
Acenaphthoimidazolium chloride-enabled nickel-catalyzed amination of bulky aryl tosylates†‡
Jian
Jiang
,
Haibo
Zhu
,
Yajing
Shen
and
Tao
Tu
*
Department of Chemistry, Fudan University, 220 Handan Road, 200433, Shanghai, China. E-mail: taotu@fudan.edu.cn
First published on 15th September 2014
Abstract
A robust catalyst derived from NiCl2·(DME) and acenaphthoimidazolium chloride exhibited high activity towards the challenging amination of bulky and heterocyclic aryl tosylates with secondary amines under mild reaction conditions at 2 mol% catalyst loading within 3 hours. Further, a wide range of primary amines were also successfully mono-aminated by our newly developed protocol.
1. Introduction
Carbon–nitrogen bonds exist in numerous bioactive, conductive and fluorescent compounds, which have broad applications in pharmaceutical and materials sciences.1 It is generally accepted that transition metal-catalyzed amination reactions are recognized as one of the most powerful and practical approaches to construct C–N bonds,2,3 as represented by the Buchwald–Hartwig amination of aryl halides4 and triflates.5 Along with these significant contributions and ever-growing developments, however, more and more attention is currently focused on the amination of inactive phenolic derivatives in the presence of inexpensive metal catalysts.6 Among the family of phenolic derivatives, aryl tosylates are very attractive and promising partners in amination reactions due to their ready accessibilities, pronounced stabilities and low toxicities.7 On the other hand, the low activities of aryl tosylates renders their amination elusive, probably because of the difficulty in the selective oxidative addition of the Caryl–O bond instead of the Ssulfonyl–O bond.8 According to these previous findings, the use of electron-rich and bulky σ-donor ligands may facilitate the oxidative addition of inert electrophiles. Therefore, alkylphosphines with electron-rich and bulky σ-donor functional groups9 were considered as efficient ligands in the cross-coupling reactions of inactive electrophiles, including aryl tosylates. However, such amination reactions using inexpensive nickel catalysts always suffered from low yields, even when high catalyst loadings, elevated reaction temperatures and extended reaction times were employed.10 Therefore, one of the particular emphases in organic synthesis is put on addressing the limitations of aryl tosylates as substrates in amination reactions.As we know, N-heterocyclic carbenes (NHCs) have been well-accepted as ligands with strong σ-electron-donor and weak π-acceptor properties,11 and many NHC-enabled coupling reactions have been developed so far.13 For example, the first nickel-catalyzed amination of aryl tosylates was initiated by Yang and co-workers by using trans-arylbis(triphenylphosphine)nickel(II) as a precatalyst and 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride (IPr·HCl, an N-heterocyclic carbene precursor) as an additional ligand.12 Control experiments indicated that a catalytic system devoid of NHCs did not work well for the above amination reactions. By switching the precatalyst to an air-sensitive styrene-coordinated Ni(0) complex derived from IPr·HCl, Nicasio and co-workers realized the amination of aryl tosylates without any reductant addition.14 Despite these advances in nickel-catalyzed amination for C–N bond formation in recent years, known methodologies still suffer from: (1) the air-sensitivity of the precatalysts; (2) relatively high catalyst loading (at least 5 mol% catalyst was required); and (3) limited substrate scope. Electron-rich, bulky and heterocyclic aryl tosylates were unfavorable for the transformation. As a consequence, we conceived that the amination of aryl tosylates could be realized efficiently with a broad functional-group tolerance in the presence of a nickel and π-extended NHC catalyst.
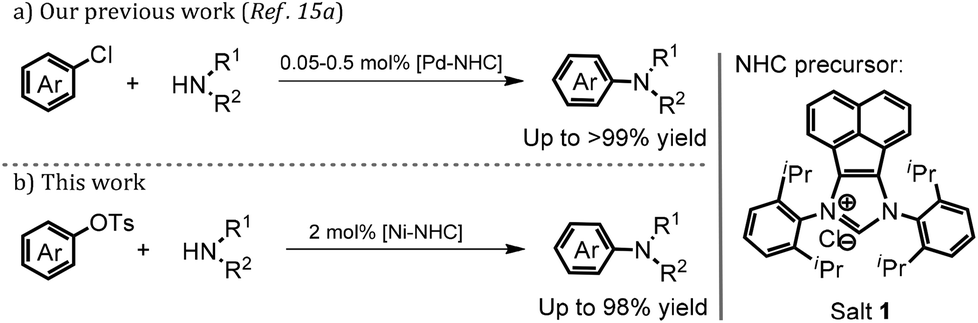
Recently our group accomplished the amination of inactive (hetero)aryl chlorides with diverse secondary and primary amines by using a robust Pd–NHC complex derived from imidazolium salt 1 as the precatalyst. The reactions proceeded smoothly to offer the desired amination products in good to excellent yields. Particularly, the sterically-hindered and heterocyclic substrates were well-tolerated even in the presence of 0.5 mol% catalyst loading (Scheme 1).15 Inspired by what is mentioned above and our continuous interest in the synthesis, characterization and applications of novel NHC-based transition metal complexes in catalysis and soft matter,15–17 we would like to further explore the feasibility of the amination of bulky (hetero)aryl tosylates with diverse amines by using the nickel and imidazolium salt 1 directly.
 | ||
| Scheme 1 Representative examples of imidazolium salt-enabled amination reactions with different transition metals. | ||
2. Results and discussion
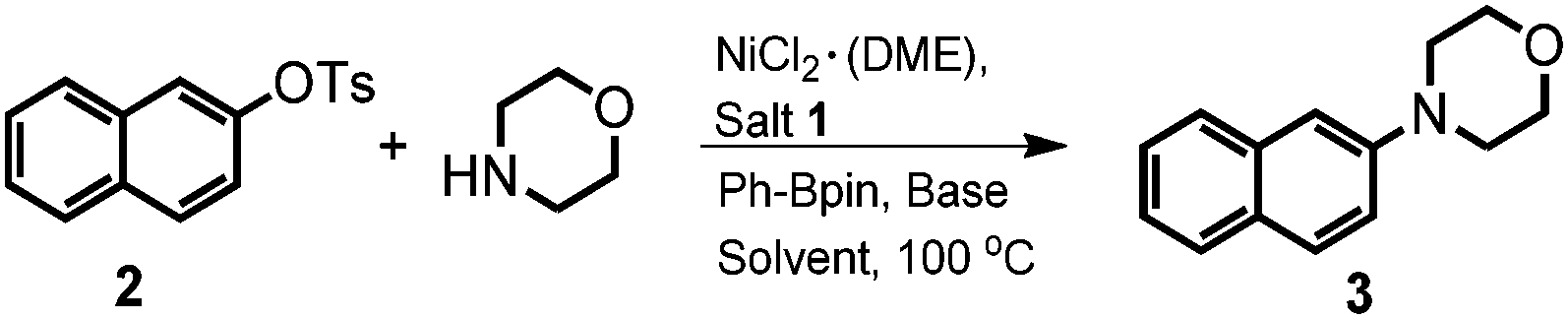
Initially, the model reaction of bulky β-naphthyltosylate 2 and morpholine was carried out with 2 mol% NiCl2·(DME), 4 mol% imidazolium salt 1, 2.0 equiv. phenylboronic acid pinacol ester (Ph-Bpin), and 3.0 equiv. t-BuOK in dioxane at 100 °C. As expected, it was pleasing to find that the amination product 3 was afforded in a 68% isolated yield. Other inorganic salts were also evaluated accordingly. From the screening results, it seemed that t-BuONa was the best choice (Table 1, entry 2). When a relatively weak base, such as K3PO4, was employed, only an 18% isolated yield was obtained (Table 1, entry 4). The control experiments with bases suggested the base was still critical for the transformation.15b More details on the evaluation of bases were illustrated in the ESI.‡ Decreases in the catalyst loading to 1 mol% or the base amount to 1.6 equiv. were unfavorable for the amination, and the desired amine 3 was produced at yields of 82% and 51%, respectively (Table 1, entries 5 and 6, respectively). Other result-affecting factors such as solvent and temperature were also optimized. The yields of the reactions decreased when toluene (78% yield) and m-xylene (86% yield) were used as the solvent (Table 1, entries 7 and 8, respectively). Ether solvents like THF and DME afforded worse outcomes than that of dioxane (72% and 59% vs. 92%, respectively, Table 1, entries 9 and 10 vs. 2, respectively). In order to improve the amination efficiency, solvent mixtures were also involved in the optimization, however, no better results were found. When dioxane and toluene were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, an excellent yield was found, which was similar to that obtained from dioxane (91%, Table 1, entry 11). Furthermore, other classical NHC precursors, such as IPr·HCl and IMes·HCl were also involved the optimization, however, only inferior yields were observed (51% and 13%, respectively, Table 1, entries 12 and 13, respectively), which was also consistent with previous reports.12
:1 ratio, an excellent yield was found, which was similar to that obtained from dioxane (91%, Table 1, entry 11). Furthermore, other classical NHC precursors, such as IPr·HCl and IMes·HCl were also involved the optimization, however, only inferior yields were observed (51% and 13%, respectively, Table 1, entries 12 and 13, respectively), which was also consistent with previous reports.12
|
|
|||
|---|---|---|---|
| Entry | Solvent | Base (equiv.) | Yieldb (%) |
|
a 0.5 mmol reaction scale, with 2 mol% NiCl2·(DME) and imidazolium salt 1 at 100 °C for 3 hours.
b Isolated yield.
c With 1 mol% NiCl2·(DME) and imidazolium salt 1.
d Dioxane and toluene in 1:1 ratio.
e IPr·HCl was used.
f IMes·HCl was used.
|
|||
| 1 | Dioxane | t-BuOK (3.0) | 68 |
| 2 | Dioxane | t-BuONa (3.0) | 92 |
| 3 | Dioxane | t-BuOLi (3.0) | 41 |
| 4 | Dioxane | K3PO4 (3.0) | 18 |
| 5 | Dioxane | t-BuONa (3.0) | 82c |
| 6 | Dioxane | t-BuONa (1.6) | 51 |
| 7 | Toluene | t-BuONa (3.0) | 78 |
| 8 | m-Xylene | t-BuONa (3.0) | 86 |
| 9 | THF | t-BuONa (3.0) | 72 |
| 10 | DME | t-BuONa (3.0) | 59 |
| 11 | Dioxane–toluene | t-BuONa (3.0) | 91d |
| 12 | Dioxane | t-BuONa (3.0) | 51e |
| 13 | Dioxane | t-BuONa (3.0) | 13f |
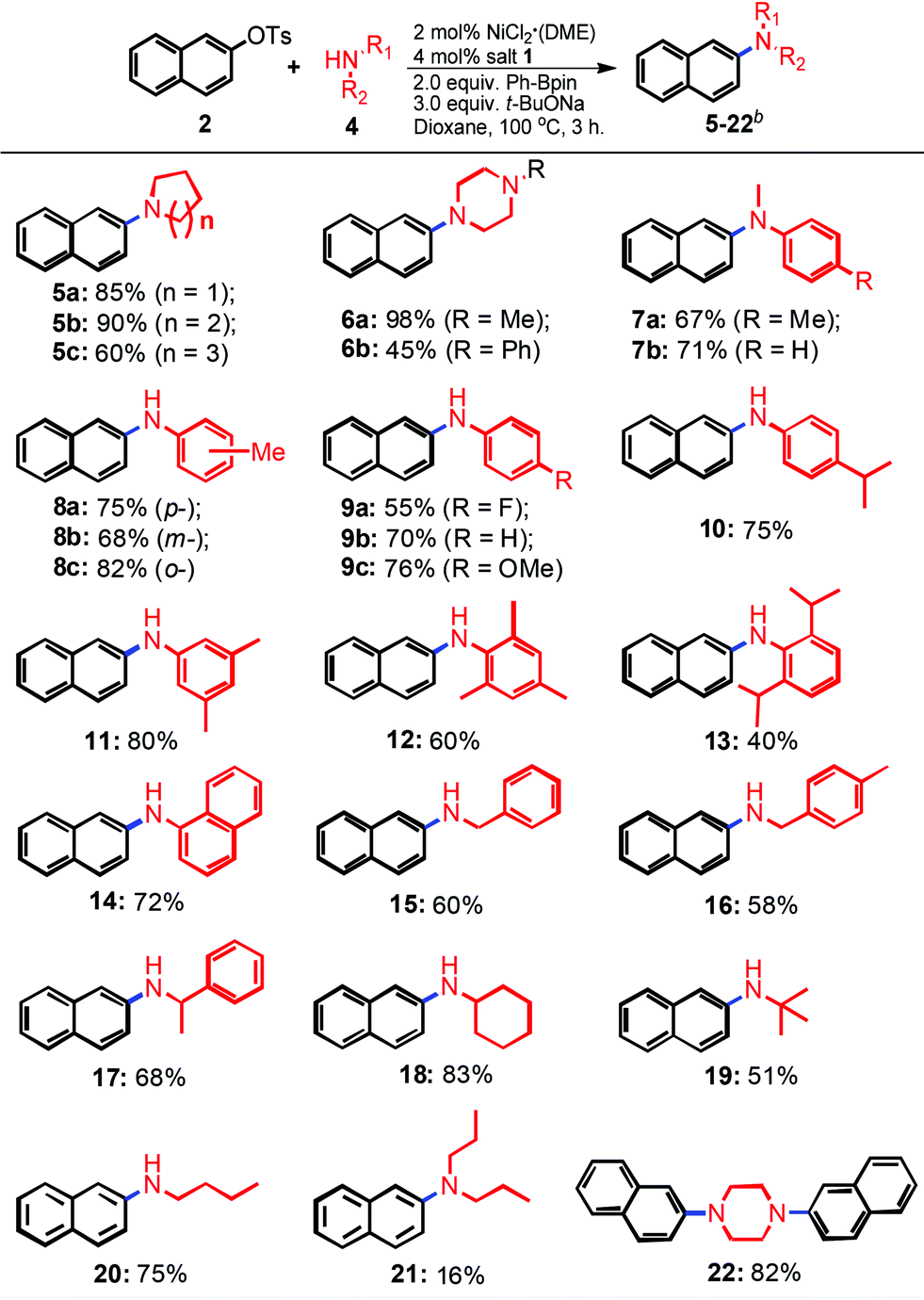
With the optimized reaction conditions in hand (2 mol% NiCl2·(DME), 4 mol% imidazolium salt 1, 2.0 equiv. Ph-Bpin, and 3.0 equiv. t-BuONa in dioxane at 100 °C), the scope of the reaction was then explored. As shown in Table 2, various primary and secondary amines were used to evaluate the protocol efficiency. In the case of secondary amines, whenever cyclic and linear amines were employed, the reaction worked well for the formation of the products 5–7 in moderate to excellent yields. The ring size of the cyclic amine slightly influenced the amination process, piperidine revealed the best outcome compared to pyrrolidine and azepane (5bvs.5a and 5c, 90% vs. 85% and 60%, respectively). The amination using N-methylpiperazine as the substrate gave an almost quantitative yield (98%, 6a), however, only a 45% isolated yield was observed with the use of its phenyl analogue (6b) as the reaction partner. Acyclic N-methylanilines with various electronic properties were also compatible for the amination, and similar results were obtained (67 and 71%, 7a and b, respectively). To our delight, when a series of primary amines was utilized, the corresponding mono-amination products 8–20 were found in moderate to good isolated yields. The steric hindrances and electronic effects of primary amines made a slight impact on the transformations. The results compiled in Table 2 revealed that electron-rich groups were more favorable than electron-poor ones (75% and 76% vs. 55%, 8a and 9cvs.9a, respectively). The reactions of o-substituted substrates gave rise to better outcomes than those of their p- and m-analogues (82% vs. 75% and 68%, 8cvs.8a and 8b, respectively). Interestingly, more sterically-hindered substrates, such as 2,4,6-trimethylaniline, 2,6-diisopropyl-aniline and α-naphthylamine, were also investigated, still resulting in products 12, 13 and 14 in 60%, 40% and 72% yields, respectively. Due to the potential tendency of β-hydride elimination of the resulting transition metal amide complexes, acyclic aliphatic primary amines were considered as challenging substrates for the amination reactions.18 Pleasingly, our results indicated that they are also suitable substrates for the transformation (15–20). Again, steric hindrance was well-tolerated, aliphatic primary amines with α-substituents were more favorable for the transformation, and better yields were afforded (83% and 68% vs. 60%, 18 and 17vs.15, respectively). As we expected, a 51% isolated yield was obtained when low-bowling point t-butyl amine was used as the substrate (19). When n-butyl amine, which has a higher boiling point, was involved, the yield could be increased to 75% (20). However, only a 16% yield was obtained when n-alkylated secondary amine ((n-Pr)2NH) was used (21), which is absolutely unlike the cyclic cases (5 and 6). Diamination reactions were observed when 1,4-piperazine was involved. Diarylation product (22) was afforded in an 82% yield under the standard conditions.
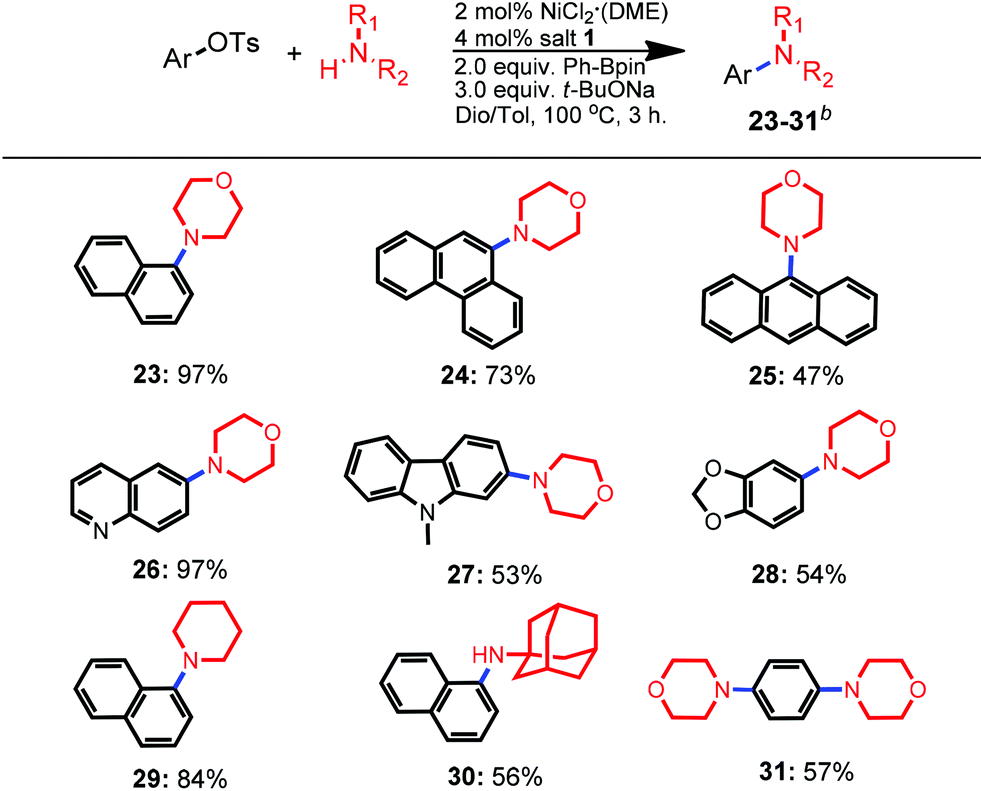
Considering that our newly developed protocol was applicable for sterically-bulky substrates for both amination partners, morpholine was selected to verify its feasibility for diverse sterically-bulky and heterocyclic tosylates (Table 3). By altering the solvent to the co-solvent mixture of dioxane and toluene, all selected bulky aryl tosylates were well-tolerated in the amination process. For instance, the amination of bulky α-naphthyl tosylate resulted in a higher yield than that of its β-analogue (97% vs. 92%, 23vs.3 in Table 1, respectively, entry 2). Tosylate derived from phenanthren-9-ol was more favorable for the amination than that of the anthryl analogue, producing the corresponding products 24 and 25 in the yields of 73% and 47%, respectively. To our delight, bulky heterocyclic tosylates containing a quinolone core, carbazole and benzodioxole rings were all tolerated and gave moderate to excellent yields (up to 97%, 26–28). When other amines such as piperidine and adamantan-1-amine were utilized in the amination of bulky α-naphthyltosylate, moderate to good yields were also obtained (84% and 56% yields, 29 and 30, respectively). Furthermore, double amination of 1,4-phenylene-bis-tosylates with morpholine also proceeded well and provided a 57% isolated yield (31).
3. Conclusions
In summary, we have discovered a robust catalyst derived from NiCl2·(DME) and acenaphthoimidazolium chloride 1, which exhibited high activity towards the amination of bulky and heterocyclic aryl tosylates with various secondary amines under mild reaction conditions at 2 mol% catalyst loading within 3 hours. Furthermore, a wide range of primary amines were also successfully mono-aminated. The protocol presented herein may provide a practical approach for the synthesis of disubstituted aryl amines. The reaction results mentioned above confirm that the NHC ligand derived from acenaphthoimidazolium chloride 1 does enhance the catalytic efficiency of the resulted Ni–NHC complex, enabling the challenging amination of inactive bulky and heterocyclic aryl tosylates. More detailed studies on the applications of acenaphthoimidazolium chloride in other challenging coupling reactions are under exploration in our laboratory, and the results will be reported in due course.Acknowledgements
Financial support from the National Natural Science Foundation of China (no. 91127041 and 21172045), the Changjiang Scholars and Innovative Research Team in University (IRT1117), the Shanghai Shuguang Program and Pujiang Programs, and Department of Chemistry Fudan University is gratefully acknowledged.Notes and references
- (a) M. Negwer, Organic-Chemical Drugs and their Synonyms (An International Survey), Akademie Verlag GmbH, Berlin, 7th edn, 1994 Search PubMed; (b) A. R. Muci and S. L. Buchwald, Practical Palladium Catalysts for C-N and C-O Bond Formation, in Top. Curr. Chem., ed. N. Miyaura, Springer, Berlin, 2001, vol. 219, p. 131 Search PubMed.
- (a) H. Bader, A. R. Hansen and F. J. Mccart, J. Org. Chem., 1966, 31, 2319 CrossRef CAS; (b) J. E. Shaw, D. C. Kunerth and S. B. Swanson, J. Org. Chem., 1976, 41, 732 CrossRef CAS; (c) N. Chéron, R. Ramozzi, L. E. Kaïm, L. Grimaud and P. Fleurat-Lessard, J. Org. Chem., 2012, 77, 1361 CrossRef PubMed; (d) G. W. Gribble, P. D. Lord, J. Skotnicki, S. E. Dietz, J. T. Eaton and J. L. Johnson, J. Am. Chem. Soc., 1974, 96, 7812 CrossRef CAS; (e) P. Marchini, G. Liso and A. Reho, J. Org. Chem., 1975, 40, 3453 CrossRef CAS.
- (a) J. P. Wolfe, S. Wagaw, J.-F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805 CrossRef CAS; (b) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046 CrossRef CAS. In the case of C–N bonds formation, see: (c) M. H. Ali and S. L. Buchwald, J. Org. Chem., 2001, 66, 2560 CrossRef CAS PubMed; (d) D. W. Old, J. P. Wolfe and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 9722 CrossRef CAS; (e) J. P. Wolfe and S. L. Buchwald, Tetrahedron Lett., 1997, 38, 6359 CrossRef CAS.
- (a) J. Louie and J. F. Hartwig, Tetrahedron Lett., 1995, 36, 3609 CrossRef CAS; (b) A. S. Guram, R. A. Rennels and S. L. Buchwald, Angew. Chem., Int. Ed. Engl., 1995, 34, 1348 CrossRef CAS; (c) J. P. Wolfe and S. L. Buchwald, J. Org. Chem., 1996, 61, 1133 CrossRef CAS.
- (a) J. P. Wolfe and S. L. Buchwald, J. Org. Chem., 1997, 62, 1264 CrossRef CAS; (b) S. Cacchi, P. G. Ciattini, E. Morera and G. Ortar, Tetrahedron Lett., 1986, 27, 3931 CrossRef CAS; (c) J. Louie, M. S. Driver, B. C. Hamann and J. F. Hartwig, J. Org. Chem., 1997, 62, 1268 CrossRef CAS.
- (a) T. Mesganaw, A. L. Silberstein, S. D. Ramgren, N. F. Fine Nathel, X. Hong, P. Liu and N. K. Garg, Chem. Sci., 2011, 2, 1766 RSC; (b) T. Mesganaw and N. K. Garg, Org. Process Res. Dev., 2013, 17, 29 CrossRef CAS.
- (a) T. A. Jensen, X. Liang, D. Tanner and N. Skjaerbaek, J. Org. Chem., 2004, 69, 4936 CrossRef CAS PubMed; (b) J. Ahman and S. L. Buchwald, Tetrahedron Lett., 1997, 38, 6363 CrossRef CAS.
- (a) A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 8704 CrossRef CAS PubMed; (b) T. Ogata and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 13848 CrossRef CAS PubMed.
- B. P. Fors, D. A. Watson, M. R. Biscoe and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 13552 CrossRef CAS PubMed.
- L. Ackermann, R. Sandmann and W. Song, Org. Lett., 2011, 13, 1784 CrossRef CAS PubMed.
- (a) C. A. Wheaton and M. Stradiotto, Can. J. Chem., 2013, 91, 755 CrossRef CAS; (b) A. Chartoire, A. Boreux, A. R. Martin and S. P. Nolan, RSC Adv., 2013, 3, 3840 RSC.
- C.-Y. Gao and L.-M. Yang, J. Org. Chem., 2008, 73, 1624 CrossRef CAS PubMed.
- L. Hie, S. D. Ramgren, T. Mesganaw and N. K. Garg, Org. Lett., 2012, 14, 4182 CrossRef CAS PubMed.
- M. J. Iglesias, J. F. Blandez, M. R. Fructos, A. Prieto, E. Alvarez, T. R. Belderrain and M. C. Nicasio, Organometallics, 2012, 31, 6312 CrossRef CAS.
- (a) T. Tu, W. Fang and J. Jiang, Chem. Commun., 2011, 47, 12358 RSC; (b) T. Tu, Z. Sun, W. Fang, M. Xu and Y. Zhou, Org. Lett., 2012, 14, 4250 CrossRef CAS PubMed; (c) W. Fang, J. Jiang, Y. Xu, J. Zhou and T. Tu, Tetrahedron, 2013, 69, 673 CrossRef CAS PubMed; (d) W. Fang, Q. Deng, M. Xu and T. Tu, Org. Lett., 2013, 15, 3678 CrossRef CAS PubMed; (e) Z. Liu, N. Dong, M. Xu, Z. Sun and T. Tu, J. Org. Chem., 2013, 78, 7436 CrossRef CAS PubMed.
- (a) T. Tu, J. Malineni and K. H. Dötz, Adv. Synth. Catal., 2008, 350, 1791 CrossRef CAS; (b) T. Tu, J. Malineni, X. Bao and K. H. Dötz, Adv. Synth. Catal., 2009, 351, 1029 CrossRef CAS; (c) T. Tu, H. Mao, C. Herbert, M. Xu and K. H. Dötz, Chem. Commun., 2010, 46, 7796 RSC; (d) T. Tu, X. Feng, Z. Wang and X. Liu, Dalton Trans., 2010, 39, 10598 RSC; (e) Z. Wang, X. Feng, W. Fang and T. Tu, Synlett, 2011, 951 CAS; (f) M. Xu, X. Li, Z. Sun and T. Tu, Chem. Commun., 2013, 49, 11539 RSC.
- (a) T. Tu, W. Assenmacher, H. Peterlik, R. Weisbarth, M. Nieger and K. H. Dötz, Angew. Chem., Int. Ed., 2007, 46, 6368 CrossRef CAS PubMed; (b) T. Tu, W. Assenmacher, H. Peterlik, G. Schnakenburg and K. H. Dötz, Angew. Chem., Int. Ed., 2008, 47, 7127 CrossRef CAS PubMed; (c) T. Tu, X. Bao, W. Assenmacher, H. Peterlik, J. Daniels and K. H. Dötz, Chem. – Eur. J., 2009, 15, 1853 CrossRef CAS PubMed; (d) T. Tu, W. Fang, X. Bao, X. Li and K. H. Dötz, Angew. Chem., Int. Ed., 2011, 50, 6601 CrossRef CAS PubMed; (e) T. Tu, W. Fang and Z. Sun, Adv. Mater., 2013, 25, 5304 CrossRef CAS PubMed; (f) W. Fang, Z. Sun and T. Tu, J. Phys. Chem. C, 2013, 117, 25185 CrossRef CAS; (g) W. Fang, X. Liu, Z. Lu and T. Tu, Chem. Commun., 2014, 50, 3313 RSC; (h) W. Fang, C. Liu, Z. Lu, Z. Sun and T. Tu, Chem. Commun., 2014, 50, 3313 RSC.
- L. Ackermann, J. H. Spatz, C. J. Gschrei, R. Born and A. Althammer, Angew. Chem., Int. Ed., 2006, 45, 7627 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Li-Xin Dai on the occasion of his 90th birthday. |
| ‡ Electronic supplementary information (ESI) available: General experimental methods and the characterization data of compounds. See DOI: 10.1039/c4qo00233d |
| This journal is © the Partner Organisations 2014 |