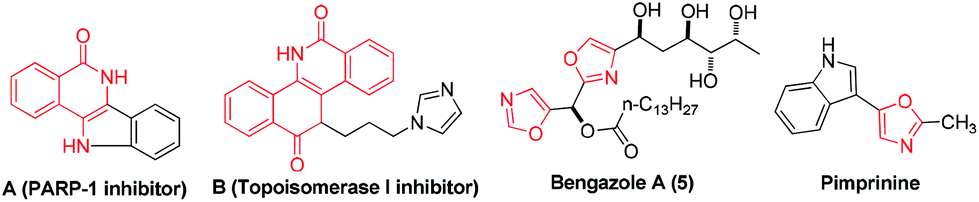
Isoquinolin-1(2H)-ones are probably the most ubiquitous heterocycles that widely exist in natural products, medicinal agents and functional materials.1 Notably, 4-amino-isoquinolin-1(2H)-ones are also prevalent core structural units. Many compounds containing 4-amino-isoquinolin-1(2H)-one motifs exhibit potent biological activities.1a–e For example, molecule A is a new scaffold that can be used as a kind of PARP-1 inhibitor (Scheme 1). Compound B is designed as a topoisomerase I (Top1) inhibitor as shown in Scheme 1. Owing to their indispensable biomedical value, the exploration of their synthetic method has been carried out during the past few years. However, the methods that have been explored for the synthesis of 4-amino-isoquinolin-1(2H)-one frameworks are very limited and have some obvious drawbacks,1a–e,2 such as harsh reaction conditions, multiple-step sequences and a longer reaction time. Therefore, it is quite urgent to develop simpler, general, and convenient processes for the synthesis of 4-amino-isoquinolin-1(2H)-one derivatives using easily available starting materials.
 | ||
| Scheme 1 Biologically active natural products containing 4-amino-isoquinolin-1(2H)-ones and oxazoles. | ||
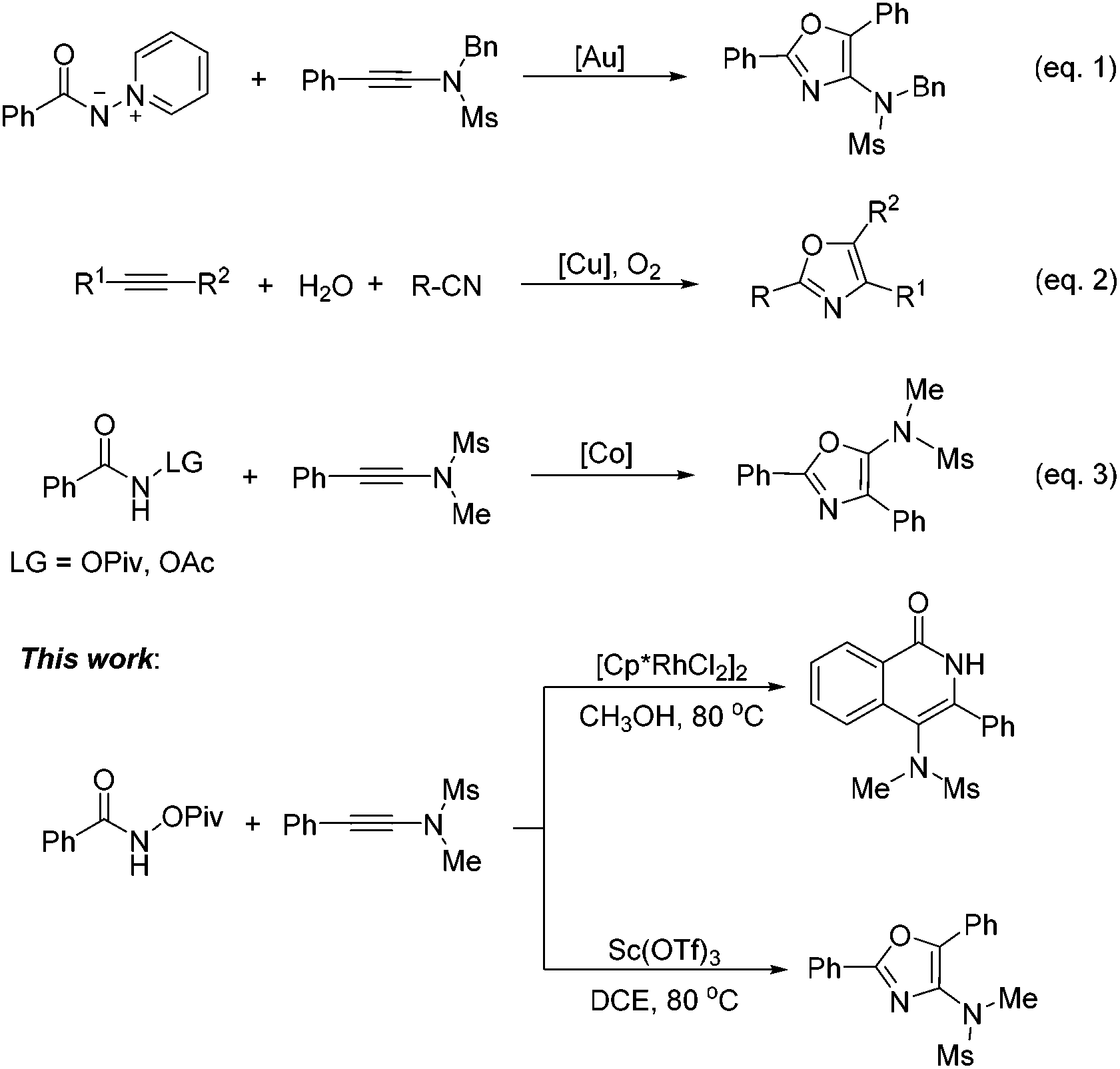
On the other hand, oxazoles are of great importance due to their presence in a number of biologically active compounds and pharmaceuticals,3 such as bengazole A (5) and pimprinine (Scheme 1). They are also versatile synthetic blocks in organic synthesis.4 During the past few decades, numerous outstanding studies to access oxazole molecules have been accomplished.5,6 Among these synthetic methods, transition metal catalyses with Au,5a–d Pd,5e Ag,5f Cu,5g–i and Co7 play a predominant role. For example, in 2011, Davies and co-workers employed conjugated N-ylides and ynamides to directly produce highly substituted and functionalized 1,3-oxazoles (Scheme 2, eqn (1)).5c In 2012, Jiang and co-workers developed a highly efficient copper-catalyzed aerobic transformation of internal alkynes and nitriles to functionalized 1,3-oxazoles (Scheme 2, eqn (2)).5i More recently, Wang's group explored a new method for the synthesis of 5-aminooxazoles by using Cp*Co(III) as a catalyst (Scheme 2, eqn (3)).7 However, these procedures are often accompanied by complex metal catalysts, vigorous reaction conditions, and expensive or toxic reagents. Therefore, the development of straightforward and convenient methods for the synthesis of oxazoles is also of great significance and highly pursued in synthetic chemistry.
 | ||
| Scheme 2 Approaches for the synthesis of oxazoles. | ||
In this paper, we wish to report our new findings in the synthesis of 4-amino-isoquinolin-1(2H)-one and oxazole derivatives from the reaction of easily available N-(pivaloyloxy)-amides and ynamides using different metal catalysts (Scheme 2, this work).
In the past few decades, transition-metal-catalyzed direct C–H bond functionalization has emerged as a powerful tool for the formation of carbon–carbon and carbon–heteroatom bonds.8 It is a classical reaction pattern that alkenes, alkynes, allenes or arenes serve as active counterparts to cross-coupling with metallacyclic complexes derived from C–H bond activation to give various functionalized molecules (Fig. 1). We envisioned whether ynamides could be taken as compatible counterparts to directly form 4-amino-isoquinolin-1(2H)-one frameworks.
 | ||
| Fig. 1 The general reaction pattern of C–H bond activation with the unsaturated bond. | ||
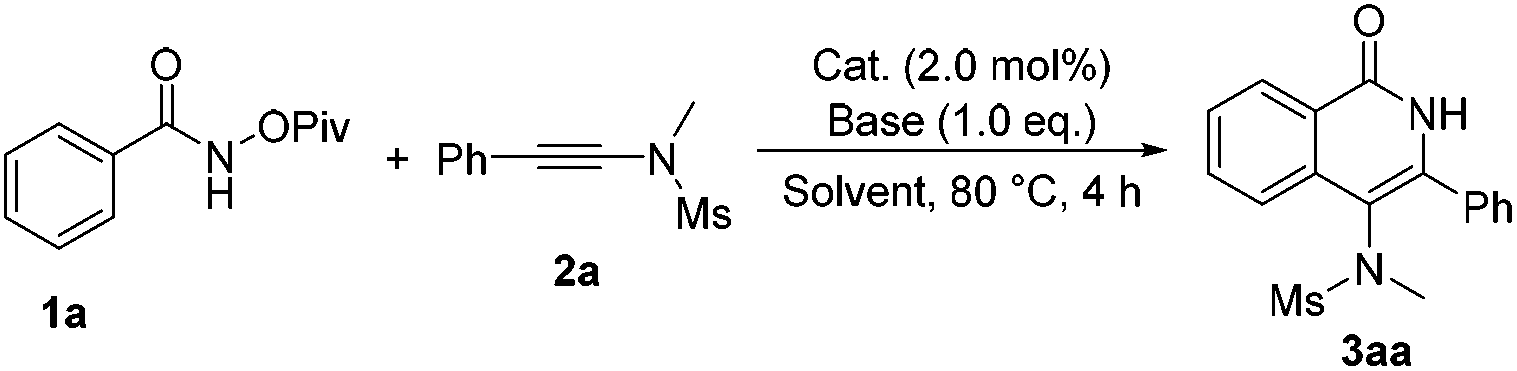
To test our hypothesis, we commenced our study with N-(pivaloyloxy)benzamide 1a and N-methyl-N-(phenylethynyl)methanesulfonamide 2a in the presence of Cp*Rh(III) (2 mol%) and CsOAc (1.0 eq.) in CH3OH at 80 °C. To our delight, the desired product 3aa was obtained in 85% yield (Table 1, entry 1). Various bases, such as LiOAc or NaOAc, were tested in the reaction, but they did not further improve the yield (Table 1, entries 2 and 3). Afterwards, we carried out this reaction in different solvents, such as acetonitrile, 1,2-dichloroethane and toluene (Table 1, entries 4–6), but none of the desired product was observed. Other metal catalysts such as [Cp*IrCl2]2, [Cp*RuCl2]2 and [(p-cymene)RuCl2]2 did not catalyze the reaction (Table 1, entries 7–9). When CsF (1.0 eq.) was used as an additive, the reaction also performed very well, giving the desired product 3aa in 85% yield (Table 1, entry 10). Next, we found that this reaction proceeded more smoothly when PivOH (1.0 eq.) was used as an additive under otherwise identical conditions, affording 3aa in 87% yield (Table 1, entry 11). Based on the above results, we then performed this reaction at different temperatures, but no better results were obtained (entries 12–15). Therefore, we identified that the reaction should be carried out in MeOH and [Cp*RhCl2]2 should be used as the catalyst with CsOAc as a base in the presence of PivOH (Table 1, entry 11).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Cat. | Additive | Base | Solvent | Yieldb, % |
| a The reactions were carried out using 1a (0.2 mmol), 2a (0.2 mmol), cat. (2.0 mol%), base (1.0 eq.) and solvent (2.0 mL) in a Schlenk tube. b Isolated yields. c T = RT. d T = 40 °C. e T = 60 °C. f T = 100 °C. | |||||
| 1 | [Cp*RhCl2]2 | — | CsOAc | MeOH | 85 |
| 2 | [Cp*RhCl2]2 | — | LiOAc | MeOH | 82 |
| 3 | [Cp*RhCl2]2 | — | NaOAc | MeOH | 69 |
| 4 | [Cp*RhCl2]2 | — | CsOAc | DCE | nr |
| 5 | [Cp*RhCl2]2 | — | CsOAc | MeCN | nr |
| 6 | [Cp*RhCl2]2 | — | CsOAc | Toluene | nr |
| 7 | [Cp*IrCl2]2 | — | CsOAc | MeOH | nr |
| 8 | [Cp*RhCl2]2 | — | CsOAc | MeOH | nr |
| 9 | [(p-Cymene)RuCl2]2 | — | CsOAc | MeOH | nr |
| 10 | [Cp*RhCl2]2 | CsF | CsOAc | MeOH | 85 |
| 11 | [Cp*RhCl 2 ] 2 | PivOH | CsOAc | MeOH | 87 |
| 12c | [Cp*RhCl2]2 | PivOH | CsOAc | MeOH | 59 |
| 13d | [Cp*RhCl2]2 | PivOH | CsOAc | MeOH | 67 |
| 14e | [Cp*RhCl2]2 | PivOH | CsOAc | MeOH | 79 |
| 15f | [Cp*RhCl2]2 | PivOH | CsOAc | MeOH | 73 |
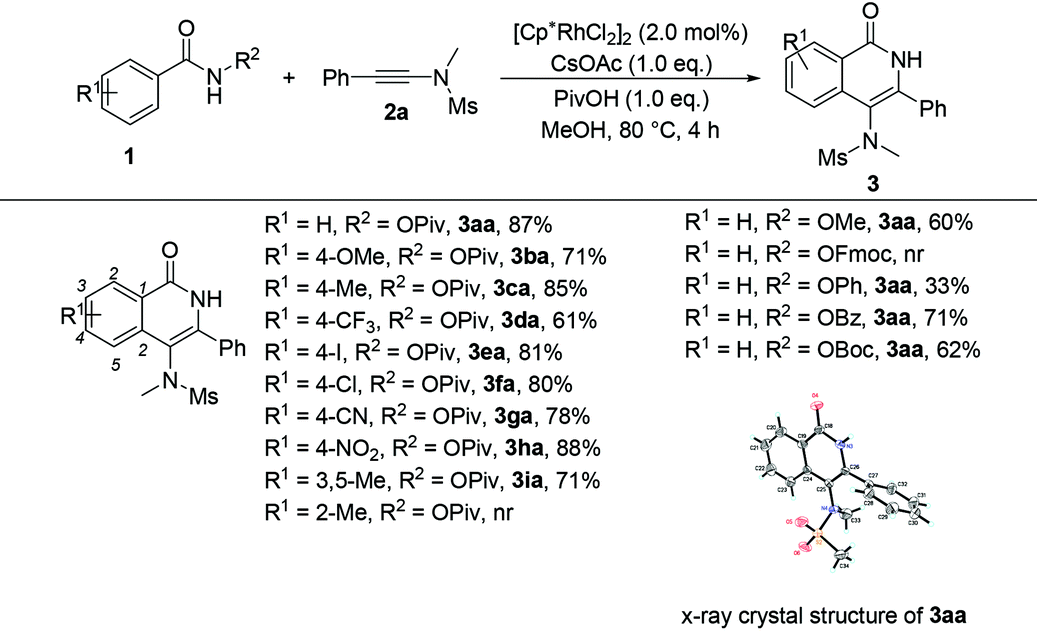
Having the optimized reaction conditions in hand, we examined the scope of benzamides bearing diverse substituents on the aromatic ring and the results are shown in Table 2. When the leaving group R2 is the OPiv anion, we first examined the electronic effect at the para-position of the benzene ring. As for substrates 1b–1h, regardless of whether they have electron-rich or electron-poor aromatic rings, the reactions could proceed smoothly to furnish the desired products 3ba–3ha in 61–88% yields. The corresponding disubstituted substrate (3,5-dimethyl) was also tolerated under the identical conditions, providing the desired product 3ia in 71% yield. However, the ortho-substituted substrate could not be transformed into the desired product. When R2 was replaced by other leaving groups, including OMe, OPh, OBz, and OBoc, the yield of 3aa was inferior to that of the OPiv leaving group. The use of OFmoc as the leaving group did not give the desired product. The structure of 3aa has been unequivocally determined by X-ray diffraction and its ORTEP drawing is shown in Table 2.9
| a Reaction conditions: 1 (0.2 mmol), 2a (0.2 mmol), [RhCp*Cl2]2 (2.0 mol%), CsOAc (1.0 eq.) and PivOH (1.0 eq.) in MeOH at 80 °C for 4 h. |
|---|
|
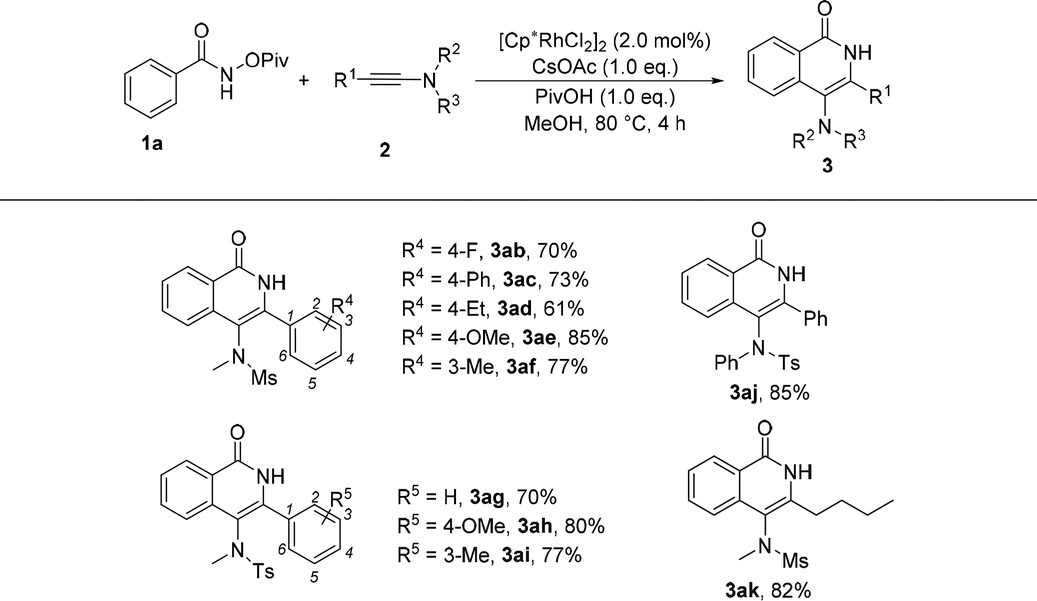
Subsequently, the substrate scope of ynamides was also investigated and the results are summarized in Table 3. First, ynamides containing aryls were investigated. When R2 and R3 were mesly and methyl groups, the effect of R4 group at the benzene ring was first examined. We found that a variety of substituents were well tolerated, regardless of their electronic nature and the substituted position at the benzene ring, affording the corresponding products 3ab–3af in good yields. Moreover, the R2 and R3 groups at the nitrogen atom were also explored and the reaction of N-tosyl ynamide and N-phenyl ynamide with 1a proceeded efficiently, providing the desired products 3ag–3aj in 70%–85% yields. Furthermore, alkyl-substituted ynamide also exhibited high reactivity in this reaction, and the corresponding product was obtained in 82% yield (3ak).
| a Reaction conditions: 1a (0.2 mmol), 2 (0.2 mmol), [Cp*RhCl2]2 (2.0 mol%), CsOAc (1.0 eq.) and PivOH (1.0 eq.) in MeOH at 80 °C for 4 h. |
|---|
|
Based on previous studies,10 ynamides could be activated by acids to form the corresponding keteniminium species. Therefore, we envisaged whether a Lewis acid could catalyze this reaction in a totally different reaction mode. Therefore, we turned our attention to investigate the reaction of N-(pivaloyloxy)benzamide 1a with N-methyl-N-(phenylethynyl)methanesulfonamide 2a in the presence of a catalytic amount of the Lewis acid. Initially, we performed the reaction in DCE using AgSbF6 as the catalyst, and found that the desired cycloadduct 4aa was obtained in 55% yield (Table 4, entry 1). This result encouraged us to further optimize the reaction conditions for the synthesis of product 4aa. Next, we examined a set of Lewis acids and found that Sc(OTf)3 afforded 4aa in higher yield (78%) and inferior results were obtained when In(OTf)3, Yb(OTf)3 or Eu(OTf)3 was used as the catalyst under identical conditions (Table 4, entries 2–5). However, a Brønsted acid such as CF3COOH failed to transform the corresponding substrates into the desired product (Table 4, entry 6). We also screened several different solvents, such as CH3OH and CH3CN, but the desired product was not acquired (Table 4, entries 7 and 8). Reducing the catalyst loading to 0.1 eq. improved the yield of 5aa to 80% (Table 4, entry 9). Finally, we identified that carrying out the reaction at room temperature afforded the desired product 5aa in 35% yield, which is different from the previous work reported by Wang and Li (Table 4, entry 10).7
|
|
|||
|---|---|---|---|
| Entrya | Lewis acid | Solvent | Yield (%) |
| a The reactions were carried out using 1a (0.2 mmol), 2a (1.3 eq.), Lewis acid (0.2 eq.) and solvent (2.0 mL) in a Schlenk tube. b Lewis acid (0.1 eq.). c At room temperature. | |||
| 1 | AgSbF6 | DCE | 55 |
| 2 | Sc(OTf)3 | DCE | 78 |
| 3 | In(OTf)3 | DCE | 70 |
| 4 | Eu(OTf)3 | DCE | 21 |
| 5 | Yb(OTf)3 | DCE | 17 |
| 6 | CF3COOH | DCE | nr |
| 7 | Sc(OTf)3 | CH3OH | nr |
| 8 | Sc(OTf)3 | CH3CN | Trace |
| 9 | Sc(OTf) 3 | DCE | 80 |
| 10c | Sc(OTf)3 | DCE | 35 |
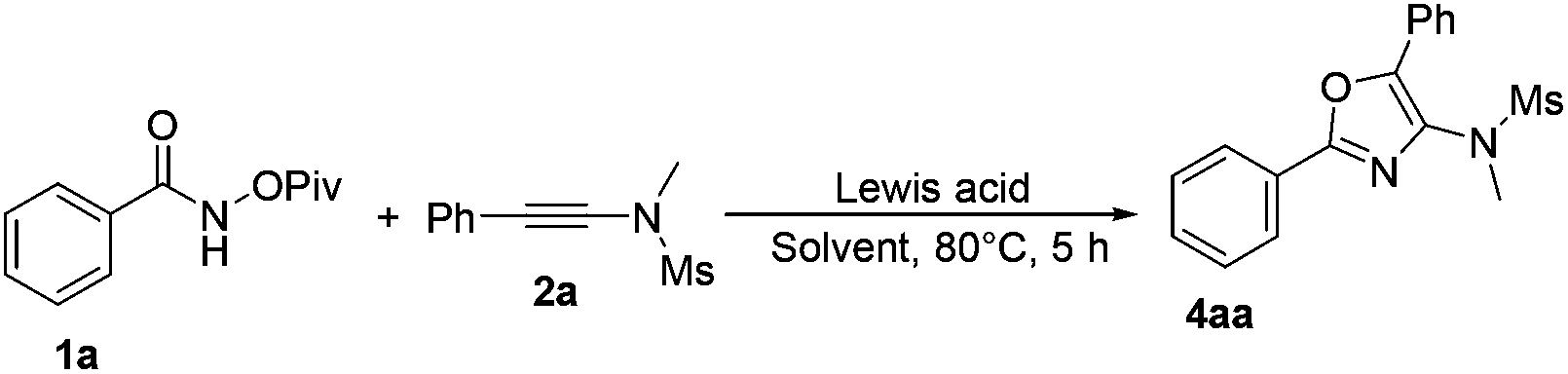
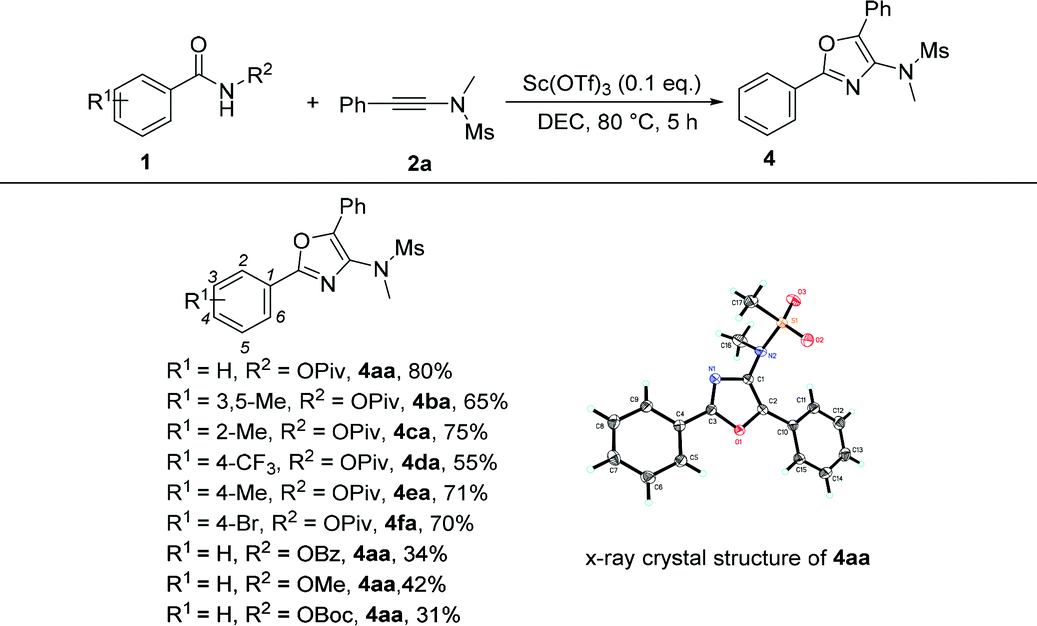
Under the optimized conditions, we next surveyed the substrate scope of benzamides and the results are shown in Table 5. When the leaving group R2 is the OPiv anion, we first examined the disubstituted and ortho-substituted substrates and found that they were well tolerated under the optimal reaction conditions, providing the desired products 4ba and 4ca in 65% and 75% yields, respectively. The electronic effect at the para-position of the benzene ring was then examined. Regardless of whether they have electron-rich or electron-poor substitution groups, all of the reactions proceeded smoothly to furnish the desired products 4da–4fa in 55–71% yields. The structure of 4aa has been unambiguously determined by X-ray diffraction and its ORTEP drawing is shown in Table 5.11 However, benzamides bearing other leaving groups, including OMe, OBz, and OBoc, afforded the corresponding products in poor yields.
| a The reactions were carried out using 1a (0.2 mmol), 2a (1.3 eq.), Sc(OTf)3 (0.1 eq.) and DCE (2.0 mL) in a Schlenk tube. |
|---|
|
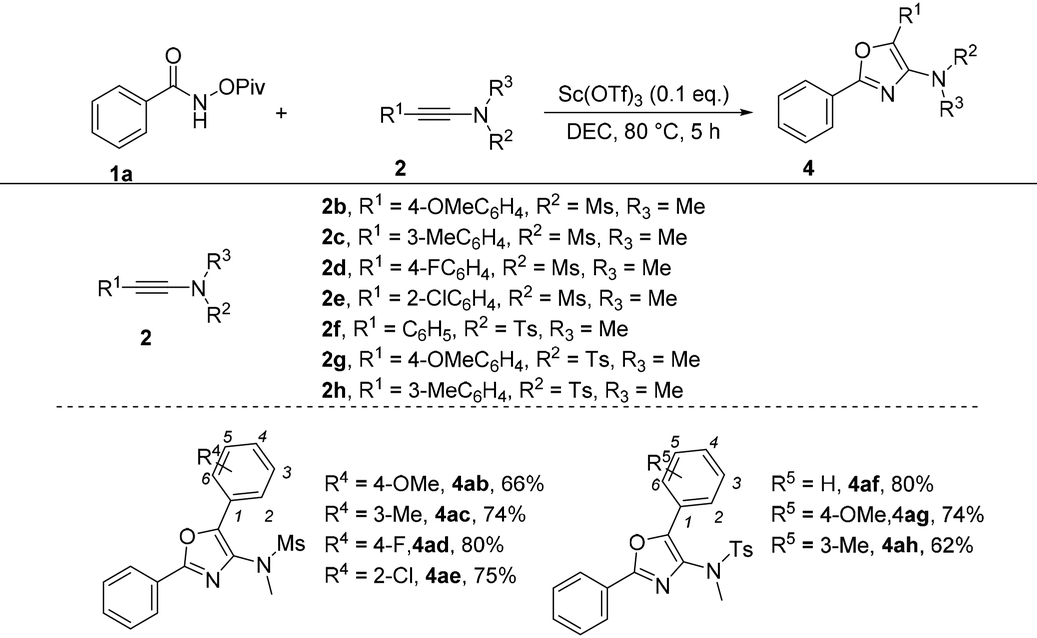
Subsequently, the optimized reaction conditions were then challenged with the diversity of ynamides by taking 1a as the reaction partner (Table 6). First, when R2 and R3 were Me and Ms groups, a diversified range of substituents on the benzene ring including both electron-donating and electron-withdrawing groups at the para-position were tolerated, giving the corresponding products 4ab and 4ad in 66 and 80% yields, respectively. In the case of the meta-substituted substrate (3-methyl) or ortho-substituted substrate (2-chloro), the reactions also performed very well, providing the corresponding products 4ac and 4ae in 74% and 75% yields, respectively. When R2 and R3 were tosyl and methyl groups, the desired products 4af–4ah were achieved in 62%–80% yields.
| a The reactions were carried out using 1a (0.2 mmol), 2 (1.3 eq.), Sc(OTf)3 (0.1 eq.) and DCE (2.0 mL) in a Schlenk tube. |
|---|
|
A plausible reaction mechanism for the formation of 4aa is proposed in Scheme 3. Intermediate A is formed upon the treatment of 1a with Sc(OTf)3, which undergoes an isomerization to give intermediate B. The reaction of intermediate B with intermediate C, generated upon the activation of 2a with Sc(OTf)3, provides intermediate D. Next, the desired product 4aa is formed through an intramolecular nucleophilic attack along with the release of the OPiv anion.
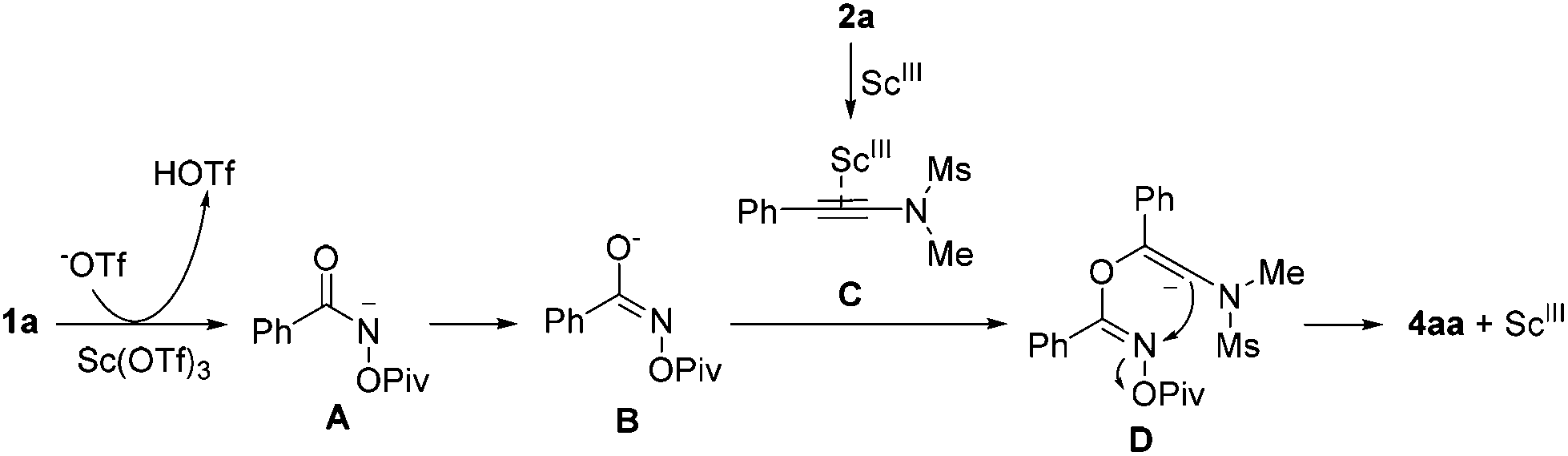 | ||
| Scheme 3 A plausible reaction mechanism. | ||
In summary, we have developed an efficient and convenient synthetic protocol to produce oxazole or 4-amino-isoquinolin-1(2H)-one derivatives from the reaction of N-(pivaloyloxy)-amides with ynamides by employing different metal catalysts. In the presence of Cp*Rh(III) catalyst, 4-amino-isoquinolin-1(2H)-one derivatives could be produced in good yields under mild reaction conditions. The use of Sc(OTf)3 as the catalyst gave oxazole derivatives in good yields. The reaction can be performed with easily available starting materials and exhibits a broad substrate scope along with moderate to good yields and good functional group tolerance.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful for the financial support from the National Basic Research Program of China (973)-2015CB856603, the Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDB20000000 and sioczz201808, the National Natural Science Foundation of China (20472096, 21372241, 21572052, 20672127, 21421091, 21372250, 21121062, 21302203, 20732008, 21772037 and 21772226), and the Fundamental Research Funds for the Central Universities 222201717003.Notes and references
- (a) L. h. Li and W. K. S. Chua, Tetrahedron Lett., 2011, 52, 1574 CrossRef CAS; (b) V. Tyagi, S. Khan, A. Giri, H. M. Gauniyal, B. Sridhar and P. M. S. Chauhan, Org. Lett., 2012, 14, 3126 CrossRef CAS PubMed; (c) J. Chen, H. Peng, J. He, X. Huan, Z. Miao and C. Yang, Bioorg. Med. Chem. Lett., 2014, 24, 2669 CrossRef CAS PubMed; (d) E. Kiselev, N. Empey, K. Agama, Y. Pommier and M. Cushman, J. Org. Chem., 2012, 77, 5167 CrossRef CAS PubMed; (e) P. G. Jagtap, E. Baloglu, G. Southan, W. Williams, A. Roy, A. Nivorozhkin, N. Landrau, K. Desisto, A. L. Salzman and C. Szabó, Org. Lett., 2005, 7, 1753 CrossRef CAS PubMed; (f) A. Saeed and Z. Ashraf, Pharm. Chem. J., 2008, 42, 277 CrossRef CAS; (g) J. H. Rigby, U. S. M. Maharoof and M. E. Mateo, J. Am. Chem. Soc., 2000, 122, 6624 CrossRef CAS; (h) V. A. Glushkov and Y. V. Shklyaev, Chem. Heterocycl. Compd., 2001, 37, 663 CrossRef CAS.
- G. Tan, X. Huang, Q. Wu, L.-Q. Zhang and J. You, RSC Adv., 2014, 4, 49186 RSC . You's work revealed that more reaction time (16 h) was required when the methoxyl group was used as the leaving group.
- (a) D. Davyt and G. Serra, Mar. Drugs, 2010, 8, 2755 CrossRef CAS PubMed; (b) Z. Jin, Nat. Prod. Rep., 2006, 23, 464 RSC; (c) S. Heng, K. R. Gryncel and E. R. Kantrowitz, Bioorg. Med. Chem., 2009, 17, 3916 CrossRef CAS PubMed; (d) J. A. Bull, E. P. Balskus, R. A. J. Horan, M. Langner and S. V. Ley, Angew. Chem., Int. Ed., 2006, 45, 6714 CrossRef CAS PubMed; (e) I. E. Kibriz, M. Sacmaci, E. Sahin and I. P. Yildirim, Tetrahedron, 2017, 73, 1795 CrossRef CAS; (f) X. Wu, X. Geng, P. Zhao, J. Zhang, Y. Wu and A. Wu, Chem. Commun., 2017, 53, 3438 RSC; (g) C. Wan, L. Gao, Q. Wang, J. Zhang and Z. Wang, Org. Lett., 2010, 12, 3902 CrossRef CAS PubMed; (h) G. Jégou and S. A. Jenekhe, Macromolecules, 2001, 34, 7926 CrossRef; (i) H. Tong, L. Wang, X. Jing and F. Wang, Macromolecules, 2003, 36, 2584 CrossRef CAS; (j) V. S. C. Yeh, Tetrahedron, 2004, 60, 11995 CrossRef CAS.
- (a) J. M. Atkins and E. Vedejs, Org. Lett., 2005, 7, 3351 CrossRef CAS PubMed; (b) E. Vedejs and D. A. Barda, Org. Lett., 2000, 2, 1033 CrossRef CAS PubMed; (c) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084 CrossRef CAS PubMed.
- (a) Y. Luo, K. Ji, Y. Li and L. Zhang, J. Am. Chem. Soc., 2012, 134, 17412 CrossRef CAS PubMed; (b) I. Cano, E. Alvarez, M. C. Nicasio and P. J. Perez, J. Am. Chem. Soc., 2011, 133, 191 CrossRef CAS PubMed; (c) P. W. Davies, A. Cremonesi and L. Dumitrescu, Angew. Chem., Int. Ed., 2011, 50, 8931 CrossRef CAS PubMed; (d) A. D. Gillie, R. J. Reddy and P. W. Davies, Adv. Synth. Catal., 2016, 358, 226 CrossRef; (e) J. Zheng, M. Zhang, L. Huang, X. Hu, W. Wu, H. Huang and H. Jiang, Chem. Commun., 2014, 50, 3609 RSC; (f) Y. Hu, R. Yi, F. Wu and B. Wan, J. Org. Chem., 2013, 78, 7714 CrossRef CAS PubMed; (g) W. He, C. Li and L. Zhang, J. Am. Chem. Soc., 2011, 133, 8482 CrossRef CAS PubMed; (h) C. Wan, J. Zhang, S. Wang, J. Fan and Z. Wang, Org. Lett., 2010, 12, 2338 CrossRef CAS PubMed; (i) X. Li, L. Huang, H. Chen, W. Wu, H. Huang and H. Jiang, Chem. Sci., 2012, 3, 3463 RSC; (j) D. R. Williams and L. Fu, Org. Lett., 2010, 12, 808 CrossRef CAS PubMed; (k) D. Bonne, M. Dekhane and J. P. Zhu, Angew. Chem., Int. Ed., 2007, 46, 2485 CrossRef CAS PubMed.
- For some selected reviews, see: (a) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084 CrossRef CAS PubMed; (b) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed.
- X.-L. Han, C.-J. Zhou, X.-G. Liu, S.-S. Zhang, H. Wang and Q. Li, Org. Lett., 2017, 19, 6108 CrossRef CAS PubMed . According to Li and co-workers, they did not find that Sc(OTf)3 could catalyze this transformation at room temperature.
- (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed; (b) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef PubMed; (c) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (d) B. Liu, F. Hu and B.-F. Shi, ACS Catal., 2015, 5, 1863 CrossRef CAS; (e) J.-W. Xu, Z.-Z. Zhang, W.-H. Rao and B.-F. Shi, J. Am. Chem. Soc., 2016, 138, 10750 CrossRef CAS PubMed; (f) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 121, 5196 CrossRef; (g) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308 CrossRef CAS PubMed; (h) J. Wencel-Delord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (i) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 125, 11942 CrossRef; (j) Y. Zhao, S. Li, X. Zheng, J. Tang, Z. She, G. Gao and J. You, Angew. Chem., Int. Ed., 2017, 56, 4286 CrossRef CAS PubMed; (k) Z. Long, Y. Yang and J. You, Org. Lett., 2017, 19, 2781 CrossRef CAS PubMed.
- The crystal data of 3aa have been deposited in CCDC with number 1545434.†.
- G. Evano, M. Lecomte, P. Thilmany and C. Theunissen, Synthesis, 2017, 3183 CrossRef CAS.
- The crystal data of 4aa have been deposited in CCDC with number 1545423.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds. CCDC 1545423 and 1545434. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qo00125a |
| This journal is © the Partner Organisations 2018 |