
N-heterocyclic carbene-based ruthenium-catalyzed direct amidation of aldehydes with amines†
Cheng
Chen
a,
Min Ha
Kim
b and
Soon Hyeok
Hong
*b
aCollege of Chemistry, Central China Normal University, 152 Luoyu Road, Wuhan 430079, P. R. China
bDepartment of Chemistry, College of Natural Sciences, Seoul National University, Seoul 151-747, Republic of Korea. E-mail: soonhong@snu.ac.kr; Fax: (+82)2-889-1568
First published on 13th January 2015
Abstract
The direct amide synthesis from alcohols and amines is a highly environmentally-benign and atom economic process. In situ generated NHC-based Ru halide catalytic systems are active for the amidation of alcohols with amines. However, these systems show poor activities in the amidation of aldehydes with amines, though aldehydes are the proposed reaction intermediates formed by dehydrogenation of alcohols; imines are obtained as major byproducts in this case. In this study, an improved method was developed by incorporating the idea of using a hemiaminal intermediate as the precatalyst activator, for the direct amide synthesis from aldehydes and amines using a commercially available Ru complex [Ru(p-cymene)Cl2]2, an N-heterocyclic carbene ligand, a base, and pyridine. Using this method, various amides were synthesized from several aldehydes and amines.
Introduction
The amide bond is an important functionality that plays a pivotal role in both organic and biological chemistry.1 Aldehydes are versatile starting materials because of their availability and non-toxicity. The transition-metal-catalyzed direct amidation of aldehydes with amines has been highlighted as an attractive alternative to traditional amide synthesis,2 and several groups have reported Ru-,3 Pd-,4 Cu-,5 Rh-,6 Au-,7 and lanthanide-based8 catalytic systems for this transformation. The general mechanism of this reaction is shown in Scheme 1. An aldehyde reacts with an amine to produce a hemiaminal intermediate, which is further oxidized to the corresponding amide. A major advantage of this approach is that it provides efficient and fast access to amides without the isolation of carboxylic acid intermediates. Therefore, it is especially useful for synthesizing compounds containing functional groups that are either unstable in the presence of carboxylic acids or incompatible with the reaction conditions used in the classical amide formation.2b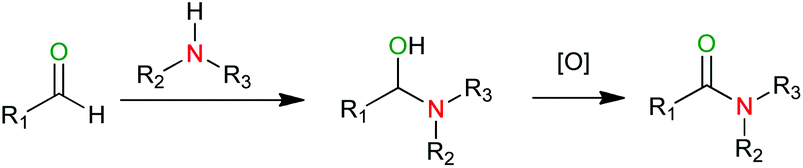 | ||
| Scheme 1 Amide synthesis from aldehydes and amines. | ||
Recently, the transition-metal-catalyzed direct amide synthesis from alcohols and amines has been highlighted as an attractive protocol for constructing the amide bond.9,10 Ru-,3,11 Rh-,12 and Ag-,13 based catalytic systems have been reported for this transformation. In our previous studies, the catalytic systems 1a and 1b showed excellent activities for the amidation of alcohols with amines.11o However, when we tested 1a and 1b for the reaction of benzaldehyde with benzyl amine, the amide was obtained with only 48% and 11% yields, respectively, with the corresponding imine as the major byproduct (Scheme 2).11o Later, our group developed well-defined NHC-based Ru catalysts for the amidation of alcohols with amines that also showed low activity for direct amide synthesis from aldehydes and amines.11m Interestingly, the efficient amidation of aldehydes can be realized by the addition of a catalytic amount of a primary alcohol (Scheme 3). It was demonstrated that the formation of an active Ru hydride catalytic species from precatalyst 1c, by the addition of an alcohol, is essential for the amidation of aldehydes with amines. The effective formation of a Ru-hydride was crucial for amide synthesis from both alcohols and aldehydes.11j
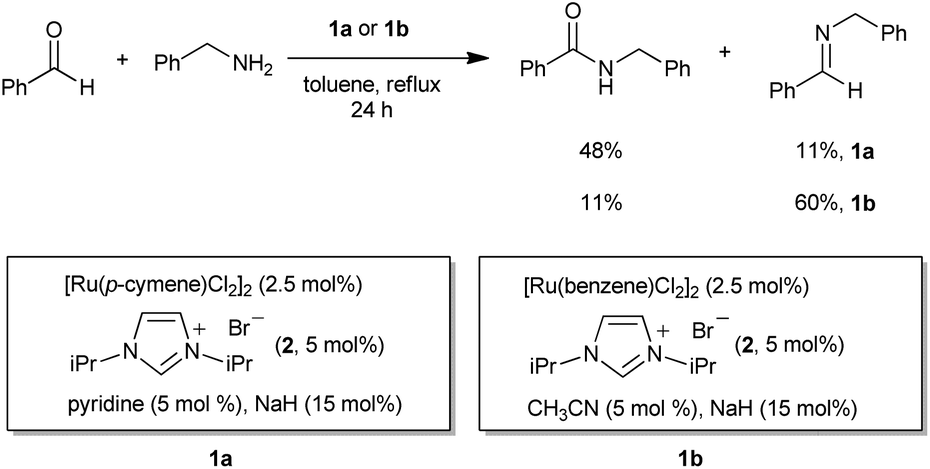 | ||
| Scheme 2 Reaction of benzaldehyde with benzyl amine catalyzed by 1a and 1b. | ||
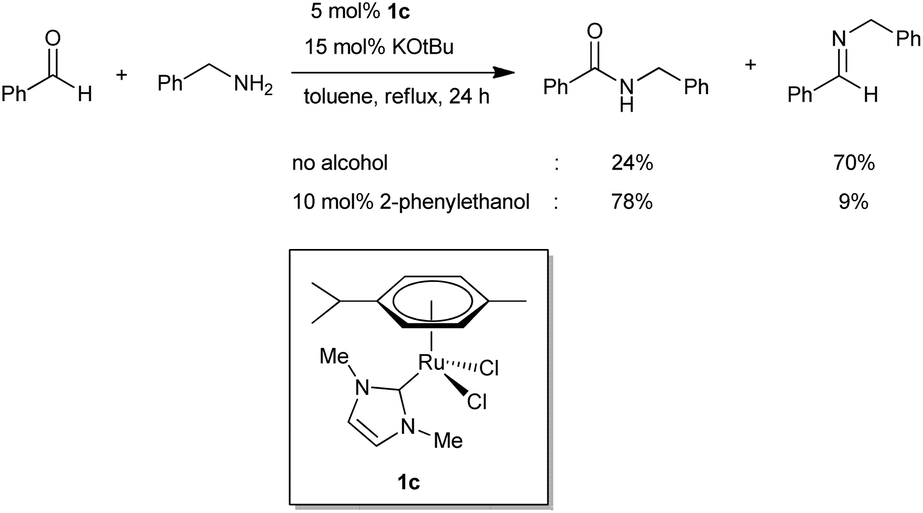 | ||
| Scheme 3 Reaction of benzaldehyde with benzyl amine catalyzed by 1c. | ||
Based on a mechanistic understanding of the essential role of an alcohol in activating a Ru-halide precatalyst, to the Ru-hydride form, we envisioned that a hemiaminal, an important alcoholic intermediate for the formation of amides from aldehydes and amines, could be used to activate the Ru halide precatalyst to generate the key Ru hydride species as shown in Scheme 4. With this hypothesis, herein, we report the direct amide synthesis from aldehydes and amines by modifying the reaction conditions based on 1a.
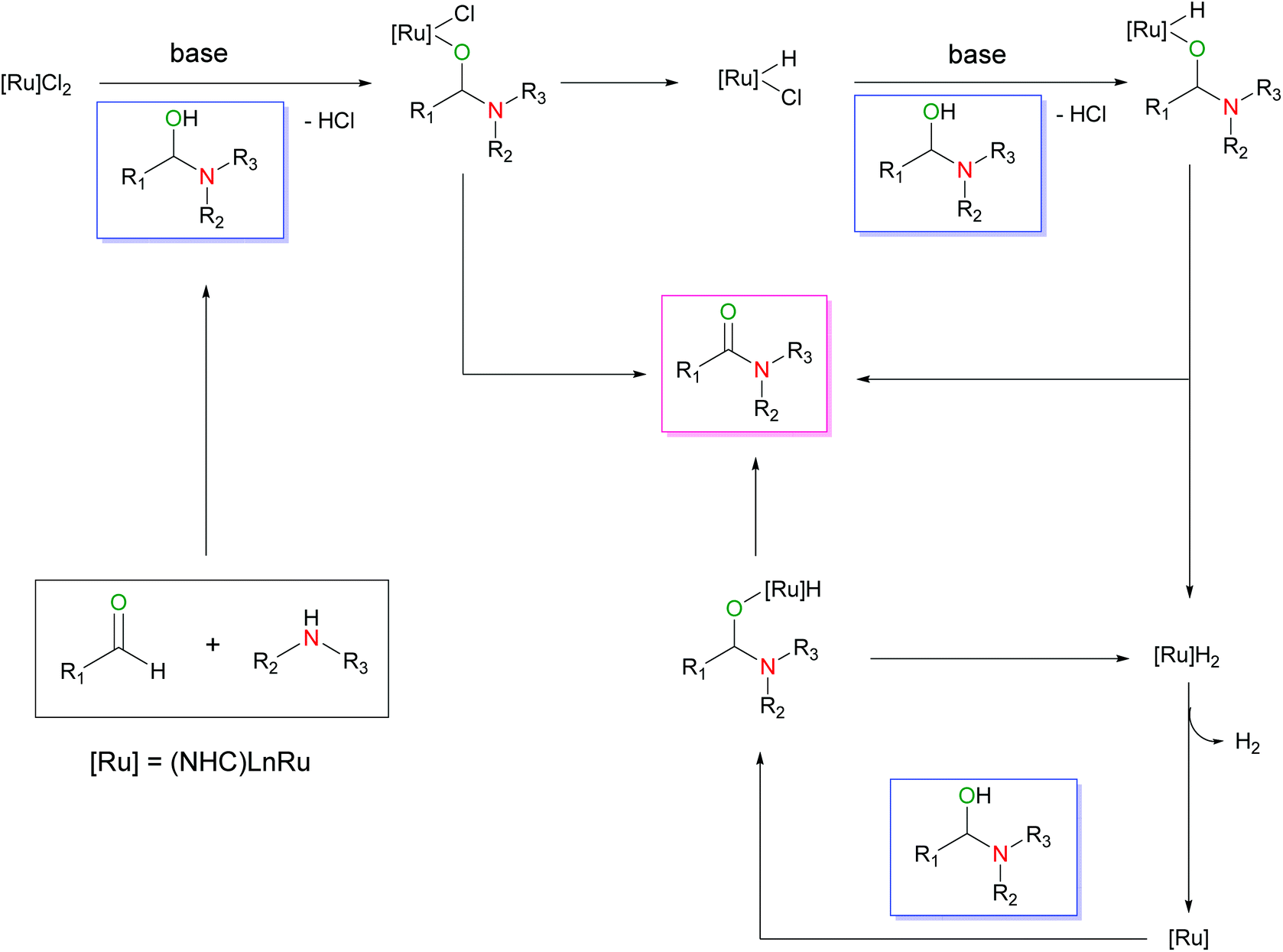 | ||
| Scheme 4 Proposed mechanism of aldehyde amidation with amines using a hemiaminal intermediate as an activator. | ||
Results and discussion
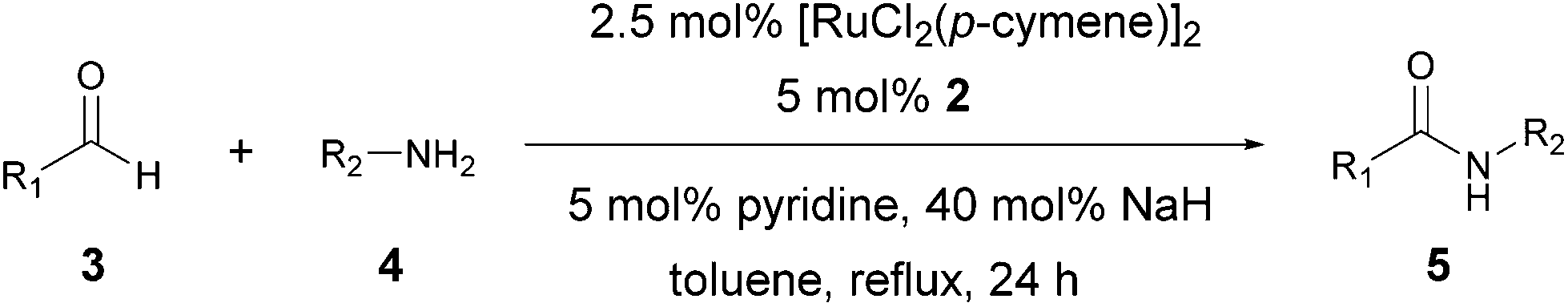
A reaction between benzaldehyde and benzylamine was selected as the model reaction for the optimization of reaction conditions (Table 1). In our previous report, when the catalytic system 1a was used for the reaction, only a limited yield of amide 5a was obtained (48%, entry 1) with the corresponding imine as the major byproduct. Inspired by the reported work that used an alcohol to activate precatalyst 1c, we added a catalytic amount of readily available primary and secondary alcohols. Disappointingly, yields of 5a (entries 2–4) were comparable to or even lower than that of entry 1. Subsequently, we turned our attention to the role of NaH to facilitate the binding of the in situ generated hemiaminal intermediate to the Ru species by forming the corresponding alkoxide. To our delight, a dramatic improvement was achieved on changing the amount of NaH from 15 mol% to 40 mol% (87% for entry 7 vs. 48% for entry 1, entries 1, 5–7). However, further increasing the amount of NaH resulted in reduced yields of the amide (entries 8–10). Milder KOtBu was not as effective as NaH (entries 11–16). The optimized catalytic system, identified as [Ru(p-cymene)Cl2]2 (2.5 mol%), NHC precursor 2 (5 mol%), pyridine (5 mol%), NaH (40 mol%), was used for further study.|
|
|||
|---|---|---|---|
| Entry | Base (mol%) | Additive (10 mol%) | Yieldb (%) |
| a [Ru(p-cymene)Cl2]2 (2.5 mol%), NHC precursor 2 (5 mol%), base, pyridine (5 mol%), additive (10 mol%) in toluene at reflux for 24 h. b GC yields using n-dodecane as an internal standard. | |||
| 1 | NaH (15) | — | 48 |
| 2 | NaH (15) | MeOH | 35 |
| 3 | NaH (15) | iPrOH | 25 |
| 4 | NaH (15) | PhCH2CH2OH | 40 |
| 5 | NaH (20) | — | 51 |
| 6 | NaH (30) | — | 63 |
| 7 | NaH (40) | — | 87 |
| 8 | NaH (50) | — | 80 |
| 9 | NaH (60) | — | 62 |
| 10 | NaH (100) | — | 50 |
| 11 | KOtBu (20) | — | 25 |
| 12 | KOtBu (30) | — | 29 |
| 13 | KOtBu (40) | — | 42 |
| 14 | KOtBu (50) | — | 42 |
| 15 | KOtBu (60) | — | 54 |
| 16 | KOtBu (100) | — | 53 |

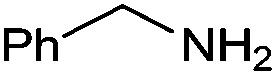
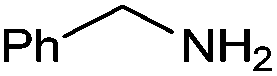
With the optimized reaction conditions in hand, the substrate scope and limitations of this method were investigated. A range of amides was obtained in fair to good yields (Table 2). Aromatic aldehydes are good substrates for this reaction. Benzaldehyde reacted smoothly with sterically nonhindered primary amines (entries 1, 2 and 15–16). A reduced yield was observed for a moderately hindered amine 4c (entry 3). Less basic aniline was converted to the corresponding amide in only 31% yield even at elevated temperature with p-xylene as the solvent (entry 4). Furan was more tolerant than pyridine under our reaction conditions (entries 5 and 6). Electronic effects of substituents on aldehydes were explored using different benzaldehyde derivatives and benzyl amine, and a slightly lower yield was obtained for an electron-deficient substrate (entries 1, 7 and 8). In the case of cyclic secondary amines, piperidine showed good reactivity with both electron-deficient and electron-rich aromatic aldehydes (entries 9–11). Aliphatic aldehydes were also employed, giving the desired amides in 40–50% yields (entries 12–14).
|
|
||||
|---|---|---|---|---|
| Entry | Aldehyde | Amine | Amide | Yieldb (%) |
| a [Ru(p-cymene)Cl2]2 (2.5 mol%), NHC precursor 2 (5 mol%), NaH (40 mol%), pyridine (5 mol%) in toluene at reflux for 24 h. b Isolated yields. c In p-xylene at reflux for 24 h. | ||||
| 1 |

|

|
|
75 |
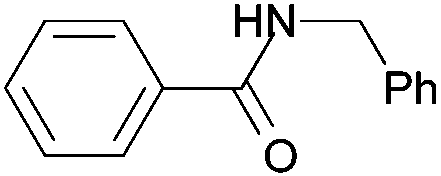
| 3a | 4a | 5a | ||
| 2 |

|

|
|
80 |
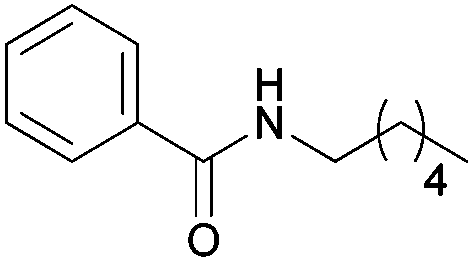
| 4b | 5b | |||
| 3 |
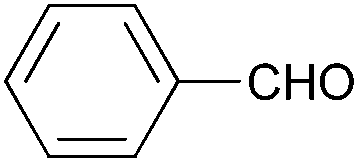
|
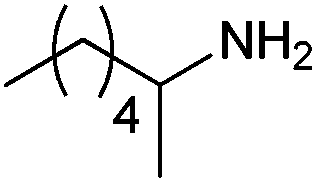
|
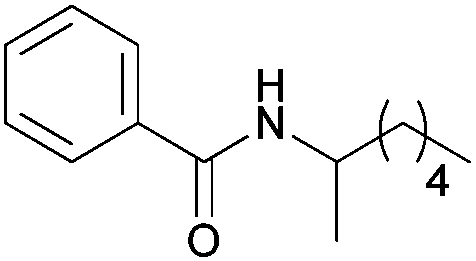
|
57 |
| 4c | 5c | |||
| 4 |

|

|

|
31c |
| 4d | 5d | |||
| 5 |

|

|
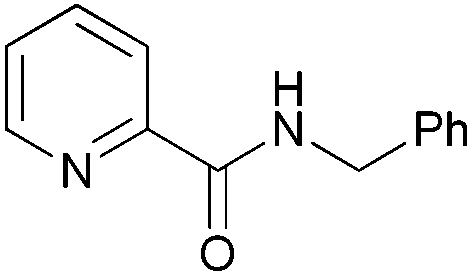
|
38c |
| 3e | 5e | |||
| 6 |
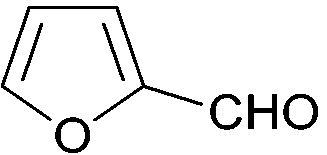
|

|

|
60 |
| 3f | 5f | |||
| 7 |
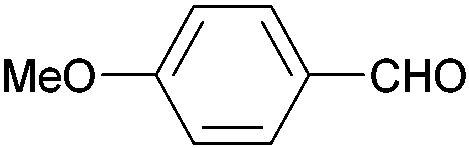
|
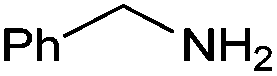
|
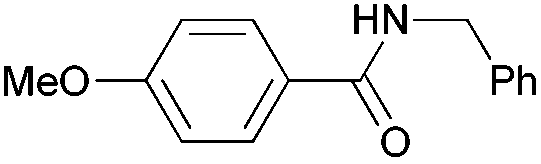
|
81 |
| 3g | 5g | |||
| 8 |

|

|
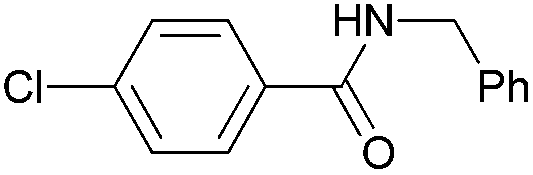
|
64 |
| 3h | 5h | |||
| 9 |

|
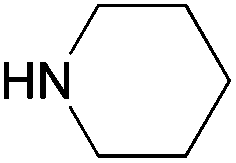
|
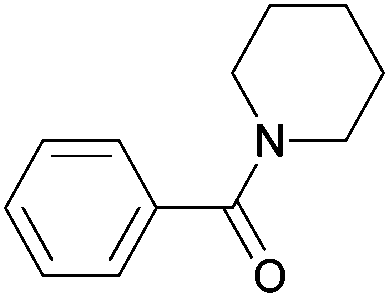
|
67 |
| 4i | 5i | |||
| 10 |

|
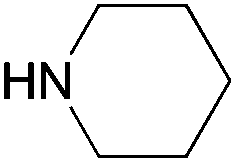
|
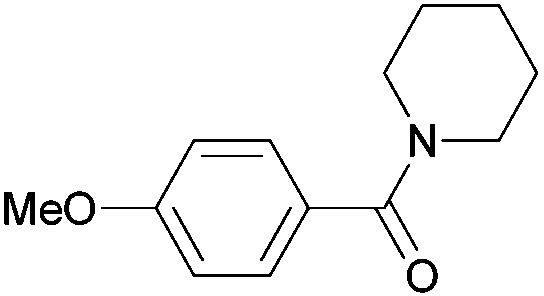
|
77 |
| 5j | ||||
| 11 |

|

|
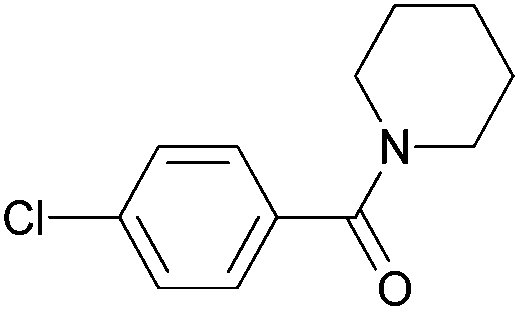
|
70 |
| 5k | ||||
| 12 |

|
|
|
42 |
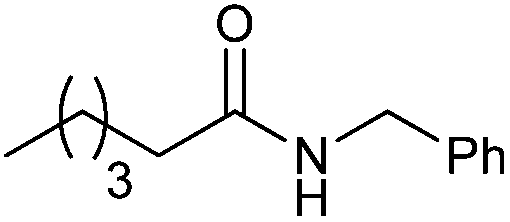
| 3l | 5l | |||
| 13 |

|
|

|
52 |
| 3m | 5m | |||
| 14 |

|

|

|
40 |
| 3n | 5n | |||
| 15 |

|
|
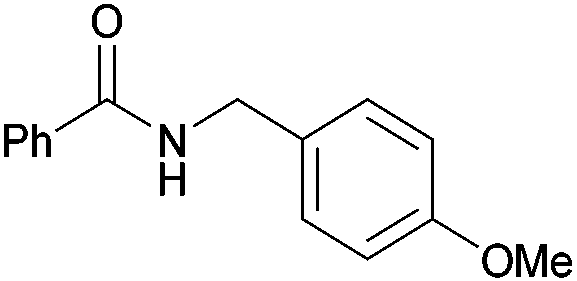
|
75 |
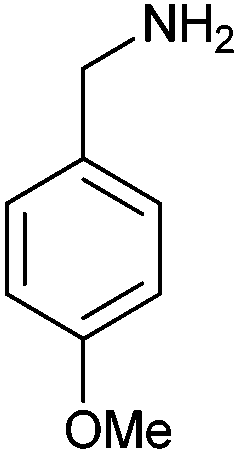
| 4o | 5o | |||
| 16 |

|

|

|
57 |
| 4p | 5p | |||
Conclusions
The Ru-catalyzed direct amide synthesis from aldehydes and amines was performed by incorporating the idea of using a hemiaminal intermediate for generating the active Ru-hydride species. This was achieved by using a higher amount of a strong base compared to the previously reported catalytic conditions, which was conducive for direct amide synthesis from alcohols and amines, but not for the synthesis from aldehydes and amines. A range of amides was synthesized directly from aldehydes and amines in fair to good yields.Experimental section
General information
All reactions were carried out using standard Schlenk techniques or in an argon-filled glove box unless otherwise mentioned. Toluene, THF, and dichloromethane were dried over the PureSolv solvent purification system. NMR spectra were obtained on an Agilent 400-MR DD2 Magnetic Resonance System (400 MHz). Chemical shift values were recorded in ppm relative to tetramethylsilane (TMS) as an internal standard, and coupling constants in hertz (Hz). GC analyses were carried out using the 7980A GC system from Agilent Technologies, equipped with an HP-5 column using dodecane as an internal standard. The high resolution mass spectra were recorded with Waters Q-Tof from Premier Micromass instrument, using the electrospray ionization (ESI) mode. 1,3-Diisopropylimidazolium bromide (2)11o was prepared using the procedure reported in the literature. Most substrates were obtained from commercial suppliers and used as received without further purification.General procedure
[Ru(p-cymene)Cl2]2 (7.7 mg, 0.0125 mmol), 1,3-diisopropylimidazolium bromide (2, 5.8 mg, 0.025 mmol), NaH (4.8 mg, 0.2 mmol), pyridine (2 μL, 0.025 mmol), and toluene (0.6 mL) were added to an oven-dried Schlenk tube placed inside an argon-filled glovebox. The reaction mixture was heated to reflux under an argon atmosphere for 30 min. The flask was taken out of the oil bath before the aldehyde (0.5 mmol) and the amine (0.55 mmol) were added, and the mixture was heated to reflux in an open condition under an argon atmosphere for 24 h. The reaction mixture was cooled down to room temperature and the solvent was removed under reduced pressure. The residue was purified by silica gel flash column chromatography to afford the amide. N-Benzylbenzamide (5a),11oN-hexylbenzamide (5b),14N-benzyl-4-methoxybenzamide (5g),15N-benzyl-4-chlorobenzamide (5h),16 phenyl(piperidin-1-yl)methanone (5i),11o (4-methoxyphenyl)(piperidin-1-yl)methanone (5j),8d (4-chlorophenyl)(piperidin-1-yl)methanone (5k),8dN-benzylhexanamide (5l),11oN-benzyl-3-methylbutanamide (5n),17N-(4-methoxybenzyl)benzamide (5o),18 and N-(4-chlorobenzyl)benzamide (5p)18 were identified by spectral comparison with the literature data.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to afford it as a white solid, mp: 64–65 °C. Isolated yield: 57%. 1H NMR (CDCl3): δ = 7.76–7.74 (m, 2H), 7.51–7.38 (m, 3H), 6.02 (d, 1 H, J = 7.3 Hz), 4.23–4.13 (m, 1H), 1.65–1.10 (m, 11H), 0.87 (t, 3H, J = 7.1 Hz); 13C NMR (CDCl3): 167.0, 135.2, 131.4, 128.7, 127.0, 46.0, 37.2, 31.9, 26.0, 22.8, 21.2, 14.2; HR-MS (ESI): m/z = 220.1697 [MH+], calcd for C14H22NO: 220.1701.
:1) to afford it as a white solid, mp: 161–163 °C. Isolated yield: 31%. 1H NMR (CDCl3): δ = 7.88 (bs, 1H), 7.89–7.77 (m, 2H), 7.69–7.60 (m, 2H), 7.58–7.50 (m, 1H), 7.48–7.42 (m, 2H), 7.40–7.30 (m, 2H), 7.20–7.11 (m, 1H); 13C NMR (CDCl3): 166.0, 138.1, 135.2, 132.0, 129.3, 129.0, 127.2, 124.8, 120.4; HR-MS (ESI): m/z = 198.0925 [MH+], calcd for C13H12NO: 198.0919.
:1) to afford it as a white solid, mp: 83–85 °C. Isolated yield: 38%. 1H NMR (CDCl3): δ 8.52 (d, 1H, J = 5.0 Hz), 8.39 (bs, 1H), 8.24 (d, 1H, J = 8.2 Hz), 7.90–7.75 (m, 1H), 7.50–7.20 (m, 6H), 4.67 (d, 2H, J = 6.0 Hz); 13C NMR (CDCl3): 164.4, 150.0, 148.2, 138.4, 137.5, 128.9, 128.0, 127.6, 126.4, 122.5, 43.6; HR-MS (ESI): m/z = 213.1028 [MH+], calcd for C13H13N2O: 213.1028.
:1) to afford it as a white solid, mp: 111–113 °C. Isolated yield: 60%. 1H NMR (CDCl3): δ 7.40 (s, 1H), 7.38–7.20 (m, 5H), 7.13 (m, 1H), 6.74 (bs, 1H), 6.49 (m, 1H), 4.60 (d, 2H, J = 5.9 Hz); 13C NMR (CDCl3): 158.4, 148.0, 144.1, 138.2, 128.9, 128.0, 127.8, 114.5, 112.3, 43.3; HR-MS (ESI): m/z = 202.0872 [MH+], calcd for C12H12NO2: 202.0868.
:1) to afford it as a white solid, mp: 66 °C. Isolated yield: 52%. 1H NMR (CDCl3): δ 7.40–7.15 (m, 5H), 5.75 (bs, 1H), 4.44 (d, 2H, J = 5.5 Hz), 2.21 (t, 2H, J = 7.8 Hz), 1.65 (m, 2H), 1.40–1.10 (m, 10H), 0.88 (t, 3H, J = 6.9 Hz); 13C NMR (CDCl3): 173.2, 138.6, 128.9, 128.0, 127.7, 43.8, 37.0, 32.0, 29.5, 29.4, 26.0, 22.9, 14.3; HR-MS (ESI): m/z = 248.2020 [MH+], calcd for C16H26NO: 248.2014.
:1) to afford it as a white solid, mp: 64–65 °C. Isolated yield: 57%. 1H NMR (CDCl3): δ = 7.76–7.74 (m, 2H), 7.51–7.38 (m, 3H), 6.02 (d, 1 H, J = 7.3 Hz), 4.23–4.13 (m, 1H), 1.65–1.10 (m, 11H), 0.87 (t, 3H, J = 7.1 Hz); 13C NMR (CDCl3): 167.0, 135.2, 131.4, 128.7, 127.0, 46.0, 37.2, 31.9, 26.0, 22.8, 21.2, 14.2; HR-MS (ESI): m/z = 220.1697 [MH+], calcd for C14H22NO: 220.1701.
:1) to afford it as a white solid, mp: 161–163 °C. Isolated yield: 31%. 1H NMR (CDCl3): δ = 7.88 (bs, 1H), 7.89–7.77 (m, 2H), 7.69–7.60 (m, 2H), 7.58–7.50 (m, 1H), 7.48–7.42 (m, 2H), 7.40–7.30 (m, 2H), 7.20–7.11 (m, 1H); 13C NMR (CDCl3): 166.0, 138.1, 135.2, 132.0, 129.3, 129.0, 127.2, 124.8, 120.4; HR-MS (ESI): m/z = 198.0925 [MH+], calcd for C13H12NO: 198.0919.
:1) to afford it as a white solid, mp: 83–85 °C. Isolated yield: 38%. 1H NMR (CDCl3): δ 8.52 (d, 1H, J = 5.0 Hz), 8.39 (bs, 1H), 8.24 (d, 1H, J = 8.2 Hz), 7.90–7.75 (m, 1H), 7.50–7.20 (m, 6H), 4.67 (d, 2H, J = 6.0 Hz); 13C NMR (CDCl3): 164.4, 150.0, 148.2, 138.4, 137.5, 128.9, 128.0, 127.6, 126.4, 122.5, 43.6; HR-MS (ESI): m/z = 213.1028 [MH+], calcd for C13H13N2O: 213.1028.
:1) to afford it as a white solid, mp: 111–113 °C. Isolated yield: 60%. 1H NMR (CDCl3): δ 7.40 (s, 1H), 7.38–7.20 (m, 5H), 7.13 (m, 1H), 6.74 (bs, 1H), 6.49 (m, 1H), 4.60 (d, 2H, J = 5.9 Hz); 13C NMR (CDCl3): 158.4, 148.0, 144.1, 138.2, 128.9, 128.0, 127.8, 114.5, 112.3, 43.3; HR-MS (ESI): m/z = 202.0872 [MH+], calcd for C12H12NO2: 202.0868.
:1) to afford it as a white solid, mp: 66 °C. Isolated yield: 52%. 1H NMR (CDCl3): δ 7.40–7.15 (m, 5H), 5.75 (bs, 1H), 4.44 (d, 2H, J = 5.5 Hz), 2.21 (t, 2H, J = 7.8 Hz), 1.65 (m, 2H), 1.40–1.10 (m, 10H), 0.88 (t, 3H, J = 6.9 Hz); 13C NMR (CDCl3): 173.2, 138.6, 128.9, 128.0, 127.7, 43.8, 37.0, 32.0, 29.5, 29.4, 26.0, 22.9, 14.3; HR-MS (ESI): m/z = 248.2020 [MH+], calcd for C16H26NO: 248.2014.
Acknowledgements
This work was supported by the National Research Foundation funded by the Korean Government (NRF-2014R1A2A1A11050028; NRF-2014S1A2A2028156; NRF-2014R1A5A1011165, Center for New Directions in Organic Synthesis) and the Natural Science Foundation of Hubei Province funded by Science and Technology Department of Hubei Province (2014CFB280) and the China Postdoctoral Science Foundation (2014M562044).Notes and references
- (a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed; (b) M. A. Ali and T. Punniyamurthy, Adv. Synth. Catal., 2010, 352, 288–292 CrossRef CAS; (c) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC; (d) T. Cupido, J. Tulla-Puche, J. Spengler and F. Albericio, Curr. Opin. Drug Discovery Dev., 2007, 10, 768–783 CAS; (e) J. W. Bode, Curr. Opin. Drug Discovery Dev., 2006, 9, 765–775 CAS; (f) J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243–2266 CrossRef CAS PubMed.
- (a) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC; (b) K. Ekoue-Kovi and C. Wolf, Chem. – Eur. J., 2008, 14, 6302–6315 CrossRef CAS PubMed.
- (a) S. Muthaiah, S. C. Ghosh, J. E. Jee, C. Chen, J. Zhang and S. H. Hong, J. Org. Chem., 2010, 75, 3002–3006 CrossRef CAS PubMed; (b) T. Naota and S. I. Murahashi, Synlett, 1991, 693–694 CrossRef CAS.
- (a) Y. Suto, N. Yamagiwa and Y. Torisawa, Tetrahedron Lett., 2008, 49, 5732–5735 CrossRef CAS PubMed; (b) Y. Tamaru, Y. Yamada and Z. Yoshida, Synthesis, 1983, 474–476 CrossRef CAS PubMed.
- W. J. Yoo and C. J. Li, J. Am. Chem. Soc., 2006, 128, 13064–13065 CrossRef CAS PubMed.
- (a) W. Dai, Y. C. Liu, T. Tong, X. W. Li and F. Luo, Chin. J. Catal., 2014, 35, 1012–1016 CrossRef CAS; (b) R. E. Rodriguez-Lugo, M. Trincado and H. Grützmacher, ChemCatChem, 2013, 5, 1079–1083 CrossRef CAS; (c) J. Chan, K. D. Baucorn and J. A. Murry, J. Am. Chem. Soc., 2007, 129, 14106–14107 CrossRef CAS PubMed; (d) A. Tillack, I. Rudloff and M. Beller, Eur. J. Org. Chem., 2001, 523–528 CrossRef CAS.
- G. L. Li, K. K. Y. Kung and M. K. Wong, Chem. Commum., 2012, 48, 4112–4114 RSC.
- (a) Z. Q. Guo, Q. Liu, X. H. Wei, Y. B. Zhang, H. B. Tong, J. B. Chao, J. P. Guo and D. S. Liu, Organometallics, 2013, 32, 4677–4683 CrossRef CAS; (b) J. A. Thomson and L. L. Schafer, Dalton Trans., 2012, 41, 7897–7904 RSC; (c) J. F. Wang, J. M. Li, F. Xu and Q. Shen, Adv. Synth. Catal., 2009, 351, 1363–1370 CrossRef CAS; (d) C. W. Qian, X. M. Zhang, J. M. Li, F. Xu, Y. Zhang and Q. Shen, Organometallics, 2009, 28, 3856–3862 CrossRef CAS; (e) J. M. Li, F. Xu, Y. Zhang and Q. Shen, J. Org. Chem., 2009, 74, 2575–2577 CrossRef CAS PubMed; (f) S. Seo and T. J. Marks, Org. Lett., 2008, 10, 317–319 CrossRef CAS PubMed.
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed.
- C. Chen and S. H. Hong, Org. Biomol. Chem., 2011, 9, 20–26 CAS.
- (a) B. Kang, Z. Q. Fu and S. H. Hong, J. Am. Chem. Soc., 2013, 135, 11704–11707 CrossRef CAS PubMed; (b) D. Srimani, E. Balaraman, P. Hu, Y. Ben-David and D. Milstein, Adv. Synth. Catal., 2013, 355, 2525–2530 CrossRef CAS; (c) N. Ortega, C. Richter and F. Glorius, Org. Lett., 2013, 15, 1776–1779 CrossRef CAS PubMed; (d) I. S. Makarov, P. Fristrup and R. Madsen, Chem. – Eur. J., 2012, 18, 15683–15692 CrossRef CAS PubMed; (e) Z. Fu, J. Lee, B. Kang and S. H. Hong, Org. Lett., 2012, 14, 6028–6031 CrossRef CAS PubMed; (f) N. D. Schley, G. E. Dobereiner and R. H. Crabtree, Organometallics, 2011, 30, 4174–4179 CrossRef CAS; (g) C. Chen, Y. Zhang and S. H. Hong, J. Org. Chem., 2011, 76, 10005–10010 CrossRef CAS PubMed; (h) J. H. Dam, G. Osztrovszky, L. U. Nordstrøm and R. Madsen, Chem. – Eur. J., 2010, 16, 6820–6827 CrossRef CAS PubMed; (i) A. Nova, D. Balcells, N. D. Schley, G. E. Dobereiner, R. H. Crabtree and O. Eisenstein, Organometallics, 2010, 29, 6548–6558 CrossRef CAS; (j) C. W. Qian, X. M. Zhang, Y. Zhang and Q. Shen, J. Organomet. Chem., 2010, 695, 747–752 CrossRef CAS PubMed; (k) S. C. Ghosh and S. H. Hong, Eur. J. Org. Chem., 2010, 4266–4270 CrossRef CAS; (l) J. Zhang, M. Senthilkumar, S. C. Ghosh and S. H. Hong, Angew. Chem., Int. Ed., 2010, 49, 6391–6395 CrossRef CAS PubMed; (m) Y. Zhang, C. Chen, S. C. Ghosh, Y. X. Li and S. H. Hong, Organometallics, 2010, 29, 1374–1378 CrossRef CAS; (n) A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, Org. Lett., 2009, 11, 2667–2670 CrossRef CAS PubMed; (o) S. C. Ghosh, S. Muthaiah, Y. Zhang, X. Y. Xu and S. H. Hong, Adv. Synth. Catal., 2009, 351, 2643–2649 CrossRef CAS; (p) L. U. Nordstrøm, H. Vogt and R. Madsen, J. Am. Chem. Soc., 2008, 130, 17672–17673 CrossRef PubMed; (q) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed.
- (a) T. Zweifel, J. V. Naubron and H. Grützmacher, Angew. Chem., Int. Ed., 2009, 48, 559–563 CrossRef CAS PubMed; (b) K. Fujita, Y. Takahashi, M. Owaki, K. Yamamoto and R. Yamaguchi, Org. Lett., 2004, 6, 2785–2788 CrossRef CAS PubMed.
- K. Shimizu, K. Ohshima and A. Satsuma, Chem. – Eur. J., 2009, 15, 9977–9980 CrossRef CAS PubMed.
- T. Ohshima, T. Iwasaki, Y. Maegawa, A. Yoshiyama and K. Mashima, J. Am. Chem. Soc., 2008, 130, 2944–2945 CrossRef CAS PubMed.
- J. R. Martinelli, T. P. Clark, D. A. Watson, R. H. Munday and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 8460–8463 CrossRef CAS PubMed.
- X. X. Shen, Q. Liu, R. G. Xing and B. Zhou, Catal. Lett., 2008, 126, 361–366 CrossRef CAS PubMed.
- P. Starkov and T. D. Sheppard, Org. Biomol. Chem., 2011, 9, 1320–1323 CAS.
- C. Chen and S. H. Hong, Org. Lett., 2012, 14, 2992–2995 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of compound 5. See DOI: 10.1039/c4qo00319e |
| This journal is © the Partner Organisations 2015 |