
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA large spin-crossover [Fe4L4]8+ tetrahedral cage†‡
Li
Li
a,
Naoto
Saigo
b,
Yingjie
Zhang
c,
Daniel J.
Fanna
a,
Nicholas D.
Shepherd
a,
Jack K.
Clegg
d,
Rongkun
Zheng
 e,
Shinya
Hayami
b,
Leonard F.
Lindoy
f,
Janice R.
Aldrich-Wright
a,
Chun-Guang
Li
a,
Jason K.
Reynolds
a,
David G.
Harman
gh and
Feng
Li
*a
e,
Shinya
Hayami
b,
Leonard F.
Lindoy
f,
Janice R.
Aldrich-Wright
a,
Chun-Guang
Li
a,
Jason K.
Reynolds
a,
David G.
Harman
gh and
Feng
Li
*a
aSchool of Science and Health, University of Western Sydney, Penrith, NSW 2751, Australia. E-mail: feng.li@uws.edu.au
bDepartment of Chemistry, Graduate School of Science and Technology, Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Japan
cAustralian Nuclear Science and Technology Organisation, Locked Bag 2001 Kirrawee DC, NSW 2232, Australia
dSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane St Lucia, QLD 4072, Australia
eSchool of Physics, The University of Sydney, NSW 2006, Australia
fSchool of Chemistry, The University of Sydney, NSW 2006, Australia
gMolecular Medicine Research Group, School of Medicine, University of Western Sydney, Building 30, Goldsmith Avenue, Campbelltown, NSW 2560, Australia
hOffice of the Deputy Vice-Chancellor (Research and Development), University of Western Sydney, Penrith, NSW 2751, Australia
First published on 20th May 2015
Abstract
A large discrete face-capped tetranuclear iron(II) cage, [Fe4L4](BF4)8·n(solvent), was synthesised via metal-ion directed self-assembly. The cage is formed from a rigid tritopic ligand that incorporates chelating imidazole-imine functional groups. The cage displays temperature induced spin-crossover and LIESST effects and is amongst the largest iron(II) tetrahedral cages with such properties reported. The synthesis, structure and magnetic properties of this new metallo-cage are presented.
Introduction
Polynuclear Fe(II) coordination cages have received considerable attention and have been demonstrated to display interesting host–guest chemistry1–7 due to the presence of their well-defined central cavities. The vast majority of these systems have been designed such that their Fe(II) centres are in the low-spin (LS) state at ambient temperatures; there are only a few examples of Fe(II) cages that show spin-crossover (SCO) or high-spin (HS) behaviour.8–12 The successful construction of larger supramolecular Fe(II) cages, especially those containing spin-crossover Fe(II) centres, still remains a challenge. Clearly the design of the organic ligand is a key element for achieving such SCO molecular cage systems.In the present study, building on design principles demonstrated to be successful by Kruger,11 Nitschke10 and Gu;8 imidazole-imine sites were included in the organic ligand component since these groups show weaker ligand strength than the 2,2′-bipyridyl and pyridyl-imine sites commonly employed in tetrahedral cage syntheses. In addition, imidazole-imine groups have been well documented to be ‘classical’ coordination units for inducing SCO behaviour in Fe(II) complex systems,13–18 including the limited number of Fe(II) SCO tetrahedral cages already reported.8,10,11
Here we report the synthesis of a new face-capped tetranuclear cage 1 of type [Fe4L4](BF4)8, employing a large fully conjugated rigid tribranched framework ligand (L in Fig. 1) aimed at inducing Fe(II) spin-crossover behaviour at each of its equivalent metal centres.
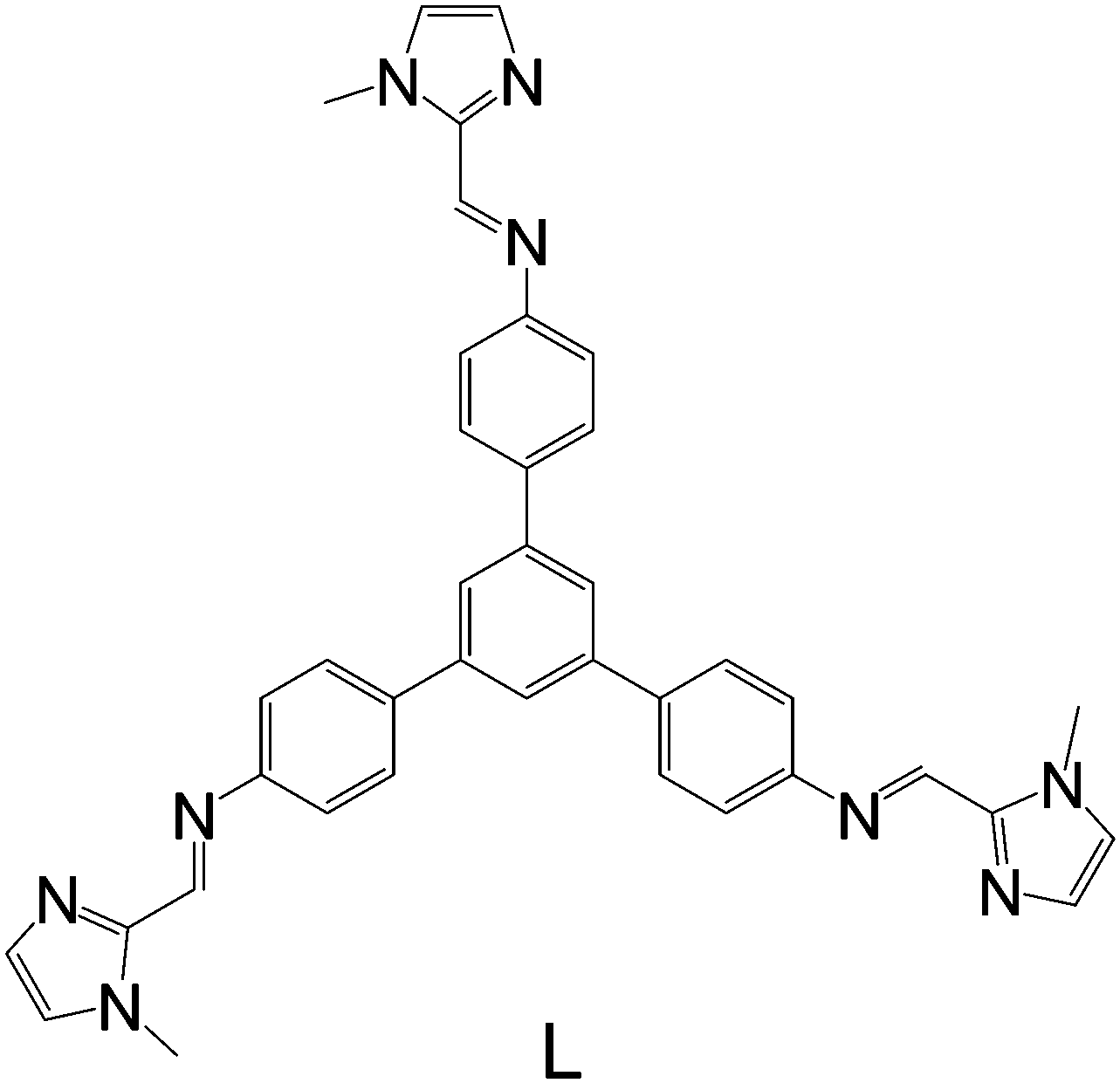 | ||
| Fig. 1 Schematic representation of rigid ligand L. | ||
Results and discussion
The reaction of 1-methyl-2-imidazolecarboxaldehyde and 1,3,5-tris(4-aminophenyl)-benzene in methanol produced the desired C3-symmetric tris-bidentate ligand L in 75% yield. 1H and 13C NMR spectra (Fig. S1 and S2, ESI‡) and high resolution electrospray ionisation (HR-ESI) mass spectrometry results were consistent with the proposed structure of L. In the HR-mass spectrum, the major peak is observed at m/z 628.2943 [L + H]+ (Fig. S3, ESI‡) and the appropriate isotope pattern for [L + H]+ was observed (Fig. S4, ESI‡).The further reaction of L with iron(II) tetrafluoroborate in acetonitrile followed by the slow diffusion of diethyl ether into the reaction mixture produced prismatic crystals of [Fe4L4][BF4]8·16MeCN suitable for X-ray diffraction studies (Fig. 2). The four homochiral facially coordinated octahedral metal centres are bridged by four of the ligands such that the ligands cover each face of a tetrahedron, and each tetrahedron has overall T-symmetry with C3-axes of symmetry passing through each metal centre and the centre of each ligand. There are three independent complexes in the asymmetric unit and the iron(II) centres within each tetrahedron are separated by between 14.5 and 15.1 Å placing this amongst the largest spin-crossover cages yet synthesised. For comparison, the two other spin-crossover cages reported have iron–iron separations of 11.85 Å10 and 14.16.11 Unlike many related complexes, this coordination cage crystallises in a chiral space group (P3) such that the cage molecules spontaneously resolve within each crystal. In the crystal examined all the metal centres displayed Δ-stereochemistry. The cage encapsulates a volume of 183 Å3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 and no significant residual electron density was located within the central cavity. The iron–nitrogen bond lengths are between 1.961(11) and 2.288(10) Å consistent with the magnetic results which suggest an intermediate spin at 100 K. Attempts to collect a crystal structure of the cage in its high-spin state (300 K) failed due to solvent loss from the crystals. In addition, scanning electron microscopy (SEM) photographs confirmed that the nanocage 1 uniformly crystallised in a polyhedral shape (Fig. 3a–c) and the crystals underwent rapid decay due to the loss of solvents (Fig. 3b and c).
19 and no significant residual electron density was located within the central cavity. The iron–nitrogen bond lengths are between 1.961(11) and 2.288(10) Å consistent with the magnetic results which suggest an intermediate spin at 100 K. Attempts to collect a crystal structure of the cage in its high-spin state (300 K) failed due to solvent loss from the crystals. In addition, scanning electron microscopy (SEM) photographs confirmed that the nanocage 1 uniformly crystallised in a polyhedral shape (Fig. 3a–c) and the crystals underwent rapid decay due to the loss of solvents (Fig. 3b and c).
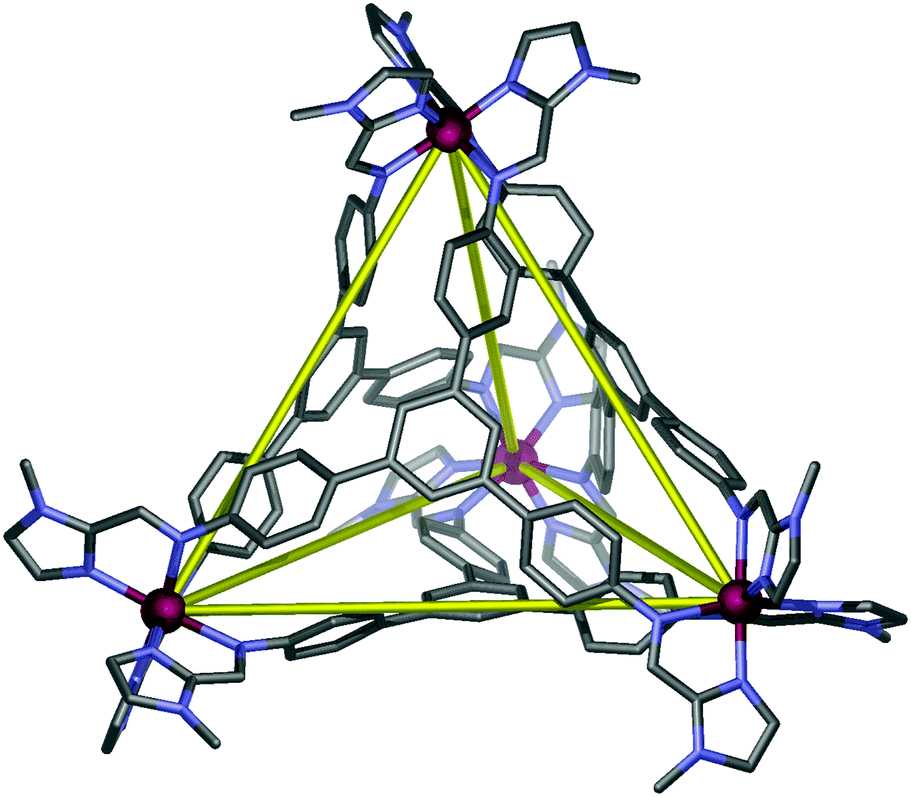 | ||
| Fig. 2 Schematic representation of the X-ray structure of cage 1. Hydrogen atoms, anions and solvent molecules are not shown for clarity. | ||
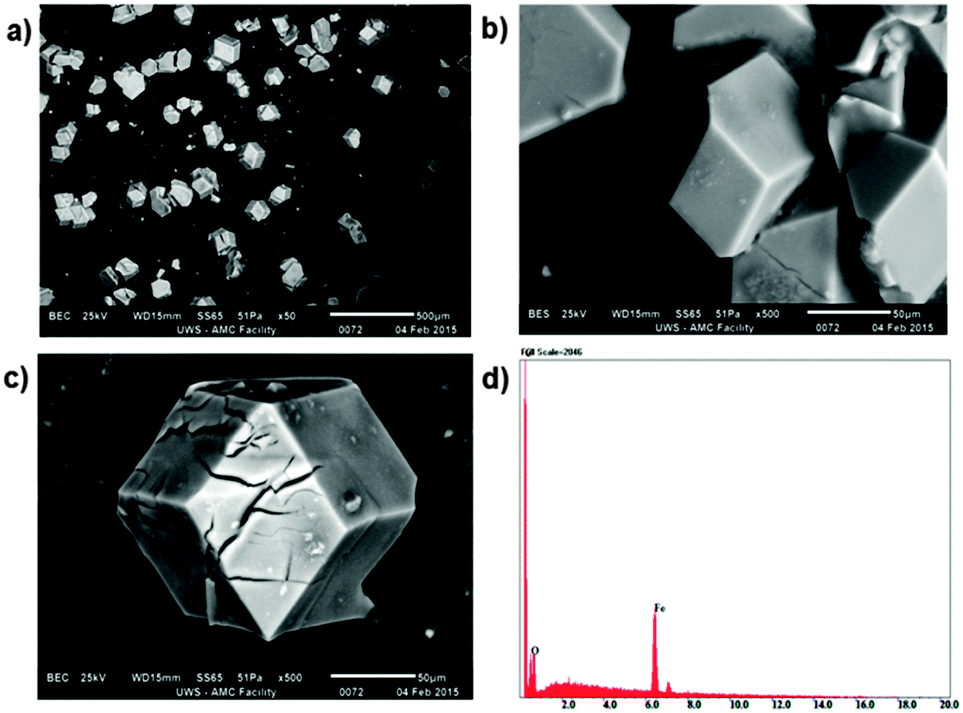 | ||
| Fig. 3 SEM images of crystals with polyhedral shape (a, b and c) and an EDS spectrum of cage 1 (d). | ||
Elemental analysis of the tetrahedral cage 1 suggested a metal:ligand ratio consistent with the crystal structure. Scanning electron microscopy-energy-dispersive spectroscopy (SEM-EDS) analysis of 1 was also carried out to support the above results. HR-ESI mass spectrometry results (Fig. S5–S12, ESI‡) clearly revealed a series of peaks of various charges corresponding to [Fe4L4][BF4](8−n)n+ (n = 1–8), which are consistent with the successive loss of [BF4]− anions.
The UV-vis spectrum of the coordination cage 1 in the solid state over the region 2000–350 nm (Fig. 4) reveals a relative low broad absorption band at 530 nm and an intense band at 410 nm. The former is attributed to a metal-to-ligand (MLCT) (d–π*) transition characteristic of an Fe(II) centre coordinated to a imidazole-imine based large aromatic ligand.8,11 The intense band at 410 nm is likely to arise from π–π* transitions.8,11
 | ||
| Fig. 4 Solid state UV-vis-NIR spectra (F(R) is the Kubelka-Munk transform) of L (red) and 1 (black). The inset shows the relatively low intensity transition in region 450–680 nm. | ||
FT-IR spectra and Raman spectra of L and 1 were recorded at room temperature (Fig. S13, S14 and S15, S16, ESI‡). Both the ligand L and coordination cage 1 show absorptions in the region 1600–1500 cm−1, these signals are typical of stretching imidazole-imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) groups. In the FT-IR spectrum of cage 1 (Fig. S14, ESI‡), it shows the existence of BF4− at 1047 cm−1. Raman spectra of L (Fig. S15, ESI‡) and 1 (Fig. S16, ESI‡) are very similar and also confirmed the presence of CN groups.
N) groups. In the FT-IR spectrum of cage 1 (Fig. S14, ESI‡), it shows the existence of BF4− at 1047 cm−1. Raman spectra of L (Fig. S15, ESI‡) and 1 (Fig. S16, ESI‡) are very similar and also confirmed the presence of CN groups.
Magnetic measurements
Magnetic susceptibility measurements reveal a gradual incomplete spin transition over the range 5–300 K (Fig. 5). The spin-crossover phenomenon between the high-spin and low-spin states for solvated [Fe4L4](BF4)8·16CH3CN and non-solvated [Fe4L4](BF4)8 was followed by measurements of the molar magnetic susceptibility χm as a function of temperature (Fig. 5a and b).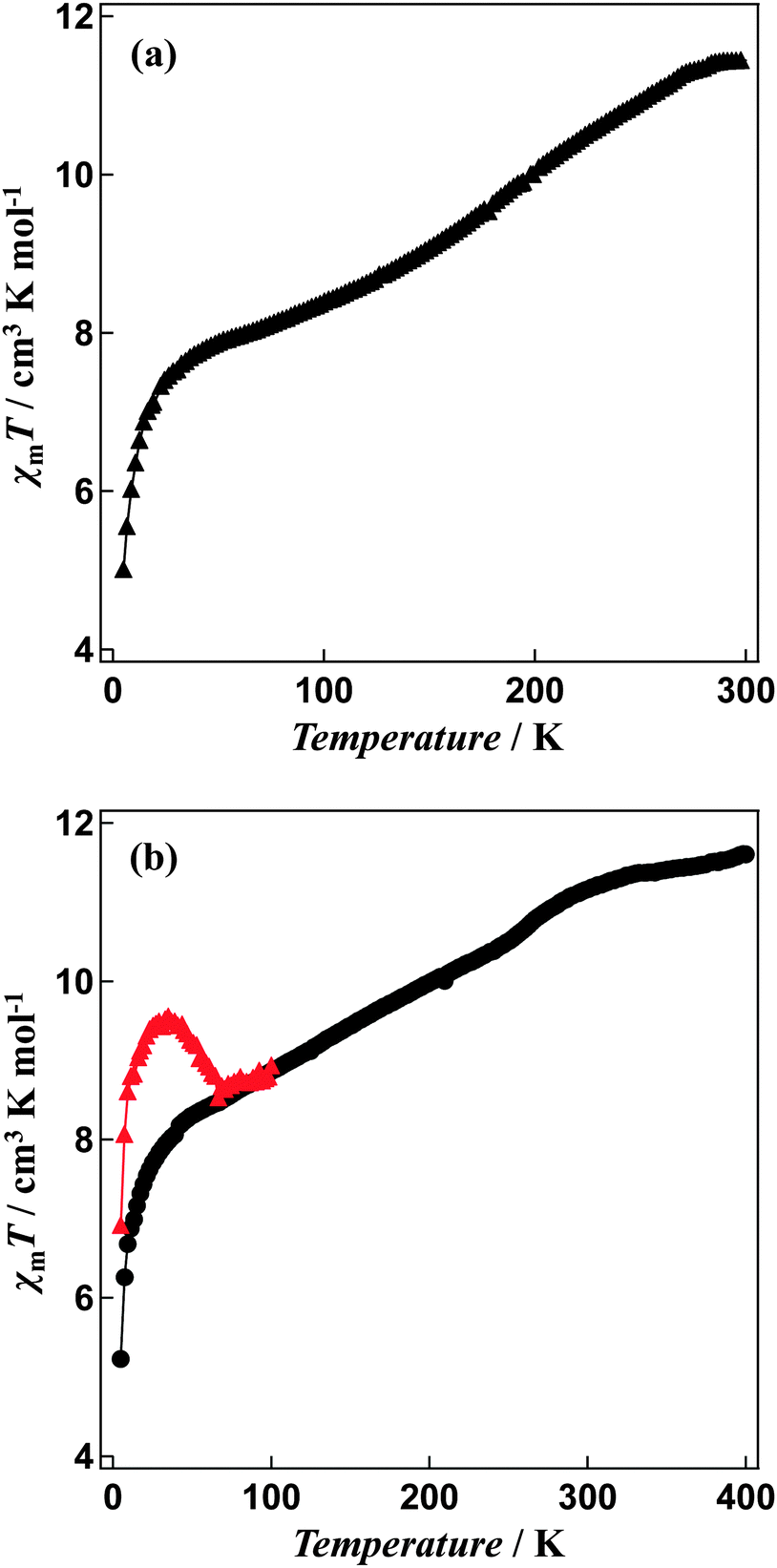 | ||
| Fig. 5 χ m T versus T plots for (a) solvated [Fe4L4](BF4)8·16CH3CN, the sample was measured in the temperature range between 5 K and 300 K and (b) non-solvated [Fe4L4](BF4)8 compound, the magnetic susceptibilities of the sample annealed at 400 K in the SQUID cavity was measured in the temperature range between 5 K and 400 K. χmT versus T plots for LIESST experiment were recorded in the warming mode after the sample was exposed to light illumination for 1 hour at 5 K. | ||
The χmT value for [Fe4L4](BF4)8·16CH3CN is equal to 11.44 cm3 K mol−1 at 300 K and 7.85 cm3 K mol−1 at 50 K consistent with a gradual spin-crossover transition (Fig. 5a). After annealing at 400 K, non-solvated compound [Fe4L4](BF4)8 was obtained. The χmT value of the desolvated material is equal to 11.60 cm3 K mol−1 at 400 K, suggesting iron(II) is in the high-spin state. On cooling, the χmT values gradually decreases (Fig. 5b). The χmT value at 50 K is equal to 8.29 cm3 K mol−1, which shows that spin-crossover from the high-spin to the low-spin states is induced in about 30% of the iron(II) ions. In addition, the Mössbauer spectrum measured at 5 K support the existence of iron(II) both in the high-spin and low-spin states (Fig. 6). The χmT values are also in agreement with the area ratios of Mössbauer absorption intensity of the high-spin and low-spin species.
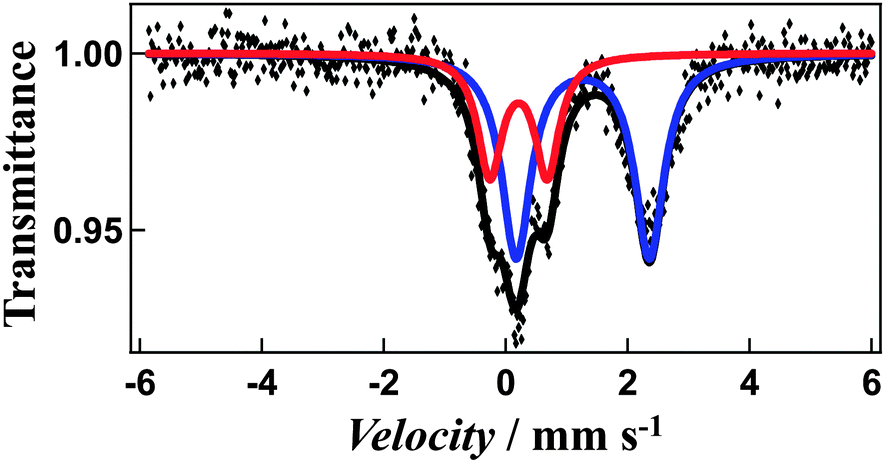 | ||
| Fig. 6 Mössbauer spectrum of non-solvated cage 1 at 5 K. The iron(II) high-spin is shown in blue, and low-spin in red. | ||
The Mössbauer spectrum measured at 5 K reveals a two quadrupole-split doublets. The first doublet is wide (quadrupole-splitting Q.S. = 2.18 mm s−1 and isomer shift I.S. = 1.26 mm s−1) and the second narrow (Q.S. = 0.94 mm s−1 and I.S. = 0.21 mm s−1), representing the high-spin and low-spin states (Fig. 6), respectively. The χmT values are also in agreement with the area ratios of Mössbauer absorption intensity of the high-spin and low-spin species (67:33). This, combined with the magnetic susceptibility studies suggests that the spin transition arises from only one of the four metal centres changing from high-spin to low-spin, while similar cage molecules showed a more complete transition, with three of the four centres switching spin states, potentially arising from changes in metal–metal distances and coordinating ligands.10,11
A green semiconductor laser (λ = 532 nm, 10 mW cm−2) was used as a light source to investigate the effects of illumination. The passed light was guided via an optical fibre into the SQUID. The sample was placed on the edge of the optical fibre. When the annealed samples were illuminated at 5 K, an increase in the susceptibility by illumination was observed (Fig. 5b). The change in the χmT value persisted for several hours, even after the illumination was halted. This suggests that the transition from the low-spin state to the high-spin state can be induced by illumination i.e. Light-Induced Excited Spin-State Trapping (LIESST).20 The χmT value decreases with the increase in temperature and that the thermal relaxation to the ground state occurs. The LIESST effect in the non-solvated compound occurs with T(LIESST) of 55 K.
Conclusions
In summary, we describe the efficient synthesis of a new discrete tetranuclear SCO Fe(II) cage incorporating rigid tris-bidentate ligands with suitable ligand strength arising from the imidazole-imine functional units. The structure has been unambiguously characterised by X-ray crystallography, ESI mass spectrometry, UV-vis-NIR, FT-IR and Raman spectroscopy. SCO behaviour of the coordination cage 1 has been investigated. Further studies on these and related other Fe(II) coordination cages, including a high pressure solid state investigation, are ongoing and will be reported in due course.Experimental
Materials and synthesis
All reagents and solvents were purchased from commercial sources.Physical measurements
1H NMR and 13C NMR spectra were recorded on a Bruker 300 MHz spectrometer. High resolution ESI-MS data were acquired using a Waters Xevo QToF mass spectrometer, operating in positive ion mode. FT-IR spectra were recorded on a Bruker Tensor 27 Fourier transform infrared spectrometer using diamond single bounce ATR sampling device. The UV-vis spectra were measured at ambient temperature using a Cary 5000 spectrophotometer equipped with a Labsphere Biconical Accessory.Mössbauer experiments were carried out using a Wissel MVT-1000 Mössbauer spectrometer with a 57Co/Rh source in a constant-acceleration transmission spectrometer (Topologic Systems) equipped with a closed-cycle helium refrigerator cryostat (Iwatani Co., Ltd). All isomer shifts are given relative to α-Fe at room temperature. Measurements at low temperature were performed.
Ligand synthesis
1-Methyl-2-imidazolecarboxaldehyde (469 mg, 4.26 mmol) in methanol (20 mL) was added dropwise to a suspension of 1,3,5-tris(4-aminophenyl)-benzene (500 mg, 1.42 mmol) in methanol (50 mL). The mixture was heated at reflux for overnight leading to a clear yellow solution. The residue obtained after the removal of the solvent was recrystallised from an ethanol–acetonitrile mixture. The crystalline product was washed with acetonitrile (3 × 5 mL) to give L as a pale yellow powder (669 mg, 75%). 1H NMR (DMSO, 300 MHz) δ (ppm) 8.59 (s, 3H, aromatic ring), 7.95 (d, 9H, aromatic ring), 7.44 (d, 9H, aromatic ring), 7.19 (s, 3H), 4.09 (s, 9H, methyl); 13C NMR (DMSO, 75.5 MHz) δ (ppm) 151.21, 150.34,142.78, 141.04, 138.01, 129.80, 128.07, 126.61, 123.87, 121.62, 35.24 (–CH3); FT-IR ATR νmax/cm−1: 3340(br), 1628s, 1594s, 1475s, 873s, 833s, 752s, 688s, 541m; ESI-HRMS (positive-ion detection, CH3OH/H2O): m/z = 628.2943.Complex synthesis
Crystallography
X-ray structural data were collected at beamline MX1 of the Australian Synchrotron employing silicon double crystal monochromated synchrotron radiation (0.7108 Å) at 100(2) K.21,22 Data integration and reduction were undertaken with XDS23 and subsequent computations were carried out using the WinGX-32 graphical user interface.24 The structure was solved by charge flipping using SUPERFLIP.25 Empirical absorption corrections were applied to the data set using the program SADABS26 Data were refined and extended with SHELXL-2014.27 In general non-hydrogen atoms with occupancies greater than 0.5 were refined anisotropically. Carbon-bound hydrogen atoms were included in idealised positions and refined using a riding model. The crystals employed rapidly lost solvent after removal from the mother liquor. Rapid (<10 seconds) handling at dry ice temperatures prior to quenching in the cryostream was required to collect data. Despite these measures, the use of a synchrotron source and multiple collection attempts, no reflections at better than 0.9 Å resolution were observed. In addition, data collection at the beamline was restricted to a 360° rotation around a single axis resulting in less than ideal redundancy. Reflecting the less than ideal diffraction, there is substantial disorder in a number of the anions which required the use of bond length and angle restraints to facilitate realistic modelling. Further reflecting the solvent loss and poor diffraction properties, there is a significant amount of void volume in the lattice containing smeared electron density from disordered solvent and anions. This area of density could not be successfully modelled and the SQUEEZE28 function of PLATON29 was employed resulting in significantly improved residuals. Despite these limitations the quality of the data is more than suitable for establishing the connectivity of the system. The Flack parameter refined to 0.05(3).30–33Formula C188H180B8F32Fe4N52, M 4085.71, trigonal, space group P3(#143), a 31.590(5) Å, b 31.590(5) Å, c 18.320(4) Å, γ 120°, V 15833(6) Å3, Dc 1.286 g cm−3, Z 3, crystal size 0.05 by 0.05 by 0.05 mm, colour orange, habit prism, temperature 100(2) K, λ(Synchrotron) 0.7108 Å, μ(Synchrotron) 0.359 mm−1, T(SADABS)min,max 0.3657, 0.4272, 2θmax 43.93, hkl range −33 33, −33 33, −19 19, N 139384, Nind 25723 (Rmerge 0.0847), Nobs 23244(I > 2σ(I)), Nvar 2204, residuals R1(F) 0.0961, wR2(F2) 0.2580, GoF(all) 1.077, Δρmin,max −0.579, 1.767 e− Å−3.
Acknowledgements
The research described herein was supported by the University of Western Sydney (UWS). The authors acknowledge AMCF and MS facilities at UWS and MX1 beamline at the Australian Synchrotron. J.K.C., R.Z. and L.F.L. thank the Australian Research Council for support. L.L. also acknowledges receipt of an Australian Postgraduate Award and UWS Top-up Award.Notes and references
- D. M. Wood, W. Meng, T. K. Ronson, A. R. Stefankiewicz, J. K. M. Sanders and J. R. Nitschke, Angew. Chem., Int. Ed., 2015, 54, 3988–3992 CrossRef CAS PubMed.
- W. J. Ramsay and J. R. Nitschke, J. Am. Chem. Soc., 2014, 136, 7038–7043 CrossRef CAS PubMed.
- A. Jiménez, R. A. Bilbeisi, T. K. Ronson, S. Zarra, C. Woodhead and J. R. Nitschke, Angew. Chem., Int. Ed., 2014, 53, 4556–4560 CrossRef PubMed.
- M. M. J. Smulders, S. Zarra and J. R. Nitschke, J. Am. Chem. Soc., 2013, 135, 7039 CrossRef CAS PubMed.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS PubMed.
- C. R. K. Glasson, J. K. Clegg, J. C. McMurtrie, G. V. Meehan, L. F. Lindoy, C. A. Motti, B. Moubaraki, K. S. Murray and J. D. Cashion, Chem. Sci., 2011, 2, 540–543 RSC.
- C. R. K. Glasson, G. V. Meehan, J. K. Clegg, L. F. Lindoy, P. Turner, M. B. Duriska and R. Willis, Chem. Commun., 2008, 1190–1192 RSC.
- D.-H. Ren, D. Qiu, C.-Y. Pang, Z. Li and Z.-G. Gu, Chem. Commun., 2015, 51, 788–791 RSC.
- F. Li, N. F. Sciortino, J. K. Clegg, S. M. Neville and C. J. Kepert, Aust. J. Chem., 2014, 67, 1625–1628 CrossRef CAS.
- R. A. Bilbeisi, S. Zarra, H. L. C. Feltham, G. N. L. Jameson, J. K. Clegg, S. Brooker and J. R. Nitschke, Chem. – Eur. J., 2013, 19, 8058–8062 CrossRef CAS PubMed.
- A. Ferguson, M. A. Squire, D. Siretanu, D. Mitcov, C. Mathoniere, R. Clerac and P. E. Kruger, Chem. Commun., 2013, 49, 1597–1599 RSC.
- M. B. Duriska, S. M. Neville, B. Moubaraki, J. D. Cashion, G. J. Halder, K. W. Chapman, C. Balde, J.-F. Letard, K. S. Murray, C. J. Kepert and S. R. Batten, Angew. Chem., Int. Ed., 2009, 48, 2549–2552 CrossRef CAS PubMed.
- Y. Sunatsuki, R. Kawamoto, K. Fujita, H. Maruyama, T. Suzuki, H. Ishida, M. Kojima, S. Iijima and N. Matsumoto, Coord. Chem. Rev., 2010, 254, 1871–1881 CrossRef CAS PubMed.
- N. Bréfuel, H. Watanabe, L. Toupet, J. Come, N. Matsumoto, E. Collet, K. Tanaka and J.-P. Tuchagues, Angew. Chem., Int. Ed., 2009, 48, 9304–9307 CrossRef PubMed.
- D. Pelleteret, R. Clerac, C. Mathoniere, E. Harte, W. Schmitt and P. E. Kruger, Chem. Commun., 2009, 221–223 RSC.
- N. Brefuel, C. Duhayon, S. Shova and J.-P. Tuchagues, Chem. Commun., 2007, 5223–5225 RSC.
- Y. Sunatsuki, Y. Ikuta, N. Matsumoto, H. Ohta, M. Kojima, S. Iijima, S. Hayami, Y. Maeda, S. Kaizaki, F. Dahan and J.-P. Tuchagues, Angew. Chem., Int. Ed., 2003, 42, 1614–1618 CrossRef CAS PubMed.
- I. Katsuki, Y. Motoda, Y. Sunatsuki, N. Matsumoto, T. Nakashima and M. Kojima, J. Am. Chem. Soc., 2002, 124, 629–640 CrossRef CAS PubMed.
- G. J. Kleywegt and T. A. Jones, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1994, 50, 178–185 CrossRef CAS PubMed.
- J.-F. Létard, L. Capes, G. Chastanet, N. Moliner, S. Létard, J.-A. Real and O. Kahn, Chem. Phys. Lett., 1999, 313, 115–120 CrossRef.
- T. M. McPhillips, S. E. McPhillips, H.-J. Chiu, A. E. Cohen, A. M. Deacon, P. J. Ellis, E. Garman, A. Gonzalez, N. K. Sauter, R. P. Phizackerley, S. M. Soltis and P. Kuhn, J. Synchrotron Radiat., 2002, 9, 401–406 CrossRef CAS PubMed.
- N. P. Cowieson, D. Aragao, M. Clift, D. J. Ericsson, C. Gee, S. J. Harrop, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Williamson and T. Caradoc-Davies, J. Synchrotron Radiat., 2015, 22, 187–190 CrossRef CAS PubMed.
- W. Kabsch, J. Appl. Crystallogr., 1993, 26, 795–800 CrossRef CAS.
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- G. M. Sheldrick, SADABS: Empirical Absorption and Correction Software, 1996-2008, University of Göttingen, Germany, 1996–2008 Search PubMed.
- G. M. Sheldrick, SHELXS 2014, Program for the solution of crystal structures, University of Göttingen, Germany, 2014 Search PubMed.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- H. D. Flack and G. Bernardinelli, J. Appl. Crystallogr., 2000, 33, 1143–1148 CAS.
- H. D. Flack and G. Bernardinelli, Acta Crystallogr., Sect. A: Found. Crystallogr., 1999, 55, 908–915 CrossRef PubMed.
- G. Bernardinelli and H. D. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 1985, 41, 500–511 CrossRef.
- H. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 1983, 39, 876–881 CrossRef.
Footnotes |
| † The themed issue on ‘Spin-State switches in Molecular Materials Chemistry’. |
| ‡ Electronic supplementary information (ESI) available: NMR, ESI-Mass, FT-IR and Raman spectra and TGA. CCDC 1057843. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5tc00991j |
| This journal is © The Royal Society of Chemistry 2015 |