
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regio- and stereoselective synthesis of α-hydroxy-β-azido tetrazoles†
Pierre
Quinodoz
a,
Cheikh
Lo
a,
Mikhail
Kletskii
b,
Oleg
Burov
b,
Jérôme
Marrot
a and
François
Couty
*a
aInstitut Lavoisier de Versailles, UMR 8180. Université de Versailles St-Quentin en Yvelines. 45, av. des Etats-Unis, 78035 Versailles Cedex, France. E-mail: couty@chimie.uvsq.fr; Web: http://www.ilv.uvsq.fr/ Fax: +33 (0) 1 39 25 44 52
bDepartment of Chemistry, Southern Federal University, 7, Zorge St., 344090 Rostov-on-Don, Russian Federation
First published on 16th March 2015
Abstract
Unreported α-hydroxy-β-azido tetrazoles were prepared in one step from readily available α,β-epoxy nitriles. This reaction involves a dibutyltin oxide-catalyzed cycloaddition of the nitrile reacting with TMSN3 leading to the tetrazole moiety, and opening of the epoxide by the azide anion. High levels of regio- and stereoselectivity are obtained in this reaction and are discussed, also by means of quantum mechanical DFT calculations. The azido group in these compounds could be uneventfully reduced to the corresponding amine thus leading to an α-hydroxy-β-amino tetrazole, surrogate of the corresponding carboxylic acid, while reaction with triphenylphosphine led to propargylic amines.
Introduction
Tetrazoles have found applications in various domains including energetic materials, owing to their high nitrogen content, and medicinal chemistry, due to the fact that 5-substituted tetrazoles (5-ST) are bioisosteres of carboxylic acids.1 This last property has been notably popularized with the release of the antihypertensive drug Losartan,2 which soon became a blockbuster. The synthesis of tetrazoles has been extensively studied, cycloaddition of azides (anion or derivatives) with nitriles being the most popular way to efficiently produce this heterocycle. Many key improvements in this reaction, which can be promoted either by Brønsted or Lewis acids have appeared, including the use of sodium azide or TMSN3 with NH4Cl,3 ZnBr2,4 Me3Al,5 I2,6 or AgNO3.7 Microwave irradiation8 has also been used with much success, but this reaction still requires elevated temperatures (typically above 100 °C) to proceed, thus narrowing its scope to quite robust nitriles. Another possibility lies in the use of dibutyltin oxide as a catalyst, in conjunction with TMSN3.9 In contrast to the above methods, this reaction involves neutral reaction medium and a weak Lewis acid, thus allowing cycloaddition of nitriles fitted with a Lewis base, such as amino nitriles.10 We therefore decided to study this cycloaddition with α,β-epoxy nitriles, aiming at the preparation of functionalized tetrazoles suitable for further synthetic transformations. Our findings are exposed in the next section.Results
We first studied reaction depicted in Scheme 1 with epoxide 1, readily prepared by a Darzens reaction.11 Thus, reacting this epoxide (18 h) in toluene at 60 °C with TMSN3 (5 equiv.) and a substoichiometric amount of Bu2SnO (0.5 equiv.) led, after acidic hydrolysis, to the α-hydroxy-β-azido tetrazole 2 in good yield. Much to our delight, this reaction, involving both cycloaddition and epoxide opening, occurred with high regioselectivity and complete inversion at the β-carbon.12 The structure of 2 was verified by X-ray crystallography (see ESI†).13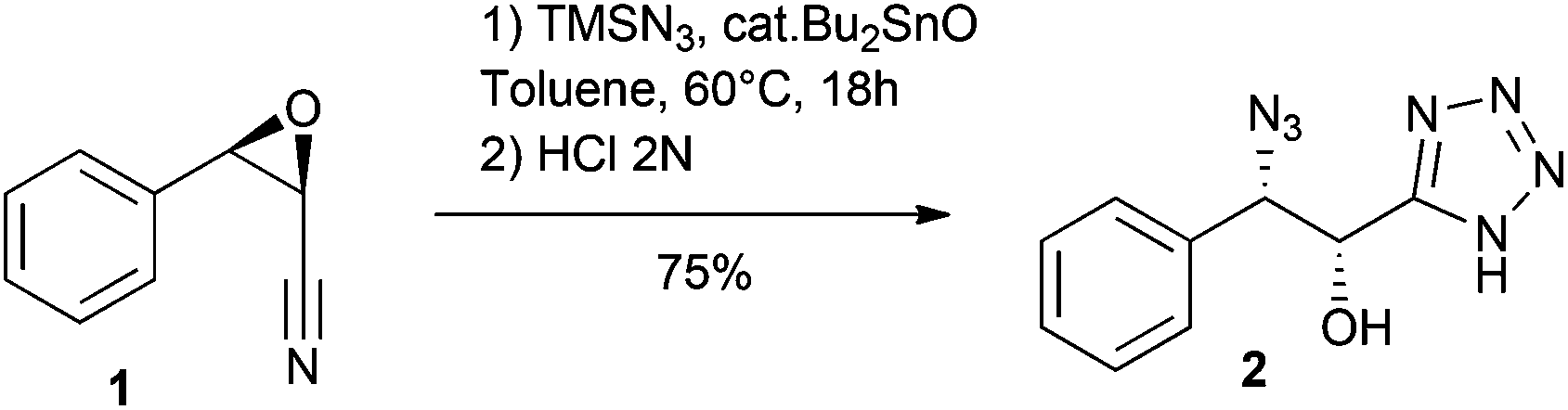 | ||
| Scheme 1 Bu2SnO-catalyzed reaction of epoxynitrile 1 with TMSN3 leads regioselectively to α-hydroxy β-azido tetrazole 2. | ||
We were surprised by the low temperature required for completion of this reaction and we first screened the amount of catalyst and TMSN3 needed in order to maintain a high yield. Reactions were run in toluene at 60 °C for 18 h. (Table 1).
Increasing the amount of Bu2SnO (entry 2) gave an excellent yield of 2, and lowering the amount of TMSN3 to three equiv. (entry 3) maintained a high yield, but decreasing of the amount of catalyst to 20 mol% lowered the yield significantly (entry 5). In order to determine which event first occurred (cycloaddition or epoxide opening), we also used one equivalent of TMSN3 and Bu2SnO (preheated until dissolution) and isolated after reaction and acidic workup β-chloro-α-hydroxy tetrazole, resulting from the opening of the epoxide by the chloride anion (entry 6), suggesting that cycloaddition first occurs. Thus, we chose to examine the scope of this reaction with other epoxides (shown in Fig. 1) with 3 equiv. of TMSN3, using 50 mol% of Bu2SnO (conditions of entry 3). Structures of the isolated compounds are shown in Fig. 2.
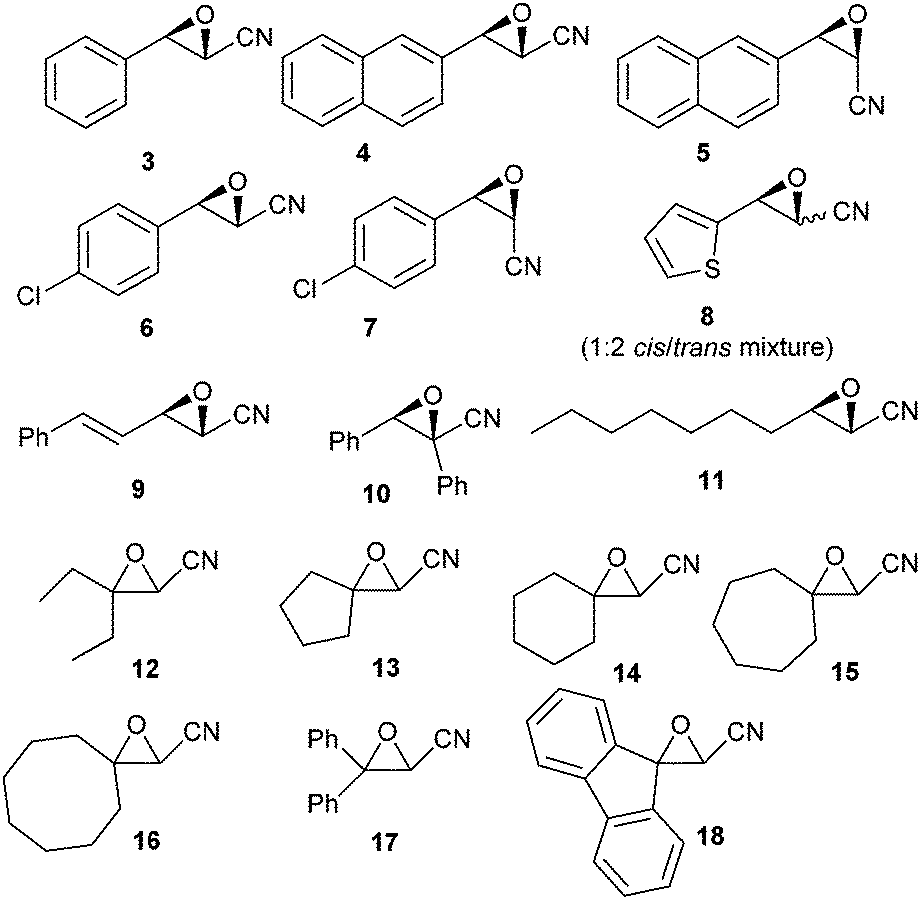 | ||
| Fig. 1 Structures of the cyanoepoxides used to examine the scope of this reaction. | ||
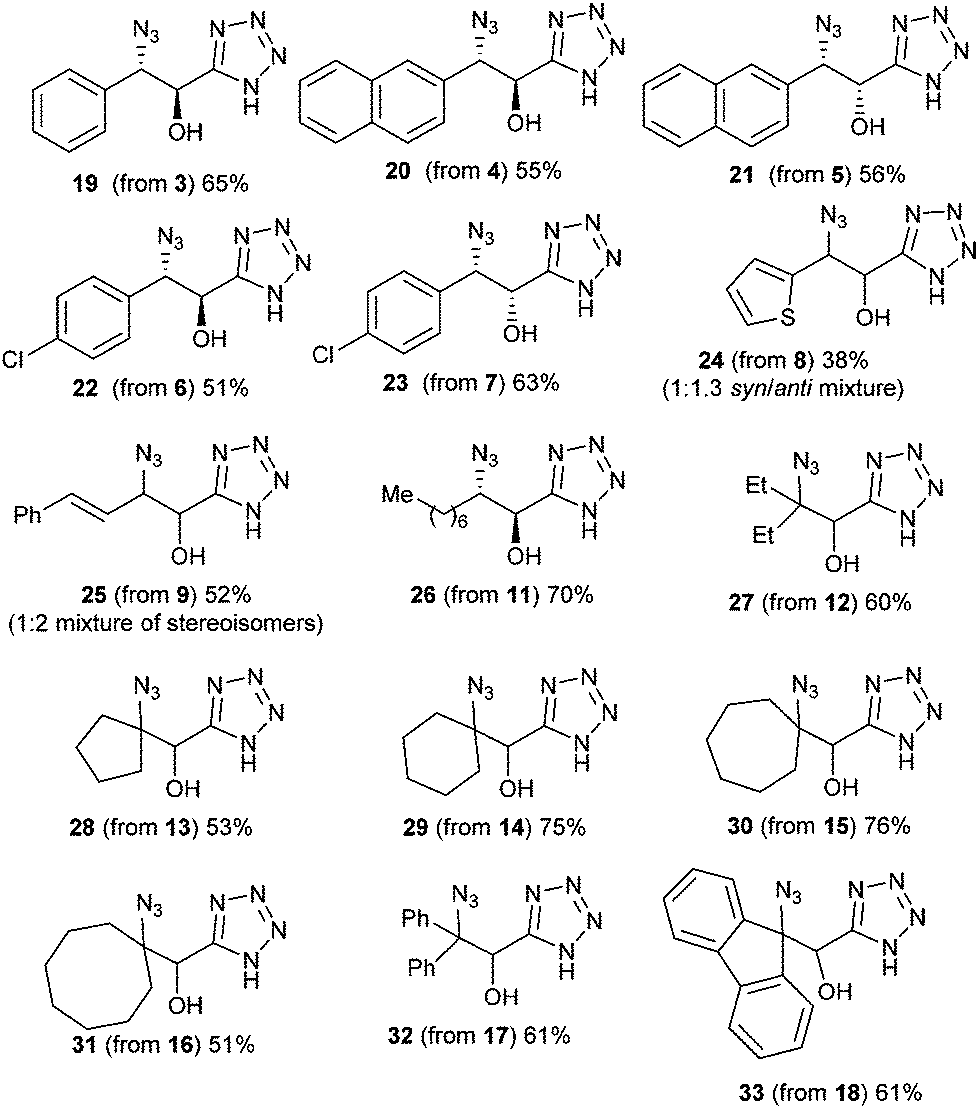 | ||
| Fig. 2 Structures and yields of the produced isolated α-hydroxy-β-azido tetrazoles. | ||
As depicted in these figures, the scope of this reaction was very good, tolerating aryl, alkenyl and alkyl groups at the β-position of the epoxide. Yields are modest to good (38–76%) and these compounds are isolated as crystalline materials.‡ β-Disubstituted epoxides 12–18 also reacted very well and tertiary azides were obtained in good yields. Only α-disubstituted epoxide 10 failed to react in these conditions, leaving unreacted starting material. Regioselectivity was constantly excellent, leading to a unique β-azido regioisomer whatever the degree and the nature of substitution at the β-carbon. The stereoselectivity of this reaction could be evaluated with the cis or trans epoxides 1, 3–9 and 11. For 1, 3–7 and 11 the reaction appears to be stereospecific and involves an inversion at the β-carbon as shown from the X-ray structure of 2 and slightly different NMR data for diastereoisomers 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19, 20:21 and 22:23 (see ESI†). Starting with 8 (1:2 cis–trans mixture), a 1:1.3 mixture of isomers 24 was obtained, albeit in low yield, which also might reflect the stereospecificity of this reaction. Only the starting trans epoxide 9 reacted with epimerization at the β-carbon, leading to 25 as a 1:2 mixture of diastereoisomers.
:19, 20:21 and 22:23 (see ESI†). Starting with 8 (1:2 cis–trans mixture), a 1:1.3 mixture of isomers 24 was obtained, albeit in low yield, which also might reflect the stereospecificity of this reaction. Only the starting trans epoxide 9 reacted with epimerization at the β-carbon, leading to 25 as a 1:2 mixture of diastereoisomers.
Next we briefly examined the reactivity of these α-hydroxy-β-azido tetrazoles. We first tried to reduce the azido moiety into an amino group, in order to access α-hydroxy-β-amino tetrazoles,14 which can be viewed as surrogates of the corresponding α-hydroxy-β-amino acid.15 The latter compounds occupy a very important place within the family of β-amino acids, being constituents of several drugs and natural molecules such as inter alia Taxol,16 Taxotere,17 Bestatin,18 Microginin19 and the HIV protease inhibitor R-87366.20 Moreover, α-hydroxy-β-amino tetrazoles have found uses in the design of bioactive peptides with cis-conformationally restricted peptide bonds.21
Thus, aiming to reduce the azido through Staudinger conditions, 2 was reacted with triphenylphosphine in refluxing THF for 2 h, but the crude reaction mixture unexpectedly showed formation of propargylic amine 34, together with phosphine oxide. This compound was acetylated for easier purification and 35 was isolated with an overall yield of 52% (Scheme 2).
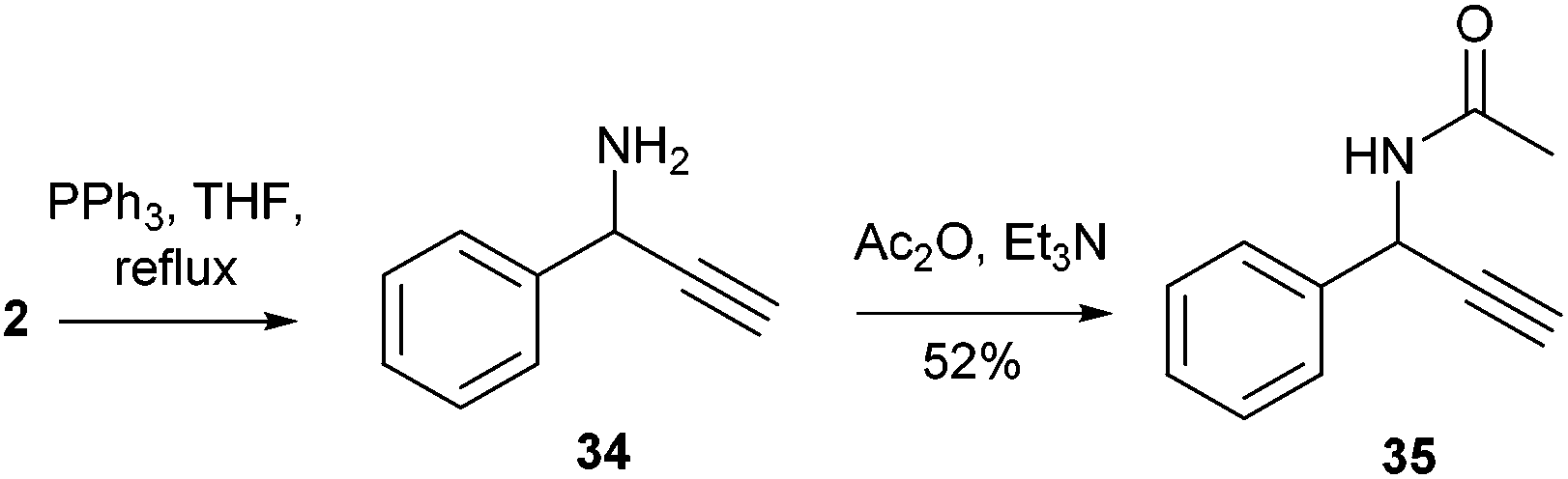 | ||
| Scheme 2 Reaction of α-hydroxy-β-azido tetrazole 2 with triphenylphosphine leads to the formation of propargylamine 34. | ||
The scope of this reaction, conducted in one pot without isolation of the intermediate amine, was briefly screened (Table 2 and Fig. 3) and it was found to be general, with yields varying from 23 to 71% in the case of secondary azides. However, no trace of acetylenic compound could be detected in the crude reaction mixture starting from tertiary azides 28 or 32.
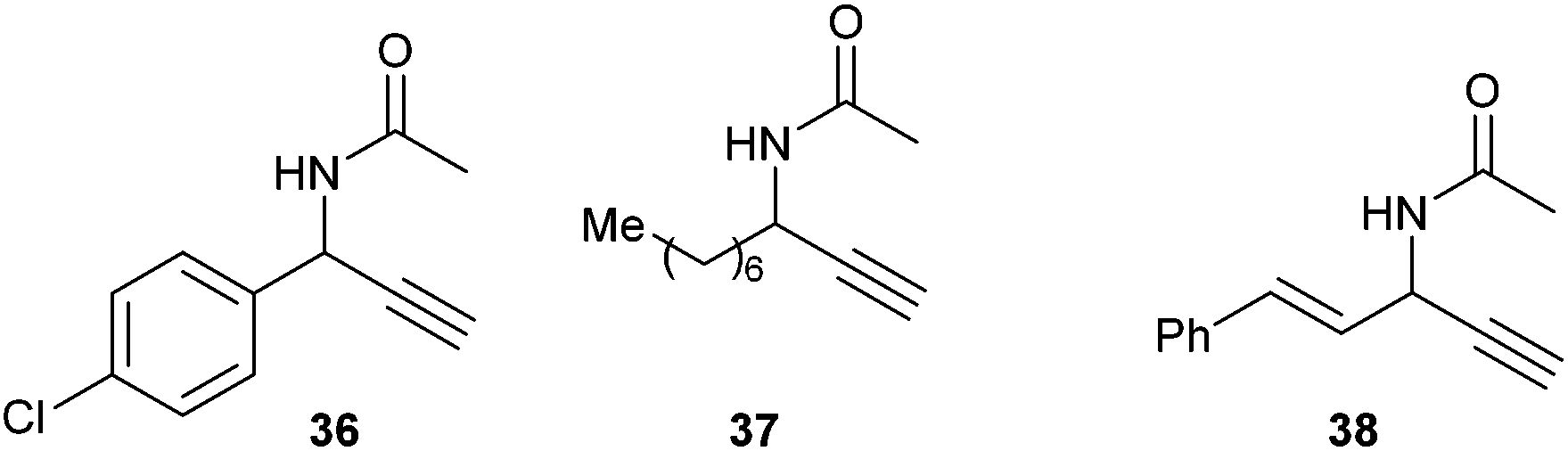 | ||
| Fig. 3 Structure of propargylamines 36–38. | ||
Alternatively, Pd/C-catalyzed hydrogenation of 19 led quantitatively to the α-hydroxy-β-amino tetrazole 39 as its chlorohydrate salt (Scheme 3).
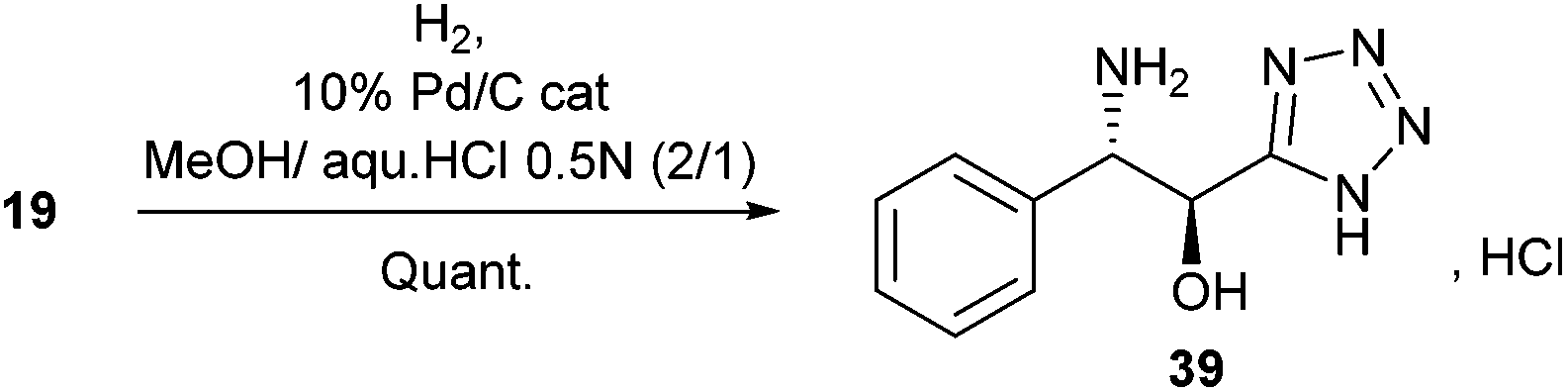 | ||
| Scheme 3 Reduction of the azide is conveniently achieved by Pd/C-catalyzed hydrogenation. | ||
Discussion
The unexpected formation of propargylic amines during the reaction of α-hydroxy-β-amino tetrazoles with triphenylphosphine deserves comment. The Blum-Ittah aziridine synthesis is an established procedure to convert 1,2-azido alcohols into aziridines by treatment with a tertiary phosphine.22 It is accepted that it goes through an oxazaphospholidine 40 that collapses to the aziridine 41 with release of phosphine oxide. A possible mechanism that would explain our results is the following. In our starting compounds, proton transfer from the acidic tetrazole to the nitrogen of the intermediate oxazaphospholidine 42 would produce 5-methylene-5H-tetrazole 43 with release of triphenylphosphine oxide, that could further decompose to vinylic carbene 44.23 This intermediate would ultimately lead to the propargylic amine 45 through a Fritsch–Buttenberg–Wiechell rearrangement (Scheme 4). Recent precedents in the literature have demonstrated the possibility of smoothly generating vinylic carbenes from α-hydroxy tetrazoles upon activation with DCC.24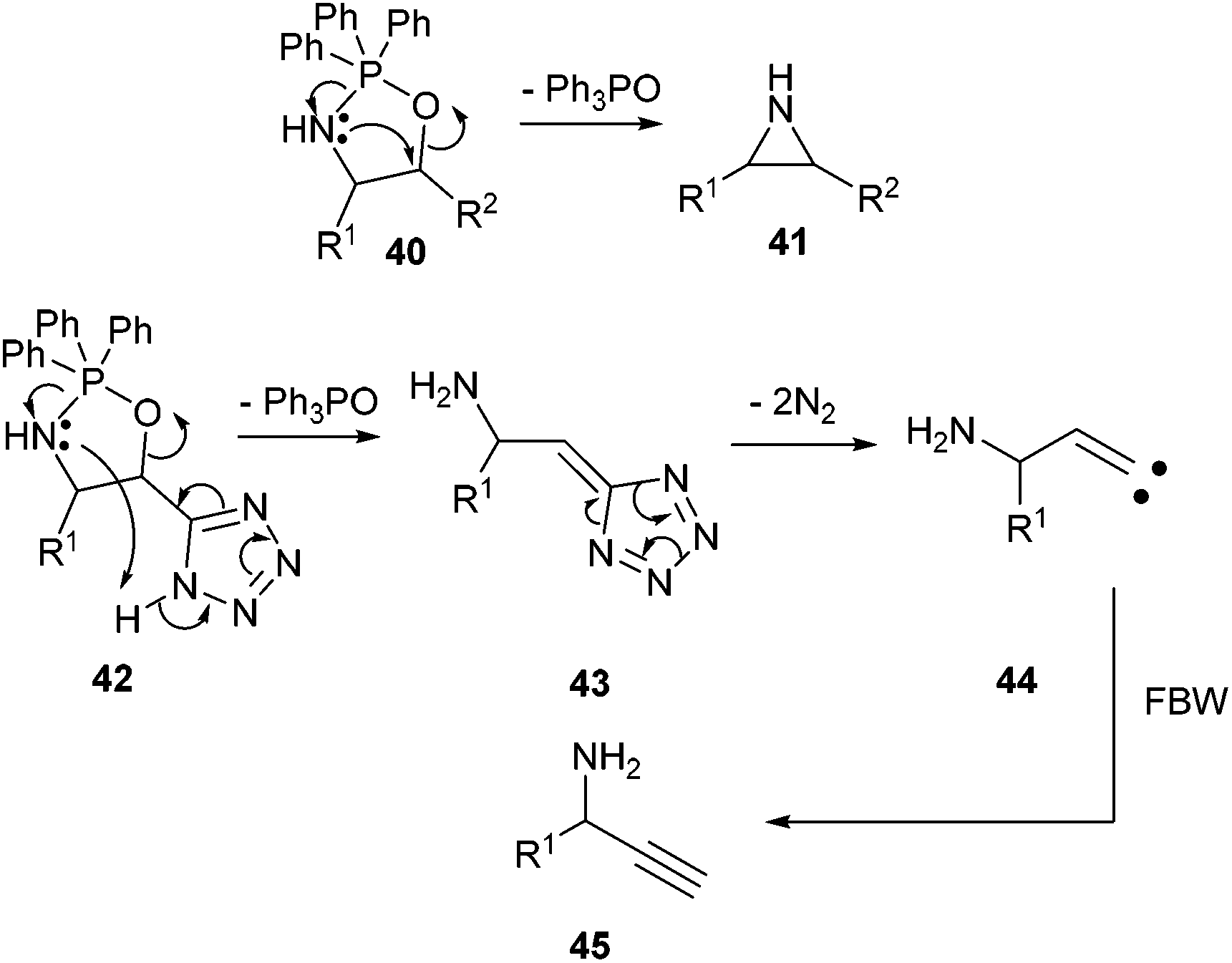 | ||
| Scheme 4 A plausible mechanism accounting for the formation of propargylic amine upon the reaction of α-hydroxy-β-azido tetrazoles with triphenylphosphine. | ||
The second point which is worth discussing is the high regio- and stereoselectivity observed during ring-opening of the epoxide. All substrates, except allylic azide 25, were obtained as single compounds, and the erosion of stereoselectivity in that particular case might be due to a dynamic [3,3] equilibration process of the allylic azide which reflects thermodynamic control.25 Though uncatalyzed ring opening of glycidates with TMSN3 has been reported to proceed with varying regio- and stereoselectivity (SN2 or SNi), depending on the stereochemistry of the epoxide,12 in our case, no reaction occurred in the absence of Bu2SnO suggesting the crucial role of the tin catalyst for both cycloaddition and epoxide opening. The mechanism of Bu2SnO-catalyzed cycloaddition of TMSN3 with alkynes has been studied in details.26 It was demonstrated that the active catalytic species is Bu2Sn(OTMS)N3, and that regeneration of this catalyst occurs through a SN2 displacement at the silicon atom, which was calculated to require only 28 kcal mol−1, followed by fast ligand exchange at the tin atom (Scheme 5). In our case, the tin atom in the produced tetrazole 46 is ideally located to assist in the opening of the epoxide by the azide anion at the β-position. This concerted reaction would account for the regioselectivity of the opening and the SN2 process. In this case, regeneration of the catalyst would then imply reaction of the produced tin alkoxide 47 with TMSN3, to produce OTMS derivative 48, an exchange that can be promoted from a thermodynamic viewpoint considering the much stronger O–Si bond (190 kcal mol−1) compared to the O–Sn bond (130 kcal mol−1).
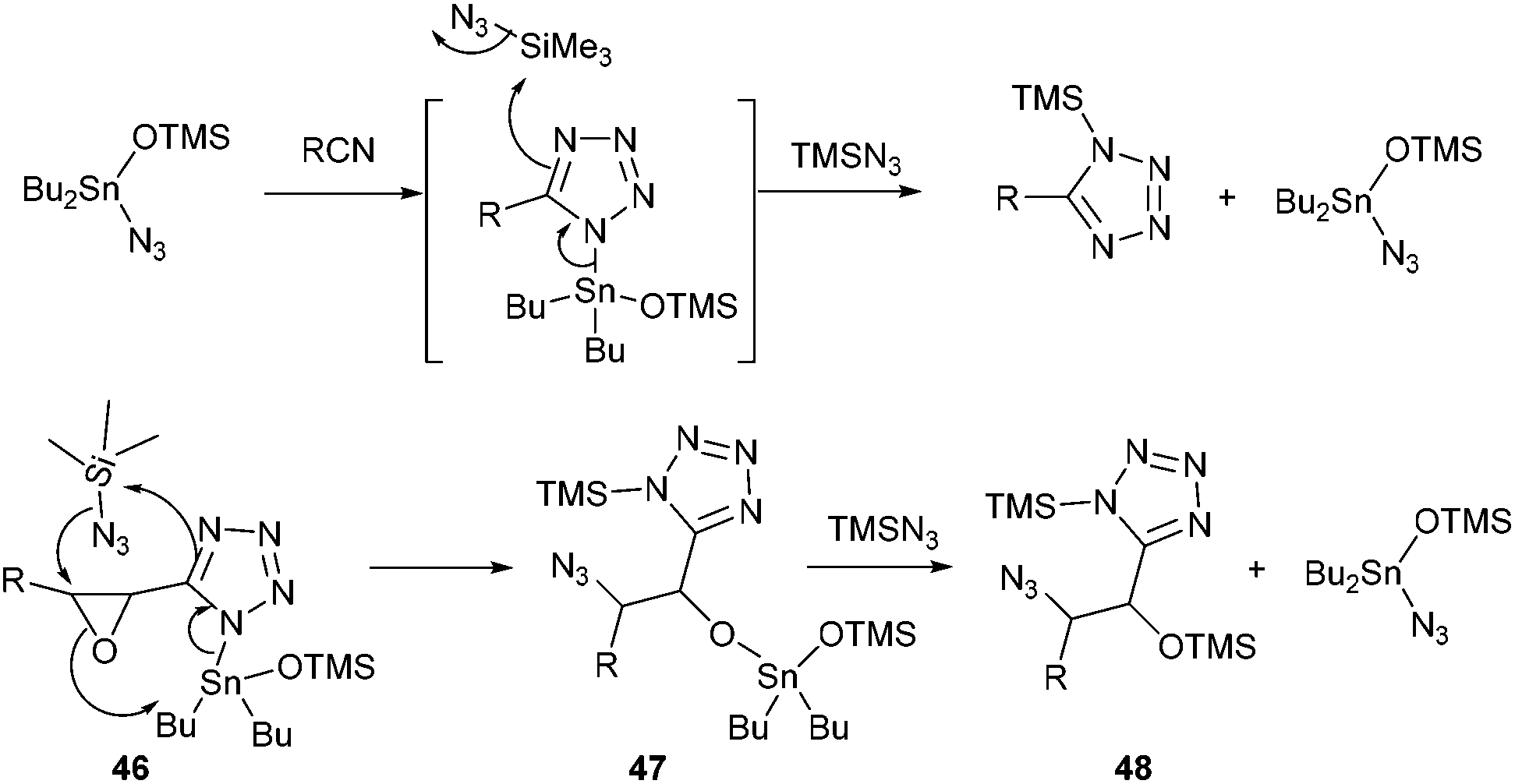 | ||
| Scheme 5 A plausible mechanism accounting for the regio- and stereoselectivity of the ring opening process. | ||
In order to evaluate the feasibility of this tin to silicon exchange (47→48), simplified reaction depicted in Scheme 6 was considered. Quantum mechanical calculations at the B3LYP level of theory [with LANL2DZ ECP for tin atom27 and 6-31G** basis set28 for other atoms] were performed with the Firefly 8.0.1 package of programs.29 The structure of the optimized transition state (TS) of this concerted reaction, located at only 17.9 kcal mol−1, together with the structures RC1 and RC2 of pre- and post-reaction complexes are shown in Fig. 4, while Fig. 5 outlines the energetic profile of this reaction.
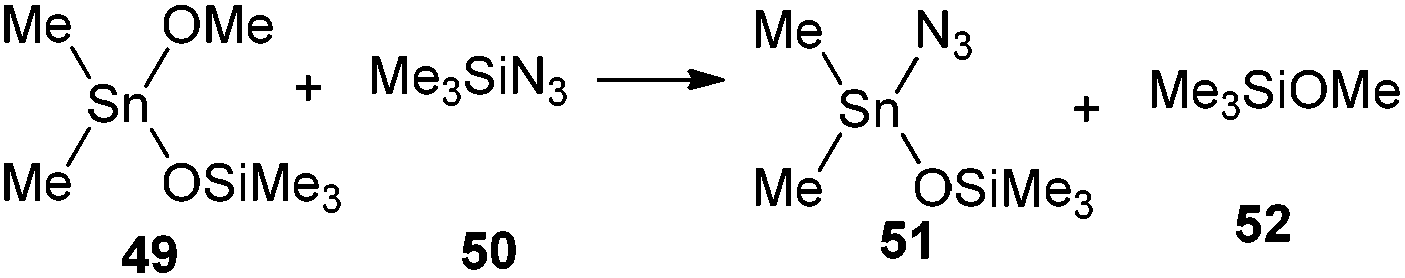 | ||
| Scheme 6 Simplified computed reaction for the evaluation of the tin to silicon exchange leading to 48. | ||
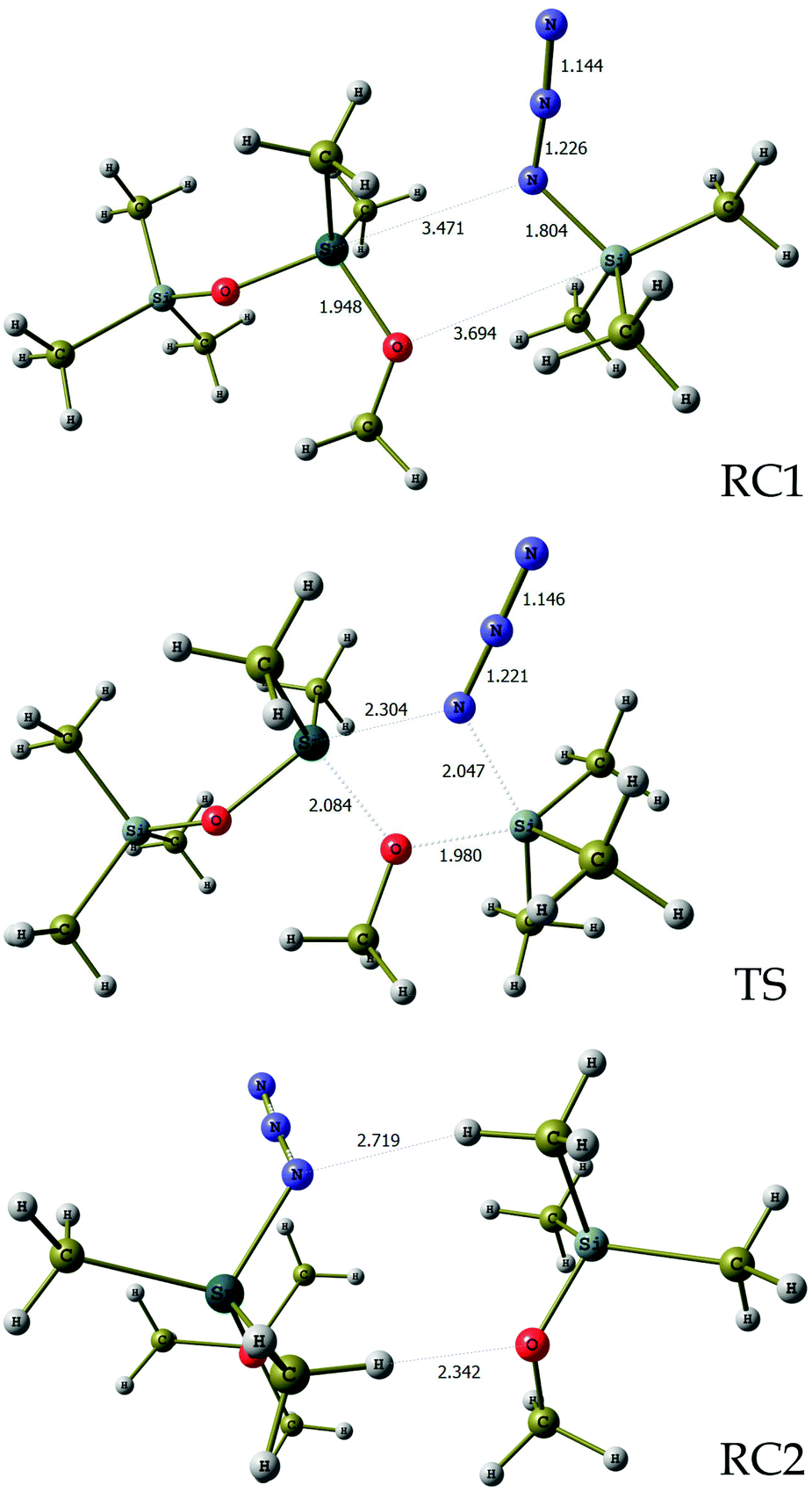 | ||
| Fig. 4 Geometrical details of reaction complexes RC1, RC2 and transition state TS. Bond lengths are in angstroms. Imaginary vibration frequency for TS is 92.0 cm−1. | ||
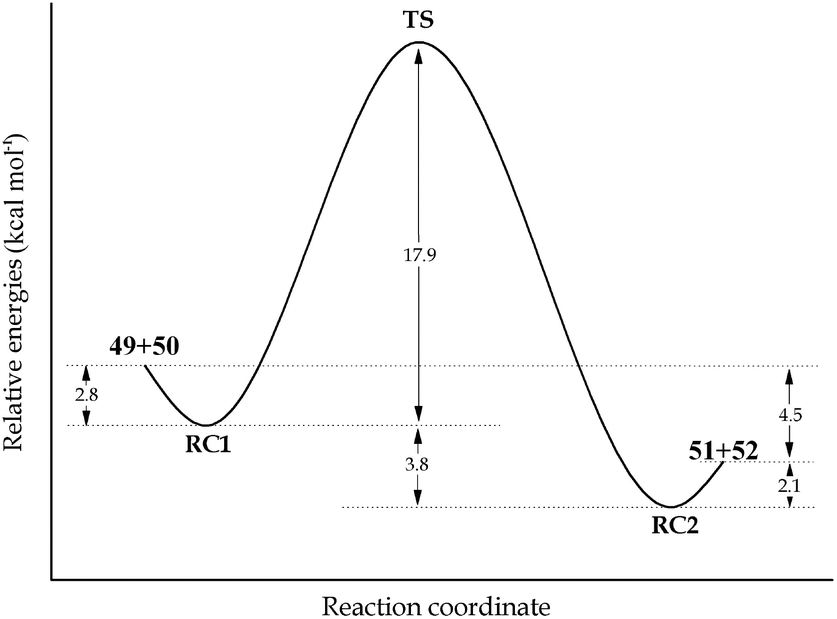 | ||
| Fig. 5 Minimum energy path for the concerted exchange process hold in reaction complex with the modified structure (RC1 and RC2). All values were obtained with zero point energies corrections. | ||
Indeed, calculations demonstrate that this exchange is favored both from kinetic and thermodynamic viewpoints, thus reinforcing our hypothesis.
Conclusions
In conclusion, we have described a straightforward entry to so far unreported α-hydroxy-β-azido tetrazoles, together with a brief examination of their reactivity. Considering the stereospecificity of the opening of the epoxide, this process should allow the preparation of non-racemic molecules starting from readily available enantiopure epoxy nitriles.30 Further work is in progress to extend the scope and applications of this reaction.Acknowledgements
University of Versailles St-Quentin-en-Yvelines and CNRS are acknowledged for funding. This work was partially supported by a public grant (PQ) overseen by the French National Research Agency (ANR) as part of the “Investissements d'Avenir” program n° ANR-11-IDEX-0003-02 and CHARMMMAT ANR-11-LABX-0039. The authors are also grateful for the support by the RSF (Russian Scientific Foundation), project no. 14-13-00103.Notes and references
- J. Roh, K. Vávrová and A. Hrabálek, Eur. J. Org. Chem., 2012, 6101 CrossRef CAS
.
- D. J. Carini, J. V. Duncia, P. E. Aldrich, A. T. Chiu, A. L. Johnson, M. E. Pierce, W. A. Price, J. B. Santella, G. J. Wells, R. R. Wexler, P. C. Wong, S. E. Yoo and P. B. M. W. M. Timmermans, J. Med. Chem., 1991, 33, 1186 Search PubMed
- W. G. Finnegan, R. A. Henry and R. Lofquist, J. Am. Chem. Soc., 1958, 80, 2395 CrossRef
- P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945 CrossRef PubMed
- B. E. Huff and M. A. Starzak, Tetrahedron Lett., 1993, 34, 8011 CrossRef CAS
- B. Das, C. R. Reddy, D. N. Kumar, M. Krishnaiah and R. Narender, Synlett, 2010, 391 CrossRef CAS
- P. Mani, A. K. Sing and S. K. Awasthi, Tetrahedron Lett., 2014, 55, 1879 CrossRef CAS
-
(a) M. Alterman and A. Hallberg, J. Org. Chem., 2000, 65, 7984 CrossRef CAS PubMed
- S. J. Wittenberger and B. G. Donner, J. Org. Chem., 1993, 58, 4139 CrossRef CAS
-
A. Yanagisawa, T. Kuboyama, S. Aratake, K. Hemmi, K. Ueno, M. Suzuki, M. Matsubara, K. Yao, A. Hamaguchi and Y. Tsukumo, Kyowa Hakko Kogyo Co., Ltd, Patent EP, 1988091 A1, 2008 Search PubMed
-
(a) A. Jończyk, M. Fedoryński and M. Makosza, Tetrahedron Lett., 1972, 23, 2395 CrossRef
- An isolated report of uncatalyzed epoxide opening by TMSN3 was found to occur with retention through a SNi mechanism. See: B. Alcaide, C. Blurrun, A. Martinez and J. Plumet, Tetrahedron Lett., 1995, 36, 5417 CrossRef CAS
- X-ray structure of 2 has been deposited on the Cambridge database and has been assigned CCDC number 1036722. See ESI† for details.
- For prior syntheses of these compounds through cycloadditions with nitriles or amides see:
(a) M. Tao, R. Bihovsky and J. C. Kauer, Bioorg. Med. Chem. Lett., 1996, 6, 3009 CrossRef CAS
- G. Cardillo and C. Tomasini, Chem. Soc. Rev., 1996, 117 RSC
- M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, J. Am. Chem. Soc., 1971, 93, 2325 CrossRef CAS PubMed
- D. Guenard, F. Gueritte-Voegelein and P. Potier, Acc. Chem. Res., 1993, 26, 160 CrossRef CAS
- H. Umezawa, T. Aoyagi, H. Suda, M. Hamada and T. Takeuchi, J. Antibiot., 1976, 29, 97 CrossRef CAS PubMed
- T. Okino, H. Matsuda, M. Murakami and K. Yamaguchi, Tetrahedron Lett., 1993, 34, 501 CrossRef CAS
- T. Mimoto, J. Imai, S. Kisanuki, H. Enomoto, N. Attori, K. Akaji and K. Kiso, Chem. Pharm. Bull., 1991, 39, 3088 CrossRef CAS PubMed
-
(a) J. Zabrocki, G. D. Smith, J. B. Dunbar, H. Iijima and G. R. Marshall, J. Am. Chem. Soc., 1988, 110, 5875 CrossRef CAS
- Y. Ittah, Y. Susson, S. Tsaroom and J. Blum, J. Org. Chem., 1978, 43, 4271 CrossRef
- R. Knorr, Chem. Rev., 2004, 104, 3795 CrossRef CAS PubMed
- D. J. Wardrop and J. P. Komenda, Org. Lett., 2012, 14, 1548 CrossRef CAS PubMed
- A. K. Feldman, B. Colasson, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 13444 CrossRef CAS PubMed
- D. Cantillo, B. Gutmann and C. O; Kappe, J. Am. Chem. Soc., 2011, 133, 4465 CrossRef CAS PubMed
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS
-
(a) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS
- A. A. Granovsky, Firefly version 8.0. http://classic.chem.msu.su/gran/firefly/index.html Search PubMed.
- See inter alia:
(a) I. Yamakawa, H. Urabe, Y. Kobayashi and F. Sato, Tetrahedron Lett., 1991, 32, 2045 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures for all compounds, copies of 1H and 13C NMR data for all compounds, X-ray data for 2. CCDC 1036722. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00345d |
| ‡ Due to the high nitrogen content of these compounds, hazards resulting from violent decomposition must be anticipated, though we have never observed such behaviour with these compounds. |
| This journal is © the Partner Organisations 2015 |