
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent developments in protease activity assays and sensors
Irvine Lian Hao
Ong
 and
Kun-Lin
Yang
*
and
Kun-Lin
Yang
*
National University of Singapore, 4 Engineering Drive 4, Singapore 117585. E-mail: cheyk@nus.edu.sg; Fax: +65 6779-1936; Tel: +65 6516-6614
First published on 19th April 2017
Abstract
Proteases play a pivotal role in regulating important physiological processes from food digestion to blood clotting. They are also important biomarkers for many diseases such as cancers. The importance of proteases has led to extensive efforts in the screening of proteases and their inhibitors as potential drug molecules. For example, human immunodeficiency virus (HIV) patients have been treated with HIV-1 protease inhibitors to prolong the life expectancy of patients. Such a close relationship between diseases and proteases provides a strong motivation for developing sensitive, selective, and robust protease assays and sensors, which can be exploited to discover new proteases and inhibitors. In this aspect, protease assays based on levels of proteolytic activities are more relevant than protease affinity assays such as immunoassays. In this review, recent developments of protease activity assays based on different detection principles are discussed and compared. For homogenous assays, fluorescence-based techniques are the most popular due to their high sensitivity and quantitative results. However, homogeneous assays have limited multiplex sensing capabilities. In contrast, heterogeneous assays can be employed to detect multiple proteases simultaneously, given the microarray technology that is already available. Among them, electrochemical methods, surface spectroscopy techniques, and enzyme-linked peptide protease assays are commonly used. Finally, recent developments in liquid crystal (LC)-based protease assays and their applications for detecting proteases and their inhibitors are discussed.
 Irvine Lian Hao Ong | Irvine received his PhD and BEng in Chemical and Biomolecular Engineering from the National University of Singapore and Nanyang Technological University, respectively. His PhD research areas include the development of biosensors for monitoring protease activity and applications of liquid crystals as soft templates for the self-assembly of colloids and biosensors. He is currently a Research Scientist at Matralix, an NUS startup company. His present research involves the use of fluidic devices for the precise manufacturing of products in the pharmaceutical and cosmetic industries. |
 Kun-Lin Yang | Dr Yang is an associate professor in the Department of Chemical and Biomolecular Engineering at the National University of Singapore (NUS). Before joining NUS, he was a post-doctoral researcher in the Chemical and Biomolecular Department at the University of Wisconsin – Madison working on liquid crystal sensors. He received his BS and MS degrees in Chemical Engineering from National Taiwan University and PhD degree from Georgia Tech. He is the recipient of the Defense Innovation Research Program Award and the A*Star Research Grant Award for his work on liquid crystal sensors. His present research interests include biosensors, liquid crystals and microfluidics. |
Introduction
Proteases are enzymes that catalyse the hydrolysis of peptide bonds in peptide chains. Some proteases are classified based on their catalytic sites, namely aspartic, glutamic, cysteine, serine, and threonine proteases. On the other hand, matrix metalloproteinases (MMPs) refer to a group of proteases that require metal ions for their catalytic activites.3 In humans, 1.7% of human genes is coded for proteases, and proteases are responsible for basic functions such as protein digestion, wound healing, and the activation of immune systems.5 Furthermore, protease activities are linked to diseases such as cancers,6 cardiovascular diseases,8 Alzheimer's disease,9 human immunodeficiency virus (HIV),10 thrombosis,11 and diabetics.12 Inhibitions of disease-related proteases can potentially be used as drug molecules to treat these diseases. One canonical example is the case of HIV, where medication with protease inhibitors has been proven to prolong the life expectancy of HIV-positive patients.13 Given such compelling motivations, extensive efforts have been devoted to develop sensitive, selective, and robust protease assays for the quantitative detection of proteases and their inhibitors. The primary function of proteases is to hydrolyse a peptide bond between two amino acids (endopeptidase) or terminal amino acids at the amino (N-) or carboxyl (C-) terminals (exopeptidase).3 This results in fragmentation of a single peptide chain into two shorter peptide chains. Scheme 1 describes the hydrolysis at positions P1 and P1′ of a peptide chain. The degree of substrate specificity can vary widely. For example, serine proteases such as trypsin only require positively-charged lysine and arginine at the P1 position.14 On the other hand, prostate-specific antigen (PSA) activities require a sequence of HSSKLQ (P6-P1).2 Another example is the optimized sequence of LVPR||GS (P4-P2′) for thrombin activities,4 while an aspartic acid at the P10 position can enhance the peptide's specificity towards thrombin activities.7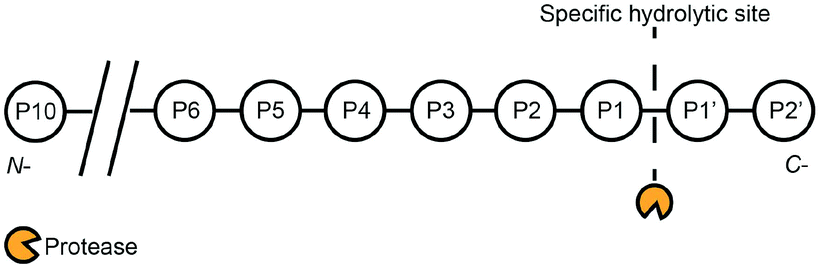 | ||
| Scheme 1 Proteases exhibit varying degrees of substrate specificity. For example, trypsin essentially only requires P1 to be lysine or arginine.1 On the other hand, prostate-specific antigen (PSA) requires a sequence of HSSKLQ (P6-P1).2 Another example is the optimized sequence of LVPR||GS (P4-P2′) for thrombin activities,4 while an aspartic acid at the P10 position can enhance the peptide's specificity towards thrombin activities.7 | ||
Mechanistically, the hydrolysis of peptide chains by proteases involves specific interactions between catalytic sites of proteases and complementary amino acid sequences through covalent bond formation, electrostatic attraction, hydrogen bonding, or van der Waal's forces.14,15 Specificities of such protease activities result in the hydrolysis of peptide chains that contain specific amino acid sequences. Given this understanding, custom-made peptide substrates are often used in protease assays to detect the presence of a target protease. This is complemented by the advent of the solid-phase synthesis of peptides with predetermined amino acid sequences.16 This approach provides tremendous advantages over conventional, non-specific protease assays using casein as a non-specific substrate.17–19
In terms of assay classification, protease assays can be classified as activity and affinity assays. For affinity assays, protease detection and quantification is based on the presence of proteases, regardless of their activity levels. In contrast, activity assays are more relevant to detect protease functions. Such activity assays are of great interest, given the direct relationship between protease activities and diseases. Therefore, we shall focus on protease activity assays in this review. For assay operation modes, protease assays can be classified into homogeneous and heterogeneous assays. For homogeneous assays, both substrates and samples are present in an aqueous phase. Conversely, heterogeneous assays consist of protease substrates that are immobilised on a solid platform, while samples are in the aqueous phase. On the basis of detection methods, homogeneous assays can be further classified into colorimetric assays,20,21 mass spectrometry-based assays,22–24 and fluorescence resonance energy transfer (FRET) assays.25–34 More recently, nanomaterials such as noble metal nanoparticles,35–46 quantum dots (QDs),47–59 and graphene oxide (GO)60–64 are also used in the homogeneous protease assays with impressive detection limits. However, homogenous assays generally have a limited multiplexed sensing capability. This limitation arises due to the limited number of probes that can produce distinct signals. For example, in a single sample readout, fluorogenic probes are constrained by their emission profiles, and only a few probes can be used to provide well-resolved emission signals from the sample. Although high throughput microtiter plates can provide a platform for automated and high throughput screening,65 the homogenous assay inherently requires more liquid sample handling during screening. This limitation can be circumvented by using heterogeneous assays, where a large number of substrates can be immobilised at discrete locations on a solid surface. This approach allows the simultaneous and multiplexed detection of proteases present in a single sample, since detection results will depend on signal levels at discrete locations on the solid surface. Heterogeneous assays can be further classified into electrochemical assays,66–79 surface-enhanced Raman scattering (SERS) assays,80–83 and surface plasmon resonance (SPR) assays.84–87
Recently, liquid crystal (LC)-based assays are emerging as label-free assays for many applications. These LC assays are simple to use and highly sensitive. They also generate optical signals that are visible to the naked eye. Molecules which form a LC phase are typically rod-shaped molecules with long-range interactions. When they are subjected to an electric or magnetic field, they re-orient to a preferential direction. Because these molecules are also birefringent, they display unique optical textures when viewed under crossed polarizers.88 This phenomenon has been ubiquitously applied in liquid crystal displays. Another interesting property is their ability to influence anchoring of LCs by engineering the chemical composition or morphology of surfaces supporting the LC phase.89,90 This phenomenon was rigorously studied by Abbott and co-workers, who established principles to detect the adsorption of biomolecules on solid surfaces or LC/water interfaces.91–99 These events were visualised directly by using changes in the optical textures of LCs. Later on, Yang and co-workers demonstrated excellent sensitivity, selectivity, and multiplex capability of LC-based protease activity assays.100,101 In this review, we discuss recent developments of protease assays according to their assay formats (homogenous or heterogeneous) and detection principles. We also assess the performance of LC-based assays compared to other protease assays.
Homogenous protease assays
Conventionally, peptide fragments resulting from homogeneous protease assays can be separated using liquid chromatography and then detected by UV,102 fluorescence,103,104 or mass spectrometry.105 The advantage of this approach is that different peptide fragments can be identified and analysed accurately. However, only end-pointed measurements are allowed and expensive instrumentation is needed in this approach. To develop real-time protease assays, recent efforts have been focused on the use of FRET-based detection techniques. This research field is also fuelled by emerging fluorescent nanomaterials like QDs and gold nanoclusters (AuNCs).FRET principles
FRET-based assays often consist of a short peptide linked to a FRET donor and a FRET quencher in close proximity (1–10 nm), such that the emission of the fluorescence donor is quenched by the acceptor.106 Thus, hydrolysis of the FRET peptide substrate will cause the FRET pair to separate and the fluorescence signal to increase (Scheme 2a). For the homogeneous assays, both proteases and their substrates are in an aqueous phase. The main difference among these assays lies in the donors and acceptors, which range from organic molecules to nanomaterials. | ||
| Scheme 2 Principles of FRET-based protease sensors. (a) FRET sensors based on organic FRET donors and acceptors. (b) FRET sensors based on nanomaterials as FRET acceptors and FRET donor-labelled protease substrates. (c) FRET sensors based on nanomaterials as FRET donors and FRET acceptor-labelled protease substrates. | ||
Organic FRET donors and acceptors
Traditionally, FRET-based protease sensors use an organic FRET acceptor and donor covalently linked to a peptide chain (Scheme 2a). For example, Matayoshi et al. synthesized HIV-1 protease substrates with an organic fluorescent donor, 5-[(2-aminoethyl)amino]naphthalene-1-sulfonic acid (EDANS), and a fluorescent quencher, 4-(4-dimethylaminophenylazo)benzoic acid (DABCYL), at the C- and N-terminals of the peptide substrates, ((DABCYL)-(γ-aminobutyric acid, GABA)-SQNY||PIVQ-(EDANS) and (DABCYL)-(GABA)-SVVY||PVVQ-(EDANS)),† respectively.107 GABA was inserted to prevent steric hindrance that arises from the bulky DABCYL during the binding of peptide substrates to HIV-1 protease. Hydrolysis of the peptide substrate at Tyr-Pro increased fluorescence emissions of EDANS, and protease activities were monitored by the emission spectrum of EDANS. This approach forms the basis of most FRET-based protease assays. To improve detection limits, Zauner et al. incorporated an enzyme cascade to improve the sensitivity of the FRET-based protease assay.108 They introduced the trypsin precursor, trypsinogen, which was activated by enteropeptidase to form trypsin. This simple enzymatic cascade system greatly amplified enteropeptidase activities, and 0.2 pM of enteropeptidase could be detected using an optimised peptide sequence labelled with fluorescein (FAM, FRET donor) and 5(6)-carboxytetramethylrhodamine (TAMRA, FRET acceptor) ((TAMRA)-GSR||C-(FAM)). However, reliance on organic FRET acceptors and donors is susceptible to long-term stability and photo-bleaching issues. The interferences from assay compounds may also affect the fluorescence signals of such assays.109 Thus, the focus has been shifted to nanomaterials which can overcome these stability issues.Nanomaterials as FRET acceptors or donors
Nanomaterials like carbon nanotubes (CNTs), GO, and gold nanoparticles (AuNPs) have been successfully employed as effective FRET acceptors in biosensing applications.110,111 Furthermore, peptides can be immobilised on CNTs or GO through bioconjugation to form a FRET-based protease sensor (Scheme 2b).112 Consequently, these carbon nanomaterials can serve the dual function of a surface for facile bioconjugation and a fluorescence quencher due to the strong π-stacking. For example, Huang et al. developed a multiplex protease assay for matrix metalloproteinase-7 (MMP-7), MMP-2, and urokinase-type plasminogen activator (uPA) by immobilisation of protease-specific peptide substrates that were labelled with different organic dyes (MMP-7: GVPLSLTMGK-(fluorescein isothiocyanate (FITC)); MMP-2: GCGPLGVRGK-(indocarbocyanine (Cy3)); uPA: GGSGRSAAAK-(indocarbocyanine (Cy5))).113 The specific detection of the three proteases was based on fluorescence emission of the respective organic dye linked to each peptide substrate. A similar approach was adopted by Zhu et al., who used a CNT-based material (FRET acceptor) to electrostatically attract FAM-labelled peptide substrates (FRET donor, (FAM)-GGLVPR||GSG).114 Hydrolysis of the peptide substrate by 0.1 nM of thrombin resulted in the observable emission of FAM. This was due to the removal of a FAM-labelled peptide fragment from the CNT-based substrate by thrombin activities.Similar detection strategies involving GO (FRET acceptor) and fluorescence-labelled peptide substrates (FRET donor) were also demonstrated by other groups.62,63,115–117 For example, Feng et al. used GO (FRET acceptor) and a fluorescence-labelled peptide substrate (KALQLKSSHE-(FITC), FRET donor) to detect PSA.63 Fluorescence increased in the presence of PSA at 0.3 nM. Gu et al. also demonstrated the detection of trypsin using a similar concept of GO (FRET acceptor) and a fluorescence-labelled peptide substrate (R6-(FAM), FRET donor).115 Interestingly, GO was exploited as a FRET donor by Kwak et al. for the detection of chymotrypsin and MMP-2.64 In this case, QXL570 (FRET acceptor)-labelled peptide substrates were immobilised on GO, and the hydrolysis of these peptides by proteases caused an increase in the fluorescence from the GO. In general, carbon nanomaterials can be used as generic quenchers for fluorescent molecules in close proximity. This strategy is very straightforward as there is no need to match the excitation–emission profiles of FRET donors and acceptors. However, reliance on organic dyes as FRET donors is still subject to the limitations mentioned above.
AuNPs are also effective FRET acceptors and they have been exploited to develop protease sensors.118–120 This is because the localised surface plasmon resonance of AuNPs leads to strong absorbance within the visible light spectrum. In the literature, peptides with cysteine residues were covalently immobilised on AuNPs as protease substrates. For example, Wang et al. used peptides with terminal cysteine residues to immobilise fluorescence-labelled peptides ((FITC)-ALNNGGGGHAKRRLIFGGGC, FRET donor) on AuNPs (FRET acceptor).121 The hydrolysis of this peptide by trypsin, chymotrypsin, or thermolysin removed FITC-labelled peptide fragments from the AuNPs, leading to an increase in the fluorescence signal. Park et al. also demonstrated the immobilisation of fluorescence-labelled peptides ((TAMRA)-GPLGMRGLH6, FRET donor) on carboxyl-modified AuNPs (FRET acceptor) in the presence of nickel. The detection of MMP-7 was achieved when the hydrolysis of fluorescence-labelled peptide increased the fluorescence signal.122
These examples show that nanomaterials can have dual functions in protease assays as FRET acceptors and a robust platform for immobilisation of protease substrates. However, because organic dyes are used as FRET donors, these assays are subject to the limitations mentioned above.
Quantum dots (QDs) as FRET donors
Recently, inorganic QDs are emerging as excellent fluorescent materials with superior photo-stability, a high molar absorption coefficient, and wide excitation–emission peak separation.123 In the past, QDs have been used as FRET donors in protease sensors, in which peptide substrates were immobilised on the surface of QDs and labelled with a FRET quencher. Such designs allow emissions of QDs to be restored upon hydrolysis of the immobilised peptide substrates (Scheme 2c). For example, Sapsford et al. used core/shell QDs (CdSe/ZnS) to detect light-chain botulinum neurotoxin serotype A (BoNT/A-Lc) using Cy3-labelled peptides ((acetyl)-(Cy3)-CSSNKTRIDQANQ||RATKMLSWGLSGG(alpha-amino butyric acid, Aib)AAA(Aib)AASLH6-(CONH2)).52 In this work, they elucidated the importance of surface capping agents of QDs in influencing the assembly and proteolytic hydrolysis of peptide substrates. Wang and Xia also developed a similar protease assay for thrombin detection with an LOD of 0.03 μM. In this case, they used CdSe/ZnS QDs and Cy5-labelled peptide ((Cy5)-DDDLVPR||GSGP9GGH6) as the donor and acceptor pair, respectively.50 Elsewhere, Serrano et al. successfully detected trypsin activity using QDs functionalised with TAMRA-labelled peptide substrates (CK||R||VK-(TAMRA)), with an LOD of 1 nM.51 However, the above-mentioned studies involving QDs still required organic dyes as FRET acceptors, which are susceptible to photobleaching or interferences from assay compounds. To overcome this issue, Lowe et al. used AuNPs as FRET acceptors and QDs as FRET donors for the detection of protease uPA. However, extensive surface modifications of QDs with streptavidin and the peptide substrate with terminal cysteine and biotin (uPA: (biotin)-SGR||SANC-(CONH2)) are required.53 To simplify the process, a label-free method was proposed by He and Ma.49 They showed that a dithiol peptide (GCK||GCG) inhibited the formation of cadmium-telluride (CdTe) QDs, while a monothiol peptide promoted the formation of QDs. Thus, trypsin activities that hydrolysed dithiol peptides to monothiol peptides resulted in the formation of QDs with fluorescent emission. In this system, they reported an LOD of 0.8 pM. This work presented a label-free method for the specific detection of protease activities by using peptides to influence the synthesis of QDs. However, a more in-depth study on the role of cysteine (or other amino acids) during the synthesis of QDs is required before this detection principle can be applied to other types of proteases. Furthermore, such a mechanistic understanding is required to predict and overcome possible interferences from compounds present in the samples, which may also influence QD formations. Another label-free method was proposed by Li et al.59 They used a thrombin-specific peptide substrate (CCSGR||PVLGG) that induced the aggregation of CdTe QDs and quenched fluorescence emission. The presence of thrombin (0.2 nM) caused the dispersion of QDs and increased fluorescence signals. The use of specific peptide substrates to induce the aggregation of QDs provides a universal principle for developing label-free protease assays. In another study, Wu et al. also suggested the use of label-free substrates for serine proteases.48 They showed that the presence of native cytochrome C (Cyt C) quenched the phosphorescence of manganese (Mn)-doped ZnS QDs, and hydrolysis of Cyt C by trypsin increased the phosphorescence signals. However, the use of a protein substrate limits its application for specific protease detection. Therefore, an aggregation induced by peptide substrates would be more attractive. Nonetheless, such aggregation-induced optical responses seen in the above-mentioned studies provide a straightforward detection mechanism and optical responses that are easily quantifiable.Bioluminescence resonance energy transfer (BRET)
Some enzymatic reactions with bioluminescence can be coupled to a fluorescent protein in a so-called BRET system (Scheme 3).124 This phenomenon is caused by the overlapping of bioluminescence of the enzyme with the excitation of the fluorescent protein. Such an energy transfer mechanism is identical to FRET and requires the same criterion of close proximity (<10 nm) between the donor (light-emitting enzyme) and the quencher (fluorescent protein). Thus, cleavage of a peptide linking the enzyme and the protein leads to a decrease in the fluorescence. Using this principle, Dacres et al. detected thrombin by using a BRET substrate, (green fluorescent protein, GFP)-LQGSLVPR||GS-(Renilla luciferase, RLuc).125 The bioluminescence was produced by RLuc, which excited GFP to give fluorescence emission. Upon the hydrolysis of the specific peptide substrate by thrombin, the emission by GFP was reduced. Because no external excitation source is required, the assay is highly sensitive (LOD ∼0.22 nM). In another example, Branchini et al. incorporated an organic fluorophore in their assay, where a red fluorescent protein (RFP) was labelled with Alexa Fluor® (AF) 680 dye and linked to a luciferase (Luc) through a specific peptide substrate.126 They detected caspase 3, thrombin, and factor Xa activities with LODs of 0.41 nM, 3 nM, and 1 nM, respectively. The advantage of incorporating a second resonance energy transfer with a fluorophore was the enhanced peak separation ∼143 nm, compared to 45 nm without AF 680. Another variant of BRET-based assays was demonstrated by Yu et al., where they used a carboxyl derivative of fullerene (C60-COOH) as the acceptor of bioluminescence from humanised Gaussia luciferase (hGLuc).127 The final BRET protein system consisted of a histidine tag, a thrombin-specific peptide, a peptide linker (PL), and hGLuc (H6LVPR||GS-(PL)-(hGLuc)). The histidine tag was used, in the presence of Ni2+, for immobilisation of the BRET protein system with C60-COOH. The presence of thrombin activities increased bioluminescence measurements as C60-COOH becomes separated from the hGLuc, and they reported a detection limit of 11 pM. One major disadvantage of BRET systems is the influence of buffer conditions on the fluorescence intensity measurements, as seen in the study by Li and co-workers.128 They showed that the presence of an optimised buffer containing imidazole was required to enhance the detection limit of enterokinase by five-fold. To address the susceptibility of BRET assays towards buffer conditions, Li and co-workers recently developed a BRET system based on hGLuc and tandem dimeric Tomato (tdTomato) as a RFP.129 This system showed minimal variations in the ratio of BRET signals when buffering agents or pH values were varied. Their work clearly illustrated the strong influences of assay media on the performance of BRET assays. Lastly, a recent work by Wu et al. demonstrated the on-line detection of protease activity in a microfluidic setup, based on fluorescence measurements of a modified BRET system ((GFP)-LQGSLVPR||QGSLQ-(RLuc)).130 The use of a microfluidic assay format led to better sensitivity and a lower LOD (27 pM), thanks to the faster mass transfer and smaller sample volume required. Overall, BRET-based assays provide a low background fluorescence measurement, due to the absence of excitation sources required, and good peak separations between the bioluminescence and fluorescent protein emissions. However, the careful design of a BRET system is required to prevent erroneous measurements that may arise due to a varied assay composition. | ||
| Scheme 3 Principles of BRET for protease activity assays. (a) Diminished emission from the light emitting enzyme and increased emission from the fluorescent proteins signify the absence of protease activities, while (b) the diminished emission from the fluorescent protein signifies the hydrolysis of a peptide substrate by a target protease. | ||
Fluorescence polarisation (FP)
Besides the measurement of fluorescence intensities, the polarisation of the fluorescence signal is capable of providing information on the rotational motions of the fluorescent-labelled molecules. The polarisation of the fluorescence signal is increased for larger molecules, as the rotational motion is slower compared to a small molecule.131 In a recent study, this phenomenon was adopted by Kim et al., where they detected protease activity by monitoring the changes in the FP of fluorescence-labelled casein in a microfluidic setup. The hydrolysis of the non-specific substrate resulted in a decrease in FP, due to the smaller, fluorescence-labelled hydrolysis products (Scheme 4). Another example was demonstrated by Lee et al.,132 where they used Gag proteins for the screening of HIV-1 protease inhibitors. Zhang et al. also used native protein substrates for the monitoring of ADAM metallopeptidase domain 17 (ADAM 17).133 However, due to the large proteins that were required in these studies, the specific detection of proteases is not possible. To overcome this issue, an amplification method is required to create drastic changes in the molecular weight of fluorescence-labelled peptides that are specific to their target proteases. For instance, Huang et al. immobilised FAM-labelled peptides (FAM-GGLVPR||GSGH10) on the multiwalled CNT.134 Upon hydrolysis by thrombin, significant changes to the overall molecular weight of the FAM-labelled peptide resulted in a measurable decrease of FP. This method was also applicable for the detection of chymotrypsin activities. Such a conjugation of specific peptides on nanomaterials can potentially overcome the selectivity issues related to the use of proteins as protease substrates. Overall, FP-based protease assays provide a good format to detect protease activities with their natural protein substrates, and the measurement of FP instead of fluorescence intensity circumvents problems associated with the variety of factors (e.g. assay medium compositions or pH) that can affect fluorescence intensity. Furthermore, nanomaterials can be used as solid carriers for the immobilisation of protease substrates and amplification of FP measurements. Nonetheless, a possible shift in the fluorophore quantum yield after the binding of fluorescence-labelled substrates to proteins is expected, and the aggregation of fluorescence-labelled substrates in reaction media may cause erroneous results.109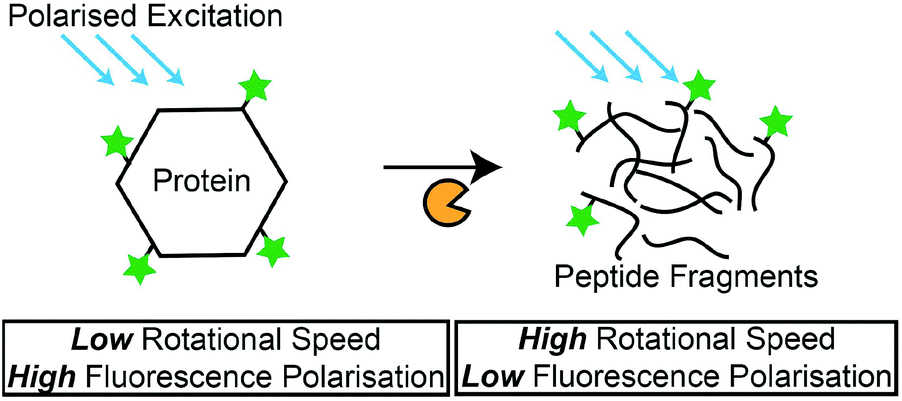 | ||
| Scheme 4 Principles of FP-based protease assays. FP measurements decrease as the fluorescence-labelled protein is hydrolysed by proteases. | ||
Gold nanoclusters (AuNCs)
AuNCs are clusters of tens of Au atoms and highly fluorescent.135 They are potential candidates for replacing organic fluorescence dyes for biosensor applications. In the literature, protein-encapsulated AuNCs can be synthesized in the presence of bovine serum albumin (BSA). This system was exploited as a platform for protease detection.136 Wang et al. reported that the fluorescence of AuNCs decreased when BSA was hydrolysed by proteases.137 They hypothesized that the hydrolysis of BSA exposed AuNCs to ambient oxygen and reduced AuNC fluorescence. Lin et al. also made use of this concept to detect cystatin C (Cys C).138 However, numerous proteases also hydrolyse BSA, resulting in poor selectivity. To overcome this issue, Gu et al. used peptide templates (CCAAA||A) for AuNC synthesis.139 This strategy allowed a selective detection of elastase at 0.03 nM. Thus, AuNCs can potentially function as a label-free, fluorescence-based protease sensor. Such an approach of introducing peptides during the synthesis process is similar to the work presented by He and Ma for the QD-based protease assay.49Interestingly, these reports show the great potential of peptides in influencing the synthesis of nano-scaled materials. As opposed to the use of small organic molecules to stabilise and influence the growth of nanomaterials, like AuNRs,141 peptides may provide a more complex influence on the nuclei formation and growth mechanism of nanomaterials. Therefore, protease assays can potentially be developed using peptides that strongly influence the synthesis process of nano-scaled materials.142 However, an established model would be required to achieve this goal. So far, there have been no reports of a generic approach to select peptide templates for AuNC synthesis, and the application of this technique for different target proteases is limited.
Gold nanoparticles (AuNPs) in colorimetric assays
AuNPs are commonly used in colorimetric assays due to their localised surface plasmon resonance phenomenon.143 The aggregation of nanoparticles can lead to larger nanoparticles which exhibit different colors.144 Making use of such principles, Xue et al. developed trypsin assays by using peptides with repeated arginine residues (R6) to induce the aggregation of AuNPs.45 Hydrolysis of this peptide substrate resulted in the dispersion of AuNPs and an observable colour change from purple to red, with an LOD of 0.07 nM. To improve selectivity, Ding et al. reported the use of customised peptides (e.g. trypsin: YHPQMNPYTK||AGGGC) that induced the aggregation of AuNPs.36 Thus, hydrolysis of peptides by target proteases (trypsin and chymotrypsin) caused the dispersion of AuNPs. Elsewhere, Chen et al. reported the use of peptide-immobilised AuNPs for the detection of MMP-7.41 Substrate peptides (NAADLEKAIEA||LEKHLEAKGPCDAAQ||LEKQLEQAFEAFERAG) prevented the aggregation of AuNPs through their secondary structures,145 and hydrolysis of the substrates by MMP-7 induced the aggregation of AuNPs. Using a similar approach, Chen et al. immobilised negatively-charged peptides (trypsin: EEEEGLLGALGK||C; MMP-2: EEEEGPLG||LAGGC) on AuNPs dispersed in aqueous solutions.140 Subsequently, hydrolysis of the immobilised peptide promoted the aggregation of AuNPs due to the removal of negatively-charged peptide fragments that were required to stabilise the AuNPs. Using this method, they reported an LOD of 5 nM for trypsin and MMP-2. These assays demonstrated the feasibility of using AuNPs for detecting proteases.146 AuNP-based detection is advantageous because the colour changes can be detected with the naked eye or spectrometry for quantitative analysis. Moreover, localised surface plasmon resonance is not susceptible to photo-bleaching as observed in the fluorescence-based methods mentioned above. Nonetheless, the colloidal stability of the AuNPs must be carefully studied to ensure a robust assay, especially in a complex matrix.Table 1 shows a summary of homogeneous protease assays for detecting trypsin and chymotrypsin, keeping in mind that the inherent activities of proteases used in these studies may vary, and the concentrations presented serve as a common unit for comparison. Among all the techniques, the fluorescence-based protease sensor developed by Fan et al. has the lowest LOD of 6 pM for chymotrypsin.32 However, this technique requires peptide substrates flanked by repeated arginine residues. This chemical modification may limit its applications for proteases that do not hydrolyse such substrates. For trypsin, the assay developed by He and Ma has the lowest LOD of 4 pM.49 It must be noted that this technique requires substrates having 2 cysteine residues to influence the formation of QDs. However, no in-depth analysis on the role of cysteine residues was provided. Thus, more studies would be required for a better understanding of such protease assays.
| Reference | Detection principle | Protease substratea | LOD |
|---|---|---|---|
| a Peptide sequences follow the one-letter code for amino acids. || indicates cleavage site. | |||
| Zauner et al.108 | Fluorescence | (FAM)-CAR||SG-TAMRA | Trypsin (2 pM) |
| Gu et al.115 | Fluorescence | R 6 -FAM | Trypsin (∼4 nM) |
| He and Ma49 | Fluorescence | GCK||GCG | Trypsin (4 pM) |
| Serrano et al.51 | Fluorescence | CK||R||VK-(TAMRA) | Trypsin (0.043 μM) |
| Fan et al.32 | Fluorescence | R 5 | Trypsin (0.011 nM) |
| RRF||FRR | Chymotrypsin (6 pM) | ||
| Wang et al.121 | Fluorescence | Bovine serum albumin | Trypsin (0.7 nM) |
| Chymotrypsin (0.4 nM) | |||
| Xue et al.45 | Colorimetric | R 6 | Trypsin (0.07 nM) |
| Ding et al.36 | Colorimetric | YHPQMNPYTK||AGGGC | Trypsin (0.5 nM) |
| Chen et al.140 | Colorimetric | EEEEGLLGALGK||C | Trypsin (5 nM) |
Heterogeneous protease assays
Heterogeneous protease assays require the immobilisation of protease substrates on solid surfaces. These surfaces allow proteases in the aqueous phase to interact with the immobilised substrates and a large number of substrates can be immobilised on the same platform for multiplex sensing applications. This is possible as optical or fluorescence signals at discrete locations can be identified and associated with a specific protease. An important consideration for the heterogeneous assay is the choice of solid platforms for signal readouts. For example, electrochemical-based sensors require conductive surfaces like metals, while colorimetric sensors usually require optically transparent platforms like glass. Thus, careful design of peptide substrates is required to ensure that functional groups are present for the covalent immobilisation of peptides on these surfaces.Electrochemical sensors
Electrochemical sensors rely on oxidation or reduction reactions which result in measurable electrical signals using electrodes.147 In these sensors, peptides are often labelled with redox reporters and immobilised on the electrodes to be cleaved by proteases. For example, Ji et al. developed a thrombin sensor using a p-aminodiphenylamine (pADA)-labelled peptide substrate (PFR||(pADA)), where pADA acted as a redox reporter.70 Hydrolysis of the peptide substrate caused the electrochemical signal to decrease due to the departure of the redox reporter (Scheme 5a). A similar approach was adopted by Shin et al. to detect MMP-9 using a methylene blue (MB)-labelled peptide substrate ((MB)-GPLG||MWSRC).148 Hydrolysis of the peptide substrate by MMP-9 decreased electrochemical signals. In another example, Ko et al. used ferrocene (Fc), a common redox reporter, to label fibrinogen for the detection of thrombin activities.73 Fibrinogen-coated surfaces were incubated with thrombin and Fc-labelled fibrinogen. Subsequently, the hydrolysis of fibrinogen and Fc-labelled fibrinogen produced fibrin and Fc-labelled fibrin that adsorbed onto the surface through electrostatic attraction. Therefore, increasing thrombin activities produced greater electrochemical signals through an increase in the Fc-labelled fibrin on the surfaces (Scheme 5b). However, their LOD (2.7 pM) was still too high compared to other sensors. The poor LOD may be attributed to the reliance on the adsorption/desorption of reporter molecules, where such events do not involve any amplification mechanism to drastically increase the signals generated. Therefore, to further amplify the presence of protease activities, signal amplification techniques were developed. For example, Wu et al. incorporated streptavidin-alkaline phosphatase (Sav-ALP) on electrodes through a biotin-labelled peptide substrate ((biotin)-ALNN(D-leucine, dL)||(dL)||KNNLAC). Sav-ALP catalysed the formation of electrochemically active phenol and increased the initial electrochemical signals.77 In the presence of a protease biomarker from Bacillus licheniformis, the biotin moiety was removed from the electrodes and that caused a reduction in the electrochemical signal. However, the need for second enzymatic reactions may limit its general applicability due to the unforeseen or undesirable interactions of the secondary enzyme with the peptide substrates used, thereby affecting the accuracy of such assays. Recently, an ultrasensitive protease was reported by Park et al.,149 with an impressive detection limit of 0.5 pg ml−1 (∼0.02 pM) for trypsin activity. This was achieved through a sequential process of capturing the target protease at the substrate interface, followed by the selective hydrolysis of the p-aminophenol (AP)-labelled peptide substrate (GPR||(AP)) and a secondary redox cycling reaction that occurs at the interface to generate an amplified electrochemical signal. This does not require any washing step and it is convenient to use. However, one important criterion is the availability of protease-specific antibodies for the initial immobilisation of target proteases at the substrate interface. Another amplification technique was demonstrated by Liu et al., who used electrodes functionalised with peptides containing repeated arginine residues (CAPGGAR5) to attract CdTe QDs.74 Fluorescence arising from QDs was then converted to measurable photocurrents. Thus, the hydrolysis of these substrates by trypsin removed CdTe attached to the arginine fragments from the electrode surfaces, and this decreased the photoelectrochemical signals. However, these assays usually required the synthetic labelling or binding of QDs to peptide substrates, both of which may adversely affect protease activities. Understandably, label-free substrates were desired to minimise such effects. For example, Cao et al. used peptides containing positively-charged arginine residues (R||GGLAC) to prevent the interaction of electrodes with cationic electrochemical species in the aqueous phase.66 Therefore, the neutral peptide fragment (GGLAC) remaining on the electrode after hydrolysis by trypsin allowed electrochemical species to access the electrodes and increased electrochemical signals. A similar approach was reported by Deng et al., who used a peptide substrate ((acetyl)-CHS||SKLQK) that electrostatically attracted electroactive species which are charged. Subsequently, charge transfer between the charged species and electrodes gave electrical signals.68 A neutral peptide fragment ((acetyl)-CHS) after the hydrolysis of peptide substrates by PSA limited the access of electrochemical species with the electrodes and decreased the measured signals, and they reported an LOD of 0.02 nM. This LOD is one order better than the FRET-based PSA assay developed by Feng et al.63 Another interesting technique was demonstrated by Xia et al. They used an oxidation reaction catalysed by the peptide–heme complex to increase electrochemical signals.78 Thus, cleavage of a peptide substrate (CPPPPGGGKTEEISEVNL||DAEFRHDSGYEVHHQK) by beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) prevented the complex formation and decreased electrochemical signals. The dual role of the peptide as a protease substrate and an enzyme is very useful. Nonetheless, the enzymatic activity requires a 16-amino acid-long sequence and may limit the specificity of such assays, since it contains hydrolysis sites for multiple proteases. Elsewhere, GO was also employed by Chen et al. to increase the binding of MB near electrodes.67 It was achieved by the immobilisation of a peptide substrate ((acetyl)-GGHDEVD||HGGGC) through C-terminal cysteine on an electrode. Upon hydrolysis by caspase-3, the remaining peptide fragment (HGGGC) presented free NH2 groups at the electrode and promoted covalent attachment of GO. Consequently, the presence of GO attracted MB to increase electrochemical signals. They reported a remarkable LOD of 0.06 pg ml−1 (∼3 fM), which was among the lowest LOD reported. This achievement may be attributed to the outstanding conductivity of GO, making them an excellent reporter for electrochemical-based assays. Electrochemical-based protease assays are not only sensitive but can also be miniaturised for point-of-care (POC) applications.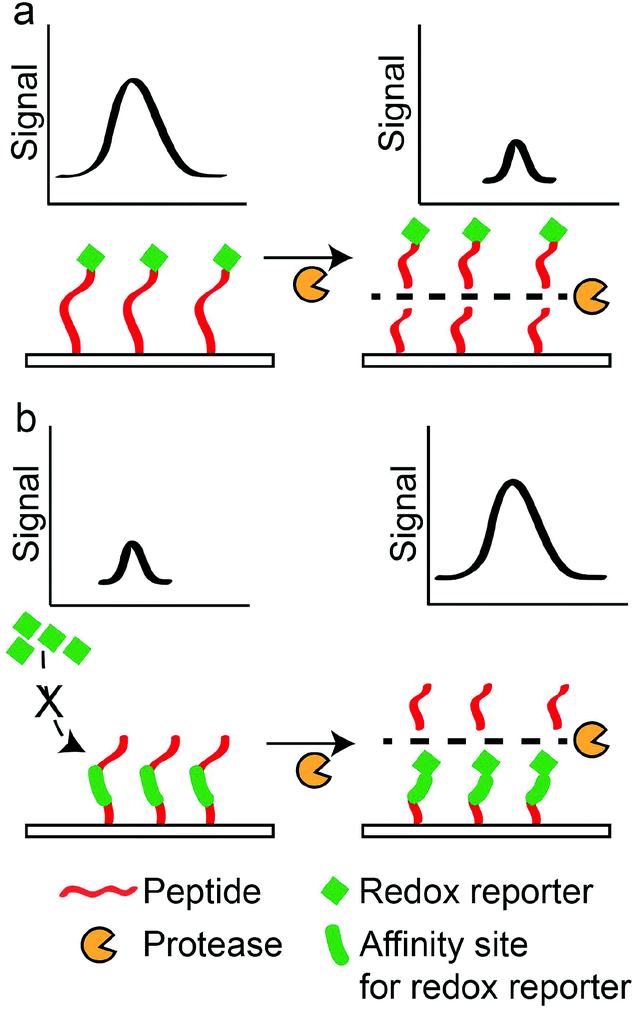 | ||
| Scheme 5 Principle for electrochemical-based protease sensors. (a) “Turn-off” detection principle, where hydrolysis of the immobilised peptide labelled with a redox reporter decreases the electrochemical signal. (b) “Turn-on” detection principle, where hydrolysis of the immobilised peptide allows the binding of a redox reporter to increase the electrochemical signal. | ||
Surface plasmon resonance (SPR) sensors
Label-free methods are attractive as native peptides without additional tags can be used as protease substrates. Thus, protease activities can be determined without influences from synthetic labels. SPR is a common label-free technique which can be used to monitor the cleavage of a peptide immobilised on a sensor surface. In SPR, a beam of light with varying incident angles impinges on a gold surface, and intensities of the reflected light are measured at different angles to determine the reflection angle with the lowest light intensity. Importantly, such measurements are used to characterise the refractive index of the sensor surface, and the concentration of analytes bound to the surface can be correlated with changes in the refractive indexes. This working principle provides a quantitative detection mechanism for developing protease assays.150 Typically, a decrease in the surface substrate density due to protease activities is monitored by using SPR. For example, an early work by Steinrücke et al. included a helical protein (78 amino acid long) after a protease specific peptide section to ensure that protease activities removed the helical protein to produce detectable SPR signals.151 More recently, Esseghaier et al. immobilised peptides that were tagged to 30 nm magnetic beads on the SPR sensor surfaces.85 HIV-1 protease activities, in addition to the magnetic field applied above the sensor surface, formed the amplification mechanism to produce SPR signals. They also showed that a longer spacer (8 vs. 4 amino acid) and smaller magnetic beads (30 nm vs. 1 μm) provided better access for the protease and higher surface density of peptides, respectively, and they reported a detection limit of 10 pg ml−1. Besides using such amplification mechanisms, SPR was also reported to be highly sensitive to changes in the surface charge.152 Therefore, Chen et al. developed a caspase-3 SPR-based protease assay using a cationic peptide substrate (K4DEVD||HH5C) immobilised on a SPR sensor surface.84 The removal of the charged fragment (K4DEVD) in the presence of caspase-3 activity drastically altered the surface charge, and they reported a good detection limit of 1 pg ml−1. Overall, the SPR technique provides a label-free platform for real-time activity monitoring. However, the SPR technique is more adapted for protein–protein interactions,150 and some form of signal amplification (as seen above) is required to detect the hydrolysis of peptide substrates for specific protease detection.Surface-enhanced Raman spectroscopy (SERS) sensors
SERS is another optical spectroscopy technique that relies on the enhancement of Raman scattering by metallic nanostructures formed on surfaces or through aggregation (termed as “SERS hot-spots”).153 SERS-based protease assays can be performed in a homogenous format, where noble metal nanoparticles are used in conjunction with a SERS reporter molecule. The dispersion/aggregation of the nanoparticles in the presence of target proteases produces SERS signals.80,81,83,154 However, the uncontrolled aggregation of noble metal nanoparticles can create reproducibility issues due to the uncontrollable formation of SERS hot-spots. To overcome this, Sun et al. fabricated heterogeneous protease assay made up of well-controlled nanoplasmonic resonator (NPR) arrays, where each NPR consisted of a sandwiched SiO2 layer (5 nm-thick) between two Ag layers (each layer is 20 nm-thick).155 The PSA-specific peptide substrate ((SERS reporter: Rhodamine 19)-HSSKLQ||LAAAC) was immobilised on the NPRs, and the lowest PSA concentration detected was 6 pM. The repeatable and highly-controlled fabrication of NPRs can provide a reliable SERS substrate for reproducible SERS readings. The authors also suggested the potential of detecting over 500 proteases (with ten peptide substrates for each target protease) in a single array, based on the microarray technology that was already available. In contrast, a less-controlled heterogeneous SERS-based assay was presented by Yazgan et al., where they immobilised protein substrates labelled with gold nanoparticles or nanorods.82 5,5-Dithiobis(2-nitrobenzoic acid) was used as a SERS reporter and covalently immobilised on the AuNPs or gold nanorods (AuNRs). The hydrolysis of the protein substrate by crude protease extracted by bovine pancreas decreased the SERS signal, and they reported an LOD of 0.43 mU ml−1 for the crude protease (1 U = 1.0 absorbance change at 345 nm in 1 min and at 25 °C and pH 7.5). They showed that the AuNRs were most efficient in enhancing the performance of the SERS-based assay, while an alternative homogenous assay format produced poorer assay performance. The use of crude proteases limited the comparison of this assay with other works, while the protein substrates used prevented the direct application of this method for specific protease activity detection. In another work, Yang et al. fabricated surfaces that were sequentially modified with a peptide substrate (CHR||DDG) that contained two oppositely charged segments and a Raman reporter molecule (4-mercaptobenzoic acid, MBA).156 Upon hydrolysis of the peptide by trypsin, the surface became positively-charged and attracted negatively-charged AgNPs. As a result of the decreased proximity between the AgNPs and MBA, SERS signals increased. This assay had an LOD of 1 nM. Despite the enhanced protease specificity compared to the work by Yazgan et al., the random process of surface roughening may cause great SERS signal variations among different locations of the supporting substrate. Overall, SERS-based assays, combined with precise nanofabrication and microarray technology, can provide a sensitive and selective protease assay.Enzyme-linked peptide protease assays
Enzyme-linked peptide protease assays typically provide measurable signals through enzymes which are linked to the peptide substrates. These enzymes catalyse the formation of coloured compounds such that they are able to report the presence of peptide substrates (Scheme 6). For example, Ding et al. immobilised biotin-labelled peptides (PPLR||INR||HILTR||GGG-(biotin)) on a glass surface, and then conjugated the peptides to Sav-ALP.157 The enzyme (ALP) was able to remove a phosphate group from ascorbic acid 2-phosphate and produced ascorbic acid, which reduced silver ions to silver particles. Based on this principle, they reported an LOD of 0.4 nM for trypsin. Cheng et al. also used silver deposition to produce optical signals for the detection of MMP-7, with a detection limit of 4.8 pM. Peptides conjugated with AuNPs (H6RPLALWRSC-AuNP) and target protease were concurrently incubated on a surface.158 Due to the hydrolysis of the peptide by MMP-7, the AuNP-containing peptide fragments are removed from the surface. Subsequently, a secondary silver enhancement reaction was carried out to quantify the amount of remaining AuNPs on the surface. Although these methods provide colorimetric responses, they require secondary reactions to generate the colorimetric response. Thus, specific conditions are required to carry out these secondary reactions, and that complicates the signal generation process. Furthermore, the stability of secondary enzymes may require special attention to prevent any denaturation or undesired interactions that may affect the accuracy of the assays. | ||
| Scheme 6 Formation of coloured products from enzyme-linked peptides. Absence of coloured products indicates the presence of a target protease. | ||
Liquid crystal- (LC)-based assays
In modern societies, LCs are known for their ubiquitous presence in TV displays, computer monitors and mobile devices. LCs also possess a wealth of unique properties that can be utilised in many other applications, ranging from directing microparticle,159 nanoparticle,160 and molecular assemblies161 to water-responsive gels162 and gas sensors.95 LCs are a unique class of materials that exhibit a certain degree of molecular orderliness while retaining the fluidity of conventional liquids. There are several phases of LCs, depending on the degree of molecular orderliness, and the nematic phase of LCs is widely used in assay developments. LCs that form the nematic phase are made up of rod-like molecules that orient in a specific direction (or director) without positional order when subjected to external fields. In addition, chemical compositions of surfaces in contact with the LC phase can influence the direction of LCs. Importantly, LCs away from the surfaces also orient under the influence of these surfaces as a consequence of the minimization of the overall free energy.88 The ability of surfaces to influence the overall orientation of LCs is an important property utilised in LC-based assays. In LC-based protease assays, LCs are used to distinguish surfaces that are functionalised with long and short peptides.100,101,163,164 The working principle of LC-based protease assays is to design peptide-immobilised surfaces that cause LCs to orient planarly (parallel to the surface) and produce a bright optical signal. In the presence of target proteases, peptides were hydrolysed and removed from the surface. The decrease in the peptide chain length resulted in a homeotropic orientation (perpendicular to surface) and produced dark optical signals. Based on this principle, Bi et al. developed an optical bar chart for the semi-quantitative detection of protease activities.100 The hydrolysis of the immobilised peptide substrate (CDR||VYIHPFHLK) by trypsin resulted in dark LC optical signals, and they reported an LOD of 2 nM (Scheme 7). Later on, Chen and Yang also used a similar approach to detect trypsin and chymotrypsin activities, where they immobilised peptide substrates (for example: CKGSNRTRIDEGNQRATRMLGGKETSAAKLKRKYWW) and detected the hydrolysis of peptides by proteases. Using this method, they reported an LOD of 0.04 nM and 4 pM for trypsin and chymotrypsin, respectively.101 However, this method is unable to distinguish between the two proteases. These studies demonstrated the potential of carrying out heterogeneous protease assays that are capable of multiplex sensing.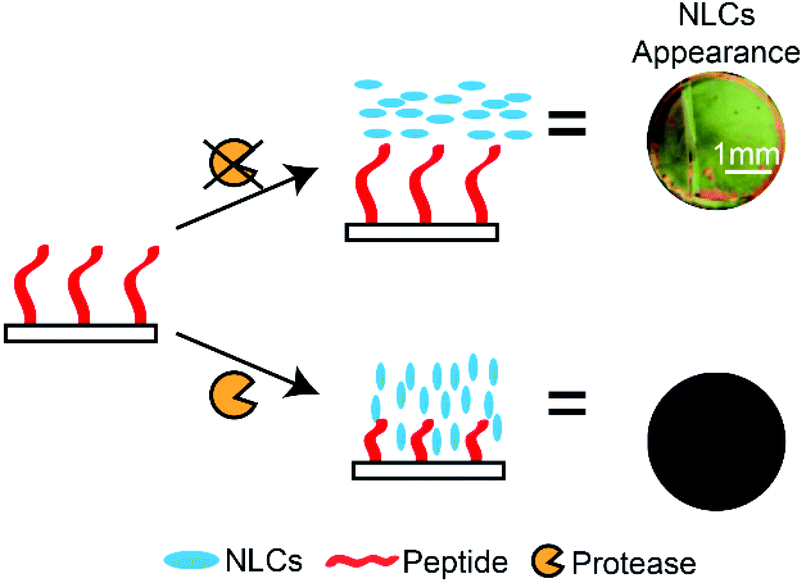 | ||
| Scheme 7 Principles of nematic liquid crystal (NLC)-based protease assays: differences in the LC optical texture in the presence of a target protease. Peptide substrates are immobilised on glass substrates, and cleavage of the peptide substrate changes surface composition. | ||
Furthermore, LC-based assays are not susceptible to long-term stability and photo-bleaching issues. However, these LC-based protease assays relied solely on immobilised peptides to influence the LC signals.100,163,165 Similar to the detection principles presented above, to improve the LOD of these assays, the incorporation of an amplification mechanism is required to enhance changes in the LC optical signals after hydrolysis of the immobilised peptides. A possible approach is shown in the literature by Ding and Yang. They exploited the enzymatic reactions and silver deposition on solid surfaces to amplify the protease activities.157 Such an approach can potentially be applied to all LC-based assays and improves detection limits.
Elsewhere, studies using LC/water interfaces were also studied as a possible sensing platform for protease activities. Such systems provide the real-time detection of protease activity, since the LC interface can be monitored in real-time with proteases in the aqueous phase. For example, Zhang and Jang reported the use of the poly-L-lysine decorated LC/water interface for the detection of trypsin activity.166 They showed that the hydrolysis of poly-L-lysine at the LC/water interface resulted in a bright-to-dark transition in the LC texture. To improve the selectivity of LC sensors at the LC/water interface, Chang and Chen reported the detection of chymotrypsin activity using customised peptide substrates (CKKKY||GSNY||TY||IDEGNQY||ATY||GGGGY||ETSAAY||GY||KKKC).167 The amphiphilic peptides were physically adsorbed and cross-linked on the LC/water interface, and the subsequent hydrolysis of the peptides by trypsin disrupted the cross-linked structure of peptides at the LC/water interface. This resulted in the peptide desorption and re-orientation of LC molecules that led to a transition in the LC optical texture, with an LOD of 0.02 μM. This work shows great potential for the development of a specific protease assay at the LC/water interface. Nonetheless, it would be desirable for the covalent immobilisation of peptide substrates at the LC/water interface. Such an approach would likely be more robust and applicable in assays with a complex matrix, given the greater stability of covalent immobilisation as compared to physical adsorption. However, the covalent immobilisation of peptides on the LC/water interface is less studied. One previous work was reported by Bi and Yang, where they doped lauric aldehyde within the LC phase.168 Subsequently, the LC phase was immersed in an aqueous solution containing the peptide substrate that reacted with the aldehyde group of the dopant. More recently, Eimura et al. doped biotin-labelled LC molecules within an unmodified LC phase and observed the self-assembly of the biotin-labelled LCs at the LC/water interface.169 These works can potentially be developed upon for the detection of protease activities in a complex matrix. Compared to detection at glass surfaces, the advantage of such LC/water interface detection is the real-time detection of protease activity. However, more work may be required to establish the robust immobilisation of peptide substrates at the LC/water interfaces.
Table 2 summarises the recently developed heterogeneous trypsin and chymotrypsin assays based on concentrations of proteases as a common unit for the comparison of LOD. Electrochemical-based detection by Park et al. provided the best LOD for trypsin (∼0.02 pM).149 However, one limitation in this method is the use of protease-specific antibodies for the capturing of target proteases, where the availability of the antibodies may limit its generic application for wide-ranging proteases. For chymotrypsin, LC-based assays by Chen and Yang provided the best LOD for chymotrypsin (4 pM).101 In this assay, peptides with multiple lysine residues were required for stable peptide immobilisation. This caused selectivity issues, and they were unable to distinguish between trypsin and chymotrypsin activities. Therefore, further work is required for the development of a heterogeneous LC-based protease assay with better selectivity and sensitivity.
| Reference | Detection principle | Protease substratea | LOD |
|---|---|---|---|
| a Peptide sequences follow the one-letter code for amino acids. b AP: p-aminophenol. || indicates cleavage site. | |||
| Liu et al.74 | Electrochemical | CAPGGAR5 | Trypsin (3 nM) |
| Cao et al.66 | Electrochemical | R||GGLAC | Trypsin (0.6 nM) |
| Park et al.149 | Electrochemical | GPR||(AP)b | Trypsin (∼0.02 pM) |
| Ding and Yang157 | Colorimetric | PPLR||INR||HILTR||GGG-(biotin) | Trypsin (0.4 nM) |
| Bi et al.100 | LC | CDR||VYIHPFHLK | Trypsin (2 nM) |
| Chen and Yang101 | LC | CK||GSNR||TR||IDEGNQR||ATR||MLGGKETSAAKLKR||K||YWW | Trypsin (0.04 nM) |
| CL||SEL||DDRADAL||QAGASQF||ESSAAKL||KRKY||W||W||KNL||K | Chymotrypsin (4 pM) | ||
Conclusions
Currently, there are many types of protease assays under development, but none of them is perfect. For homogenous assays, even though very low detection limits (∼pM) can be achieved, they suffer from issues such as limited stability of the fluorescent molecules. In addition, the interference of fluorescence signals from assay components can result in false positives or negatives.109 To overcome this, inorganic QDs and AuNCs are exploited to provide additional stability. Besides fluorescence-based techniques, colorimetric assays with AuNPs are widely studied due to their simplicity. However, due to the inherent instability of AuNPs, carefully designed peptides with specialised properties are immobilised on AuNPs to ensure better stability against aggregation.140 Such specific peptide properties will need to be carefully considered for the general design of peptide substrates for different proteases. On the other hand, heterogeneous assays provide a multiplexed platform for the detection of proteases. However, since protease substrates are typically immobilised on surfaces, only proteases near these surfaces interact with these substrates. Therefore, the sensitivity of heterogeneous assays is low. To overcome this, immobilised peptides on microparticles with a high surface-to-volume ratio can be used to improve interactions between proteases and peptides for better sensitivity. In the future, LC-based assays may emerge as an alternative platform which provides the fast, sensitive and multiplex detection of proteases. However, challenges in LC-based assays, including the stability of immobilised peptide substrates, sensitivity of the assay, and limited quantitative detection capabilities, need to be addressed.Overall, most studies presented above worked with model proteases, like trypsin and chymotrypsin, while assays were more extensively studied based on a buffered matrix. Undoubtedly, the use of model proteases in simple buffer systems is an important first-step required to proof detection concepts. The absence of possible interferences from a complex biological matrix allows researchers to unravel the intrinsic mechanistic behaviour of the assays. However, with the growing interest for understanding protease activities as potential drug targets or biomarkers for diseases, there has been a growing emphasis on the need for more extensive studies to be carried out in a complex matrix or real biological samples. Such a matrix can cause unforeseen interferences to the detection mechanisms and must be carefully studied to ensure an accurate assay platform. Additionally, the use of a complex matrix must be coupled with the targeting of disease-related proteases to provide a holistic study to assess the feasibility and performance of assays with a clear clinical relevance.
Acknowledgements
I would like to thank Dr Kung Yi and Dr Gobi for the fruitful discussion on this work.Notes and references
- J. T. T. Baird and C. S. Craik, in Handbook of Proteolytic Enzymes, ed. G. Salvesen, Academic Press, 2013, pp. 2594–2600, DOI:10.1016/B978-0-12-382219-2.00575-5.
- S. R. Denmeade, W. Lou, J. Lövgren, J. Malm, H. Lilja and J. T. Isaacs, Cancer Res., 1997, 57, 4924–4930 CAS.
- C. López-Otín and J. S. Bond, J. Biol. Chem., 2008, 283, 30433–30437 CrossRef PubMed.
- D. B. Smith and K. S. Johnson, Gene, 1988, 67, 31–40 CrossRef CAS PubMed.
- X. S. Puente, L. M. Sanchez, C. M. Overall and C. Lopez-Otin, Nat. Rev. Genet., 2003, 4, 544–558 CrossRef CAS PubMed.
- R. D. A. Wilkinson, R. Williams, C. J. Scott and R. E. Burden, Biol. Chem., 2015, 396, 867–882 CrossRef CAS PubMed.
- H. C. Marsh, Y. C. Meinwald, T. W. Thannhauser and H. A. Scheraga, Biochemistry, 1983, 22, 4170–4174 CrossRef CAS PubMed.
- Y. Hua and S. Nair, Biochim. Biophys. Acta, Mol. Basis Dis., 2015, 1852, 195–208 CrossRef CAS PubMed.
- J. Yuan and B. A. Yankner, Nature, 2000, 407, 802–809 CrossRef CAS PubMed.
- H. Haim, I. Salas and J. Sodroski, J. Virol., 2013, 87, 1884–1889 CrossRef CAS PubMed.
- A. Doucet and C. M. Overall, Mol. Aspects Med., 2008, 29, 339–358 CrossRef CAS PubMed.
- S. H. Havale and M. Pal, Bioorg. Med. Chem., 2009, 17, 1783–1802 CrossRef CAS PubMed.
- C. Flexner, Nat. Rev. Drug Discovery, 2007, 6, 959–966 CrossRef CAS PubMed.
- L. Hedstrom, Chem. Rev., 2002, 102, 4501–4524 CrossRef CAS PubMed.
- B. D. Hames and N. M. Hooper, Biochemistry, Taylor & Francis, New York, N.Y., 3rd edn, 2005 Search PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- O. Folin and V. Ciocalteu, J. Biol. Chem., 1927, 73, 627–650 CAS.
- A. Voller, A. Bartlett and D. E. Bidwell, J. Clin. Pathol., 1978, 31, 507–520 CrossRef CAS PubMed.
- S. Aldrich, Universal protease activity assay: casein as a substrate, http://www.sigmaaldrich.com/life-science/learning-center/life-science-video/universal-protease.html, (accessed 16/07/2016).
- X. Ding and K.-L. Yang, Analyst, 2015, 140, 340–345 RSC.
- Z. J. Zhou, L. Peng, X. Y. Wang, Y. Xiang and A. J. Tong, Analyst, 2014, 139, 1178–1183 RSC.
- J. Hu, F. Liu and H. Ju, Anal. Chem., 2015, 87, 4409–4414 CrossRef CAS PubMed.
- X. Yan, L. Yang and Q. Wang, Angew. Chem., 2011, 50, 5130–5133 CrossRef CAS PubMed.
- U. S. Lathia, O. Ornatsky, V. Baranov and M. Nitz, Anal. Biochem., 2011, 408, 157–159 CrossRef CAS PubMed.
- H. W. Wang, D. N. Udukala, T. N. Samarakoon, M. T. Basel, M. Kalita, G. Abayaweera, H. Manawadu, A. Malalasekera, C. Robinson, D. Villanueva, P. Maynez, L. Bossmann, E. Riedy, J. Barriga, N. Wang, P. Li, D. A. Higgins, G. H. Zhu, D. L. Troyer and S. H. Bossmann, Photochem. Photobiol. Sci., 2014, 13, 231–240 CAS.
- T. M. Feltrup and B. R. Singh, Anal. Chem., 2012, 84, 10549–10553 CrossRef CAS PubMed.
- X. H. Xia, M. X. Yang, L. K. Oetjen, Y. Zhang, Q. G. Li, J. Y. Chen and Y. N. Xia, Nanoscale, 2011, 3, 950–953 RSC.
- P. M. Shih, T. K. Liu and K. T. Tan, Chem. Commun., 2013, 49, 6212–6214 RSC.
- A. K. Dwivedi and P. K. Iyer, J. Mater. Chem. B, 2013, 1, 4005–4010 RSC.
- D. L. Liao, Y. X. Li, J. Chen and C. Yu, Anal. Chim. Acta, 2013, 784, 72–76 CrossRef CAS PubMed.
- H. J. Kang, J. H. Kim and S. J. Chung, Biosens. Bioelectron., 2015, 67, 413–418 CrossRef CAS PubMed.
- H. L. Fan, X. H. Jiang, T. Zhang and Q. H. Jin, Biosens. Bioelectron., 2012, 34, 221–226 CrossRef CAS PubMed.
- W. Q. Yang, G. Y. Zhang, W. Weng, B. Qiu, L. H. Guo, Z. Y. Lin and G. N. Chen, RSC Adv., 2014, 4, 58852–58857 RSC.
- Y. H. Wang, P. Shen, C. Y. Li, Y. Y. Wang and Z. H. Liu, Anal. Chem., 2012, 84, 1466–1473 CrossRef CAS PubMed.
- X. J. Wang, G. F. Wang, C. C. Li, J. Wang and N. Xia, Int. J. Electrochem. Sci., 2013, 8, 8210–8217 CAS.
- X. Ding, D. Ge and K.-L. Yang, Sens. Actuators, B, 2014, 201, 234–239 CrossRef CAS.
- T. Zeng, T. Zhang, W. Wei, Z. Li, D. Wu, L. Wang, J. Guo, X. He and N. Ma, ACS Appl. Mater. Interfaces, 2015, 7, 11849–11856 CAS.
- A. V. Fuchs, N. Kotman, J. Andrieu, V. Mailander, C. K. Weiss and K. Landfester, Nanoscale, 2013, 5, 4829–4839 RSC.
- D. W. Deng, D. Y. Zhang, Y. Li, S. Achilefu and Y. Q. Gu, Biosens. Bioelectron., 2013, 49, 216–221 CrossRef CAS PubMed.
- C. J. Kim, D. I. Lee, C. Kim, K. Lee, C. H. Lee and I. S. Ahn, Anal. Chem., 2014, 86, 3825–3833 CrossRef CAS PubMed.
- P. Chen, R. Selegard, D. Aili and B. Liedberg, Nanoscale, 2013, 5, 8973–8976 RSC.
- D. V. Le, V. T. Nguyen, L. J. Tang, J. H. Jiang, R. Q. Yu and Y. Z. Wang, Talanta, 2013, 107, 233–238 CrossRef CAS PubMed.
- B. B. Chen, H. Liu, C. Z. Huang, J. Ling and J. Wang, New J. Chem., 2015, 39, 1295–1300 RSC.
- L.-J. Ou, X.-Y. Li, L.-J. Li, H.-W. Liu, A.-M. Sun and K.-J. Liu, Analyst, 2015, 140, 1871–1875 RSC.
- W. X. Xue, G. X. Zhang and D. Q. Zhang, Analyst, 2011, 136, 3136–3141 RSC.
- W. T. Zhao, C. L. Yao, X. T. Luo, L. Lin and I. M. Hsing, Electrophoresis, 2012, 33, 1288–1291 CrossRef CAS PubMed.
- G. B. Kim and Y. P. Kim, Theranostics, 2012, 2, 127–138 CrossRef CAS PubMed.
- P. Wu, T. Zhao, J. Y. Zhang, L. Wu and X. D. Hou, Anal. Chem., 2014, 86, 10078–10083 CrossRef CAS PubMed.
- X. W. He and N. Ma, Small, 2013, 9, 2527–2531 CrossRef CAS PubMed.
- J. H. Wang and J. Xia, Anal. Chim. Acta, 2012, 709, 120–127 CrossRef CAS PubMed.
- I. C. Serrano, G. Stoica, A. M. Adams and E. Palomares, Nanoscale, 2014, 6, 13623–13629 RSC.
- K. E. Sapsford, J. Granek, J. R. Deschamps, K. Boeneman, J. B. Blanco-Canosa, P. E. Dawson, K. Susumu, M. H. Stewart and I. L. Medintz, ACS Nano, 2011, 5, 2687–2699 CrossRef CAS PubMed.
- S. B. Lowe, J. A. G. Dick, B. E. Cohen and M. M. Stevens, ACS Nano, 2012, 6, 851–857 CrossRef CAS PubMed.
- W. R. Algar, A. P. Malanoski, K. Susumu, M. H. Stewart, N. Hildebrandt and I. L. Medintz, Anal. Chem., 2012, 84, 10136–10146 CrossRef CAS PubMed.
- M. Wu, E. Petryayeva and W. R. Algar, Anal. Chem., 2014, 86, 11181–11188 CrossRef CAS PubMed.
- H. Kim, C. Y. W. Ng and W. R. Algar, Langmuir, 2014, 30, 5676–5685 CrossRef CAS PubMed.
- X. Li, D. W. Deng, J. P. Xue, L. Z. Qu, S. Achilefu and Y. Q. Gu, Biosens. Bioelectron., 2014, 61, 512–518 CrossRef CAS PubMed.
- T. T. Zheng, R. Zhang, Q. F. Zhang, T. T. Tan, K. Zhang, J. J. Zhu and H. Wang, Chem. Commun., 2013, 49, 7881–7883 RSC.
- L. J. Li, H. Lin, C. Y. Lei, Z. Nie, Y. Huang and S. Z. Yao, Biosens. Bioelectron., 2014, 54, 42–47 CrossRef CAS PubMed.
- L. L. Jin, K. Yang, K. Yao, S. Zhang, H. Q. Tao, S. T. Lee, Z. Liu and R. Peng, ACS Nano, 2012, 6, 4864–4875 CrossRef CAS PubMed.
- J. Li, C. H. Lu, Q. H. Yao, X. L. Zhang, J. J. Liu, H. H. Yang and G. N. Chen, Biosens. Bioelectron., 2011, 26, 3894–3899 CrossRef CAS PubMed.
- E. Q. Song, D. Cheng, Y. Song, M. D. Jiang, J. F. Yu and Y. Y. Wang, Biosens. Bioelectron., 2013, 47, 445–450 CrossRef CAS PubMed.
- T. T. Feng, D. Feng, W. Shi, X. H. Li and H. M. Ma, Mol. BioSyst., 2012, 8, 1441–1445 RSC.
- S. Y. Kwak, J. K. Yang, S. J. Jeon, H. I. Kim, J. Yim, H. Kang, S. Kyeong, Y. S. Lee and J. H. Kim, Adv. Funct. Mater., 2014, 24, 5119–5128 CrossRef CAS.
- D. M. Chau, D. Shum, C. Radu, B. Bhinder, D. Gin, M. L. Gilchrist, H. Djaballah and Y. M. Li, Comb. Chem. High Throughput Screening, 2013, 16, 415–424 CrossRef CAS PubMed.
- Y. Cao, J. C. Yu, B. Bo, Y. Q. Shu and G. X. Li, Biosens. Bioelectron., 2013, 45, 1–5 CrossRef CAS PubMed.
- H. Chen, J. Zhang, Y. Gao, S. Liu, K. Koh, X. Zhu and Y. Yin, Biosens. Bioelectron., 2015, 68, 777–782 CrossRef CAS PubMed.
- D. H. Deng, Y. F. Shi, H. M. Feng, Q. Q. Chen, D. X. Li and L. Liu, Int. J. Electrochem. Sci., 2013, 8, 6933–6940 CAS.
- G. C. Fan, L. Han, H. Zhu, J. R. Zhang and J. J. Zhu, Anal. Chem., 2014, 86, 12398–12405 CrossRef CAS PubMed.
- J. Ji, J. R. Gan, J. L. Kong, P. Y. Yang, B. H. Liu and C. Ji, Electrochem. Commun., 2012, 16, 53–56 CrossRef CAS.
- R. P. Liang, X. C. Tian, P. Qiu and J. D. Qin, Anal. Chem., 2014, 86, 9256–9263 CrossRef CAS PubMed.
- M. La, X. Y. Zhao, Q. L. Peng, C. D. Chen and G. Q. Zhao, Int. J. Electrochem. Sci., 2015, 10, 3329–3339 CAS.
- H. Ko, S. Park and K. Kim, J. Electroanal. Chem., 2015, 742, 70–73 CrossRef CAS.
- Y. F. Liu, J. X. Chen, M. Q. Xu and G. C. Zhao, Int. J. Electrochem. Sci., 2014, 9, 4014–4023 Search PubMed.
- M. A. Sowole and H. B. Kraatz, Analyst, 2012, 137, 1120–1124 RSC.
- L. Z. Swisher, A. M. Prior, S. Shishido, T. A. Nguyen, D. H. Hua and J. Li, Biosens. Bioelectron., 2014, 56, 129–136 CrossRef CAS PubMed.
- H. W. Wu, S. C. Liu, J. H. Jiang, G. L. Shen and R. Q. Yu, Analyst, 2012, 137, 4829–4833 RSC.
- N. Xia, Y. Zhang, P. Guan, Y. Hao and L. Liu, Sens. Actuators, B, 2015, 213, 111–115 CrossRef CAS.
- J. Zhang, Y. Liu, J. Lv, Y. Cao and G. X. Li, Microchim. Acta, 2015, 182, 281–288 CrossRef CAS.
- L. X. Chen, X. L. Fu and J. H. Li, Nanoscale, 2013, 5, 5905–5911 RSC.
- Z. T. Wu, Y. Z. Liu, X. D. Zhou, A. G. Shen and J. M. Hu, Biosens. Bioelectron., 2013, 44, 10–15 CrossRef CAS PubMed.
- N. N. Yazgan, I. H. Boyaci, E. Temur, U. Tamer and A. Topcu, Talanta, 2010, 82, 631–639 CrossRef CAS PubMed.
- Z. T. Wu, Y. F. Liu, Y. Z. Liu, H. M. Xiao, A. G. Shen, X. D. Zhou and J. M. Hu, Biosens. Bioelectron., 2015, 65, 375–381 CrossRef CAS PubMed.
- H. X. Chen, Q. H. Mei, Y. F. Hou, X. L. Zhu, K. Koh, X. X. Li and G. X. Li, Analyst, 2013, 138, 5757–5761 RSC.
- C. Esseghaier, A. Ng and M. Zourob, Biosens. Bioelectron., 2013, 41, 335–341 CrossRef CAS PubMed.
- G. Ferracci, S. Marconi, C. Mazuet, E. Jover, M. P. Blanchard, M. Seagar, M. Popoff and C. Leveque, Anal. Biochem., 2011, 410, 281–288 CrossRef CAS PubMed.
- H. Lee, T. Zhu, K. Patel, Y. Y. Zhang, L. Truong, K. E. Hevener, J. L. Gatuz, G. Subramanya, H. Y. Jeong, S. L. Uprichard and M. E. Johnson, PLoS One, 2013, 8, e75144 CAS.
- P. G. de Gennes and J. Prost, The Physics of Liquid Crystals, Clarendon Press, 1993 Search PubMed.
- F. J. Kahn, Appl. Phys. Lett., 1973, 22, 386–388 CrossRef CAS.
- F. J. Kahn, G. N. Taylor and H. Schonhorn, Proc. IEEE, 1973, 61, 823–828 CrossRef CAS.
- V. K. Gupta, J. J. Skaife, T. B. Dubrovsky and N. L. Abbott, Science, 1998, 279, 2077–2080 CrossRef CAS PubMed.
- J. J. Skaife and N. L. Abbott, Langmuir, 2000, 16, 3529–3536 CrossRef CAS.
- Y. Y. Luk, M. L. Tingey, K. A. Dickson, R. T. Raines and N. L. Abbott, J. Am. Chem. Soc., 2004, 126, 9024–9032 CrossRef CAS PubMed.
- M. L. Tingey, E. J. Snodgrass and N. L. Abbott, Adv. Mater., 2004, 16, 1331–1336 CrossRef CAS.
- K. L. Yang, K. Cadwell and N. L. Abbott, J. Phys. Chem. B, 2004, 108, 20180–20186 CrossRef CAS.
- J. M. Brake, M. K. Daschner and N. L. Abbott, Langmuir, 2005, 21, 2218–2228 CrossRef CAS PubMed.
- B. H. Clare and N. L. Abbott, Langmuir, 2005, 21, 6451–6461 CrossRef CAS PubMed.
- N. A. Lockwood and N. L. Abbott, Curr. Opin. Colloid Interface Sci., 2005, 10, 111–120 CrossRef CAS.
- B. H. Clare, O. Guzman, J. de Pablo and N. L. Abbott, Langmuir, 2006, 22, 7776–7782 CrossRef CAS PubMed.
- X. Bi, S. L. Lai and K. L. Yang, Anal. Chem., 2009, 81, 5503–5509 CrossRef CAS PubMed.
- C.-H. Chen and K.-L. Yang, Sens. Actuators, B, 2014, 204, 734–740 CrossRef CAS.
- S. Pazhanisamy, C. M. Stuver and D. J. Livingston, Anal. Biochem., 1995, 229, 48–53 CrossRef CAS PubMed.
- P. P. Tamburini, R. N. Dreyer, J. Hansen, J. Letsinger, J. Elting, A. Gore-Willse, R. Dally, R. Hanko, D. Osterman, M. E. Kamarck and H. Yoo-Warren, Anal. Biochem., 1990, 186, 363–368 CrossRef CAS PubMed.
- Q. Zhu, Z. Yu, T. Kabashima, S. Yin, S. Dragusha, A. F. M. El-Mahdy, V. Ejupi, T. Shibata and M. Kai, Sci. Rep., 2015, 5, 10323 CrossRef CAS PubMed.
- N. H. Schebb, T. Vielhaber, A. Jousset and U. Karst, J. Chromatogr. A, 2009, 1216, 4407–4415 CrossRef CAS PubMed.
- E. A. Jares-Erijman and T. M. Jovin, Nat. Biotechnol., 2003, 21, 1387–1395 CrossRef CAS PubMed.
- E. D. Matayoshi, G. T. Wang, G. A. Krafft and J. Erickson, Science, 1990, 247, 954–958 CAS.
- T. Zauner, R. Berger-Hoffmann, K. Muller, R. Hoffmann and T. Zuchner, Anal. Chem., 2011, 83, 7356–7363 CrossRef CAS PubMed.
- P. Gribbon and A. Sewing, Drug Discovery Today, 2003, 8, 1035–1043 CrossRef CAS PubMed.
- M. S. Dresselhaus and M. Terrones, Proc. IEEE, 2013, 101, 1522–1535 CrossRef CAS.
- Y. Huang, K. Hu, S. L. Zhao, M. Li, Z. F. Chen, Q. J. Lv and H. Liang, Chem. – Asian J., 2014, 9, 87–92 CrossRef CAS PubMed.
- G. T. Hermanson, in Bioconjugate Techniques, ed. G. T. Hermanson, Academic Press, Boston, 3rd edn, 2013, pp. 741–755, DOI:10.1016/B978-0-12-382239-0.00016-9.
- Y. Huang, M. Shi, K. Hu, S. L. Zhao, X. Lu, Z. F. Chen, J. Chen and H. Liang, J. Mater. Chem. B, 2013, 1, 3470–3476 RSC.
- S. Y. Zhu, Z. Y. Liu, L. Z. Hu, Y. L. Yuan and G. B. Xu, Chem. – Eur. J., 2012, 18, 16556–16561 CrossRef CAS PubMed.
- X. G. Gu, G. Yang, G. X. Zhang, D. Q. Zhang and D. B. Zhu, ACS Appl. Mater. Interfaces, 2011, 3, 1175–1179 CAS.
- H. B. Wang, Q. Zhang, X. Chu, T. T. Chen, J. Ge and R. Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 7065–7069 CrossRef CAS PubMed.
- M. Zhang, B.-C. Yin, X.-F. Wang and B.-C. Ye, Chem. Commun., 2011, 47, 2399–2401 RSC.
- U. H. Bunz and V. M. Rotello, Angew. Chem., 2010, 49, 3268–3279 CrossRef CAS PubMed.
- J. M. Obliosca, P.-C. Wang and F.-G. Tseng, J. Colloid Interface Sci., 2012, 371, 34–41 CrossRef CAS PubMed.
- B. S. Guirgis, C. Sa e Cunha, I. Gomes, M. Cavadas, I. Silva, G. Doria, G. L. Blatch, P. V. Baptista, E. Pereira, H. M. Azzazy, M. M. Mota, M. Prudencio and R. Franco, Anal. Bioanal. Chem., 2012, 402, 1019–1027 CrossRef CAS PubMed.
- X. Wang, J. Geng, D. Miyoshi, J. Ren, N. Sugimoto and X. Qu, Biosens. Bioelectron., 2010, 26, 743–747 CrossRef CAS PubMed.
- S. Y. Park, S. M. Lee, G. B. Kim and Y. P. Kim, Gold Bull., 2012, 45, 213–219 CrossRef CAS.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- J. Bacart, C. Corbel, R. Jockers, S. Bach and C. Couturier, Biotechnol. J., 2008, 3, 311–324 CrossRef CAS PubMed.
- H. Dacres, M. M. Dumancic, I. Horne and S. C. Trowell, Biosens. Bioelectron., 2009, 24, 1164–1170 CrossRef CAS PubMed.
- B. R. Branchini, J. C. Rosenberg, D. M. Ablamsky, K. P. Taylor, T. L. Southworth and S. J. Linder, Anal. Biochem., 2011, 414, 239–245 CrossRef CAS PubMed.
- J. P. Yu, M. R. Guan, F. Y. Li, Z. P. Zhang, C. R. Wang, C. Y. Shu, H. P. Wei and X. E. Zhang, Chem. Commun., 2012, 48, 11011–11013 RSC.
- F. Y. Li, J. P. Yu, Z. P. Zhang, Z. Q. Cui, D. B. Wang, H. P. Wei and X. E. Zhang, Anal. Chim. Acta, 2012, 724, 104–110 CrossRef CAS PubMed.
- F. Y. Li, J. P. Yu, Z. P. Zhang, Z. Q. Cui, D. B. Wang, H. P. Wei and X. E. Zhang, Talanta, 2013, 109, 141–146 CrossRef CAS PubMed.
- N. Wu, H. Dacres, A. Anderson, S. C. Trowell and Y. Zhu, PLoS One, 2014, 9, e88399 Search PubMed.
- H. Maeda, Anal. Biochem., 1979, 92, 222–227 CrossRef CAS PubMed.
- S.-K. Lee, N. Cheng, E. Hull-Ryde, M. Potempa, C. A. Schiffer, W. Janzen and R. Swanstrom, Biochemistry, 2013, 52, 4929–4940 CrossRef CAS PubMed.
- C. Zhang, L. Zheng, J. Nurnberg, B. M. Vacari, J. Zhou and Y. Wang, Anal. Biochem., 2014, 445, 14–19 CrossRef CAS PubMed.
- Y. Huang, K. Hu, S. Zhao, M. Li, Z.-F. Chen, Q. Lv and H. Liang, Chem. – Asian J., 2014, 9, 87–92 CrossRef CAS PubMed.
- J. Zheng, P. R. Nicovich and R. M. Dickson, Annu. Rev. Phys. Chem., 2007, 58, 409–431 CrossRef CAS PubMed.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS PubMed.
- Y. C. Wang, Y. Wang, F. B. Zhou, P. Kim and Y. N. Xia, Small, 2012, 8, 3769–3773 CrossRef CAS PubMed.
- H. Lin, L. J. Li, C. Y. Lei, X. H. Xu, Z. Nie, M. L. Guo, Y. Huang and S. Z. Yao, Biosens. Bioelectron., 2013, 41, 256–261 CrossRef CAS PubMed.
- Y. Gu, Q. Wen, Y. Q. Kuang, L. J. Tang and J. H. Jiang, RSC Adv., 2014, 4, 13753–13756 RSC.
- G. C. Chen, Y. S. Xie, H. T. Zhang, P. Wang, H. Y. Cheung, M. S. Yang and H. Y. Sun, RSC Adv., 2014, 4, 6560–6563 RSC.
- S. E. Lohse and C. J. Murphy, Chem. Mater., 2013, 25, 1250–1261 CrossRef CAS.
- Z. E. Hughes, M. A. Nguyen, Y. Li, M. T. Swihart, T. R. Walsh and M. R. Knecht, Nanoscale, 2017, 9, 421–432 RSC.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS PubMed.
- J. J. Storhoff, A. A. Lazarides, R. C. Mucic, C. A. Mirkin, R. L. Letsinger and G. C. Schatz, J. Am. Chem. Soc., 2000, 122, 4640–4650 CrossRef CAS.
- D. Aili, R. Selegård, L. Baltzer, K. Enander and B. Liedberg, Small, 2009, 5, 2445–2452 CrossRef CAS PubMed.
- R. A. Sperling and W. J. Parak, Philos. Trans. R. Soc. London, Ser. A, 2010, 368, 1333–1383 CrossRef CAS PubMed.
- R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155–196 CrossRef CAS.
- D. S. Shin, Y. Liu, Y. D. Gao, T. Kwa, Z. Matharu and A. Revzin, Anal. Chem., 2013, 85, 220–227 CrossRef CAS PubMed.
- S. Park, G. Kim, J. Seo and H. Yang, Anal. Chem., 2016, 88, 11995–12000 CrossRef CAS PubMed.
- J. Homola, Anal. Bioanal. Chem., 2003, 377, 528–539 CrossRef CAS PubMed.
- P. Steinrücke, U. Aldinger, O. Hill, A. Hillisch, R. Basch and S. Diekmann, Anal. Biochem., 2000, 286, 26–34 CrossRef PubMed.
- H. Chen, Y.-S. Gal, S.-H. Kim, H.-J. Choi, M.-C. Oh, J. Lee and K. Koh, Sens. Actuators, B, 2008, 133, 577–581 CrossRef CAS.
- M. Moskovits, Rev. Mod. Phys., 1985, 57, 783–826 CrossRef CAS.
- A. N. Ramya, M. M. Joseph, J. B. Nair, V. Karunakaran, N. Narayanan and K. K. Maiti, ACS Appl. Mater. Interfaces, 2016, 8, 10220–10225 CAS.
- C. Sun, K.-H. Su, J. Valentine, Y. T. Rosa-Bauza, J. A. Ellman, O. Elboudwarej, B. Mukherjee, C. S. Craik, M. A. Shuman, F. F. Chen and X. Zhang, ACS Nano, 2010, 4, 978–984 CrossRef CAS PubMed.
- L. Yang, T. Wu, C. Fu, G. Chen, S. Xu and W. Xu, RSC Adv., 2016, 6, 90120–90125 RSC.
- X. Ding and K.-L. Yang, Part. Part. Syst. Charact., 2014, 31, 1300–1306 CrossRef CAS.
- W. Cheng, Y. L. Chen, F. Yan, L. Ding, S. J. Ding, H. X. Ju and Y. B. Yin, Chem. Commun., 2011, 47, 2877–2879 RSC.
- L. H. Ong and K.-L. Yang, J. Phys. Chem. B, 2016, 120, 825–833 CrossRef CAS PubMed.
- M. Bagiński, A. Szmurło, A. Andruszkiewicz, M. Wójcik and W. Lewandowski, Liq. Cryst., 2016, 43, 2391–2409 CrossRef.
- X. Wang, D. S. Miller, E. Bukusoglu, J. J. de Pablo and N. L. Abbott, Nat. Mater., 2016, 15, 106–112 CrossRef CAS PubMed.
- J. E. Stumpel, D. J. Broer and A. P. H. J. Schenning, RSC Adv., 2015, 5, 94650–94653 RSC.
- C. H. Chen and K. L. Yang, Biosens. Bioelectron., 2012, 35, 174–179 CrossRef CAS PubMed.
- L. H. Ong, X. Ding and K.-L. Yang, Colloids Surf., B, 2014, 122, 166–174 CrossRef CAS PubMed.
- L. S. Birchall, R. V. Ulijn and S. J. Webb, Chem. Commun., 2008, 2861–2863, 10.1039/B805321A.
- M. Zhang and C.-H. Jang, Bull. Korean Chem. Soc., 2013, 34, 2973–2977 CrossRef CAS.
- C.-Y. Chang and C.-H. Chen, Chem. Commun., 2014, 50, 12162–12165 RSC.
- X. Y. Bi and K. L. Yang, Biosens. Bioelectron., 2010, 26, 107–111 CrossRef CAS PubMed.
- H. Eimura, D. S. Miller, X. Wang, N. L. Abbott and T. Kato, Chem. Mater., 2016, 28, 1170–1178 CrossRef CAS.
Footnote |
| † || indicates cleavage site. |
| This journal is © The Royal Society of Chemistry 2017 |