
Thienobenzene-fused perylene bisimide as a non-fullerene acceptor for organic solar cells with a high open-circuit voltage and power conversion efficiency†
Chen
Zhang‡
a,
Tao
Liu‡
b,
Weixuan
Zeng
a,
Dongjun
Xie
a,
Zhenghui
Luo
a,
Yanming
Sun
*b and
Chuluo
Yang
*a
aHubei Collaborative Innovation Center for Advanced Organic Chemical Materials, Hubei Key Lab on Organic and Polymeric Optoelectronic Materials, Department of Chemistry, Wuhan University, Wuhan, 430072, People's Republic of China. E-mail: clyang@whu.edu.cn
bHeeger Beijing Research and Development Center, School of Chemistry and Environment, Beihang University, Beijing 100191, People's Republic of China. E-mail: sunym@buaa.edu.cn
First published on 1st November 2016
Abstract
Perylene bisimide (PBI) based molecules have recently attracted tremendous interest as acceptors in non-fullerene organic solar cells. However, most PBI-based acceptors possess deep LUMO energy levels (−3.9 ∼ −4.0 eV) and show an open-circuit voltage (Voc) below 0.90 V, thus limiting the improvement of device efficiency. Here, we report two novel ring-fused PBI dimers, SdiPBI-BT and diPBI-BT, with thienobenzene fused to the bay region of the PBI subunits. Conventional bulk-heterojunction (BHJ) solar cells based on SdiPBI-BT show a power conversion efficiency (PCE) of 6.71% with a high Voc value of 0.95 V, a short-circuit current density (Jsc) of 10.31 mA cm−2 and a high fill factor (FF) of 68.7%. Devices based on diPBI-BT show a PCE of 5.84% with a high Voc value of 0.99 V. These results demonstrate that ring-fused PBI derivatives are promising materials for non-fullerene cells.
Introduction
Bulk-heterojunction (BHJ) organic solar cells (OSCs) have attracted extensive attention due to their advantages of low-cost, light-weight and flexibility.1–8 Fullerene derivatives have played a central role as electron acceptors owing to their large electron affinity, high electron mobility, charge-transport isotropy and favorable nanoscale network forming ability.9,10 However, there are several drawbacks to fullerene acceptors, including their weak absorption in the visual region and limited energy-level variability.11–13 Nowadays, small molecules and polymers as potential alternatives to replace fullerene acceptors have been actively explored, and an impressive increase in power conversion efficiency (PCE) has been witnessed.14–35For small molecular acceptors, small domain sizes and sufficient electron mobility are important criteria in order to achieve high PCEs.36,37 Non-fullerene small molecule acceptors are usually derivatives of electron-withdrawing building blocks with large conjugate structures to ensure facile exciton/charge delocalization and good charge transport.28,38 Perylene bisimide derivatives (PBIs) are the earliest and most common non-fullerene acceptors.39–50 However, traditional PBI derivatives have large planar structures and tend to form excessively large crystalline domain sizes which lead to large phase separation, reduced exciton diffusion and separation efficiency, and low PCE.16,37,41 To avoid aggregation, PBI dimers (diPBIs) with two PBI monomers linked by a single C–C bond or bulky bridge-blocks in the bay-position have been developed.41,43,49,50 The twist or bulky structures of PBI dimers reduce intermolecular interactions and aggregations. These small molecules can form smooth amorphous BHJ films with small domain sizes, resulting in relatively high PCEs compared to PBI monomers. Another challenge for PBI-based acceptors is to enhance the open-circuit voltage (Voc). The conjugation size of PBI is smaller than other high-circuit-voltage acceptors (e.g. BFI),18,20,29 and the lowest unoccupied molecular orbital (LUMO) energy levels of most PBI-based acceptors are quite deep, resulting in Voc values below 0.9 eV.16,40,41,43,46 For example, the bay-linked PBI dimer, SdiPBI (Scheme 1), shows a Voc value of 0.76–0.89 V when blended with a series of donors.41,43 Recently, a derivative of PBI-dimer with sulfur atoms incorporated into the bay region, namely SdiPBI-S, exhibited a high Voc value of 0.92 V owing to the electron-donating effect of the sulfur atoms, and the PCE of organic cells based on SdiPBI-S is up to 7.2%.49 By fusing aromatic rings to the bay-region of PBI dimers, a high-lying LUMO level and an extended conjugation system can be obtained. As a result, high Voc values and a good PCE can be anticipated.
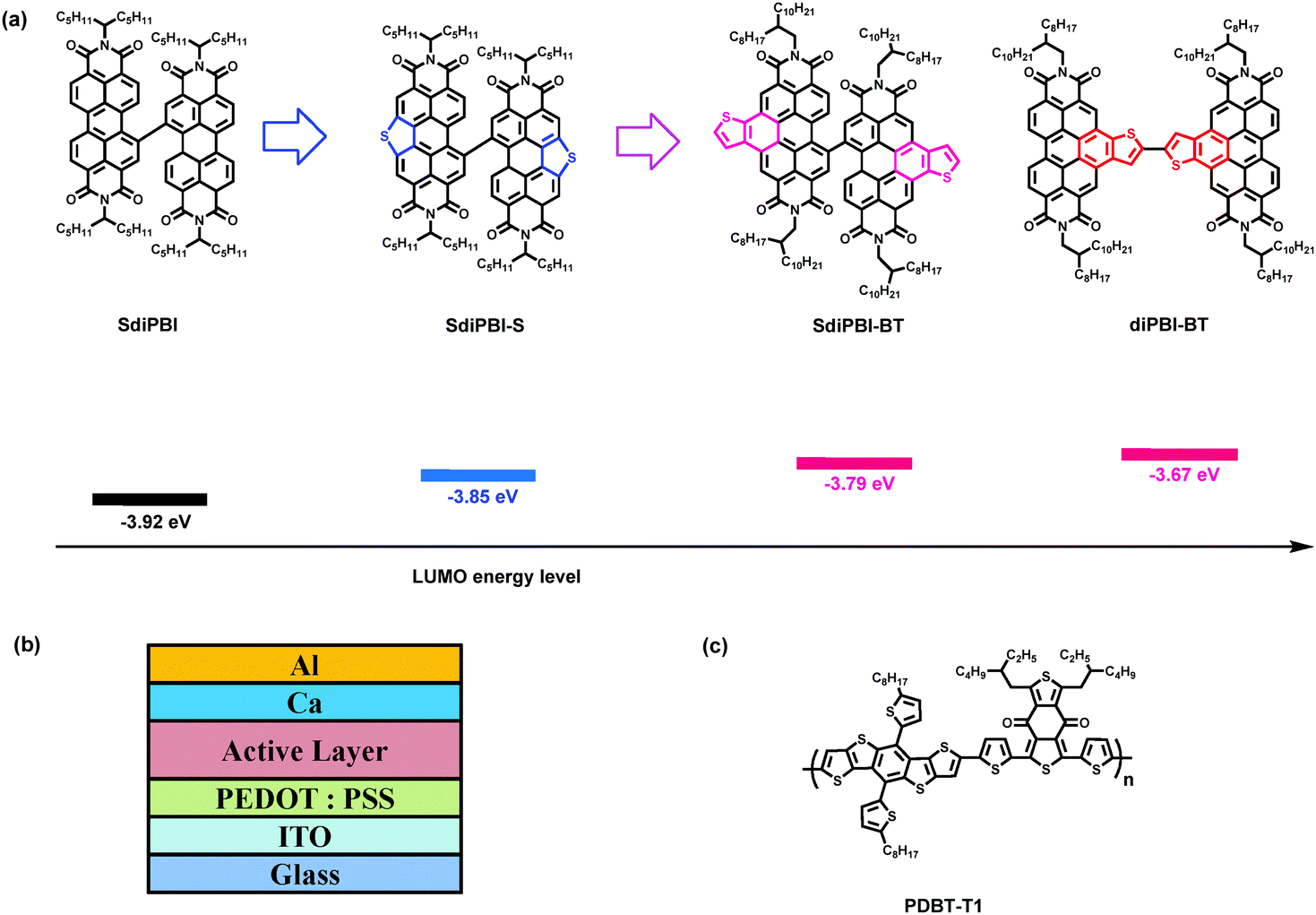 | ||
| Scheme 1 Structures and LUMO energy levels of SdiPBI, SdiPBI-S, SdiPBI-BT and diPBI-BT (a). device architecture (b), and chemical structure of PDBT-T1 (c). | ||
In this article, we reported two novel acceptors, namely SdiPBI-BT and diPBI-BT, which were modified by inserting two thienobenzenes into the bay sections of the PBI subunits (Scheme 1). SdiPBI-BT and diPBI-BT have larger conjugation structures than SdiPBI and SdiPBI-S.49 The extended conjugation systems of SdiPBI-BT/diPBI-BT and the electron-donating effect of thiophene lead to high-lying LUMO energy levels. By comparing the properties of SdiPBI-BT and diPBI-BT, the effects of the relative position of the thienobenzenes are also investigated. Since the selection of a donor plays an important role in the OSC performance,50–55 PDBT-T1 is used as the donor to blend with SdiPBI-BT. The devices based on the SdiPBI-BT/PDBT-T1 blend exhibit a relatively high PCE up to 6.71% with a high Voc value approaching 0.95 V and a high fill factor (FF) of 68.7%, and the devices of the diPBI-BT/PDBT-T1 blend show a PCE of 5.83% with a high Voc value of 0.99 V and a FF of 60.2%, indicating that the thienobenzene-fused perylene bisimide molecules are promising acceptors.
Results and discussion
Synthesis and characterization
The synthesis route of SdiPBI-BT/diPBI-BT is shown in Scheme 2. The PBI dimer (3) was prepared by Ullmann coupling of the brominated PBI (2). Thiophenes were introduced into the brominated PBI dimer (4) via the Stille reaction to get 5. SdiPBI-BT was obtained via cyclization of 5 with FeCl3 as the oxidant. diPBI-BT was prepared via Ullmann coupling from the thienobenzene-fused monomer 9. All the intermediates and the final products were characterized using NMR spectroscopy, mass spectra and elemental analysis. The two targeted molecules showed sufficient solubility in common solvents such as dichloromethane (CH2Cl2), chloroform (CHCl3) and chlorobenzene.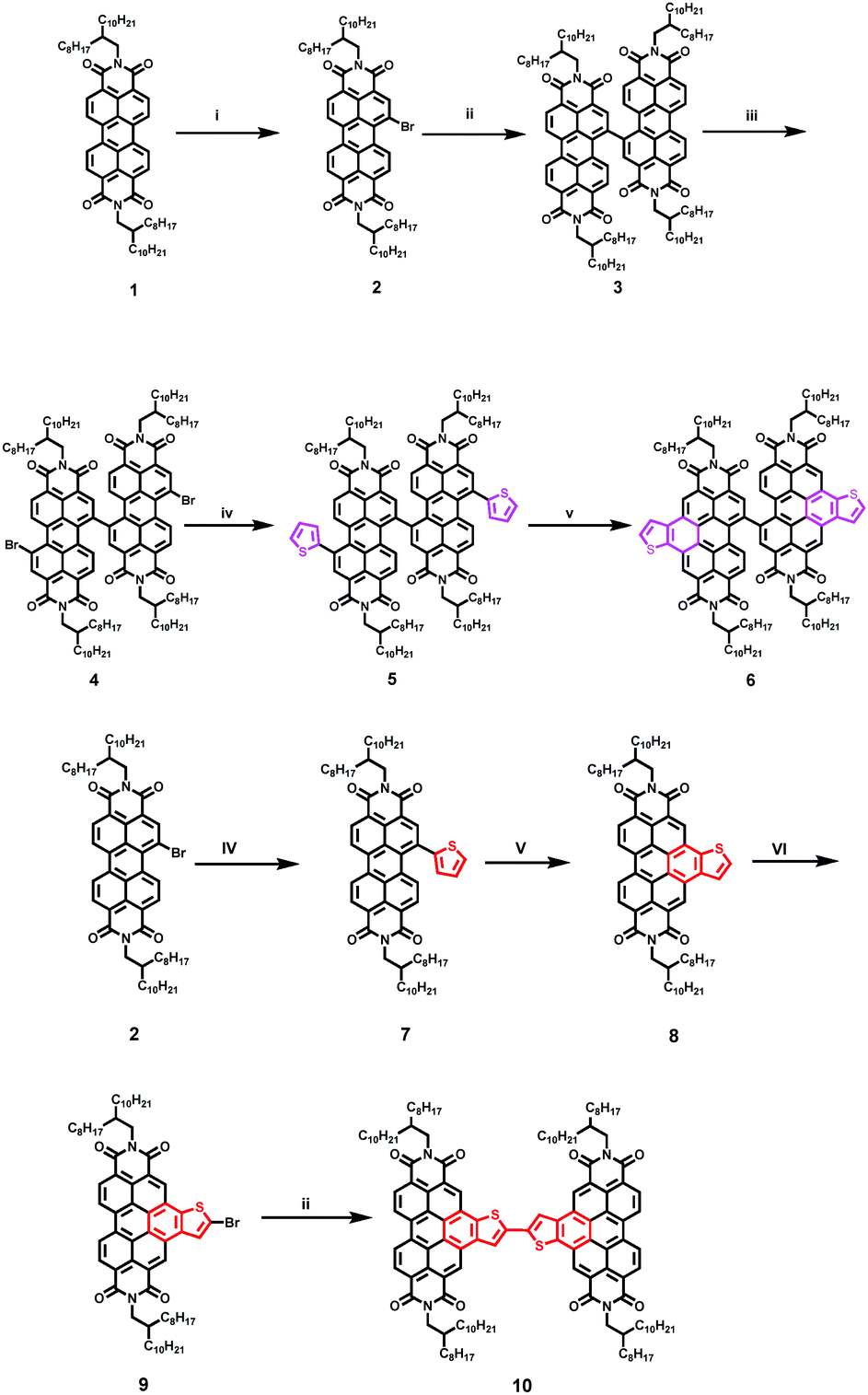 | ||
| Scheme 2 Synthesis of SdiPBI-BT. (i) K2CO3, CHCl3, Br2, 50 °C; (ii) Cu, DMSO, toluene, 85 °C; (iii) K2CO3, CHCl3, Br2, 55 °C; (iv) tributyl(thiophen-2-yl)stannane, Pd2(dba)3, P(o-tol)3, toluene, 110 °C; (v) FeCl3, toluene, CH3CN, 50 °C; and (vi) FeCl3, CHCl3, Br2, 40 °C. | ||
Theoretical calculations
To obtain the spatial configuration of SdiPBI-BT and diPBI-BT, density functional theory (DFT) methods were performed at the B3LYP/6-31G(d) level. In order to simplify the calculations, the alkyl chains of SdiPBI-BT were shortened to the methyl groups, because they do not significantly affect the equilibrium geometries or electronic properties.6 As shown in Fig. 1, the SdiPBI-BT molecule possesses a non-planar configuration due to the steric hindrance between the two PBI-based subunits. The dihedral angle between the two subunits in SdiPBI-BT is 69.6°, slightly higher than that of SdiPBI (67°),49 which indicates that the introduction of the thienobenzene unit into the PBI subunit results in a slightly more twisted molecular configuration. This could be ascribed to the electron-donating effect of thienobenzene, which increases the steric repulsion between the PBI subunits. The highly twisted configuration of SdiPBI-BT could inhibit severe aggregation of the acceptor molecules. On the other hand, only an 18.2° dihedral angle is found for diPBI-BT, illustrating that the backbone of diPBI-BT is relatively planar. The planar structure of diPBI-BT may induce a larger tendency to aggregate.16 The differences in the spatial configurations of SdiPBI-BT and diPBI-BT are due to the relative position of the thienobenzene unit, which results in a relatively large steric hindrance in the PBI subunits for SdiPBI-BT and a relatively small steric hindrance for diPBI-BT. As a result, different spatial configurations could lead to different morphologies of the active layers.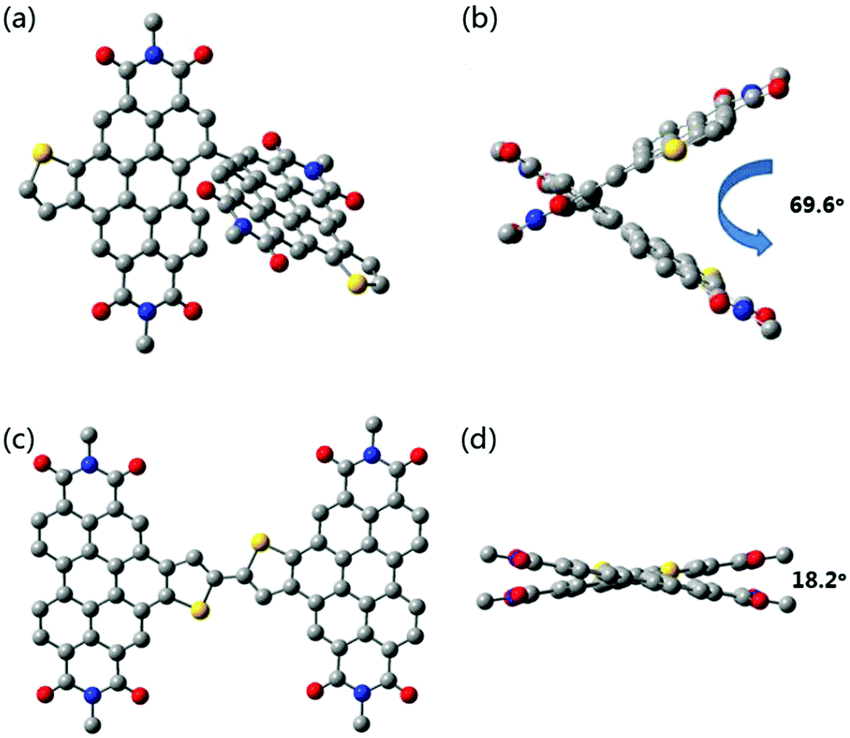 | ||
| Fig. 1 Side and top views of the optimized geometry of SdiPBI-BT (a and b) and diPBI-BT (c and d) by using DFT calculations at the B3LYP/6-31G(d) level. | ||
Optical and electrochemical properties
The UV-vis absorption spectrum of SdiPBI-BT in dilute CHCl3 solution exhibits broad absorptions in the range of 400–600 nm (Fig. 2a). Compared to the spectrum in solution, the SdiPBI-BT thin film shows a similar optical absorption spectrum in which the main absorption peaks are maintained at the same positions, indicative of weak aggregation in the solid state. The optical band gap (Eoptg) of SdiPBI-BT estimated from the film absorption edges is 2.07 eV, which is approximate to the Eoptg of SdiPBI.49 For diPBI-BT, the absorption peak at 501 nm in solution is due to the π–π* transition, while the absorption peak at 467 nm may arise from the charge transfer between the two PBI subunits. Unlike SdiPBI-BT, the absorption peaks of diPBI-BT in the films are significantly wider than those in solutions (Fig. 2b), implying that there is significant molecular aggregation in the diPBI-BT films, which is in accordance with their relatively planar configurations. Compared to the solution, the maximum absorption peak of diPBI-BT in the film is blue-shifted, which may be due to π–π stacking in the solid state caused by its planar molecular configuration. The π–π stacking in the solid state forms efficient molecular orbital overlaps that lower the energy level of the ground state. Furthermore, the molecular aggregation and π–π stacking could limit the molecular rotation along the single bond that connects the two subunits of diPBI-BT, which may increase the energy levels of the excited states (π*) of diPBI-BT. As a result, the required energy for the π–π* transition increases and the maximum absorption peak shows a blue-shift. The Eoptg of diPBI-BT is 2.12 eV, slightly higher than that of SdiPBI-BT.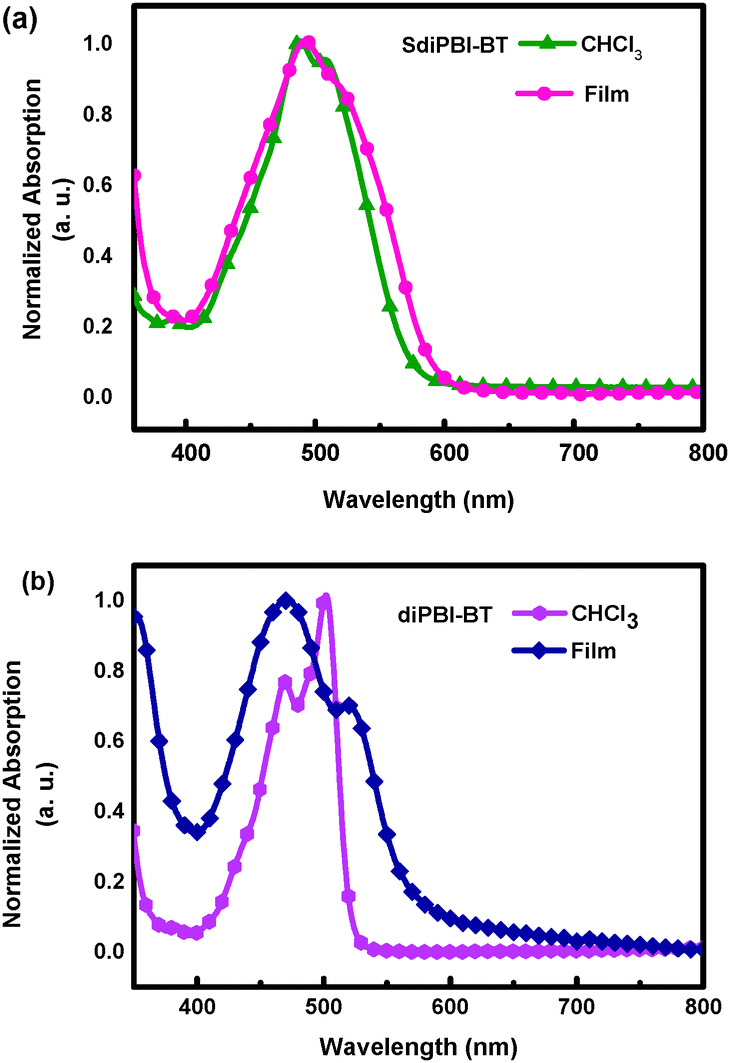 | ||
| Fig. 2 Normalized UV-Vis absorption spectra. (a) The spectra of SdiPBI-BT measured in CHCl3 solution and in thin film on a quartz plate cast from CHCl3. (b) The spectra of diPBI-BT measured in CHCl3 solution and in thin film on a quartz plate cast from CHCl3. | ||
Electrochemical cyclic voltammetry (CV) is used to evaluate the energy levels (Table 1). The LUMO energy level of SdiPBI-BT extracted from its onset reduction potential is close at −3.79 eV. Compared to the LUMO energy level of SdiPBI (−3.92 eV) and SdiPBI-S (−3.85 eV),49 the LUMO energy level of SdiPBI-BT is significantly higher due to its extended conjugate structure and the electron-donating effect of thiophene. The highest occupied molecular orbital (HOMO) energy of SdiPBI-BT calculated from the LUMO energy level and optical band gap is −5.86 eV. The energy offset between the HOMO of the donor polymer (−5.36 eV) and the LUMO of SdiPBI-BT (−3.79 eV) is 1.57 eV, indicating that a high Voc could be obtained. The LUMO energy level of diPBI-BT (−3.67 eV) is higher than SdiPBI-BT, and a higher Voc is anticipated. Since the dihedral angle of SdiPBI-BT is much larger than that of diPBI-BT, the conjugation effect between the two subunits of SdiPBI-BT is weaker than that in diPBI-BT. Besides, the thienobenzenes of diPBI-BT are in the central position of the molecule and the thienobenzenes of SdiPBI-BT are isolated on the two edges of the molecule, so the electronic structure of one subunit in diPBI-BT is significantly influenced by the electron-donating effect of its adjacent bonded thienobenzene in the other subunit of the same molecule, while the electronic structure of one subunit in SdiPBI-BT is slightly affected by the electron-donating effect of the thienobenzene in the other subunit of the same molecule. As a result, the LUMO energy level of diPBI-BT is obviously higher than that of SdiPBI-BT, but the difference between the HOMO energy levels of the two acceptor molecules is relatively small, which explains why diPBI-BT has a larger bandgap than that of SdiPBI-BT. The bandgaps of the two molecules are consistent with the DFT calculated results (2.539 eV for SdiPBI-BT and 2.548 eV for diPBI-BT) (Fig. S2, ESI†).
Photovoltaic properties
In order to estimate the photovoltaic properties, organic solar cells were fabricated with a conventional device architecture of ITO/PEDOT:PSS/PDBT-T1:SdiPBI-BT (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)/Ca/Al, where ITO (indium tin oxide) was the anode, PEDOT:PSS (poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate)) served as the hole transporting layer, PDBT-T1:SdiPBI-BT was the active layer and Ca/Al was used as the cathode. The active layer was prepared by spin-coating the blend solution of donor (D) and acceptor (A) in o-DCB. Since the high boiling point addictive of 1,8-diiodoactane (DIO) has a significant influence on the performance of solar cells, DIO with a volume fraction from 0 to 1% was investigated (Fig. S1, ESI†). Without the DIO additive, the device based on SdiPBI-BT exhibited a PCE up to 5.41% with a Jsc value of 10.20 mA cm−2 and a FF of 55.9%. When the concentration of DIO was 0.5%, the PCE was increased to 6.71% with an enhanced Jsc value of 10.31 mA cm−2 and an improved FF of 68.7%. The high FF is among the best FF values of non-fullerene solar cells.31–43,47 As the DIO concentration increases to 1%, the Jsc value slightly increases to 10.65 mA cm−2, but the FF drops to 60.7%, resulting in a decreased PCE of 6.10%.
:1)/Ca/Al, where ITO (indium tin oxide) was the anode, PEDOT:PSS (poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate)) served as the hole transporting layer, PDBT-T1:SdiPBI-BT was the active layer and Ca/Al was used as the cathode. The active layer was prepared by spin-coating the blend solution of donor (D) and acceptor (A) in o-DCB. Since the high boiling point addictive of 1,8-diiodoactane (DIO) has a significant influence on the performance of solar cells, DIO with a volume fraction from 0 to 1% was investigated (Fig. S1, ESI†). Without the DIO additive, the device based on SdiPBI-BT exhibited a PCE up to 5.41% with a Jsc value of 10.20 mA cm−2 and a FF of 55.9%. When the concentration of DIO was 0.5%, the PCE was increased to 6.71% with an enhanced Jsc value of 10.31 mA cm−2 and an improved FF of 68.7%. The high FF is among the best FF values of non-fullerene solar cells.31–43,47 As the DIO concentration increases to 1%, the Jsc value slightly increases to 10.65 mA cm−2, but the FF drops to 60.7%, resulting in a decreased PCE of 6.10%.
On the basis of the best DIO ratio, we continued to optimize the donor:acceptor (D:A) ratio of SdiPBI-BT-based devices (Table 2). When the D:A ratio is 1.5:1, the PCE drops to 6.13% (Fig. 3a). And when the D:A ratio is 1:1.5, the PCE drops to 5.73%. These results demonstrate that 1:1 is the best D/A ratio. It is noticeable that the Voc under different device preparing conditions varies from 0.94–0.95 eV, which is higher than those of SdiPBI (0.87 V) and SdiPBI-S (0.90 V). Besides, the optimal PCE (6.71%) is close to the best result of SdiPBI-S (7.16%). The IPCE plots for solar cells with various D/A ratios with 0.5% DIO additive are displayed in Fig. 3b. Solar cells based on the combination of PDBT-T1 and SdiPBI-BT show broad IPCE spectra from 300 to 700 nm, which are in accordance with the absorption positions of the PDBT-T1 and SdiPBI-BT films.49 The EQE values in the wavelength range of 520 to 634 nm are higher than 60%, indicating efficient photon harvesting and charge collection. The mismatch between the integral values and the measured Jsc values are within 4% (as shown in Table S1, ESI†).
| Active layer | D:A ratio |
V oc (V) | J sc , (mA cm−2) | FFa (%) | PCEavga (%) | PCEmax (%) |
|---|---|---|---|---|---|---|
| a The reported values are the average PCEs from ten devices. b The values in parentheses are calculated from the EQE data. | ||||||
| PDBT-T1/SdiPBI-BT | 1.5:1 |
0.95 ± 0.005 | 10.37 ± 0.21 (10.24) | 60.2 ± 0.9 | 5.93 ± 0.11 | 6.13 |
| 1:1 |
0.95 ± 0.004 | 10.32 ± 0.11 (10.26) | 67.6 ± 1.0 | 6.61 ± 0.10 | 6.71 | |
| 1:1.5 |
0.94 ± 0.004 | 10.12 ± 0.13 (10.17) | 58.0 ± 0.8 | 5.56 ± 0.13 | 5.73 | |
| PDBT-T1/diPBI-BT | 1.5:1 |
0.99 ± 0.005 | 9.03 ± 0.11 (9.42) | 56.2 ± 0.7 | 5.04 ± 0.10 | 5.12 |
| 1:1 |
0.99 ± 0.003 | 9.67 ± 0.14 (9.89) | 59.8 ± 0.4 | 5.73 ± 0.11 | 5.84 | |
| 1:1.5 |
0.99 ± 0.002 | 8.92 ± 0.12 (9.37) | 58. 0 ± 0.5 | 5.10 ± 0.10 | 5.20 | |
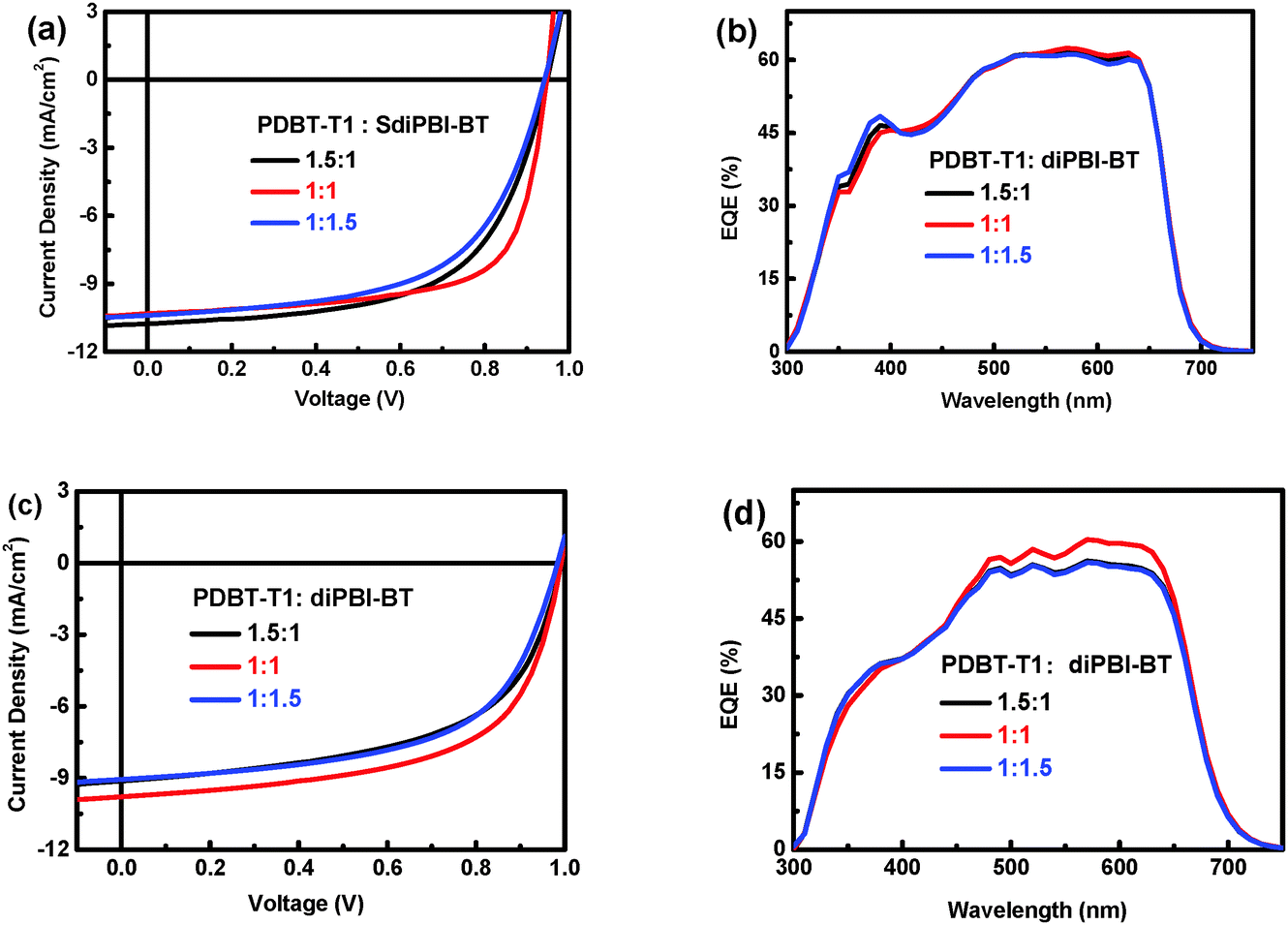 | ||
| Fig. 3
J–V curves of the PDBT-T1:SdiPBI-BT (1:1) and PDBT-T1:diPBI-BT (1:1) solar cells with different D/A ratios with 0.5% DIO additive and the corresponding IPCE spectra. | ||
The optimal results of diPBI-BT were also achieved on the condition of a 1:1 ratio and 0.5% DIO (Fig. 3c). The optimal devices show a PCE of up to 5.84%, with a Voc value of 0.99 V, a Jsc value of 9.78 mA cm−2 and a FF of 60.2%. The devices of diPBI-BT have a higher Voc value (0.99 V) than the SdiPBI-BT devices,49 resulting from the high-lying LUMO levels of diPBI-BT. However, the Jsc value and FF of diPBI-BT are lower than those of SdiPBI-BT, leading to its lower PCE. diPBI-BT also shows a broad IPCE spectra from 300 to 700 nm, but the EQE values in the range of 500–700 nm are lower than those of SdiPBI-BT (Fig. 3d), owing to their different active layer morphologies.
The morphologies were studied using atomic force microscopy (AFM). Without the DIO additive, the PDBT-T1/SdiPBI-BT (1:1) blend film exhibits a fibrous morphology (Fig. 4a). When 0.5% DIO additive was used, the fibrous feature remained unchanged, but the domain size slightly increased and the RMS also increased from 1.09 nm to 1.31 nm (Fig. 4b). The changes in RMS and the different domain sizes may be two reasons for the effects of DIO on the solar cell performance. According to the phase images (Fig. S4, ESI†), the blend films show phase separations with the scale of 10–20 nm, which is favorable for exciton diffusion and separation efficiency.37,43 The PDBT-T1/diPBI-BT (1:1) blend film using 0.5% DIO also have a fibrous morphology, but the domain sizes are significantly larger than that of SdiPBI-BT (Fig. 4c). According to the phase images (Fig. S4, ESI†), the phase separation scales of the PDBT-T1/diPBI-BT blend films are also higher than those of PDBT-T1/SdiPBI-BT. Since severe phase separation could inhibit the charge separation efficiency, the morphology of the PDBT-T1/diPBI-BT blend films could explain the relative low Jsc value and FF of diPBI-BT. Besides, the RMS of the PDBT-T1/diPBI-BT blend film is 3.11 nm, which is much higher than the PDBT-T1/SdiPBI-BT blend film. Since the high RMS may result in larger internal resistance of the solar cells, the higher RMS is another reason for the relatively low FF of diPBI-BT.
 | ||
| Fig. 4 AFM height images (2 μm × 2 μm) of the PDBT-T1/SdiPBI-BT (1:1) blend films (a), the PDBT-T1/SdiPBI-BT (1:1) blend films with 0.5% DIO (b), and the PDBT-T1/diPBI-BT (1:1) films with 0.5% DIO (c). | ||
The electron transport ability of the D/A blend films of the two acceptors was also investigated using a space-charge-limited-current (SCLC) method. The device structure is ITO/Al/PDBT-T1:acceptor/Al. The electron mobility of the PDBT-T1:SdiPBI-BT blend film is 2.21 × 10−3 cm2 V−1 s−1, which is higher than that of the PDBT-T1:diPBI-BT blend film (1.87 × 10−3 cm2 V−1 s−1). Compared to the PDBT-T1:diPBI-BT blend film, the higher electron mobility of the PDBT-T1:SdiPBI-BT blend film, together with its better morphology, contributes to its higher Jsc value and FF.
Conclusions
In summary, we have designed and synthesized two new thienobenzene-fused PBI dimers, SdiPBI-BT and diPBI-BT, as non-fullerene acceptors for organic solar cells. The theoretical calculations show that SdiPBI-BT has a highly twisted structure with an angle of 69.6°, while diPBI-BT has a relatively planar configuration. The photovoltaic devices based on PDBT-T1/SdiPBI-BT exhibit good performances, with PCEs up to 6.71% and Voc values up to 0.95 V. The optimal devices of diPBI-BT show PCEs of up to 5.84% and Voc values of up to 0.99 V. The Voc values of SdiPBI-BT and diPBI-BT are among the highest for non-fullerene organic solar cells. The PDBT-T1/SdiPBI-BT blend films exhibit favorable phase separation and the PDBT-T1/diPBI-BT blend films have relatively large phase separations. The morphology differences arise from the spatial configurations of the two acceptors. These results illustrate that the relative positions of thienobenzene have remarkable influences on the properties of non-fullerene acceptors. The good performance of SdiPBI-BT indicates that ring-fused-PBI based acceptors are potential materials for high-performance solar cells.Experiments
Material synthesis
The synthetic routes towards SdiPBI-BT and diPBI-BT are displayed in Scheme 2. 1 was synthesized according to a procedure reported in the literature.54:petroleum ether (PE) = 3:2, v/v) and 2 was obtained (1.5 g, 1.46 mmol, yield: 68%). 1H NMR (300 MHz, CDCl3) δ (ppm): 9.72 (d, J = 7.5 Hz, 1H), 8.84 (s, 1H), 8.63 (m, 3H), 8.50 (d, J = 7.5 Hz, 2H), 4.12 (t, J = 7.2 Hz, 4H), 2.00 (m, 2H), 1.60–1.06 (m, 64H), 0.86 (m, 12H). MS (MALDI, m/z): [M+] calcd for C64H89N2O4Br, 1030.3; found, 1031.7.
:1 (v/v) solution of acetone and concentrated hydrochloric acid. After stirring for 10 min, the solution was filtrated and the remaining Cu powder was washed with acetone and dried in a vacuum desiccator. Under an Ar atmosphere, 2 (1.5 g, 1.46 mmol) and the treated Cu powder (2.0 g, 31.3 mmol) were added into a flask, 40 mL of dry toluene and 80 mL of dry DMSO were added. The mixture was stirred at 85 °C for 24 h and then poured into 200 mL water. The crude product was extracted using CHCl3 and washed with water. The organic layer was concentrated via vacuum evaporation, and purified using column chromatography (CH2Cl2:PE = 3:1, v/v) to obtain the pure product (1.0 g, 0.53 mmol, yield: 71%). 1H NMR (300 MHz, CDCl3) δ (ppm): 8.80 (d, J = 8.1 Hz, 8H), 8.46 (d, J = 8.1 Hz, 2H), 8.22 (s, 2H), 8.17 (d, J = 8.1 Hz, 2H), 4.05 (m, 8H), 1.90 (m, 4H), 1.60–1.06 (m, 128H), 0.82 (m, 24H). 13C NMR (75 MHz, CDCl3) δ (ppm): 163.51, 163.39, 163.10, 163.05, 141.69, 134.69, 134.13, 134.00, 132.92, 131.57, 130.68, 129.18, 128.72, 128.58, 127.48, 127.32, 124.15, 123.96, 123.48, 123.34, 123.21, 44.65, 44.51, 36.49, 31.77, 31.49, 29.87, 29.49, 29.18, 26.31, 26.12, 22.54, 13.98. MS (MALDI-TOF, m/z): [M+] calcd for C128H178N4O8: 1900.8, found: 1901.6. Elementary analysis (%): calcd: C, 80.88; H, 9.44; N, 2.95; found: C, 80.66; H, 9.27; N, 2.92.
:PE = 2:1, v/v) and 4 was obtained (280 mg, 0.14 mmol, yield: 26%). 1H NMR (300 MHz, CDCl3) δ (ppm): 9.68 (d, J = 8.1 Hz, 2H), 8.97 (m, 2H), 8.79 (d, J = 8.1 Hz, 2H), 8.31 (m, 2H), 8.17 (m, 4H), 4.04 (m, 8H), 1.90 (m, 4H), 1.60–1.06 (m, 128H), 0.83 (m, 24H). 13C NMR (75 MHz, CDCl3) δ (ppm): 163.06, 162.99, 162.75, 162.26, 140.79, 138.13, 133.80, 133.48, 133.32, 133.02, 132.44, 130.60, 129.97, 129.42, 129.25, 128.70, 127.92, 127.70, 127.38, 124.12, 123.25, 122.96, 122.65, 121.19, 44.63, 36.48, 31.79, 31.44, 29.89, 29.52, 29.21, 26.26, 26.15, 22.56, 13.99. MS (MALDI-TOF, m/z): [M+] calcd for C128H176N4O8Br2: 2058.6; found: 2057.9. Elementary analysis (%): calcd: C, 74.68; H, 8.62; N, 2.72; found: C, 74.72; H, 8.82; N, 2.78.
:PE = 2:1, v/v) to obtain the pure product (260 mg, 0.13 mmol, yield: 93%). 1H NMR (300 MHz, CDCl3) δ (ppm): 8.79 (m, 2H), 8.39–8.13 (m, 10H), 7.57 (d, J = 5.1 Hz, 2H), 7.39 (m, 2H), 7.23 (m, 2H), 4.07–4.01 (m, 8H), 1.91 (m, 4H), 1.60–1.06 (m, 128H), 0.86 (m, 24H).13C NMR (75 MHz, CDCl3) δ (ppm): 163.34, 163.25, 163.18, 163.10, 143.43, 140.79, 136.25, 134.73, 134.45, 134.12, 133.97, 133.26, 133.05, 130.21, 130.14, 130.08, 129.99, 129.76, 129.00, 128.88, 128.42, 128.20, 128.12, 127.88, 127.83, 127.71, 127.36, 123.94, 123.89, 123.05, 122.28, 122.23, 44.57, 44.54, 44.51, 36.52, 31.95, 31.79, 31.51, 29.91, 29.52, 29.23, 29.19, 27.86, 27.76, 27.65, 27.05, 26.74, 26.39, 26.28, 26.09, 22.56, 17.39, 13.99, 13.49. MS (MALDI-TOF, m/z): [M+] calcd for C136H182N4O8S2: 2065.1, found: 2064.8. Elementary analysis (%): calcd: C, 79.10; H, 8.88; N, 2.71; found: C, 79.22; H, 8.76; N, 2.64.
:PE = 2:1, v/v) to obtain the pure product (112 mg, 0.05 mmol, yield: 43%). 1H NMR (300 MHz, CDCl3) δ (ppm): 9.99 (br, 2H), 9.27 (br, 2H), 8.79 (m, 4H), 8.59–7.75 (m, 6H), 4.21–3.86 (m, 8H), 1.96 (m, 4H), 1.69–1.06 (m, 128H), 0.82 (m, 24H). 13C NMR (75 MHz, CDCl3) δ (ppm): 164.16, 139.87, 137.71, 133.52, 130.80, 129.39, 129.05, 127.64, 127.41, 126.64, 126.24, 123.73, 123.14, 121.71, 48.58, 45.14, 44.48, 36.96, 32.19, 32.08, 30.37, 30.23, 29.96, 29.81, 29.50, 26.80, 22.96, 22.85, 14.40, 14.32. MS (MALDI-TOF, m/z): [M+] calcd for C136H178N4O8S2: 2061.0, found: 2060.6. Elementary analysis (%): calcd: C, 79.25; H, 8.71; N, 2.72; found: C, 79.22; H, 8.97; N, 2.85.
:PE = 3:1, v/v) to obtain the pure product (1.10 g, 1.12 mmol, yield: 97%). 1H NMR (300 MHz, CDCl3, δ (ppm)): 8.65 (m, 3H), 8.49 (m, 2H),8.21 (d, J = 8.1 Hz, 1H), 8.01 (d, J = 8.1 Hz, 1H), 7.52 (d, J = 4.8 Hz, 1H), 7.20 (m, 2H), 4.13 (m, 4H), 2.01 (m, 2H), 1.60–1.06 (m, 64H), 0.86 (m, 12H). MALDI-TOF (m/z): [M+] calcd: 1033.5; found: 1033.8.
:PE = 3:1, v/v) to obtain the pure product (720 mg, 6.98 mmol, yield: 75%). 1H NMR (300 MHz, CDCl3) δ (ppm): 8.48 (s, 1H), 8.33 (m, 3H), 8.12 (d, J = 4.8 Hz, 2H), 7.66 (d, J = 4.5 Hz, 1H), 7.58 (d, J = 4.5Hz, 1H), 4.17 (m, 4H), 2.01 (m, 2H), 1.60–1.06 (m, 64H), 0.86 (m, 12H). MALDI-TOF (m/z): [M+] calcd: 1031.5; found: 1031.2. Elementary analysis (%): calcd for C68H90N2O4S: C, 79.18; H, 8.79; N, 3.11. found: C, 79.04; H, 8.77; N, 3.04.
:PE = 3:1, v/v) to obtain the pure product (530 mg, 0.26 mmol, yield: 83%). 1H NMR (300 MHz, CDCl3, δ (ppm)): 8.73 (m, 5H), 8.46 (s, 1H), 7.74 (s, 1H), 4.22 (m, 4H), 2.02 (m, 2H), 1.60–1.06 (m, 64H), 0.86 (m, 12H). MALDI-TOF (m/z): [M+] calcd: 1110.4; found: 1110.2. Elementary analysis (%): calcd for C68H89N2O4SBr: C, 73.55; H, 8.08; N, 2.52. found: C, 73.54; H, 8.12; N, 2.33.
:1 (v/v) solution of acetone and concentrated hydrochloric acid. After stirring for 10 min, the solution was filtrated and the remaining Cu powder was washed with acetone and dried in a vacuum desiccator. Under an Ar atmosphere, 10 (300 mg, 0.29 mmol) and the treated Cu powder (1.0 g, 15.6 mmol) were added into a flask. 80 mL of dry toluene and 100 mL of dry DMSO were added. The mixture was stirred at 85 °C for 24 h and then poured into 200 mL of water. The crude product was extracted using CHCl3 and washed with water. The organic layer was concentrated via vacuum evaporation, and purified using column chromatography (CH2Cl2:PE = 3:1, v/v) to obtain the pure product (170 mg, 0.08 mmol, yield: 65%). 1H NMR (300 MHz, CDCl3) δ (ppm): 8.56 (m, 4H), 8.47 (m, 4H), 8.34 (m, 2H), 8.07 (m, 2H), 7.41(s, 2H), 4.16 (m, 8H), 2.03 (m, 4H), 1.60–0.90 (m, 128H), 0.83 (m, 24H). MALDI-TOF (m/z): [M+] calcd: 2061.0; found: 2060.8. Elementary analysis (%): calcd for C136H178N4O8S2: C, 79.25; H, 8.71; N, 2.72. Found: C, 79.22; H, 8.69; N, 2.78.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21572171), the National Basic Research Program of China (973 Program 2013CB834805), the Innovative Research Group of Hubei Province (no. 2015CFA014), and the International Science & Technology Cooperation Program of China (no. 2014DFA52820).Notes and references
- C. B. Nielsen, S. Holliday, H.-Y. Chen, S. J. Cryer and I. McCulloch, Acc. Chem. Res., 2005, 48, 2803–2812 CrossRef PubMed.
- J. T. Bloking, T. Giovenzana, A. T. Higgs, A. J. Ponec, E. T. Hoke, K. Vandewal, S. Ko, Z. Bao, A. Sellinger and M. D. McGehee, Adv. Energy Mater., 2014, 4, 1301426 CrossRef.
- W. Li, W. S. C. Roelofs, M. Turbiez, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2014, 26, 3304–3309 CrossRef CAS PubMed.
- Y. Zhou, T. Kurosawa, W. Ma, Y. Guo, L. Fang, K. Vandewal, Y. Diao, C. Wang, Q. Yan, J. Reinspach, J. Mei, A. L. Appleton, G. I. Koleilat, Y. Gao, S. C. B. Mannsfeld, A. Salleo, H. Ade, D. Zhao and Z. Bao, Adv. Mater., 2014, 26, 3767–3772 CrossRef CAS PubMed.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- M. Hao, G. Luo, K. Shi, G. Xie, K. Wu, H. Wu, G. Yu, Y. Cao and C. Yang, J. Mater. Chem. A, 2015, 3, 20516–20526 CAS.
- J. A. Love, I. Nagao, Y. Huang, M. Kuik, V. Gupta, C. J. Takacs, J. E. Coughlin, L. Qi, T. S. van der Poll, E. J. Kramer, A. J. Heeger, T.-Q. Nguyen and G. C. Bazan, J. Am. Chem. Soc., 2014, 136, 3597–3606 CrossRef CAS PubMed.
- S. Liu, X. Bao, W. Li, K. Wu, G. Xie, R. Yang and C. Yang, Macromolecules, 2015, 48, 2948–2957 CrossRef CAS.
- Y. Lin, J. Wang, S. Dai, Y. Li, D. Zhu and X. Zhan, Adv. Energy Mater., 2014, 4, 1400420 CrossRef.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- J. D. Servaites, B. M. Savoie, J. B. Brink, T. J. Marks and M. A. Ratner, Energy Environ. Sci., 2012, 5, 8343–8350 CAS.
- Y. He and Y. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- J. Huang, X. Wang, X. Zhang, Z. Niu, Z. Lu, B. Jiang, Y. Sun, C. Zhan and J. Yao, ACS Appl. Mater. Interfaces, 2014, 6, 3853–3862 CAS.
- L. Ye, K. Sun, W. Jiang, S. Zhang, W. Zhao, H. Yao, Z. Wang and J. Hou, ACS Appl. Mater. Interfaces, 2015, 7, 9274–9280 CAS.
- Y. Cai, L. Huo, X. Sun, D. Wei, M. Tang and Y. Sun, Adv. Energy Mater., 2015, 5, 1500032 CrossRef.
- J. D. Douglas, M. S. Chen, J. R. Niskala, O. P. Lee, A. T. Yiu, E. P. Young and J. M. J. Fréchet, Adv. Mater., 2014, 26, 4313–4319 CrossRef CAS PubMed.
- Y.-J. Hwang, H. Li, B. A. E. Courtright, S. Subramaniyan and S. A. Jenekhe, Adv. Mater., 2016, 28, 124–131 CrossRef CAS PubMed.
- O. K. Kwon, J.-H. Park, D. W. Kim, S. K. Park and S. Y. Park, Adv. Mater., 2015, 27, 1951–1956 CrossRef CAS PubMed.
- H. Li, Y.-J. Hwang, B. A. E. Courtright, F. N. Eberle, S. Subramaniyan and S. A. Jenekhe, Adv. Mater., 2015, 28, 3266–3272 CrossRef PubMed.
- H. Lin, S. Chen, Z. Li, J. Y. L. Lai, G. Yang, T. McAfee, K. Jiang, Y. Li, Y. Liu, H. Hu, J. Zhao, W. Ma, H. Ade and H. Yan, Adv. Mater., 2015, 27, 7299–7304 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- J. W. Jung and W. H. Jo, Chem. Mater., 2015, 27, 6038–6043 CrossRef CAS.
- S. Li, W. Liu, M. Shi, J. Mai, T.-K. Lau, J.-H. Wan, X. Lu, C.-Z. Li and H. Chen, Energy Environ. Sci., 2016, 9, 604–610 CAS.
- P. Josse, C. Dalinot, Y. Jiang, S. Dabos-Seignon, J. Roncali, P. Blanchard and C. Cabanetos, J. Mater. Chem. A, 2016, 4, 250–256 CAS.
- L. R. Rutledge, S. M. McAfee and G. C. Welch, J. Phys. Chem. A, 2014, 118, 7939–7951 CrossRef CAS PubMed.
- K. Cnops, G. Zango, J. Genoe, P. Heremans, M. V. Martinez-Diaz, T. Torres and D. Cheyns, J. Am. Chem. Soc., 2015, 137, 8991–8997 CrossRef CAS PubMed.
- H. Li, T. Earmme, G. Ren, A. Saeki, S. Yoshikawa, N. M. Murari, S. Subramaniyan, M. J. Crane, S. Seki and S. A. Jenekhe, J. Am. Chem. Soc., 2014, 136, 14589–14597 CrossRef CAS PubMed.
- D. Meng, D. Sun, C. Zhong, T. Liu, B. Fan, L. Huo, Y. Li, W. Jiang, H. Choi, T. Kim, J. Y. Kim, Y. Sun, Z. Wang and A. J. Heeger, J. Am. Chem. Soc., 2016, 138, 375–380 CrossRef CAS PubMed.
- Y. Lin and X. Zhan, Mater. Horiz., 2014, 1, 470–488 RSC.
- Y. Lin and X. Zhan, Adv. Energy Mater., 2015, 5, 1501063 CrossRef.
- Y. Lin and X. Zhan, Acc. Chem. Res., 2016, 49, 175–183 CrossRef CAS PubMed.
- X. Zhan, Z. a. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246–7247 CrossRef CAS PubMed.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- Y. Lin, Q. He, F. Zhao, L. Huo, J. Mai, X. Lu, C.-J. Su, T. Li, J. Wang, J. Zhu, Y. Sun, C. Wang and X. Zhan, J. Am. Chem. Soc., 2016, 138, 2973–2976 CrossRef CAS PubMed.
- L. Dou, J. You, Z. Hong, Z. Xu, G. Li, R. A. Street and Y. Yang, Adv. Mater., 2013, 25, 6642–6671 CrossRef CAS PubMed.
- Y. Liu, C. Mu, K. Jiang, J. Zhao, Y. Li, L. Zhang, Z. Li, J. Y. L. Lai, H. Hu, T. Ma, R. Hu, D. Yu, X. Huang, B. Z. Tang and H. Yan, Adv. Mater., 2015, 27, 1015–1020 CrossRef CAS PubMed.
- R. D. Pensack, C. Guo, K. Vakhshouri, E. D. Gomez and J. B. Asbury, J. Phys. Chem. C, 2012, 116, 4824–4831 CAS.
- J. Lee, R. Singh, D. H. Sin, H. G. Kim, K. C. Song and K. Cho, Adv. Mater., 2016, 28, 69–76 CrossRef CAS PubMed.
- Y. Lin, Y. Wang, J. Wang, J. Hou, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2014, 26, 5137–5142 CrossRef CAS PubMed.
- Y. Zang, C.-Z. Li, C.-C. Chueh, S. T. Williams, W. Jiang, Z.-H. Wang, J.-S. Yu and A. K. Y. Jen, Adv. Mater., 2014, 26, 5708–5714 CrossRef CAS PubMed.
- X. Zhang, Z. Lu, L. Ye, C. Zhan, J. Hou, S. Zhang, B. Jiang, Y. Zhao, J. Huang, S. Zhang, Y. Liu, Q. Shi, Y. Liu and J. Yao, Adv. Mater., 2013, 25, 5791–5797 CrossRef CAS PubMed.
- W. Jiang, L. Ye, X. Li, C. Xiao, F. Tan, W. Zhao, J. Hou and Z. Wang, Chem. Commun., 2014, 50, 1024–1026 RSC.
- J. Yi, Y. Wang, Q. Luo, Y. Lin, H. Tang, H.-Y. Wang and C. Ma, Chem. Commun., 2016, 52, 1649–1652 RSC.
- Q. Yan, Y. Zhou, Y.-Q. Zheng, J. Pei and D. Zhao, Chem. Sci., 2013, 4, 4389–4394 RSC.
- Z. Lu, B. Jiang, X. Zhang, A. Tang, L. Chen, C. Zhan and J. Yao, Chem. Mater., 2014, 26, 2907–2914 CrossRef CAS.
- Y. Liu, J. Y. L. Lai, S. Chen, Y. Li, K. Jiang, J. Zhao, Z. Li, H. Hu, T. Ma, H. Lin, J. Liu, J. Zhang, F. Huang, D. Yu and H. Yan, J. Mater. Chem. A, 2015, 3, 13632–13636 CAS.
- J. Zhao, Y. Li, J. Zhang, L. Zhang, J. Y. L. Lai, K. Jiang, C. Mu, Z. Li, C. L. C. Chan, A. Hunt, S. Mukherjee, H. Ade, X. Huang and H. Yan, J. Mater. Chem. A, 2015, 3, 20108–20112 CAS.
- D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156–11162 CrossRef CAS PubMed.
- T. Liu, D. Meng, Y. Cai, X. Sun, Y. Li, L. Huo, F. Liu, Z. Wang, T. P. Russel and Y. Sun, Adv. Sci., 2016, 3, 1600117 CrossRef PubMed.
- L. Huo, T. Liu, X. Sun, Y. Cai, A. J. Heeger and Y. Sun, Adv. Mater., 2015, 27, 2938–2944 CrossRef CAS PubMed.
- L. Lu and L. Yu, Adv. Mater., 2014, 26, 4413–4430 CrossRef CAS PubMed.
- D. Qian, L. Ye, M. Zhang, Y. Liang, L. Li, Y. Huang, X. Guo, S. Zhang, Z. a. Tan and J. Hou, Macromolecules, 2012, 45, 9611–9617 CrossRef CAS.
- J. Zhao, Y. Li, H. Lin, Y. Liu, K. Jiang, C. Mu, T. Ma, J. Y. Lin Lai, H. Hu, D. Yu and H. Yan, Energy Environ. Sci., 2015, 8, 520–525 CAS.
- C. W. Ge, C. Y. Mei, J. Ling, J. T. Wang, F. G. Zhao, L. Liang, H. J. Li, Y. S. Xie and W. S. Li, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1200–1215 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental information; device fabrication details; DFT calculated frontier orbital distributions; other OSC device data and AFM phase images. See DOI: 10.1039/c6qm00194g |
| ‡ These two authors contributed equally to this work. |
| This journal is © the Partner Organisations 2017 |