
Development of endogenous enzyme-responsive nanomaterials for theranostics
Jing
Mu
 ab,
Jing
Lin
a,
Peng
Huang
*a and
Xiaoyuan
Chen
*b
ab,
Jing
Lin
a,
Peng
Huang
*a and
Xiaoyuan
Chen
*b
aGuangdong Key Laboratory for Biomedical Measurements and Ultrasound Imaging, Laboratory of Evolutionary Theranostics, School of Biomedical Engineering, Health Science Center, Shenzhen University, Shenzhen 518060, China. E-mail: peng.huang@szu.edu.cn
bLaboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, Maryland 20892, USA. E-mail: shawn.chen@nih.gov
First published on 1st June 2018
Abstract
The development of stimuli-responsive nanomaterials provides great potential for accurate diagnosis, effective treatment and precision theranostics. Among the sources of endogenous stimuli (e.g. enzymes, pH, redox, hypoxia, etc.) and exogenous stimuli (e.g. temperature, light, magnetic field, ultrasound, light, etc.), enzymes with intrinsic merits such as high relevance for numerous diseases, specific substrate selectivity and high catalytic efficiency have been widely employed for the design of responsive materials. The catalytic mechanisms mainly include the reduction/oxidation of substrates and the formation/cleavage of chemical bonds. So far, many enzymes such as proteases, phosphatases, kinases and oxidoreductases have been used in stimuli-responsive nanomaterials for theranostics. This tutorial review summarizes the recent progress in endogenous enzyme-responsive nanomaterials based on different building blocks such as polymers, liposomes, small organic molecules, or inorganic/organic hybrid materials; their design principles are also elaborated. In the end, the challenges and prospects of enzyme-responsive biomaterials-based theranostics are also discussed.
 Jing Mu | Jing Mu received her PhD in Chemistry and Biological Chemistry from Nanyang Technological University, Singapore in 2017. She then worked with Prof. Peng Huang and Prof. Xiaoyuan (Shawn) Chen at Shenzhen University (SZU) and National Institutes of Health (NIH) as a postdoctoral fellow. Her research interest focuses on the design, synthesis, and biomedical applications of enzyme-responsive theranostics. |
 Jing Lin | Jing Lin received her PhD in Organic Chemistry from the Donghua University and Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences in 2010. She then joined the PharmaResources (Shanghai) Co., Ltd as a group leader. After two years, she moved to the United States of America and spent 4 years as a postdoctoral fellow at the University of Maryland and the National Institutes of Health (NIH). She joined the faculty of Shenzhen University (SZU) in 2016 and was promoted to Distinguished Professor in 2018. Her research focuses on the self-assembly of functional nanomaterials for diagnosis, treatment, and theranostics of diseases. |
 Peng Huang | Peng Huang received his PhD in Biomedical Engineering from the Shanghai Jiao Tong University in 2012. He then joined the Laboratory of Molecular Imaging and Nanomedicine (LOMIN) at the National Institutes of Health (NIH) as a postdoctoral fellow. In 2015, he moved to Shenzhen University (SZU) as a Distinguished Professor, Chief of the Laboratory of Evolutionary Theranostics (LET), and Director of the Department of Molecular Imaging. His research focuses on the design, synthesis, and biomedical applications of molecular imaging contrast agents, stimuli-responsive programmed targeting drug delivery systems, and activatable theranostics. |
 Xiaoyuan Chen | Xiaoyuan (Shawn) Chen received his PhD in Chemistry from the University of Idaho in 1999. He joined the University of Southern California as an Assistant Professor of Radiology in 2002. He then moved to Stanford University in 2004 and was promoted to Associate Professor in 2008. In the summer of 2009, he joined the Intramural Research Program of the NIBIB as a tenured Senior Investigator and Chief of the LOMIN. He has published over 650 papers and numerous books and book chapters. He is the founding editor-in-chief of the journal Theranostics. He is interested in developing molecular imaging tools for the early diagnosis of disease, monitoring therapy response, and guiding nanodrug discovery/development. |
Key learning points(1) Overview of the main endogenous enzymes as stimuli and their essential functions under pathological conditions.(2) Guidelines for the construction of enzyme-responsive nanomaterials based on different building blocks such as polymers, liposomes, small organic molecules, and inorganic/organic hybrid materials. (3) The principles of endogenous enzyme-responsive nanosystems for theranostics. (4) Directions to achieve enzyme-based multiple responsive systems for theranostics. (5) The challenges and opportunities of enzyme-responsive biomaterials-based theranostics. |
1. Introduction
Over the past decades, tremendous efforts have been made to fight against cancer. With the development of science and technology, significant progress has been achieved in cancer diagnosis and treatment.1,2 However, there are still questions as to how to precisely distinguish tumour cells from normal cells and effectively eliminate all cancer cells while leaving normal cells unharmed.In recent years, the flourishing field of nanotechnology has brought about a burst of research activities in nanomedicine. Various nanomedicines have been investigated in attempts to address biomedical challenges faced by traditional medicine.3,4 These have been constructed based on different nanomaterials, such as polymeric nanoparticles (NPs), liposomes, inorganic NPs, and so on.5–9 Due to their unique physicochemical properties, a variety of organic or inorganic nanomaterials could be combined with multiple types of therapeutic and imaging contrast agents to serve as ideal platforms for theranostics.10 The theranostic platforms based on the ‘all-in-one’ approach aim to achieve early disease detection, optimize treatment and monitor therapeutic response in time, thus improving the outcomes and safety of patients.10–12
Although theranostic platforms have great potential for personalized medicine, their selectivity and specificity for different diseases remain a big challenge. Therefore, the development of stimuli-responsive systems, whose structure conformations or physicochemical properties can be changed in response to the corresponding milieu, could potentially overcome this challenge.13–15 These stimuli-responsive nanoplatforms can be designed not only to be sensitive to the physiological or pathological variations in the disease sites such as enzymes, pH, redox, hypoxia, etc., but also to have response to the external stimuli like temperature, light, magnetic field, ultrasound, light, etc.13,16,17 As a representative endogenous stimulus, enzymes are involved in a variety of key physiological processes and exhibit altered expression levels in many disease-associated microenvironments. For example, several enzymes such as proteases and phosphatases present high expression levels, which have been considered as biomarkers for the diagnosis and treatment of cancer, inflammation, and neurodegeneration.16 Therefore, the development of endogenous enzyme-responsive nanomaterials is a hotspot.
Enzyme-responsive nanomaterials, which combine the advantages of the specific enzyme stimuli and fascinating properties of nanomaterials, provide great opportunities to improve the on-demand delivery, tumour accumulation and efficient cellular internalization of imaging contrast agents or therapeutic agents.18–20 In general, utilizing enzymes as stimuli present a lot of advantages compared to other types of stimuli. Enzymes are endogenous and there is no need to add external stimuli such as a magnetic field, ultrasound or light, which offers the merits of inherent biocompatibility and biosafety. In addition, most enzymatic reactions are fast, highly efficient and reaction conditions are moderate (mild temperature, pH and aqueous solutions). Enzymes can also present extraordinary specificity and selectivity for their substrates, allowing for controlled chemical reactions and biological responses.21
So far, proteases, esterases, phosphatases, kinases and oxidoreductases are the most widely reported enzyme classes as stimuli. For instance, proteases are able to break down proteins or peptide substrates. Oxidoreductases can catalyse the electron transfer from the reductant to the oxidant. Kinases modulate the activities of proteins through the phosphorylation process while the opposite action, dephosphorylation, is mediated by phosphatases. With the increasing in-depth understanding of the enzyme activities, many enzymes are being applied as biomarkers for the diagnosis or prognosis of tumours.22,23 The diagnostic probes can be designed by linking the enzyme-recognizing substrate with the imaging reporters. After enzymatic processing of the substrate by the specific enzyme, the probes can recover their signals (like fluorescence, photoacoustic, etc.) from the “off” to “on” state. In another important application, enzyme-sensitive pro-drugs are explored to achieve the selective activation of therapeutic drugs in the target tissue, while reducing toxic side effects.24,25
In this review, we summarize the recent advances in the development of endogenous enzyme-responsive nanomaterials for theranostics, based on polymers, liposomes, small organic molecules, inorganic/organic hybrid materials, and others (Fig. 1). We aim to shed light on the design principles of each type of platform and their unique advantages in the diagnosis, treatment and theranostics of diseases. The challenges of the current strategies and possible future directions of enzyme-responsive biomaterials-based theranostics are also discussed.
 | ||
| Fig. 1 Enzyme-responsive nanomaterials for theranostics. | ||
2. Typical enzymes as stimuli
Several key enzymes that have been explored to design stimuli-responsive systems and their corresponding biomedical applications are summarized in Table 1. In addition, the characteristics, preferred distribution and essential functions of representative enzymes related to different diseases, especially cancer, are also discussed in this section, such as matrix metalloproteinases (MMPs), cathepsins, cysteine caspases, phospholipases, hyaluronidases, and so on.| Stimuli-responsive nanomaterials | Enzymes | Substrates | Applications | Ref. |
|---|---|---|---|---|
| Polymeric nanoparticles | MMPs | GPLGLAGGWGERDGS | Fluorescence imaging | 30 |
| GCNSGGRMSMPVSNGG | Drug delivery | 36 | ||
| Azoreductase | Azo bond | Drug delivery | 31 | |
| Cathepsin B | GFLGKGLFG | Drug delivery | 33 | |
| Esterase | Ester bond | Drug delivery | 50 | |
| Liposomes | Phospholipases (sPLA2) | Fatty ester bond of glycerophospholipids | Drug delivery | 38 |
| Small molecules | Caspase-3/-7 | DEVD | Fluorescence imaging | 39 and 40 |
| Theranostics | ||||
| Alkaline phosphatase (ALP) | Dephosphorylation | Fluorescence imaging | 41–43 | |
| Theranostics | ||||
| Furin | RVRR | Fluorescence imaging | 25 and 44 | |
| Photoacoustic imaging | ||||
| Inorganic/organic hybrids | Telomerase | DNA sequence | Fluorescence imaging | 46 |
| Cathepsin B | GFLG | Theranostics | 45 | |
2.1 Matrix metalloproteinases (MMPs)
Matrix metalloproteinases (MMPs) are the most well-established enzymes employed as stimuli for the design of enzyme-responsive systems. MMPs are a family of zinc-dependent endopeptidases, which participate in the degradation of extracellular matrixes (ECM) and the modulation of bioactive molecules on the cell surface. Most MMPs are initially secreted in the inactive state, termed as zymogens. Proteolytical cleavage by tissue, plasma proteinases or other MMPs allows the removal of the pro-peptide domain and exposure of catalytic zinc pocket.23 According to the structure and substrate specificity, the 23 known human gene families can be divided into six sub-groups. (1) Collagenases: MMP-1, -8, -13; (2) gelatinases: MMP-2, -9; (3) stromelysins: MMP-3, -10, -11; (4) matrilysins: MMP-7, -26; (5) membrane-type MMPs (MT-MMP): MMP-14 to -17, -24, -25; (6) other MMPs: the remaining MMPs.Apart from the regulatory functions under physiological conditions, MMPs are also associated with a series of biological disorders like cardiovascular disease, arthritis and tumour invasion and metastasis.23 Among these MMPs, gelatinase-A (MMP-2) and gelatinase-B (MMP-9) can efficiently degrade the basement membrane and collagens. MMP-2 and MMP-9 can be activated by MMP-7. MMP-7 can be produced by epithelial cells in the glandular structures of several types of tumours. MMP-11 is produced in the stromal component of malignant tumours. Most of the MT-MMPs are known to anchor to the cell membrane with the transmembrane domain in their C-terminus. The comprehensive review on MMPs and their significant roles in cancer can be found elsewhere.23 Due to the severe side effects of MMP inhibitors, several clinical trials have failed in cancer and arthritis treatment. The specificity of MMP inhibitors needs to be further improved.
2.2 Cysteine caspases
Caspases are intracellular cysteine proteases that are implicated in cell death and inflammation. Caspases are usually categorized by their involvement in apoptosis (caspase-3, -6, -7, -8, and -9), and in inflammation (caspase-1, -4, -5, -12). Other caspases like caspase-2, -10, and -14 are not well investigated. Apoptosis is programmed cell death, during which the cell membrane integrity is maintained to avoid inflammation and the damage to surrounding cells. The initial apoptosis process is the activation and dimerization of caspase-8 and -9. These initiator caspases then activate the inactive forms of the executioner caspases-3, -6, and -7, to get the functional mature proteases. Consecutively, the matured proteases can facilitate the activation of other executioner caspases to form the loop of the caspase activation.26 Considering their essential roles in regulating cell death, numerous apoptosis-inducing compounds have been developed in cancer therapy. However, continuing efforts are required to further improve their in vivo delivery and specificity.2.3 Cathepsins
The cysteine cathepsins, which are predominantly located in endo/lysosomal vesicles, are also validated as important drug discovery targets for infectious diseases and cancer.27 This group consists of cathepsins B, C, F, H, K, L, O, S, V, W and X. Among these cysteine cathepsins, cathepsin B is one of most well-studied lysosomal proteases due to its high expression in various types of cancers including breast, lung, prostate, colorectum and endometrium. The secreted cathepsins B and L have been known to participate in the degradation of type IV collagen, laminin and fibronectin, resulting in cell migration. It was reported that the depletion of cathepsin B can reduce cell proliferation, migration, angiogenesis and invasion in vivo. Therefore, cathepsin B may be considered as a potential biomarker in cancer therapy. Besides, cathepsin X has been shown to be associated with carcinomas and gastric cancers. Additionally, cathepsin K has high expression levels in osteoclasts and plays key roles in bone remodelling and resorption.272.4 Hyaluronidases (HAases)
The extracellular matrix (ECM) is highly relevant to the tumour cell growth, metastasis and angiogenesis. As one of the most widely distributed ECM components, hyaluronic acid (HA), a biocompatible and natural polysaccharide, is reported to participate in cell proliferation, tissue hydration, and cell motility by interacting with the cell surface receptors like the CD44 gene encoded glycoprotein and receptor for hyaluronan-mediated motility (RHAMM). It is reported that several types of cancers, including bladder, prostate, ovary, colon, lung and so on, present increased HA levels. Hyaluronidases (HAases) are a family of enzymes that catalyse the hydrolysis of HA. HAases are known to be involved in bacterial infections and toxins in various venoms. In human beings, they support tumour cell migration and help to escape immune surveillance, leading to increased tumour growth and metastasis. Also, they are associated with the expression of CD44 isoforms and increased tumour cell cycling.28 Considering the abundant distribution of HA in the ECM, it usually tends to be employed to design enzyme-activatable theranostic agents.2.5 Phospholipases
Phospholipases such as secreted phospholipase A2 (sPLA2) are gaining much interest as drug targets for their upregulation in the inflammatory and infectious diseases. The sPLA2 is a Ca2+-dependent esterase that can hydrolyse the fatty ester group at the sn-2 position of glycerophospholipids. It is overexpressed in inflammatory diseases, atherosclerosis, and several types of cancers.29 Particularly, it is reported that the expression level of PLA2 IIA in prostate cancer is an order of magnitude higher than in normal tissues. Due to the upregulated levels in the tumour environment, several sPLA2-sensitive lipoid prodrugs have been developed by the replacement of the acyl chain at the sn-2 position with lipophilic drugs; e.g. chlorambucil, prostaglandins and retinoic acid. These prodrugs are more stable and can selectively release drugs once they accumulate in the lesion location with high concentration levels of sPLA2.292.6 Other enzymes
Besides the above-mentioned enzymes, other enzymes such as esterase, urokinase, kinases, gelatinase, prostate-specific antigen (PSA), telomerase, and so on, which are irregulated in the lesion location, are also applied as stimuli for diagnosis, treatment, and theranostic purposes. More details about the design principles can be found in previous reports.18,213. Endogenous enzyme-responsive nanomaterials for theranostics
To obtain enzyme-responsive materials, three basic requirements should be considered. Firstly, enzyme-activatable moieties need to be incorporated into the structure of nanomaterials. Since many enzyme substrates are small peptides or proteins, peptide sequences as recognition elements are widely employed to construct the enzyme-catalysed systems. Secondly, in most cases, two types of reactions, chemical bond formation or cleavage and substrate reduction or oxidation, are necessary for the design of enzyme-responsive nanomaterials during the enzymatic reactions. Thirdly, the enzyme-triggered action needs to induce a change in the properties of the nanomaterials. The following sections will illustrate the recent advances in enzyme-responsive nanomaterials used in biomedical applications. By using versatile materials such as polymers, liposomes, small-molecules or hybrids as the building nanosystems, different responsive approaches with physicochemical changes in response to the corresponding stimuli are discussed.3.1 Polymer-based nanomaterials
In the past few decades, tremendous efforts have been made to develop enzyme-responsive polymer-based nanomaterials.5 Typically, enzyme-catalysed reactions can tune the physicochemical properties through the covalent bond cleavage and non-covalent interactions such as electrostatic interactions, van der Waals forces, hydrophobic interactions or hydrogen bonding, resulting in transformations or transitions of synthetic polymers. Herein, the enzyme-responsive polymeric nanomaterials are mainly categorized into three groups: enzyme-responsive polymer assembly, disassembly and hydrogels.In the design of the enzyme-responsive polymer assembly, Gianneschi and co-workers developed amphiphilic block copolymers composed of a hydrophobic backbone and a hydrophilic MMP-responsive peptide (Fig. 2A and B).30 Tumour overexpressed MMP-catalysed peptide cleavage causes the morphology change in polymeric nanoparticles from spherical micelles to micrometer aggregates. The enzyme-driven size increase of polymeric NPs with the tissues was further verified by ex vivo super-resolution fluorescence imaging, using stochastic optical reconstruction microscopy (STORM) (Fig. 2C). Moreover, when this enzyme-responsive material was intratumourally injected into the HT-1080 xenograft, long tumour retention for up to one week was observed. Conversely, non-responsive polymeric NPs showed rapid clearance within one-day post-injection (Fig. 2D).
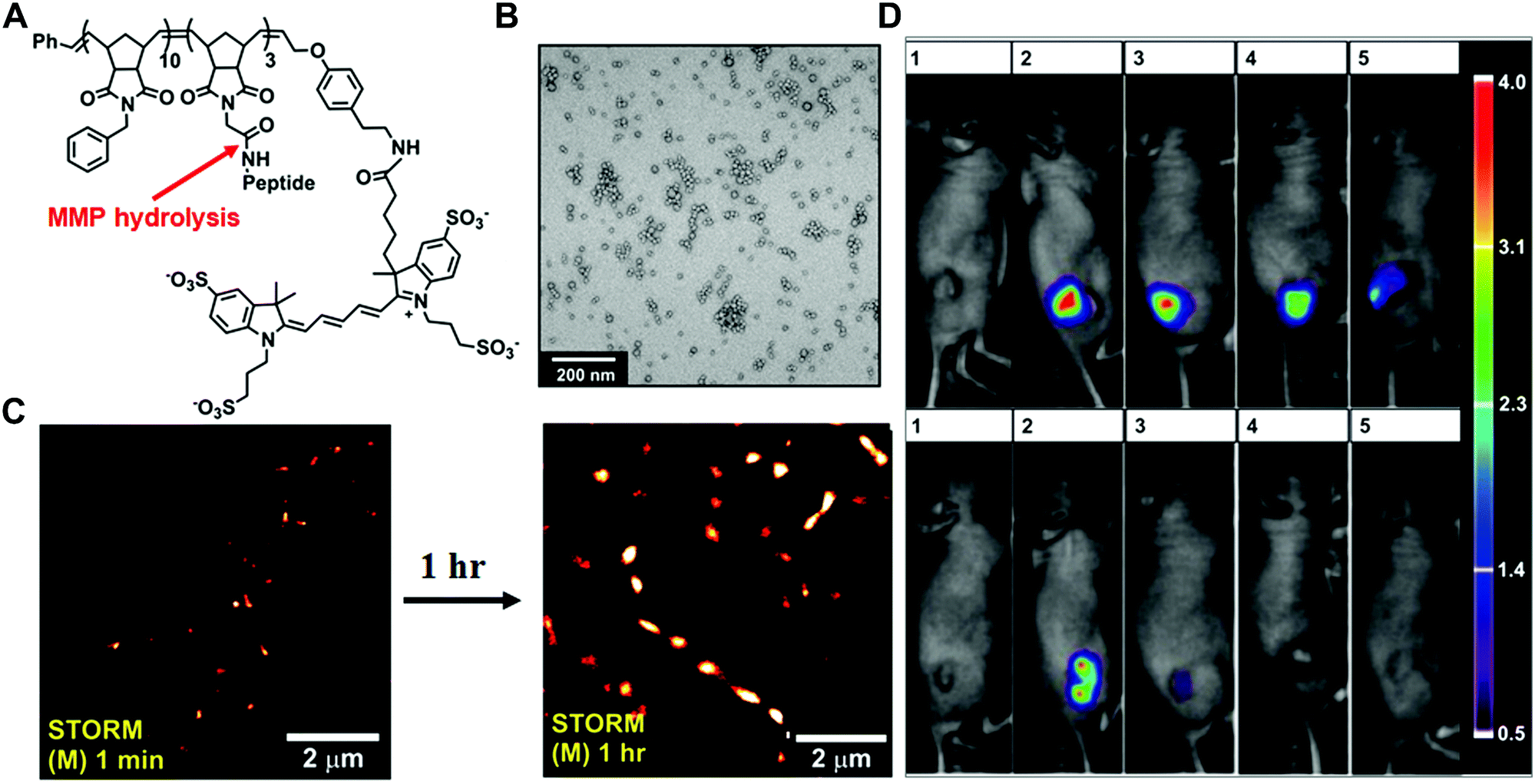 | ||
| Fig. 2 (A) The chemical structure of Alexa Fluor 647 dye terminated block copolymer; peptide sequence: GPLGLAGGWGERDGS. (B) Transmission electron microscopy (TEM) image of the micellar nanoparticles. (C) STORM images of tissue slices from micellar NPs intra-tumourally injected mice. (D). Retention of enzyme-responsive nanoparticles (upper panel) vs. non-responsive particles (lower panel) in tumours. Reprinted with permission from ref. 30. Copyright 2013, American Chemical Society. | ||
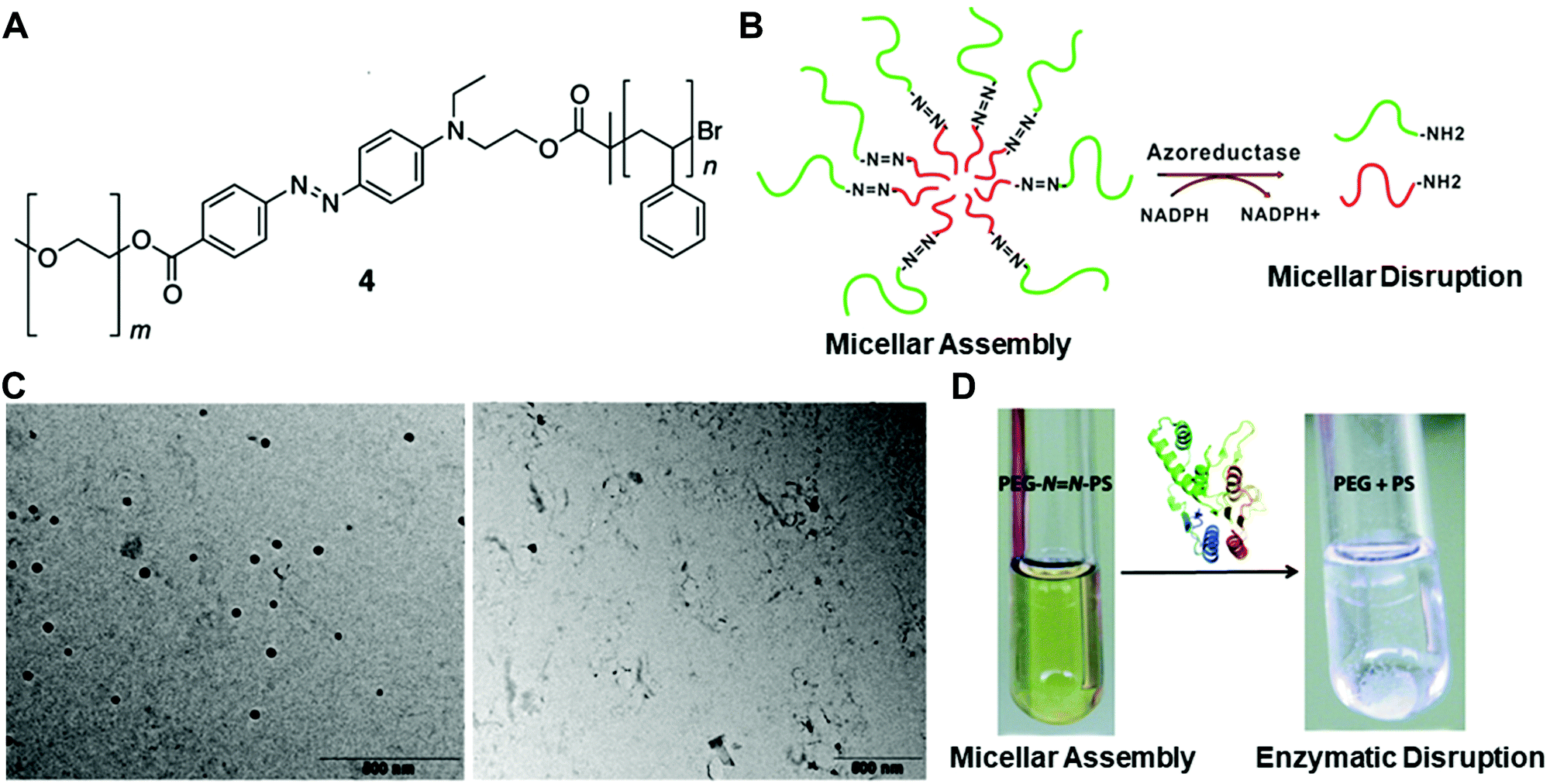 | ||
Fig. 3 (A) The chemical structure of the azobenzene-linked block copolymer. (B) Schematic illustration of the PEG–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–PS block copolymer-formed micelles and the azoreductase-induced disassembly into PEG and PS polymer segments. TEM images (C) and digital pictures (D) of the copolymer with or without the enzyme treatment. Reprinted with permission from ref. 31. Copyright 2013, American Chemical Society. N–PS block copolymer-formed micelles and the azoreductase-induced disassembly into PEG and PS polymer segments. TEM images (C) and digital pictures (D) of the copolymer with or without the enzyme treatment. Reprinted with permission from ref. 31. Copyright 2013, American Chemical Society. | ||
Besides the traditional linear and branched polymers, dendrimers with a tree-like structure and excellent monodispersity could also be designed as enzyme-responsive systems. For example, Harnoy et al. reported a smart micellar nanostructure based on a linear hydrophilic PEG and an enzyme-responsive hydrophobic dendron.32 The amphiphilic PEG–dendron hybrid could be self-assembled into micelles with a hydrophobic core, allowing the encapsulation of cargo molecules. In the presence of the penicillin G amidase (PGA) enzyme, the hydrophobic end group of phenylacetamide could be cleaved, leading to the disassembly of hybrids and the release of loaded Nile red molecules. The fluorescence intensity decrease in the PEG–dendron hybrids was observed due to the release of the dye molecules into the aqueous environment. They further investigated the mechanisms of disassembly and found that the enzyme could not penetrate the hydrophobic core to interact with the cleavable unit. Instead, the enzyme activates the monomeric form of the hybrid, which is in equilibrium with the assembled micelles. Such enzyme-responsive hybrids have great potential in drug delivery.
The enzymatic polymer disassembly can also be designed by introducing the enzyme-sensitive peptide moieties into the main chain of the polymers. For instance, Gu and co-workers prepared the amphiphilic triblock poly[N-(2-hydroxypropyl)methacrylamide] (HPMA) copolymer–doxorubicin (DOX) conjugates as drug delivery vehicles.33 The cathepsin B or papain enzyme-sensitive peptide (Gly-Phe-Leu-Gly-Lys-Gly-Leu-Phe-Gly, GFLGKGLFG) was introduced into the backbone of the polymers. The HPMA–DOX conjugate could be self-assembled into compact NPs in aqueous solution. Compared to free DOX, the polymeric NPs presented higher antitumour efficacy on the 4T1 murine breast cancer model with lower side effects to normal organs. This enzyme-responsive polymer–drug conjugate may offer a potentially useful strategy for safe and efficient drug delivery.
Several enzymes have been utilized in the in situ formation of chemically crosslinked hydrogels. For instance, Barron and co-workers reported a transglutaminase (TGase) sensitive system, which crosslinks the naturally derived polypeptide-gelatin.34 They developed a thermally induced chemical gelation system composed of calcium-loaded liposomes, hrFactor XIII (one type of TGase), thrombin, and the substrate based on a four-armed PEG. The thermally triggered liposome phase transition could release calcium ions to activate the hrFXIII, followed by the enzymatic crosslinking of diblock copolymers and hydrogel formation. Besides, another well-established strategy for the hydrogel formation is based on the oxidative coupling of phenols using horseradish peroxidase (HRP) and H2O2. A variety of polymers (such as hyaluronic acid, dextran, alginate, etc.) functionalized with tyramine, tyrosine, or aminophenol moieties have been developed for the crosslinked hydrogel triggered by enzyme reactions.35
In contrast to enzyme-responsive hydrogel formation, the degradation of polymer hydrogels is also a common approach in the enzyme triggered release of therapeutics. In a recent study, Burdick and co-workers demonstrated MMP-degradable hydrogels for the on-demand MMP inhibition in a porcine model of myocardial infarction.36 In this system, they developed an HA-based hydrogel containing the recombinant tissue inhibitor of MMPs (rTIMP-3). When the hydrogel was locally injected into the tissue where the MMP was overexpressed, the crosslinks were degraded by MMP to liberate the rTIMP-3, leading to the inhibition of MMP activity and attenuated adverse left ventricular remodelling.
Besides the enzyme-responsive hydrogel formation and degradation, the Ulijn group developed an enzyme-responsive crosslinked hydrogel that undergoes charge-induced polymer swelling when exposed to the target enzyme (Fig. 4).37 They synthesized the copolymers of polyethylene glycol and acrylamide (PEGA) with the zwitterionic enzyme-responsive peptide. The enzyme-cleavable linker (ECL) is flanked by two oppositely charged amino acids (aspartic acid and arginine) (Fig. 4A). Upon enzyme hydrolysis, the negatively charged carboxylic acid fragment is separated from the positively charged amine fragment linked with PEGA polymers, resulting in the hydrogel swelling and the release of the encapsulated molecules (Fig. 4B). They used diglycine or dialanine ECLs as examples and tested the different enzyme responsiveness by using three methods: (i) the accessibility of hydrogel beads to various sizes of fluorescently labelled dextran molecules by two-photon microscopy (TPM) (Fig. 4C); (ii) mean particle diameters of dextran molecules admitted into the beads; (iii) quantification of cleaved peptide fragments (Fig. 4D). These results indicated that the dialanine ECLs could be hydrolysed by thermolysin and elastase, while diglycine ECLs were preferentially hydrolysed by thermolysin.
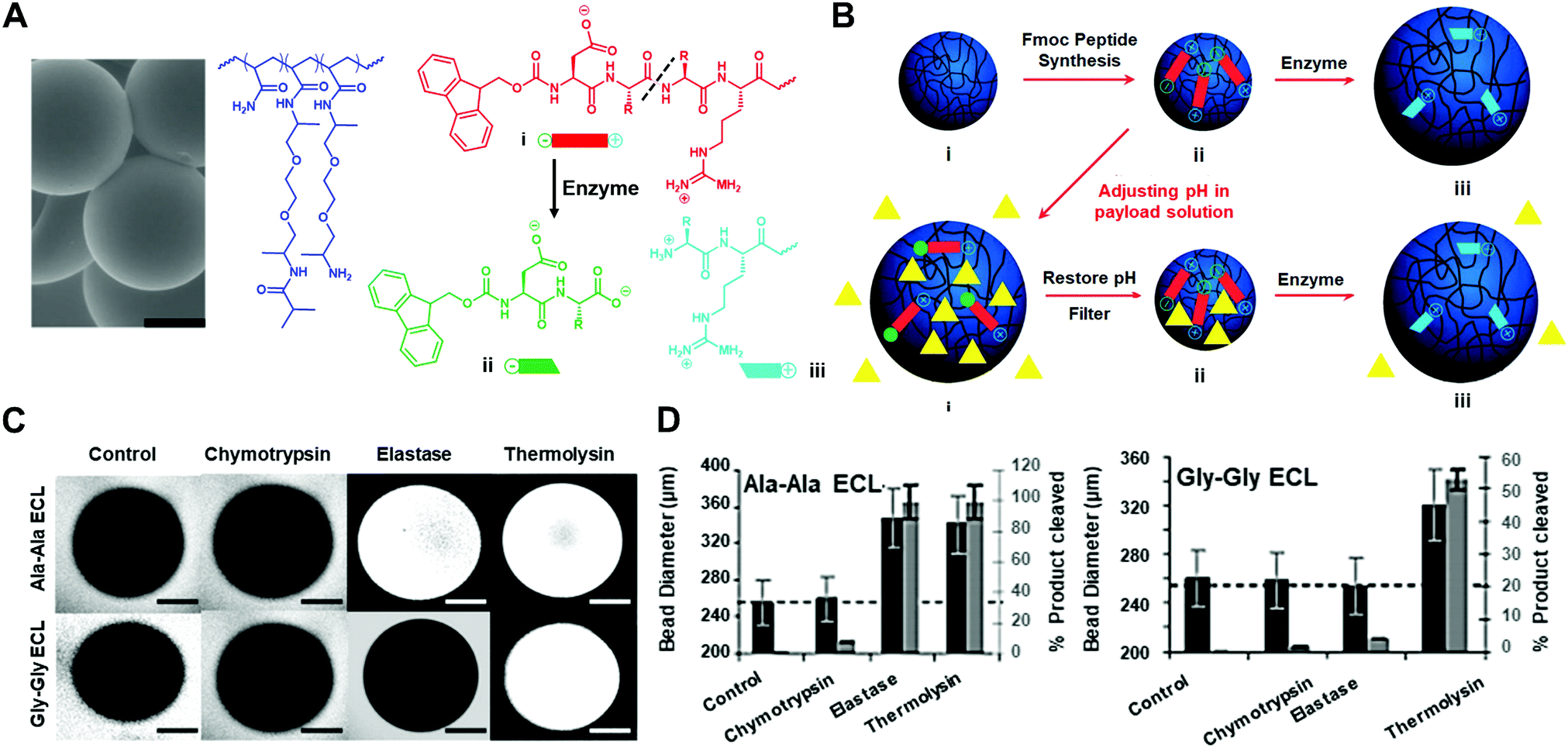 | ||
| Fig. 4 (A) The scanning electron microscope (SEM) image of PEGA hydrogel particles and the molecular structure of PEGA, enzyme-cleavable linker (ECL) and the cleavage product (scale bar: 100 μm). (B) Illustration of payload encapsulation by adjusting the pH and enzyme-responsive release behavior. (C) Two-photon microscopy images for analysis of the accessibility of fluorescently labelled dextran molecules into hydrogel particles. (D) Hydrolysis evaluation of dialanine and diglycine ECLs by different enzymes. Reprinted with permission from ref. 37. Copyright 2007, Wiley-VCH. | ||
As discussed in this section, enzyme-triggered polymeric assemblies, disassemblies and hydrogels have been developed for diverse functions including improving the retention time, controlled delivery and release. However, polymer structures with enzyme-responsive features are quite limited. More structural designs in response to various enzyme types will enrich this field. In addition, tumour targeting and the in vivo enzyme catalytic properties of these fabricated polymers have not been fully tested.
3.2 Liposome-based nanomaterials
Besides polymer-based enzyme-responsive nanomaterials, liposomes, due to their own merits of biodegradability, biocompatibility and weak immune response, are also applied for the construction of enzyme-responsive nanomaterials. Liposomes composed of natural amphiphilic phospholipids are able to encapsulate hydrophilic/lipophilic drugs or nucleic acids, thus improving their pharmacodynamics and pharmacokinetics and reducing their off-target toxicity. Doxil (doxorubicin HCl liposome injection), the first Food and Drug Administration (FDA)-approved nanodrug (in 1995) has been indicated for the treatment of ovarian cancer and Kaposi's sarcoma.12 However, the conventional liposomes usually release the loaded drugs via passive diffusion, which is slow and uncontrollable. Stimuli-responsive liposomes provide possible solutions to release payloads in a tailored manner, greatly enhancing the delivery efficiency and targeting ability.As mentioned in Section 2.5, phospholipase sPLA2 is overexpressed in several types of cancers; it can be employed as the endogenous stimulus to mediate the hydrolysis of phospholipid, thus disrupting the liposome structure to release the payloads. For example, Andersen and co-workers conjugated the model drug capsaicin to the sn-1 position of a glycerophospholipid to obtain the prodrug, which then self-assembled into small unilamellar vesicles (SUVs) (Fig. 5A and B).38 In the presence of human type IIA sPLA2 (sPLA2-IIA), the lipid structure would be hydrolysed at the sn-2 position and the liberated hydroxyl group would react with the ester bond at the sn-1 position to obtain a five-membered lactone and the liberated free drug. Their measurements showed that the amount of capsaicin released from the phospholipids after enzymatic activation could reach up to (90 ± 11)% (n = 3) after 24 h (Fig. 5C and D). Moreover, the prodrugs in the vesicles presented optimal stability in the buffer solutions and did not degrade for at least two weeks.
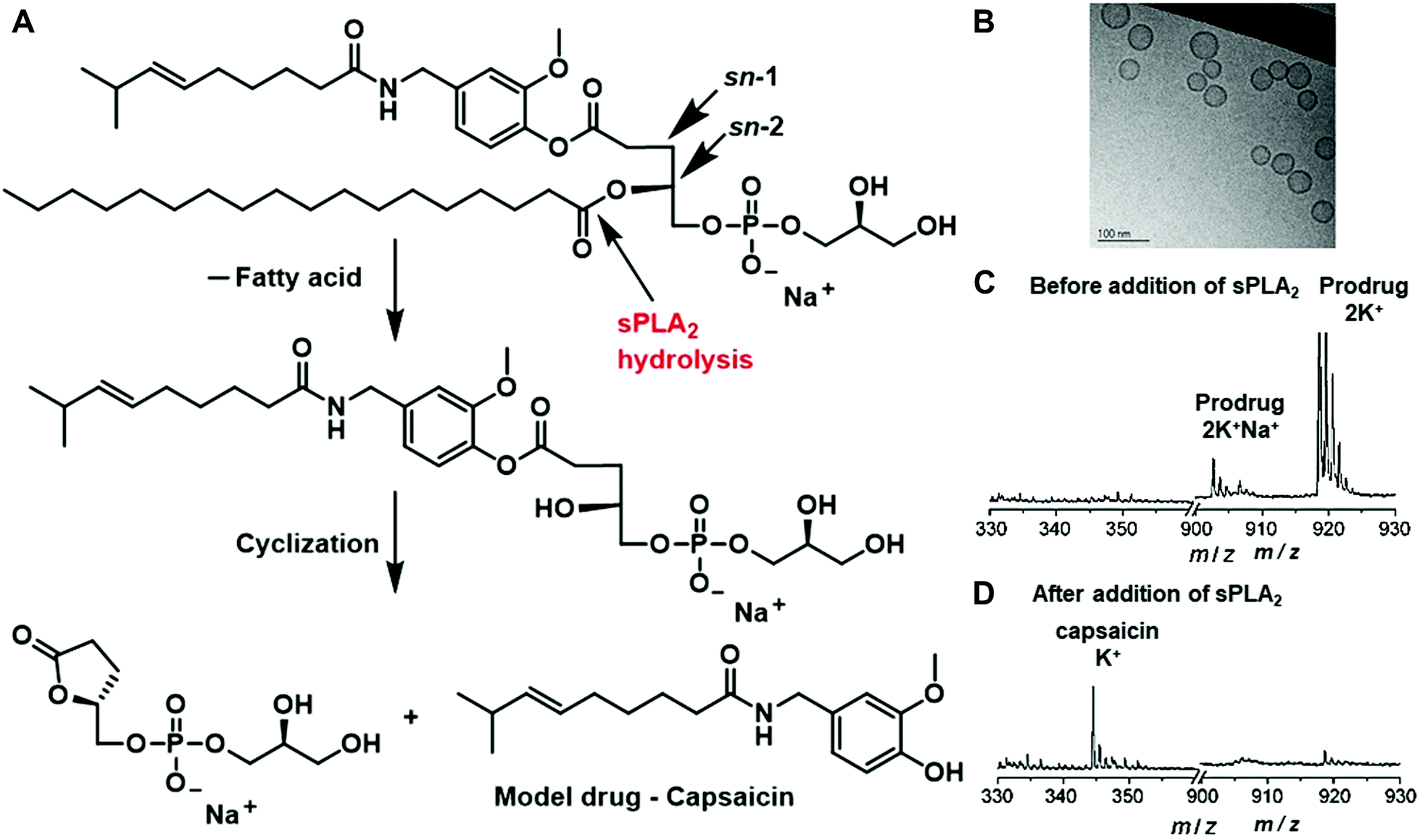 | ||
| Fig. 5 (A) Schematic of the hydrolysis of the lipid prodrug by sPLA2. (B) TEM image of the formed small unilamellar vesicles (SUVs). MALDI-TOF results of the prodrug before (C) and 24 h after (D) the addition of sPLA2-IIA. Reprinted with permission from ref. 38. Copyright 2009 Wiley-VCH. | ||
Overall, liposomes could be regarded as good nanocarriers for the delivery of prodrugs or other therapeutic agents to the diseased area, and sPLA2 could be an ideal drug activator for the site-specific release of payloads. Most of the current strategies involve the in vitro stage and more in vivo studies are necessary for evaluating the applicability and effectiveness of these materials before their translation into clinics. More information on the enzyme-induced liposomal formulations triggered by different extracellular and intracellular enzymes has been provided by the Mallik group.6
3.3 Small-molecule-based nanomaterials
The Tang and Liu groups have made huge efforts toward the design of AIE-based probes and relevant biological applications. In their earlier work, they designed the AIE fluorescence probe to visualize cell apoptosis in real time (Fig. 6).39 The probe was prepared via conjugation between a hydrophilic Asp-Glu-Val-Asp (DEVD) peptide sequence and a tetraphenylethene (TPE) unit with aggregation-induced emission characteristics. This as-prepared probe is soluble and non-fluorescent in aqueous solutions. The DEVD sequence could be specifically cleaved by the caspase-3/-7 enzyme, which is the key indicator of cell apoptosis. The released lysine-conjugated TPE (K-TPE) is very hydrophobic and fluorescence is turned on according to the AIE mechanism (Fig. 6A and B). Cell studies have demonstrated that the probe-treated apoptotic MCF-7 breast cancer cells present strong fluorescence signals whereas normal, uninduced cells or cells pre-treated with caspase inhibitors show low fluorescence signals. Moreover, the probe was applied for the in situ screening of apoptosis-inducing agents, including sodium ascorbate, cisplatin, and staurosporine (STS). The drug efficacy in living cells was quantitively evaluated and their findings showed that STS had a higher inducing efficiency for apoptosis compared to others.
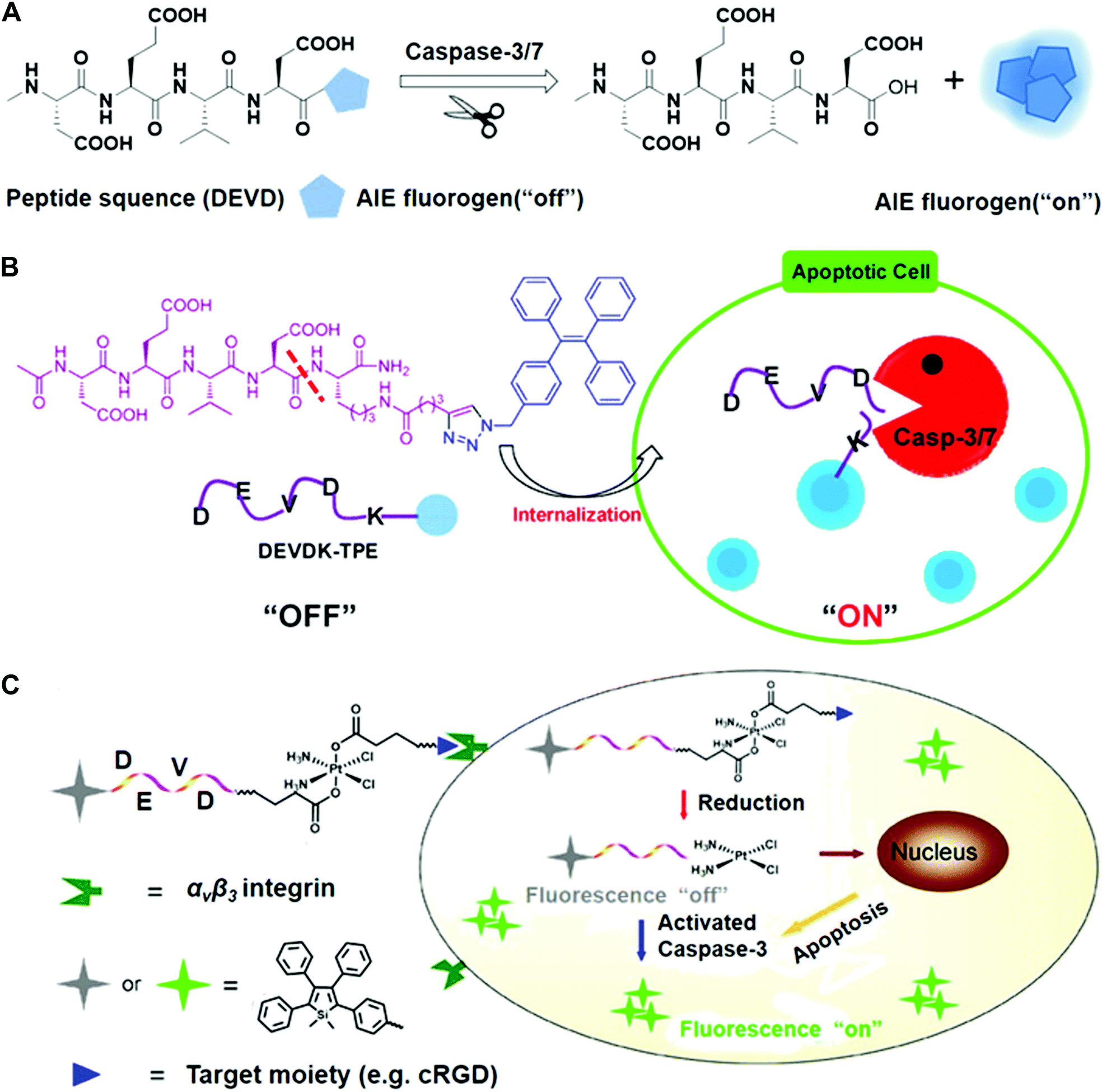 | ||
| Fig. 6 (A) Schematic of AIE fluorogen cleavage by the caspase-3/7 enzyme. (B) Schematic of the Ac-DEVDK-TPE probe for the imaging of caspase activities. Reprinted with permission from ref. 39. Copyright 2014, American Chemical Society. (C) Illustration of the AIE-activatable platinum(IV) prodrug for the evaluation of its apoptotic responses. Reprinted with permission from ref. 40. Copyright 2014, American Chemical Society. | ||
To continue their previous works, they built a targetable theranostic Pt(IV) prodrug linked to an AIE apoptosis sensor. The two axial positions of the Pt(IV) prodrug were conjugated with an apoptosis sensor and a cyclic arginine–glycine–aspartic acid (cRGD) peptide, respectively, for targeting integrin αvβ3 overexpressing cancer cells (Fig. 6C).40 The as-prepared sensor was comprised of the AIE fluorophore tetraphenylsilole (TPS) and caspase-3 enzyme specific peptide substrate (DEVD). When entering cells, the prodrug can be reduced and induce the apoptotic process, followed by the activation of the caspase-3 enzyme. The photoluminescence (PL) spectra showed that the fluorescence of the water-soluble TPS–DEVD–Pt–cRGD molecule was very weak but the aggregated TPS molecules showed intense fluorescence signals. Real-time imaging results showed that the U87-MG human glioblastoma cells treated with the prodrug achieved increased fluorescence while only weak signals were found in the inhibitor pre-treated cells. Furthermore, the generated signals from the prodrug overlapped well with the immunofluorescence signals. It should be mentioned that most developed systems are AIE-based bioprobes with emissions in the visible window. There is still a need for developing novel near-infrared emitters to create the enzyme-sensitive AIE systems.
The pioneering works on the enzyme-triggered formation of fibers or hydrogels were reported by the Xu group.41 They designed the precursor NapFFK(NBD)Yp bearing the fluorophore 4-nitro-2,1,3-benzoxadiazole (NBD) and tyrosine phosphate residue as the alkaline phosphatase (ALP) substrate (Fig. 7A and B). The NBD group is known to show strong fluorescence in a hydrophobic environment and was employed as the fluorescence indicator of the nanofiber formation (Fig. 7B). The dephosphorylation of the precursor catalysed by ALP makes the structure more hydrophobic, resulting in the assembly process and increased fluorescence. In the test of live cell fluorescence imaging, the addition of a hydrogelator precursor (500 μM) to HeLa cells afforded the fast-self-assembled nanofibers network within 5 min (Fig. 7C). Moreover, the localized fluorescence also reported the distribution of phosphatases inside cells. More detailed processes of nanofiber formation revealed that the self-assembly originated in the endoplasmic reticulum (ER) and grew from the ER towards the edge of the cells. To confirm that the protein tyrosine phosphatase (PTP1B) at the ER may play a role in the fiber formation, co-incubation of the inhibitor significantly decreased the fluorescence signals. The overall results demonstrated that PTP1B is responsible for the dephosphorylation and the nanofiber formation in the ER.
 | ||
| Fig. 7 (A) Schematic of the enzyme-triggered self-assembly in cells. (B) Molecular transformation of the precursor NapFFK(NBD)Yp and TEM image of the precursor with the treatment of ALP (scale bar: 100 nm). (C) Time course of fluorescence images incubated with 50 or 500 μM precursor (scale bar: 50 μm). Reprinted with permission from ref. 41. Copyright 2012 Nature Publishing Group. | ||
In their recent work, the Xu group combined the subcellular targeting with enzymatic self-assembly to selectively kill cells and overcome drug resistance (Fig. 8).42 They integrated the mitochondrial-targeting moiety, triphenylphosphonium (TPP), with phosphorylated tetrapeptide derivatives (L-1P or D-1P) that undergo an enzyme-instructed self-assembly (EISA) process. In the presence of phosphatases, the dephosphorylated oligomers self-assemble to form nanoscale assemblies on the cancer cell surface, followed by internalization via endocytosis (Fig. 8A and 8B). The results showed that these assemblies of L-1 (or D-1) were able to escape from late endosome/lysosome to enter the mitochondria due to the TPP targeting moiety (Fig. 8C). The intracellular localization was examined by the intense green fluorescence generated from the NBD, which overlapped with the red fluorescence signal from the Mito-Tracker. Moreover, these assemblies could effectively damage the mitochondria to release cytochrome c and ultimately kill the cancer cells (Fig. 8D). More interestingly, the multiple targeting (cell and subcellular targeting) strategy may offer one possible solution to minimize the acquired drug resistance.
 | ||
| Fig. 8 (A and B) Illustration of enzyme-instructed self-assembly for targeting mitochondria and inducing the death of cancer cells. (C) Confocal laser scanning microscopy (CLSM) images of human osteosarcoma cells (Saos2) treated with L-1P or D-1P (50 μM) for 1 or 4 h and then stained with Lyso-Tracker. (Scale bar: 10 μm.) (D) Cell viability of Saos2 cells for 48 h. Reprinted with permission from ref. 42. Copyright 2016, American Chemical Society. | ||
Another example of enzyme-responsive theranostics developed by Chen and co-workers was the co-assembly of indocyanine green (ICG, 1) and ALP-sensitive peptide NapFFKYp (2) to realize photoacoustic (PA) imaging and photothermal therapy (PTT) (Fig. 9A).43 The mixture of 1 and 2 could form micelles to prolong circulation and improve tumour accumulation. Once exposed to the overexpressed phosphatase environment, the dephosphorylation reaction could facilitate the conversion of micelles to nanofibers (5) (Fig. 9B and C). The NIR fluorescence imaging results revealed that most free ICG was excreted quickly within 4 h, while the administration of 1 + 2 could be retained in the tumour for 48 h (Fig. 9D). The in vivo tumour PA imaging upon 808 nm light excitation showed that the PA signal of 5 was much stronger than that of 1 at 24 h, most likely attributed to the improved tumour accumulation of the nanofiber (Fig. 9E). To further evaluate the PTT efficacy, mice bearing HeLa and 4T1 tumours were intravenously injected with 1 or 1 + 2, followed by PTT treatment at 24 h or 48 h post-injection. The inhibition of tumours treated with 5 was observed on day 2 without recurrence over time (Fig. 9F). The current systems mainly employ the enzyme-directed phosphorylation reactions to achieve the fiber or hydrogel transformation. More enzyme-catalysed reactions are required to be explored to diversify this area.
 | ||
| Fig. 9 (A) TEM image of the micelles (scale bar: 1 μm). (B) Illustration of enzyme-instructed self-assembly. (C) TEM image of the formed nanofibers (scale bar: 100 nm). Fluorescence images (D) and 3D PA images (E) of the ICG (1) and nanofibers (5) on tumour-bearing mice or tissues. (F) Tumour growth and survival curves from different treatments. Reprinted with permission from ref. 43. Copyright 2015, American Chemical Society. | ||
In order to apply the biorthogonal intracellular condensation reaction in living animal models and avoid the limitations of fluorescence-based imaging in the tissue penetration, an activatable PA probe for imaging the proteolytic activity in living subjects was developed (Fig. 10).44 Compared to fluorescence imaging, PA imaging could provide higher spatial resolution and deeper imaging depth. In the furin probe ESOR-PA01 (Ac-RVRRC(SEt)K-(Atto740)-CABT), the near-infrared (NIR) fluorophore, Atto740 was linked on the lysine side chain. In the meantime, the negative control probe containing the scrambled peptide sequence (cRRRVC(SEt)K(Atto740)-CABT) was also synthesized. With the uptake of the ESOR-PA probes by cells, the reductive environment (i.e., glutathione, GSH) in cells could cleave the disulfide bond of cysteine. The presence of the furin enzyme enables the removal of the RVRR substrate, leading to the biorthogonal condensation reaction between 1,2-aminothiol and the 2-cyano group of 2-cyano-6-aminobenzothiazole (CABT) to form dimers and longer oligomers (Fig. 10A). Subsequently, the formed oligomers could aggregate into nanostructures, along with the fluorescence quenching and the increase of PA signals (Fig. 10B and C). The PA imaging results in cells indicated that the probe ESOR-PA01 treated furin-overexpressing MDA-MB-231 cells produced a 2.6-fold higher PA signal compared to the furin-deficient LoVo cells, and 2.2-fold higher signal compared to inhibitor-pretreated MDA-MB-231 cells. Furthermore, when the furin activity was monitored in living mice, the PA signal of ESOR-PA01 in the MDA-MB-231 tumour was 2.7-fold higher than that treated with the negative control probe (ESOR-PA02), and was 7.1-fold higher than that in the furin-deficient LoVo tumour (Fig. 10D and E).
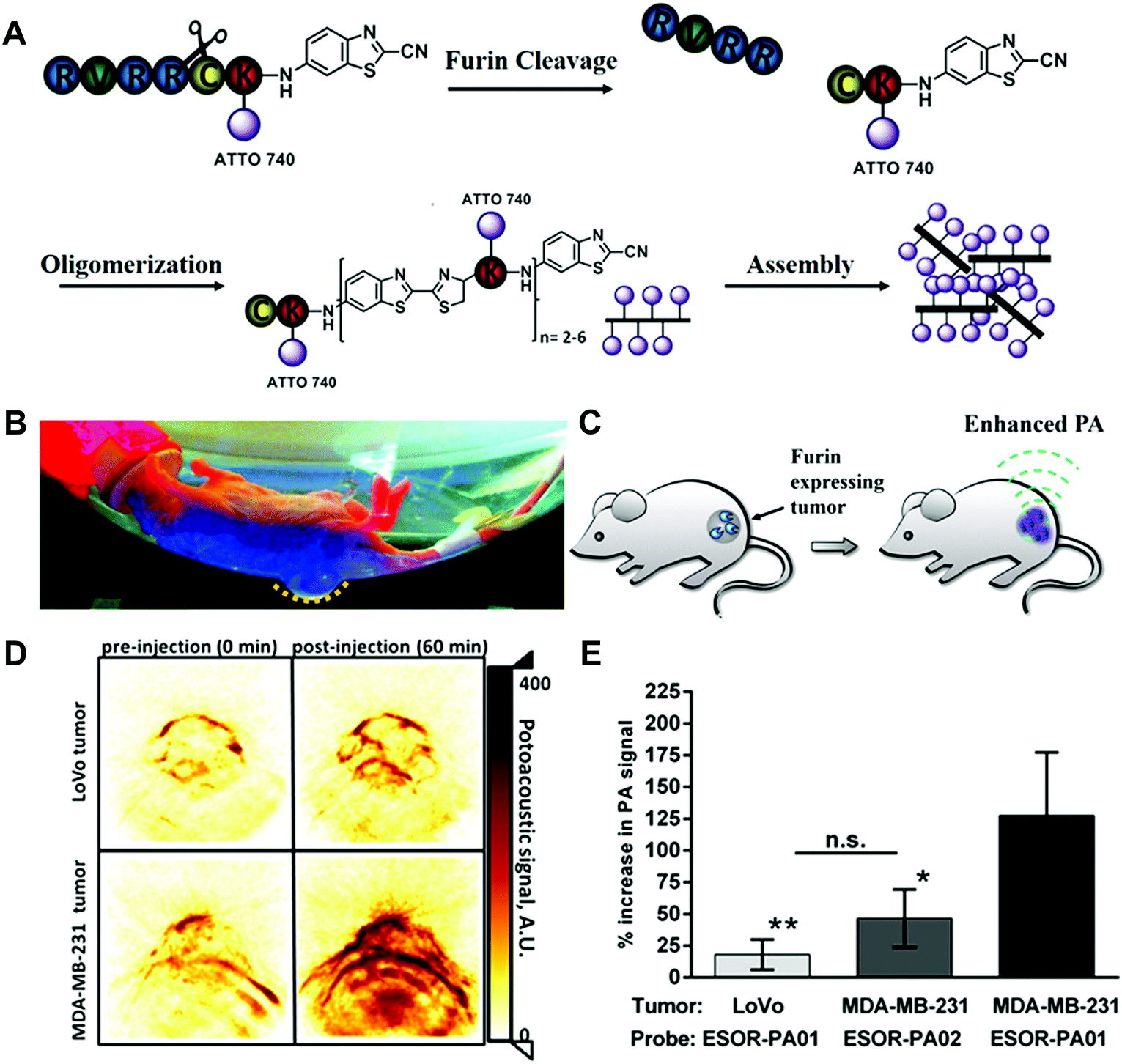 | ||
| Fig. 10 (A) Schematic of the furin-mediated formation of dimers and oligomers. Photographic image (B) and schematic (C) of tumour-bearing mice for PA imaging. (D) PA images of tumours treated with furin probe. (E) PA signal in different tumour-bearing mice. Reprinted with permission from ref. 44. Copyright 2013, American Chemical Society. | ||
Overall, the small-molecule-based in situ formation of nanomaterials can offer exceptional properties like specific localization of diagnostic or therapeutic agents at the target site, amplified fluorescence signals and even enzyme-induced assembly for therapeutic effects. Other factors like the assembly efficiency in living systems, alternative biocompatible reactions and applicability to other enzyme families should be further explored.
3.4 Inorganic/organic-based hybrid nanomaterials
Among the enzyme-responsive materials, inorganic nanomaterials have attracted extensive attention due to their superior optical, electronic, magnetic or thermal properties. A large number of studies on gold NPs, iron NPs, quantum dots (QDs), transition metals, carbon-based nanomaterials (including graphene oxide, carbon nanotubes, fullerenes, etc.) and lanthanide upconversion nanoparticles (UCNPs) have been reported for biomedical applications.8 Usually, the bare inorganic nanoparticles are required to be modified with organic materials to become enzyme responsive. According to the property changes of the materials, two basic strategies are frequently used to design the enzyme-responsive inorganic-based hybrid nanomaterials.One commonly used method is to design enzyme-responsive vehicles for the controlled delivery and release of diagnostic or therapeutic agents. The imaging agents or drugs could be physically adsorbed or covalently linked to NPs, which could greatly improve their pharmacokinetics and pharmacodynamics. Some NPs with intrinsic physical properties can be directly used as imaging agents (e.g. QDs, UCNPs), therapeutic agents (transition metals for PTT or photodynamic therapy (PDT)), or for theranostics (Au NPs, Fe3O4 NPs).
For instance, Yang and co-workers developed an effective theranostic NP formula to control the release of a chemotherapy drug and a magnetic resonance imaging (MRI) contrast agent for pancreatic cancer (Fig. 11).45 They conjugated magnetic iron oxide nanoparticles (IONPs) with amino-terminal fragment (ATF) peptides for targeting the urokinase plasminogen activator receptor (uPAR) and pancreatic cancer drug gemcitabine (Gem) via a cathepsin B-sensitive peptide (GFLG) linker (Fig. 11A and B). uPAR is an attractive target that benefits the drug delivery by the receptor-mediated cell internalization. The as-prepared NPs (ATF-IONP-Gem) exhibited 50% tumour growth inhibition in an orthotopic human pancreatic cancer xenograft model, significantly higher than the groups treated with free Gem (30%) and nontargeted IONP-Gem (23%) (Fig. 11C). The tumour-bearing mice at 48 h after the last of five administrations were imaged by T2-weighted and ultrashort echo time (UTE) MRI. Results showed that (ATF-IONP-Gem) could selectively accumulate at the targeted area (Fig. 11D) and such designed systems could monitor the tumour response to therapy by noninvasive MRI.
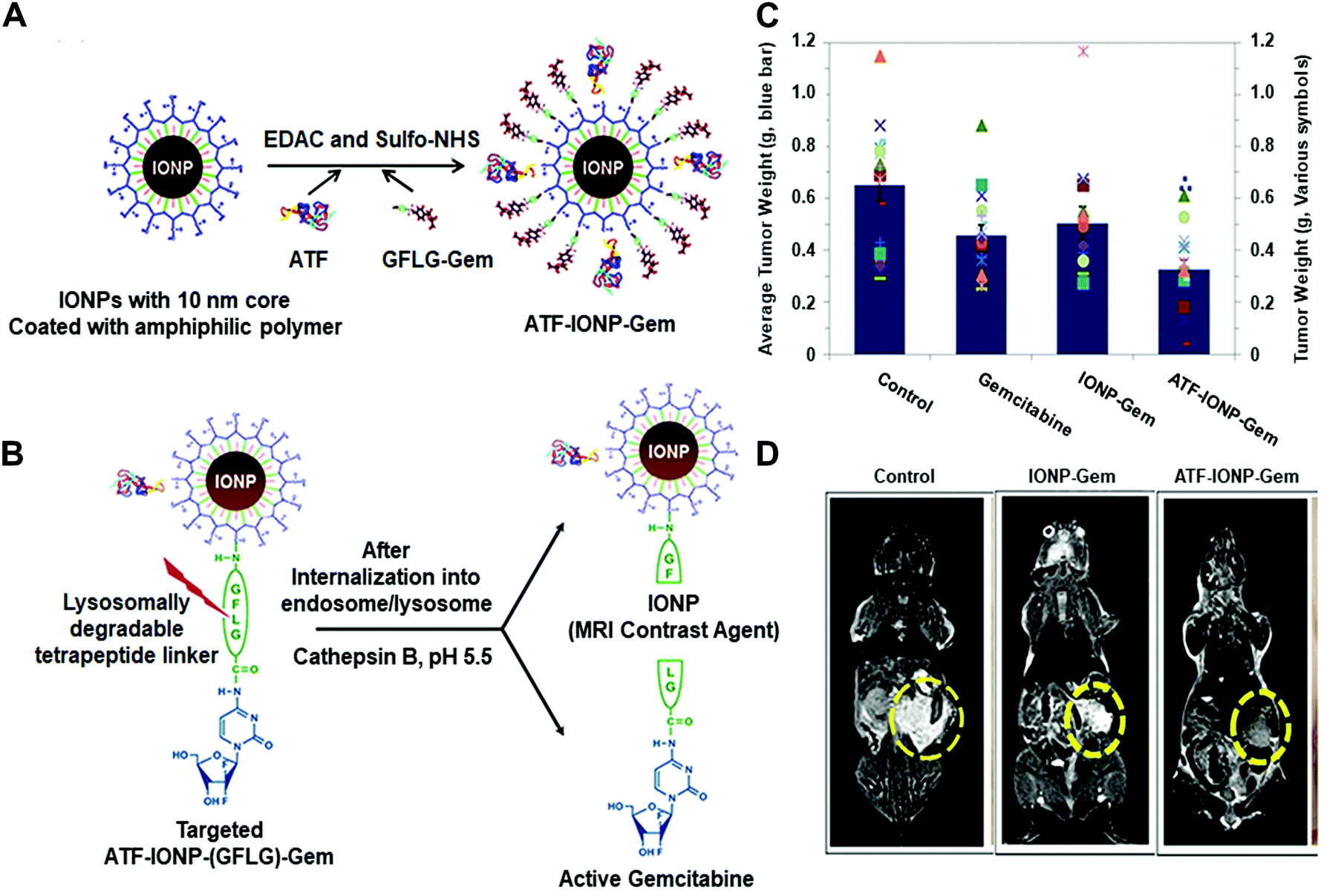 | ||
| Fig. 11 (A) Illustration of the fabrication of ATFIONP-Gem. (B) Schematic of the release of gemcitabine from ATF-IONP-Gem after cathepsin B-cleavage. (C) The mean tumour weights and individual tumour weight distribution in a pancreatic cancer xenograft model for evaluating the antitumour effect. (D) Coronal T2-weighted MR images of the tumour-bearing mice. Reprinted with permission from ref. 45. Copyright 2013, American Chemical Society. | ||
Mesoporous silica nanoparticles (MSNs) have attracted extensive attention in biomedicine, given their unique pore structure, easy functionalization, high loading capacity and favored biocompatibility. A variety of multifunctional stimuli-responsive nanocarrier systems based on MSNs have been developed.9 One typical example of the imaging of enzyme activity using MSNs was contributed by Ju and co-workers (Fig. 12).46 They prepared a telomerase-responsive MSN to achieve the switchable fluorescence imaging of the intracellular telomerase activity. Telomerase is a ribonucleoprotein reverse transcriptase enzyme that can add the DNA sequence TTAGGG to the 3′ end of telomeres. Telomerase is absent or at low levels in normal cells but becomes active in cancer cells, resulting in unlimited cellular proliferation. The fluorescein was entrapped in the MSNs and the fluorescence quencher (Black Hole Quencher, BHQ) was covalently immobilized on the inner pore surfaces. The DNA sequence (5′-(CCCTAA)nAATCCGTCGAGC AGAGTT-3′, O1) as a biogate sealed the MSNs to prevent the release of fluorescein. In the presence of telomerase and deoxy-ribonucleoside triphosphates (dNTPs), the oligonucleotide sequence formed a rigid hairpin-like DNA structure, leading to the detachment of DNA (O1) from the MSNs surface (Fig. 12A). The release of fluorescein molecules turned on the fluorescence signals and enabled the tracking of telomerase activity in living cells (Fig. 12B–D).
 | ||
| Fig. 12 (A) Schematic of MSN-based probes for the imaging of telomerase activity. (B) Fluorescence spectra of MSN probes with (a) dNTPs and with (b) dNTPs and telomerase. (C) Fluorescence images of HeLa cells treated with MSN probe. (D) TEM image of the internalization of MSN into HeLa cells. Reprinted with permission from ref. 46. Copyright 2013, American Chemical Society. | ||
Another design strategy is based on enzyme-triggered NP assembly or dispersion by changing the surface properties. In the well-established example of gold NPs, self-aggregation would make the colour change from red to blue due to the red-shift of the absorption spectrum. For the iron oxide NPs, magnetic relaxation would vary in the aggregated state.8 In a recent study, enzyme-responsive nanotheranostic agents based on rare-earth-doped upconversion nanocrystals (UCN) were reported by Xing and co-workers (Fig. 13).47 They conjugated the cathepsin B (CtsB)–sensitive peptide Ac-FKC(StBu)AC(SH)–CBT with a side-blocked cysteine and CBT on the surface of UCNs. The photosensitizer (chlorin-e6, Ce6) was coupled with the polyethylenimine (PEI) layer on the surface of UCNs. As described in Section 3.3.3, the peptide in the cross-linking of rare-earth UCNs (CRUN) was hydrolysed by lysosomal cathepsin B to initiate the CBT-based condensation reaction. Under the 808 nm laser irradiation, the enzyme-triggered cross-linking of CRUNs could enhance light converting emission and increase the generation of singlet oxygen (Fig. 13A). This design provides dual-modal (fluorescence and photoacoustic imaging) tumour imaging and efficient PDT-mediated tumour inhibition (Fig. 13B–D).
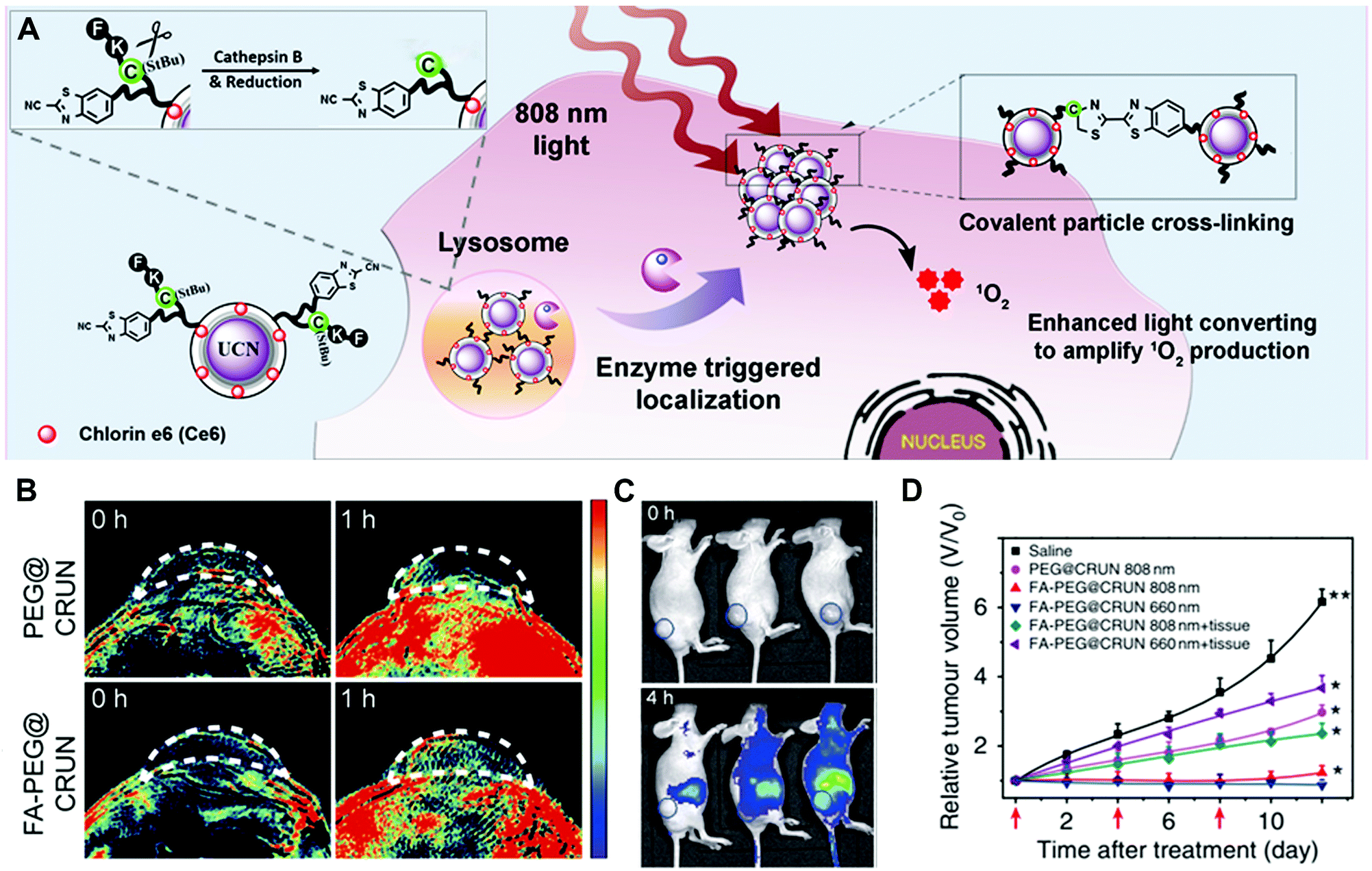 | ||
| Fig. 13 (A) Illustration of the enzyme-sensitive cross-linking of CRUNs in the tumour areas. (B) In vivo PA signals in the tumour region at different time intervals. (C) Live mice fluorescence imaging at different time intervals. (D) Changes in tumour volumes for evaluating the PDT effect. Reprinted with permission from ref. 47. Copyright 2013, Nature Publishing Group. | ||
Overall, inorganic/organic-based hybrid nanomaterials show promising results for numerous applications. The superior physical properties of inorganic nanoparticles and the versatility of surface modification allow the hybrid materials to combine different diagnostic techniques or therapies into one system, but the complexity of the fabrication process and the biosafety assessment potentially impede their bench-to-bedside translation.
4. Enzyme-based multiple responsive systems
To overcome several biological barriers in the tumour microenvironment and achieve more efficient drug/gene release, enzyme-based multiple responsive systems provide a more advanced way to obtain the improved diagnostic and therapeutic effects through the combination of two or more stimuli-responsive signals.17 By taking advantage of the special characteristics of the physiological microenvironment, the endogenous stimuli (pH, redox potentials or enzymes) and exogenous stimuli (heat, light, ultrasound or magnetic field) could be incorporated to design combined stimuli-responsive nanosystems; e.g. pH/enzyme, GSH/enzyme, thermal/enzyme or dual-enzyme triggered nanomaterials. Moreover, the spatial distribution variations of different stimuli between the extracellular and intracellular tumour cells allow one to design simultaneously or sequentially stimuli-responsive nanomaterials to achieve the desired tumour targeting, enhanced tumour retention and subsequent cellular internalization.17Huang and co-workers developed a smart polymer conjugate with both pH- and enzyme-responsive features for efficient nuclear drug delivery (Fig. 14).48 The conjugate contains the HPMA copolymer as the backbone and a biforked sub-unit with one site linked with the drug H1 peptide and the other site linked with a pH-sensitive peptide (R8NLS). The H1 peptide, derived from helix 1 (H1) of the helix-loop-helix region of c-Myc, could interrupt the transcription activation of c-Myc in the nuclear region. The fusion targeting peptide (R8NLS) is comprised of the cell penetrating peptide (octaarginine or R8) and the nuclear localization sequence (NLS, PKKKRKV), followed by the modification with anionic 2,3-dimethylmaleic anhydrides (DMA) to mask the cationic charge. The sub-units were introduced into the HPMA copolymer via a lysosomal enzyme-responsive peptide linker (GFLG) (Fig. 14A). Under the tumour tissue microenvironment (pH = 6.5), the pH-reversible DMA could be rapidly cleaved to expose the cationic peptide for improving the cellular internalization. Later, the small-molecule sub-unit was released from the copolymer backbone by the lysosomally enzymatic reactions (such as cathepsin B), leading to NLS mediated nuclear translocation. The results revealed that the DMA-masked conjugate presented better pharmacokinetic behaviour than the control conjugate without DMA modification (2-fold increase in AUC: total area under the blood concentration versus time curve). The enzyme-responsive drug release significantly improved the nuclear drug accumulation by up to 50-fold compared with the original polymer–drug conjugates (Fig. 14B and C). Moreover, the in vivo tumour treatment with the conjugates achieved much higher therapeutic efficacy with an inhibition rate of 77% (Fig. 14D).
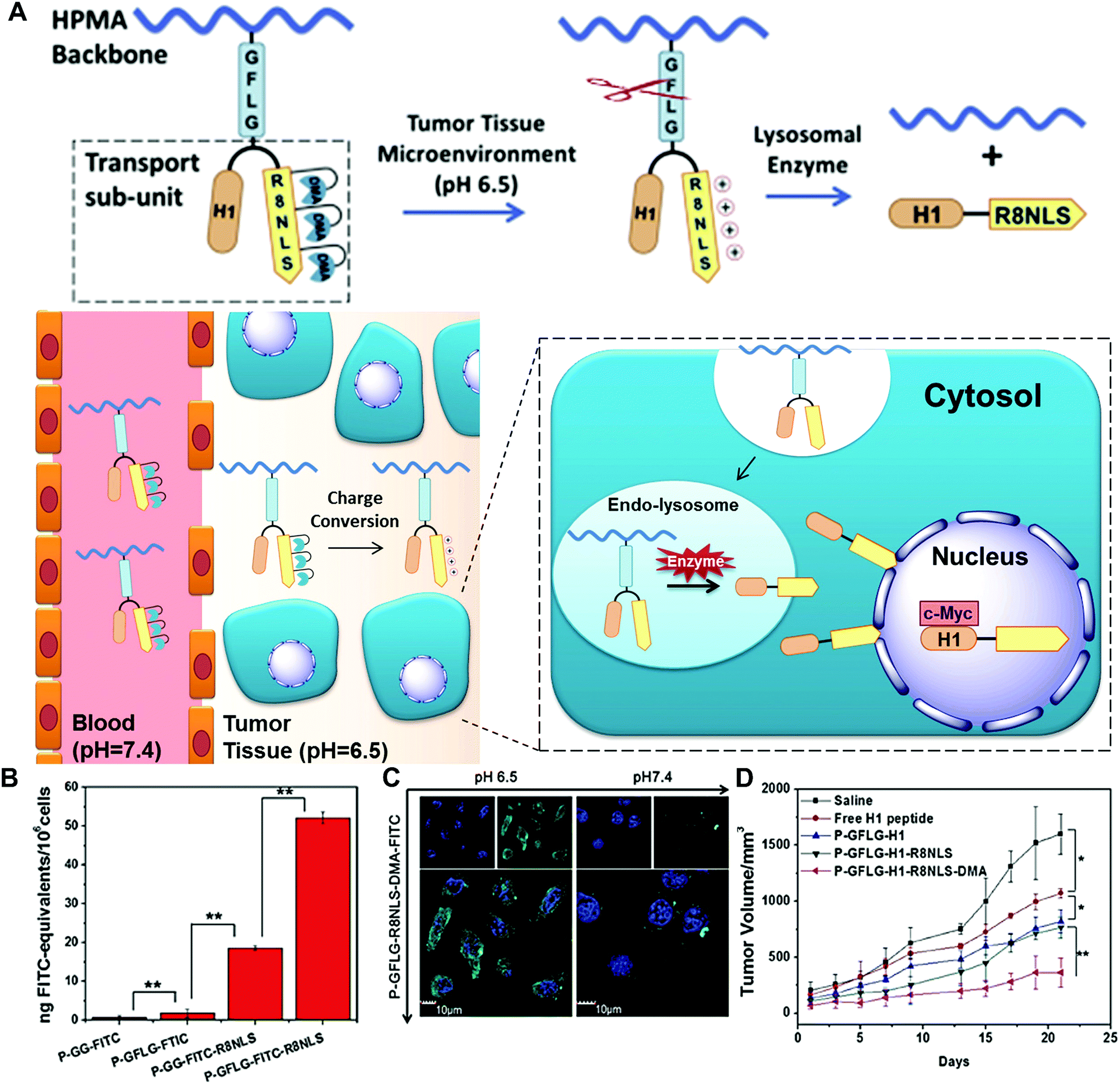 | ||
| Fig. 14 (A) Schematic of dual-responsive HPMA copolymer conjugates for multistage nuclear targeting. (B) The evaluation of nucleus transport efficiency by quantification of nuclear FITC content. (C) Subcellular distribution at different pH. (D) HeLa tumour growth curves after intravenous injection for evaluating the antitumour effect. Reprinted with permission from ref. 48. Copyright 2015, Elsevier. | ||
In another example of a dual-triggered system, Zhou and co-workers prepared an actively targeted polymeric micelle with enzyme and redox dual responsiveness for rapid intracellular drug release.49 The polymeric micelle was prepared via the assembly of the redox-responsive prodrug, monomethyl poly(ethylene glycol)-ss-camptothecin (mPEG-ss-CPT), and the phenylboronic acid (PBA)-functionalized enzyme-responsive copolymer, PBA-poly(ethyleneglycol)-4,4′-(diazene-1,2-diyl)benzoyl-poly(ε-caprolactone) (PBA-PEG-Azo-PCL). The conjugation of the hydrophobic drug camptothecin (CPT) with mPEG improved its solubility in water, and later the breakage of the disulfide bonds by the abundantly expressed glutathione (GSH) in the tumour area led to the release of active therapeutic agents. The azoreductase-responsive function was designed by linking the hydrophilic polycaprolactone (PCL) segments and hydrophobic PEG segments via azo bonds. Moreover, PBA was conjugated to the PEG segment to enable active targeting due to its interactions with overexpressed sialic acid on certain tumour cells (e.g. hepatocellular carcinoma cells). The in vitro and in vivo experiments verified that this dual-responsive nanocarrier achieved effective treatment of the tumour areas with minimum side effects to the normal tissues.
Recently, enzyme and thermal dual-responsive nanoparticles were designed by Jayakannan and co-workers.50
The hydrophobic monomer based on 3-pentadecylphenol (PDP) was copolymerized with oligoethylene glycol acrylate to produce the amphiphilic copolymer. This copolymer could self-assemble into spherical core–shell nanoparticles, and the model hydrophobic drug DOX or Nile Red could be encapsulated into this scaffold. The release kinetics revealed that only 20% of DOX was released under physiological conditions (at 37 °C) while at 43 °C (above lower critical solution temperature (LCST)), more than 90% of the loaded drugs was burst-released from the nanoparticles within 2 h. Moreover, the esterase enzyme could cleave the copolymer structure to release >95% of the drug in 12 h. This design provides the possibility for the fabrication of a temperature and enzyme dual-responsive vector for delivering therapeutic drugs in cancer cells.
Overall, compared to the single enzyme-responsive nanosystems, the multiple stimuli allow the single nanoplatform to perform diverse functions. For example, the first emerging stimuli could trigger the big NPs to disassemble into smaller ones for better penetration into deep tumour tissues. Then, subsequent stimuli could cause the charge reversal or exposure of targeting ligands for improved cellular uptake. Moreover, cellular stimuli could help to release the payloads specifically within tumour cells. Therefore, the advantages of different stimuli can be efficiently combined to amplify the diagnosis and treatment efficacy.
5. Conclusions and future outlook
Enzymes play fundamental roles in catalysing a variety of biological reactions for cell regulation and signal transduction. The biological disorders and occurrence of many diseases usually come along with the dysfunction of enzyme activities, which requires more validated tools for investigating the enzyme functions and regulations in the disease progression. On the other hand, the abnormal enzyme expression in pathological conditions offers great opportunities to drive the rational design towards early diagnosis, controlled drug/gene delivery, and monitoring of treatment efficacy. The very inspiring progress is the development of endogenous enzyme-responsive nanomaterials. By virtue of the material properties, diverse enzyme-triggered systems based on polymers, liposomes, small molecules and inorganic/organic hybrids have been built to improve the diagnostic accuracy and treatment effectiveness. Through altering the physical or chemical properties of the designed nanosystems in response to enzyme stimuli, the active imaging agents or therapeutic molecules could be selectively and specifically released at the target site, leading to enhanced theranostic efficacy and reduced side effects.Despite the enormous progress made in the design and application of enzyme-responsive nanomaterials for diagnosis, treatment and theranostics, many challenges still need to be addressed. Firstly, for many proteases, the same enzyme family such as MMPs usually have similar catalytic mechanisms and active pockets, which leads to similar substrate preferences. When the short peptide substrate is introduced into the enzyme-responsive systems, it is hard to identify whether the signals are generated from the specific enzyme or from the enzyme family. Hence, rational chemistry design in addressing the substrate specificity is required to ensure enzyme-specific response and more precise diagnosis.
Secondly, although the features of enzyme-activatable nanomaterials could provide specificity to some extent, when applied to determine the location of the lesion, more accurate imaging in the living system is still a challenge. Currently, several imaging modalities have been well established for the early detection and characterization of diseases; e.g. fluorescence imaging (FI), MRI, PA, ultrasound (US), computed tomography (CT), positron emission tomography (PET) and single photon emission computed tomography (SPECT). Each imaging modality has its inherent pros and cons. For example, the FI technique offers great advantages of high sensitivity, high-throughput capabilities, low cost and non-invasive readout, while spatial resolution and tissue penetration limit further clinical applications. On the contrary, the MRI technique has been widely used in the clinical diagnosis due to its high spatial resolution and unlimited tissue penetration, but the disadvantages of high cost, low sensitivity and time-consuming use cannot be ignored.14 Therefore, multimodality imaging should be considered for the future design of enzyme-activated nanomaterials to obtain more accurate and complementary information about the disease state of patients.
Thirdly, although nanomaterials with dual- or multi-responsive properties could respond to multiple biological environment changes, the development of multi-stimuli responsive systems is still in its infancy. Currently, only a few examples have been reported because the design principle is much more complicated than the single-stimulus sensitive system. The different functional groups in response to different stimuli have to be modified on the materials, which makes the fabrication and assembly process more difficult than for the single sensitive group. In addition, due to the heterogeneous distribution of the stimuli in the tumour microenvironment, it is hard to control the degree of the response in real situations. Sometimes, the response to one stimulus may fail, leading to the ineffectiveness of the whole system. Therefore, the efficiency of the sequential or simultaneous reactions should be carefully assessed in the living systems.
Finally, most of the current enzyme-responsive nanomaterials are still at the stage of proof-of-principle. Further investigations on their biosafety such as the evaluation of toxicity, biocompatibility, immunogenicity, pharmacokinetics and biodistribution are required before their possible clinical translation and practical use in humans. In particular, enzyme-responsive nanomaterials contain several components for multiple functions, which raise more concerns about the complicated safety issues. For different types of materials, the organic and natural materials are likely more biodegradable and less toxic than inorganic materials and are generally preferred for biomedical applications.
Overall, endogenous enzyme-responsive nanomaterials provide great potential for improving the effectiveness of disease diagnosis, treatment and theranostics. New strategies for addressing the challenges are expected to be developed, which will contribute significantly to the field of precision medicine.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the start-up fund from the Shenzhen University, the National Natural Science Foundation of China (31771036, 51703132, 51573096), the Basic Research Program of Shenzhen (JCYJ20170412111100742, JCYJ20160422091238319), Fok Ying-Tong Education Foundation for Young Teachers in the Higher Education Institutions of China (161032), the China Postdoctoral Science Foundation (2018M633104), and the Intramural Research Program, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health.References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 7–30 CrossRef PubMed.
- K. D. Miller, R. L. Siegel, C. C. Lin, A. B. Mariotto, J. L. Kramer, J. H. Rowland, K. D. Stein, R. Alteri and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 271–289 CrossRef PubMed.
- A. S. Thakor and S. S. Gambhir, Ca-Cancer J. Clin., 2013, 63, 395–418 CrossRef PubMed.
- G. Chen, I. Roy, C. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826–2885 CrossRef PubMed.
- J. Hu, G. Zhang and S. Liu, Chem. Soc. Rev., 2012, 41, 5933–5949 RSC.
- F. Fouladi, K. J. Steffen and S. Mallik, Bioconjugate Chem., 2017, 28, 857–868 CrossRef PubMed.
- G. Hong, S. Diao, A. L. Antaris and H. Dai, Chem. Rev., 2015, 115, 10816–10906 CrossRef PubMed.
- D. Aili and M. M. Stevens, Chem. Soc. Rev., 2010, 39, 3358–3370 RSC.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2013, 26, 435–451 CrossRef.
- S. S. Kelkar and T. M. Reineke, Bioconjugate Chem., 2011, 22, 1879–1903 CrossRef PubMed.
- J. H. Ryu, S. Lee, S. Son, S. H. Kim, J. F. Leary, K. Choi and I. C. Kwon, J. Controlled Release, 2014, 190, 477–484 CrossRef PubMed.
- E.-K. Lim, T. Kim, S. Paik, S. Haam, Y.-M. Huh and K. Lee, Chem. Rev., 2014, 115, 327–394 CrossRef PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef PubMed.
- X. Li, J. Kim, J. Yoon and X. Chen, Adv. Mater., 2017, 29, 1606857 CrossRef PubMed.
- J. Wang, W. Tao, X. Chen, O. C. Farokhzad and G. Liu, Theranostics, 2017, 7, 3915–3919 CrossRef PubMed.
- Y. Lu, A. A. Aimetti, R. Langer and Z. Gu, Nat. Rev. Mater., 2016, 2, 16075 CrossRef.
- B. Chen, W. Dai, B. He, H. Zhang, X. Wang, Y. Wang and Q. Zhang, Theranostics, 2017, 7, 538–558 CrossRef PubMed.
- R. de la Rica, D. Aili and M. M. Stevens, Adv. Drug Delivery Rev., 2012, 64, 967–978 CrossRef PubMed.
- Q. Hu, P. S. Katti and Z. Gu, Nanoscale, 2014, 6, 12273–12286 RSC.
- A. Razgulin, N. Ma and J. Rao, Chem. Soc. Rev., 2011, 40, 4186–4216 RSC.
- M. Zelzer, S. J. Todd, A. R. Hirst, T. O. McDonald and R. V. Ulijn, Biomater. Sci., 2013, 1, 11–39 RSC.
- C. F. Anderson and H. Cui, Ind. Eng. Chem. Res., 2017, 56, 5761–5777 CrossRef PubMed.
- G. Murphy and H. Nagase, Mol. Aspects Med., 2008, 29, 290–308 CrossRef PubMed.
- M. Gao and B. Z. Tang, Drug Discovery Today, 2017, 22, 1288–1294 CrossRef PubMed.
- G. Liang, H. Ren and J. Rao, Nat. Chem., 2010, 2, 54–60 CrossRef PubMed.
- M. Leist and M. Jäättelä, Nat. Rev. Mol. Cell Biol., 2001, 2, 589–598 CrossRef PubMed.
- M. M. Mohamed and B. F. Sloane, Nat. Rev. Cancer, 2006, 6, 764–775 CrossRef PubMed.
- J.-X. Tan, X.-Y. Wang, H.-Y. Li, X.-L. Su, L. Wang, L. Ran, K. Zheng and G.-S. Ren, Int. J. Cancer, 2011, 128, 1303–1315 CrossRef PubMed.
- E. A. Dennis, J. Cao, Y.-H. Hsu, V. Magrioti and G. Kokotos, Chem. Rev., 2011, 111, 6130–6185 CrossRef PubMed.
- M.-P. Chien, A. S. Carlini, D. Hu, C. V. Barback, A. M. Rush, D. J. Hall, G. Orr and N. C. Gianneschi, J. Am. Chem. Soc., 2013, 135, 18710–18713 CrossRef PubMed.
- J. Rao and A. Khan, J. Am. Chem. Soc., 2013, 135, 14056–14059 CrossRef PubMed.
- A. J. Harnoy, I. Rosenbaum, E. Tirosh, Y. Ebenstein, R. Shaharabani, R. Beck and R. J. Amir, J. Am. Chem. Soc., 2014, 136, 7531–7534 CrossRef PubMed.
- Y. Yang, D. Pan, K. Luo, L. Li and Z. Gu, Biomaterials, 2013, 34, 8430–8443 CrossRef PubMed.
- T. J. Sanborn, P. B. Messersmith and A. E. Barron, Biomaterials, 2002, 23, 2703–2710 CrossRef PubMed.
- M. Kurisawa, J. E. Chung, Y. Y. Yang, S. J. Gao and H. Uyama, Chem. Commun., 2005, 4312–4314 RSC.
- B. P. Purcell, D. Lobb, M. B. Charati, S. M. Dorsey, R. J. Wade, K. N. Zellers, H. Doviak, S. Pettaway, C. B. Logdon, J. Shuman, P. D. Freels, J. H. Gorman, R. C. Gorman, F. G. Spinale and J. A. Burdick, Nat. Mater., 2014, 13, 653–661 CrossRef PubMed.
- P. D. Thornton, R. J. Mart and R. V. Ulijn, Adv. Mater., 2007, 19, 1252–1256 CrossRef.
- L. Linderoth, G. H. Peters, R. Madsen and T. L. Andresen, Angew. Chem., Int. Ed., 2009, 48, 1823–1826 CrossRef PubMed.
- H. Shi, R. T. K. Kwok, J. Liu, B. Xing, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 17972–17981 CrossRef PubMed.
- Y. Yuan, R. T. K. Kwok, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2014, 136, 2546–2554 CrossRef PubMed.
- Y. Gao, J. Shi, D. Yuan and B. Xu, Nat. Commun., 2012, 3, 1033 CrossRef PubMed.
- H. Wang, Z. Feng, Y. Wang, R. Zhou, Z. Yang and B. Xu, J. Am. Chem. Soc., 2016, 138, 16046–16055 CrossRef PubMed.
- P. Huang, Y. Gao, J. Lin, H. Hu, H.-S. Liao, X. Yan, Y. Tang, A. Jin, J. Song, G. Niu, G. Zhang, F. Horkay and X. Chen, ACS Nano, 2015, 9, 9517–9527 CrossRef PubMed.
- A. Dragulescu-Andrasi, S.-R. Kothapalli, G. A. Tikhomirov, J. Rao and S. S. Gambhir, J. Am. Chem. Soc., 2013, 135, 11015–11022 CrossRef PubMed.
- G. Y. Lee, W. P. Qian, L. Wang, Y. A. Wang, C. A. Staley, M. Satpathy, S. Nie, H. Mao and L. Yang, ACS Nano, 2013, 7, 2078–2089 CrossRef PubMed.
- R. Qian, L. Ding and H. Ju, J. Am. Chem. Soc., 2013, 135, 13282–13285 CrossRef PubMed.
- X. Ai, C. J. H. Ho, J. Aw, A. B. E. Attia, J. Mu, Y. Wang, X. Wang, Y. Wang, X. Liu, H. Chen, M. Gao, X. Chen, E. K. L. Yeow, G. Liu, M. Olivo and B. Xing, Nat. Commun., 2016, 7, 10432 CrossRef PubMed.
- J. Zhong, L. Li, X. Zhu, S. Guan, Q. Yang, Z. Zhou, Z. Zhang and Y. Huang, Biomaterials, 2015, 65, 43–55 CrossRef PubMed.
- L. Zhang, Y. Wang, X. Zhang, X. Wei, X. Xiong and S. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 3388–3399 Search PubMed.
- S. Kashyap, N. Singh, B. Surnar and M. Jayakannan, Biomacromolecules, 2016, 17, 384–398 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |