
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Overcoming naphthoquinone deactivation: rhodium-catalyzed C-5 selective C–H iodination as a gateway to functionalized derivatives†
Guilherme A. M.
Jardim
ab,
Eufrânio N.
da Silva Júnior
*b and
John F.
Bower
*a
aSchool of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: john.bower@bris.ac.uk
bInstitute of Exact Sciences, Department of Chemistry, Federal University of Minas Gerais, Belo Horizonte, MG 31270-901, Brazil. E-mail: eufranio@ufmg.br
First published on 2nd March 2016
Abstract
We report a Rh-catalyzed method for the C-5 selective C–H iodination of naphthoquinones and show that complementary C-2 selective processes can be achieved under related conditions. C–C bond forming derivatizations of the C-5 iodinated products provide a gateway to previously inaccessible A-ring analogues. The present study encompasses the first catalytic directed ortho-functionalizations of simple (non-bias) naphthoquinones. The strategic considerations outlined here are likely to be applicable to C–H functionalizations of other weakly coordinating and/or redox sensitive substrates.
Introduction
1,4-Naphthoquinones function as redox cyclers and alkylating agents in a wide range of biological processes.1 For example, vitamin K encompasses a family of 2-methyl-1,4-naphthoquinones that act as cofactors for the post-translational carboxylation of glutamic acid residues, a process that is essential to blood coagulation and bone metabolism.1e Other notable naphthoquinones include the juglomycins,2a dimeric pyranonaphthoquinones, such as protoaphin-fb and protoaphin-sl2b–e and the lapachones (Scheme 1A).2f Because of their biological importance, significant efforts are devoted to the synthesis and evaluation of a wide range of naphthoquinone derivatives.3 While methods for the modification of the quinone B-ring are reasonably well established,4 flexible protocols that allow the direct functionalization of the benzenoid A-ring are rare (Scheme 1B).5 This situation is exacerbated by the limitations associated with de novo naphthoquinone construction.6 Consequently, medicinal studies on A-ring analogues are, in the main, limited to simple derivatives of natural isolates.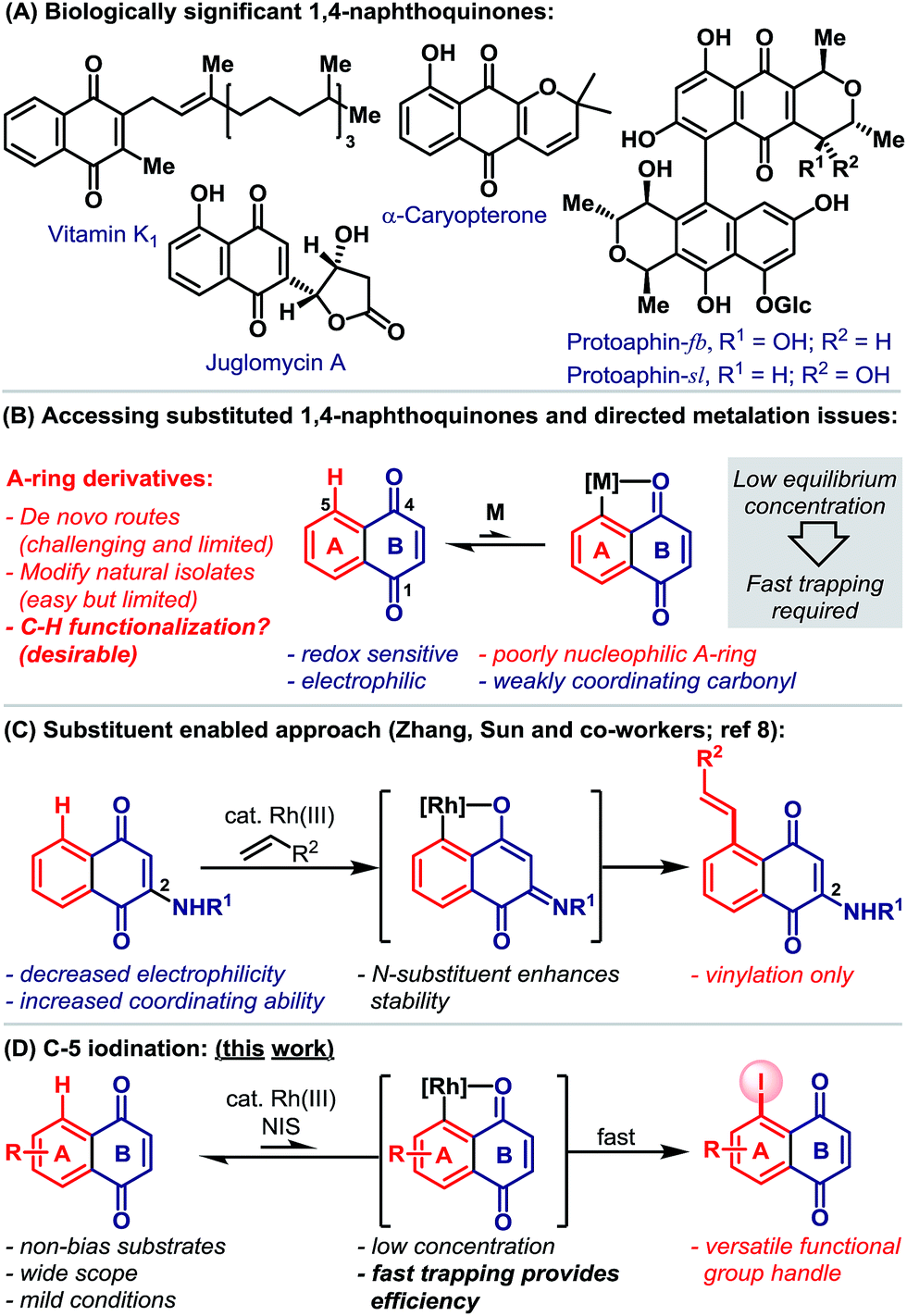 | ||
| Scheme 1 | ||
Catalytic directed ortho-C–H metalation has emerged as a powerful platform for the preparation of diverse aromatic compounds.7 However, its application to the modification of naphthoquinones is limited by (a) their susceptibility to reduction, (b) their high electrophilicity, (c) the low nucleophilicity of the benzenoid A-ring and (d) the weak coordinating ability of the B-ring carbonyls (Scheme 1B). Indeed, we are aware of only one protocol that enables catalytic carbonyl directed C–H functionalization of the naphthoquinone A-ring (Scheme 1C).8 Here, Rh(III)-catalyzed C-5 vinylation required the strategic installation of a C-2 amino substituent, which was crucial for facilitating cyclometalation.
Carbonyl directed ortho-metalation with late transition metals is usually a reversible process,7,9 where the equilibrium depends upon both the nucleophilicity of the arene and the coordinating strength of the directing group (Scheme 1B). Neither aspect is favorable for naphthoquinones, so an efficient process must necessarily rely upon fast trapping of the metalated intermediate. In considering this, we sought to introduce a synthetically versatile handle under conditions that avoid nucleophilic or reducing reagents. Consequently, we were drawn to the ortho-C–H iodination protocol reported by Glorius and co-workers,9 which involves the trapping of cyclometalated aryl-Rh(III) complexes with NIS. This reagent is highly reactive, oxidative and also non-nucleophilic, such that it seemed well suited to C-5 selective iodination. In this report, we outline the development and scope of this protocol, which provides, for the first time, a C–H activation-based gateway to diverse A-ring analogues (Scheme 1D). Additionally, we disclose that, in certain cases, fine tuning of the Rh-system enables a complete switch to C-2 selective iodination.
Results and discussion
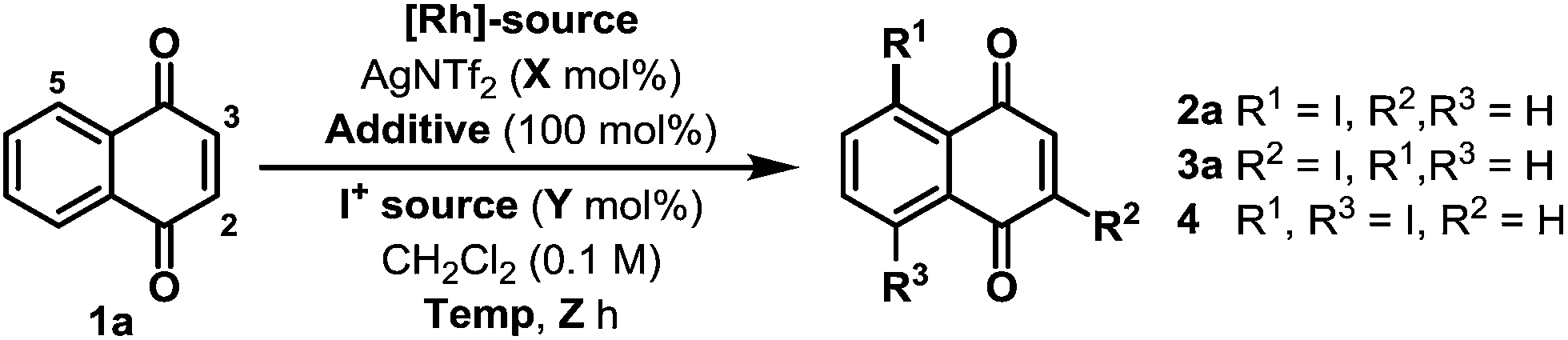
Preliminary studies involved exposing naphthoquinone 1a to a variety of electrophilic iodine sources in the presence of in situ generated cationic Rh(III)-systems (Table 1). These experiments revealed that achieving both high conversion and high C-5 regioselectivity was likely to be challenging. [RhCp*Cl2]2/AgNTf2 in combination with NIS and Cu(OAc)2 resulted in only a 39% yield of 2a, albeit with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 selectivity over C-2 regioisomer 3a (entry 1). Other electrophilic iodine sources were less effective; for example, DIH led to substantial quantities of C-2 adduct 3a and bis-iodinated product 4 (entry 2). Extensive optimization efforts were undertaken to identify an effective system, and, in part, these studies focussed on the use of other acetate ligated Lewis acids in combination with a range of Rh(III)-precatalysts (entries 3–8).10 However none of these systems were especially effective, with the key issue being competitive degradation of the iodinating agent under the reaction conditions. To resolve this, microwave conditions were investigated.11 Pleasingly, by heating at 65 W (50 °C), a 54% yield of 2a was obtained after just 2.5 hours (entry 9). Under these conditions, the major byproduct was bis-iodinated adduct 4, which was formed in 14% yield. Further refinement led to the conditions outlined in entry 10, which deliver 2a in 69% yield and with high selectivity over 3a and 4.12 The conversion of 1a to 2a has been achieved previously in 34% overall yield, but this required a substrate specific 5 step sequence via a potentially hazardous diazonium intermediate.13 Thus, the efficiency and generality (vide infra) of the new protocol described here is both notable and synthetically enabling.
:1 selectivity over C-2 regioisomer 3a (entry 1). Other electrophilic iodine sources were less effective; for example, DIH led to substantial quantities of C-2 adduct 3a and bis-iodinated product 4 (entry 2). Extensive optimization efforts were undertaken to identify an effective system, and, in part, these studies focussed on the use of other acetate ligated Lewis acids in combination with a range of Rh(III)-precatalysts (entries 3–8).10 However none of these systems were especially effective, with the key issue being competitive degradation of the iodinating agent under the reaction conditions. To resolve this, microwave conditions were investigated.11 Pleasingly, by heating at 65 W (50 °C), a 54% yield of 2a was obtained after just 2.5 hours (entry 9). Under these conditions, the major byproduct was bis-iodinated adduct 4, which was formed in 14% yield. Further refinement led to the conditions outlined in entry 10, which deliver 2a in 69% yield and with high selectivity over 3a and 4.12 The conversion of 1a to 2a has been achieved previously in 34% overall yield, but this required a substrate specific 5 step sequence via a potentially hazardous diazonium intermediate.13 Thus, the efficiency and generality (vide infra) of the new protocol described here is both notable and synthetically enabling.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Rh-source | X | Additive | I+ source | Y | Temp/°C | Z |
2a:3a:4 |
| a External temperature of the reaction vessel. NIS = N-iodosuccinimide. DIH = 1,3-diiodo-5,5-dimethylhydantoin. Cp*i-Pr = isopropyl tetramethylcyclopentadienyl. Cp*CF3 = trifluoromethyl tetramethylcyclopentadienyl. | ||||||||
| 1 | [RhCp*Cl2]2 (2.5%) | 10 | Cu(OAc)2 | NIS | 220 | 120 | 22 | 39:8:0 |
| 2 | [RhCp*Cl2]2 (2.5%) | 10 | Cu(OAc)2 | DIH | 140 | 120 | 22 | 4:14:10 |
| 3 | [RhCp*Cl2]2 (2.5%) | 10 | Cu(OPiv)2 | NIS | 140 | 120 | 22 | 14:0:0 |
| 4 | [RhCp*Cl2]2 (2.5%) | 10 | Zn(OAc)2 | NIS | 140 | 120 | 22 | 18:0:0 |
| 5 | [RhCp*Cl2]2 (4%) | 10 | Cu(OAc)2 | NIS | 120 | 100 | 16 | 44:5:0 |
| 6 | [RhCp*i-prCl2]2 (4%) | 10 | Cu(OAc)2 | NIS | 120 | 100 | 16 | 39:8:0 |
| 7 | [RhCp*CF3Cl2]2 (4%) | 10 | Cu(OAc)2 | NIS | 120 | 100 | 16 | 3:0:0 |
| 8 | [RhCp*(OAc)2] (4%) | 10 | Cu(OAc)2 | NIS | 120 | 100 | 16 | 32:2:0 |
| 9 | [RhCp*Cl2]2 (4%) | 10 | Cu(OAc)2 | NIS | 120 50a | (65 W) | 2.5 | 54:0:14 |
| 10 | [RhCp*Cl2]2 (3.75%) | 20 | Cu(OAc)2 | NIS | 100 45a | (60 W) | 2 | 69:0:5 |
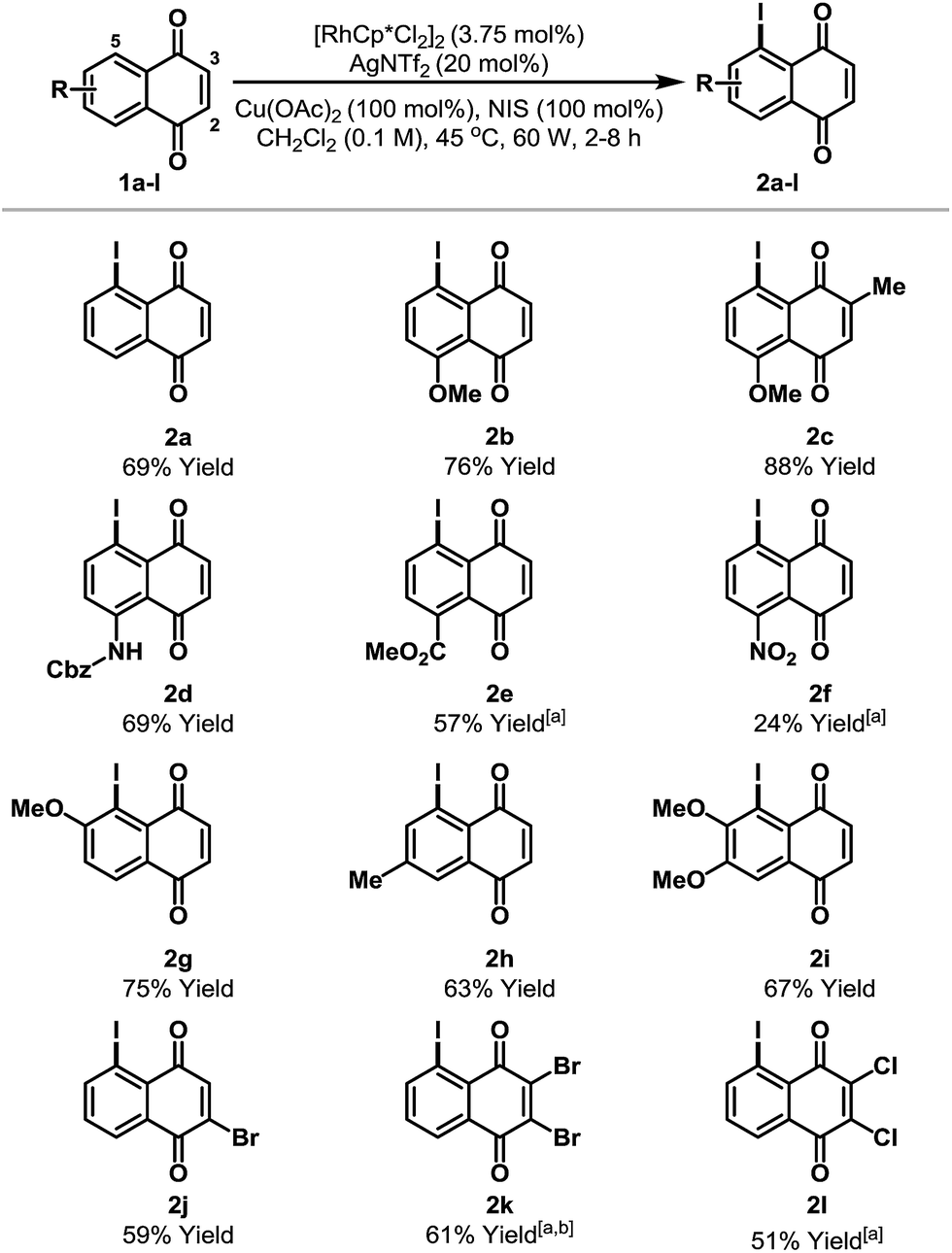
The scope of the new process is outlined in Table 2. Naphthoquinones 1b–e possessing C-8 substitution underwent efficient iodination to provide targets 2b–e in good to excellent yield. As expected, the efficiency of the process decreases as the benzenoid ring becomes more electron deficient, and, accordingly, nitro adduct 2f was generated in only 24% yield. Systems with substituents at C-6 or C-7 can potentially deliver two different regioisomeric products. Perhaps as a result of secondary coordination by the methoxy group, naphthoquinone 1g afforded selectively adduct 2g, wherein iodination has occurred at the more hindered ortho-position. Conversely, methyl-substituted system 1h favored iodination at the less hindered ortho-site to deliver iodide 2h in 63% yield. Halogen substituents are tolerated and bromo- and chloro-naphthoquinones 1j–l were converted to targets 2j–l in synthetically useful yields. For 2j, the high ortho-regioselectivity may reflect the higher basicity of the C-4 carbonyl of 1j. In all cases, only trace quantities (≤5%) of bis-iodinated and C-2/3 iodinated products were observed (cf.4 and 3a), and no significant iodination occurred in the absence of Rh-catalyst. Structural assignments were based on detailed NMR analysis (DEPT, COSY, HSQC, HMBC) and X-ray structures of 2a–c, 2f, 2j and 2k.14
| a <5% bis-iodination was observed in all cases. NIS = N-iodosuccinimide. [a] 5 mol% [RhCp*Cl2]2 and 27 mol% AgNTf2 were used. [b] Run at 65 °C and 75 W. | ||
|---|---|---|
|
During optimization we noted that the regioselectivity of iodination is strongly influenced by the nature of the Lewis acidic additive. Acetate based systems consistently provided high selectivity for C-5, likely via a Rh-acetate promoted concerted metalation–deprotonation mechanism (cf.Table 1, entry 8).15 By switching from Cu(OAc)2 to CuSO4 we were able to develop a complementary C-2 selective iodination protocol (Scheme 2).10,16 Under optimized conditions, iodination of 1a generated 3a in 90% yield and with complete regioselectivity. Perez and co-workers have shown that morpholine–iodine complex can convert 1a to 3a, but in only 35% yield and as a mixture of products.17 For unsymmetrical systems, such as 1h, C-2 vs. C-3 selectivity was not readily controlled and 3h was formed as a mixture of these two regioisomers.18 At the present stage, C-3 selective iodination of systems possessing substitution at C-2 is not feasible, perhaps due to steric inhibition of the C–H metalation event.
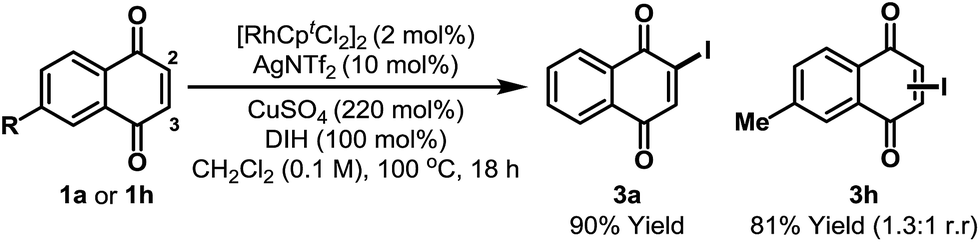 | ||
| Scheme 2 C-2 selective iodination. DIH = 1,3-diiodo-5,5-dimethylhydantoin. Cpt = 1,3-di-tert-butylcyclopentadienyl. | ||
In principle, the activation mode employed here should enable other selective naphthoquinone C–H functionalizations. However, as outlined in the introduction, efficient processes likely require highly reactive and non-reducing coupling partners. Accordingly, we have been unable to achieve direct C–H activation based C–C bond formations.19 However, the iodinated products described here provide a gateway to this important goal (Fig. 1). Because of the synthetic inaccessibility of A-ring halogenated naphthoquinones, Pd-catalyzed cross-couplings involving the benzenoid ring have not been developed. This aspect is challenging because the quinone moiety can act as an oxidant or ligand for Pd.20 For example, arylation of 2a could not be achieved under Suzuki conditions and only decomposition products were observed. After extensive investigation, we established that mild Stille cross-couplings21 are effective and, using this approach, arylated derivative 5a was isolated in high yield. Heck reactions are another promising avenue and Pd(0)-catalyzed reaction of 2a with ethyl acrylate delivered 5b in 66% yield.22 To date, alkynylation under Sonogashira conditions has not been fruitful but the use of stoichiometric alkynyl copper(I) reagents is feasible and this allowed the isolation of 5c in 64% yield.13 The studies outlined in Fig. 1 validate short and diversifiable entries to previously challenging naphthoquinone targets.
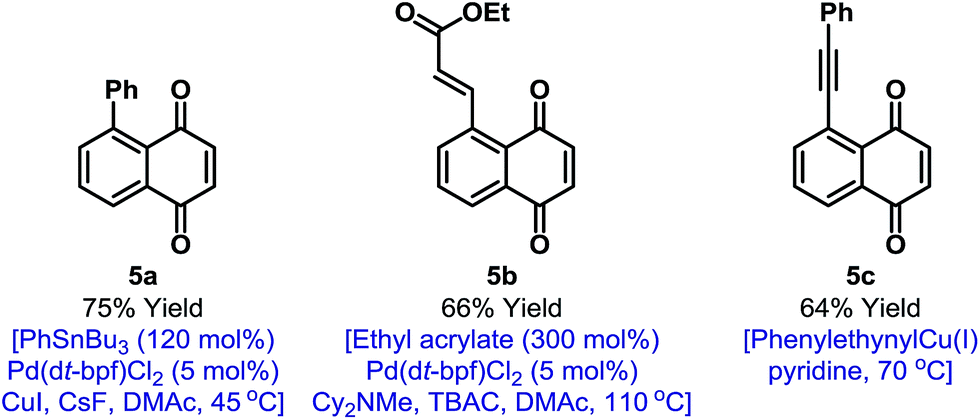 | ||
| Fig. 1 C–C bond forming derivatizations of 2a. TBAC = tetra-n-butylammonium chloride, DMAc = N,N-dimethylacetamide. | ||
Preliminary results suggest that other highly electrophilic reagents might also be effective for C-5 selective functionalization (Scheme 3). Using DBH, Rh-catalyzed C-5 selective bromination of 1a proceeded in 66% yield to afford a 7:2 mixture of C-5 and C-2 bromides 6/7; the structure of 6 was confirmed by single crystal X-ray diffraction.13 The most direct previous entry to 6 involved oxidation of 2-bromonaphthalene, but this afforded the target in only 15% yield and as a complex mixture of isomers.23 Alternative C-2 selective bromination can be achieved by adapting the conditions outlined in Scheme 2 and this enabled the selective formation of 7 in 88% yield from 1a.
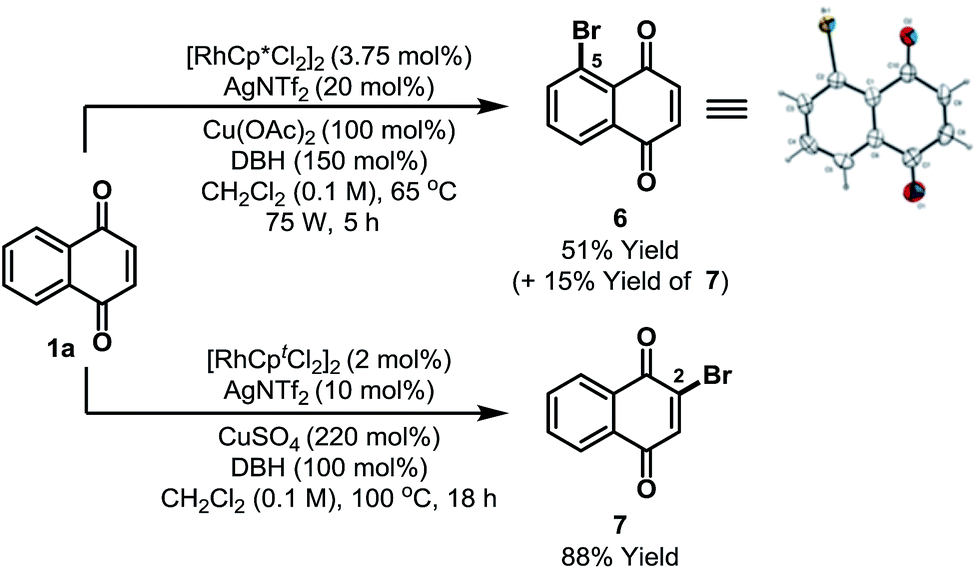 | ||
| Scheme 3 C-5 and C-2 selective bromination. DBH = 1,3-dibromo-5,5-dimethylhydantoin. Cpt = 1,3-di-tert-butylcyclopentadienyl. | ||
Conclusions
In conclusion, we report an efficient and reliable methodology for C-5 selective C–H iodination of naphthoquinoidal compounds and show that complementary C-2 selective processes can be achieved under related conditions. To the best of our knowledge, the present study provides the first method for catalytic directed ortho-functionalization of simple (non-bias) naphthoquinones. The iodinated products are amenable to C–C bond forming derivatizations and this enables flexible modifications to the naphthoquinone A-ring. The chemistry opens up new avenues for biological investigation and is likely to be of wide general interest. In broader terms, the strategic considerations outlined here may guide the development of catalytic C–H functionalizations involving other weakly coordinating and/or redox sensitive substrates.Acknowledgements
G. A. M. J. thanks the CNPq Science without Borders program for a scholarship. We thank the X-ray crystallography service at the School of Chemistry, University of Bristol, for analysis of the products described here. J. F. B. is indebted to the Royal Society for the provision of a University Research Fellowship. E. N. S. J. thanks the Royal Society of Chemistry for a JWT Jones Travelling Fellowship, CAPES and CNPq for research support.Notes and references
- (a) R. H. Thomson, Naturally Occurring Quinones, Academic Press, London, 2nd edn, 1971 Search PubMed; (b) R. H. Thomson, Naturally Occurring Quinones III Recent Advances, Chapman and Hall, London, 3rd edn, 1987 Search PubMed; (c) G. Powis, Pharmacol. Ther., 1987, 35, 57 CrossRef CAS PubMed; (d) P. J. O'Brien, Chem.-Biol. Interact., 1991, 80, 1 CrossRef; (e) E. A. Hillard, F. C. Abreu, D. C. Ferreira, G. Jaouen, M. O. F. Goulart and C. Amatore, Chem. Commun., 2008, 2612 RSC; (f) L.-O. Klotz, X. Hou and C. Jacob, Molecules, 2014, 19, 14902 CrossRef PubMed.
- (a) K. Ushiyama, N. Tanaka, H. Ono and H. Ogata, Jpn. J. Antibiot., 1971, 24, 197 CrossRef CAS PubMed; (b) J. P. E. Human, A. W. Johnston, S. F. MacDonald and A. R. Todd, J. Chem. Soc., 1950, 477 RSC; (c) H. Duewell, A. W. Johnston, S. F. MacDonald and A. R. Todd, J. Chem. Soc., 1950, 485 RSC , selected reviews on pyranonaphthoquinones; (d) M. A. Brimble, L. J. Duncalf and M. R. Nairna, Nat. Prod. Rep., 1999, 16, 267 RSC; (e) C. D. Donner, Nat. Prod. Rep., 2015, 32, 578 RSC; (f) S. Hannedouche, J. P. Souchard, I. Jacquemond-Collet and C. Moulis, Fitoterapia, 2002, 73, 520 CrossRef CAS PubMed.
- Selected studies: (a) B. C. Cavalcanti, I. O. Cabral, F. A. R. Rodrigues, F. W. A. Barros, D. D. Rocha, H. I. F. Magalhães, D. J. Moura, J. Saffi, J. A. P. Henriques, T. S. C. Carvalho, M. O. Moraes, C. Pessoa, I. M. M. de Melo and E. N. da Silva Júnior, J. Braz. Chem. Soc., 2013, 24, 145 CrossRef CAS; (b) G. A. M. Jardim, T. T. Guimarães, M. C. F. R. Pinto, B. C. Cavalcanti, K. M. Farias, C. Pessoa, C. C. Gatto, D. K. Nair, I. N. N. Namboothiri and E. N. da Silva Júnior, Med. Chem. Commun., 2015, 6, 120 RSC; (c) G. A. M. Jardim, W. J. Reis, M. F. Ribeiro, F. M. Ottoni, R. J. Alves, T. L. Silva, M. O. F. Goulart, A. L. Braga, R. F. S. Menna-Barreto, K. Salomão, S. L. de Castro and E. N. da Silva Júnior, RSC Adv., 2015, 5, 78047 RSC; (d) V. Jamier, L. A. Ba and C. Jacob, Chem.–Eur. J., 2010, 16, 10920 CrossRef CAS PubMed; (e) F. Prati, C. Bergamini, M. T. Molina, F. Falchi, A. Cavalli, M. Kaiser, R. Brun, R. Fato and M. L. Bolognesi, J. Med. Chem., 2015, 58, 6422 CrossRef CAS PubMed; (f) A. Reichstein, S. Vortherms, S. Bannwitz, J. Tentrop, H. Prinz and K. Müller, J. Med. Chem., 2012, 55, 7273 CrossRef CAS PubMed.
- Selected methodologies: (a) X. Wang, Y. Ye, G. Ji, Y. Xu, S. Zhang, J. Feng, Y. Zhang and J. Wang, Org. Lett., 2013, 15, 3730 CrossRef CAS PubMed; (b) J. Bian, X. Qian, N. Wang, T. Mu, X. Li, H. Sun, L. Zhang, Q. You and X. Zhang, Org. Lett., 2015, 17, 3410 CrossRef CAS PubMed; (c) R. Samanta, R. Narayan and A. P. Antonchick, Org. Lett., 2012, 14, 6108 CrossRef CAS PubMed; (d) C. S. Lisboa, V. G. Santos, B. G. Vaz, N. C. de Lucas, M. N. Eberlin and S. J. Garden, J. Org. Chem., 2011, 76, 5264 CrossRef CAS PubMedD. Wang, B. Ge, L. Li, J. Shan and Y. Ding, J. Org. Chem., 2014, 79, 8607 CrossRef CAS PubMed.
- Aromatic substitution approaches are generally reliant on initial C–H nitration. For example, see: H. Li, R. Liu, Y. Ji and Y. Wang, J. Chem. Pharm. Res., 2014, 6, 72 CAS.
- The assembly and oxidation of a suitable precursor is a common approach but the latter step often proceeds in low yield and/or selectivity. Representative studies: (a) P. Jacob, P. S. Callery, A. T. Shulgin and N. Castagnoli, J. Org. Chem., 1976, 41, 3627 CrossRef CAS PubMed; (b) Y. Tanoue and A. Terada, Bull. Chem. Soc. Jpn., 1988, 61, 2039 CrossRef CAS; (c) D. W. Kim, H. Y. Choi, K.-J. Lee and D. Y. Chi, Org. Lett., 2001, 3, 445 CrossRef CAS PubMed . Reviews that encompass this issue; (d) V. P. Papageorgiou, A. N. Assimopoulou, E. A. Couladouros, D. Hepworth and K. C. Nicolaou, Angew. Chem., Int. Ed., 1999, 38, 270 CrossRef; (e) V. Nair and A. Deepthi, Tetrahedron, 2009, 65, 10745 CrossRef CAS.
- Recent methodologies: (a) Y. Lu, H.-W. Wang, J. E. Spangler, K. Chen, P.-P. Cui, Y. Zhao, W.-Y. Sun and J.-Q. Yu, Chem. Sci., 2015, 6, 1923 RSC; (b) X. Qin, D. Sun, Q. You, Y. Cheng, J. Lan and J. You, Org. Lett., 2015, 17, 1762 CrossRef CAS PubMed; (c) T. Gensch, S. Vásquez-Céspedes, D.-G. Yu and F. Glorius, Org. Lett., 2015, 17, 3714 CrossRef CAS PubMed; (d) Y. Suzuki, B. Sun, K. Sakata, T. Yoshino, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2015, 54, 9944 CrossRef CAS PubMed . Selected reviews ; (e) F. Kakiuchi and S. Murai, Top. Organomet. Chem., 1999, 3, 47 CrossRef CAS; (f) Y. J. Park and C.-H. Jun, Bull. Korean Chem. Soc., 2005, 26, 871 CrossRef CAS; (g) F. Kakiuchi and T. Kochi, Synthesis, 2008, 3013 CrossRef CAS; (h) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (i) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef CAS PubMed; (j) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (k) Q.-Z. Zheng and N. Jiao, Tetrahedron Lett., 2014, 55, 1121 CrossRef CAS.
- C. Zhang, M. Wang, Z. Fan, L.-P. Sun and A. Zhang, J. Org. Chem., 2014, 79, 7626 CrossRef CAS PubMed.
- N. Schröder, J. Wencel-Delord and F. Glorius, J. Am. Chem. Soc., 2012, 134, 8298 CrossRef PubMed.
- The synthesis and utility of the modified RhCp complexes used here has been outlined previously. [RhCp*i-PrCl2]2: (a) T. Piou and T. Rovis, J. Am. Chem. Soc., 2014, 136, 11292 CrossRef CAS PubMed; (b) [RhCp*CF3Cl2]2: J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2014, 136, 2735 CrossRef CAS PubMed; (c) F. Romanov-Michailidis, K. F. Sedillo, J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2015, 137, 8892 CrossRef CAS PubMed . [RhCptCl2]2: ; (d) E. L. Dias and R. H. Grubbs, Organometallics, 1998, 17, 2758 CrossRef CAS; (e) T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846 RSC; (f) T. K. Hyster, D. M. Dalton and T. Rovis, Chem. Sci., 2015, 6, 254 RSC; (g) T. Piou and T. Rovis, Nature, 2015, 527, 86 CrossRef CAS PubMed . [RhCp*(OAc)2]: ; (h) P. M. Boyer, C. P. Roy, J. M. Bielski and J. S. Merola, Inorg. Chim. Acta, 1996, 245, 7 CrossRef CAS . For related ligand modifications, see: ; (i) Y. Hoshino, Y. Shibata and K. Tanaka, Adv. Synth. Catal., 2014, 356, 1577 CrossRef CAS; (j) M. Fukui, Y. Hoshino, T. Satoh, M. Miura and K. Tanaka, Adv. Synth. Catal., 2014, 356, 1638 CrossRef CAS; (k) T. A. Davis, C. Wang and T. Rovis, Synlett, 2015, 26, 1520 CrossRef CAS; (l) M. D. Wodrich, B. Ye, J. F. Gonthier, C. Corminboeuf and N. Cramer, Chem.–Eur. J., 2014, 20, 15409 CrossRef CAS PubMed.
- For reviews and discussion on the benefits of microwave heating for transition metal catalyzed processes, see: (a) M. R. Rosana, Y. Tao, A. E. Stiegman and G. B. Dudley, Chem. Sci., 2012, 3, 1240 RSC; (b) B. K. Singh, N. Kaval, S. Tomar, E. Van der Eycken and V. S. Parmar, Org. Process Res. Dev., 2008, 12, 468 CrossRef CAS; (c) P. Appukkuttan and E. Van der Eycken, Eur. J. Org. Chem., 2008, 1133 CrossRef CAS; (d) M. Larhed, C. Moberg and A. Hallberg, Acc. Chem. Res., 2002, 35, 717 CrossRef CAS PubMed.
- A CEM Discover microwave was used in combination with a customized, re-sealable reaction tube (see the ESI†). Under optimized conditions, a constant power input of 60 W was applied and the outer wall of the reaction tube was maintained at 45 °C using a cooling flow of N2. For the conversion of 1a to 2a, very similar results (71% yield of 2a) were achieved using a Biotage Initiator 8 microwave reactor. Heating at 45 °C under conventional thermal conditions (oil bath) gave only an 8% yield of 2a, which is consistent with microwave conditions providing more efficient heating (see ref. 11).
- N. V. Ivashkina, V. S. Romanov, A. A. Moroz and M. S. Shvartsberg, Izv. Akad. Nauk SSSR, Ser. Khim., 1984, 2561 CAS.
- CCDC 1441683–1441694† contain the supplementary crystallographic data for this paper.
- (a) J. Jiang, R. Ramozzi and K. Morokuma, Chem.–Eur. J., 2015, 21, 11158 CrossRef CAS PubMed; (b) A. P. Walsh and W. D. Jones, Organometallics, 2015, 34, 3400 CrossRef CAS.
- Rh(III)-catalyzed C-2 selective C–C bond forming processes involving quinones have been developed: Y. Moon, Y. Jeong, D. Kook and S. Hong, Org. Biomol. Chem., 2015, 13, 3918 Search PubMed . For a related process, see: X. Zhang, F. Wang, Z. Qi, S. Yu and X. Li, Org. Lett., 2014, 16, 1586 CrossRef CAS PubMed.
- A. L. Perez, G. Lamoureux and A. Herrera, Synth. Commun., 2004, 34, 3389 CrossRef CAS.
- For systems that possess electron donating groups on the B-ring (e.g.1b) this counterion effect is overridden and iodination occurs preferentially at C-5.
- For example, attempted C-5 vinylation of 1a under the conditions outlined in ref. 8 proceeded in <5% yield.
- This issue has been noted in studies on the direct functionalization of quinones: (a) Y. Fujiwara, V. Domingo, I. B. Seiple, R. Gianatassio, M. Del Bel and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 3292 CrossRef CAS PubMed; (b) S. E. Walker, J. A. Jordan-Hore, D. G. Johnson, S. A. Macgregor and A.-L. Lee, Angew. Chem., Int. Ed., 2014, 53, 13876 CrossRef CAS PubMed.
- (a) T. J. Colacot and H. A. Shea, Org. Lett., 2004, 6, 3731 CrossRef CAS PubMed; (b) S. P. H. Mee, V. Lee and J. E. Baldwin, Angew. Chem., Int. Ed., 2004, 43, 1132 CrossRef CAS PubMed.
- P. M. Murray, J. F. Bower, D. K. Cox, E. K. Galbraith, J. S. Parker and J. B. Sweeney, Org. Process Res. Dev., 2013, 17, 397 CrossRef CAS.
- M. Periasamy and M. V. Bhatt, Synthesis, 1997, 330 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for all compounds are provided. CCDC 1441683–1441694. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00302h |
| This journal is © The Royal Society of Chemistry 2016 |