Selective monofluorination of active methylene compounds: the important role of ZnCl2 in inhibiting overfluorination†
Fanzhou
Jiang
,
Yanchuan
Zhao
and
Jinbo
Hu
*
Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Ling-Ling Road, Shanghai 200032, China. E-mail: jinbohu@sioc.ac.cn; Fax: +86 21-64166128
First published on 28th April 2014
Abstract
In the presence of zinc chloride (ZnCl2), active methylene compounds can be selectively monofluorinated at room temperature, and the undesired overfluorination (gem-difluorination) can be significantly diminished. The mechanistic study shows that ZnCl2 plays an important role in selective monofluorination through its interaction with a Brønsted base to control the deprotonation of the starting methylene compounds over the corresponding monofluorinated products.
Owing to their structural diversity and peculiar reactivity, active methylene compounds have found many applications in organic synthesis such as the Michael addition and aldol and Knoevenagel reactions.1 On the other hand, compounds bearing a monofluoromethylene group (–CHF–) are important candidates in isostere-based drug design,2 and these compounds can be usually prepared by an electrophilic C–H monofluorination of active methylene compounds. However, this reaction often suffers from overfluorination,1b,3 resulting in a mixture of mono- and difluorinated products. In many cases, the similar polarity of the mono- and difluorinated products leads to difficulty in their separation (purification).3b The general solution to this problem is that equal equivalents of a base and fluorination reagent (based on that of the active methylene compounds) are incorporated at low temperatures, but the efficiency of fluorination is often unsatisfactory.4 Other methods3a,5 include the conversion of active methylenes to the corresponding enol ethers, and the latter species are selectively monofluorinated; however, this protocol is not amenable to methylene-containing sulfones. Fluorine-containing sulfones proved to be of great value in organic synthesis,6 especially in selective fluoroalkylation reactions.7 To the best of our knowledge, however, practical methods for the preparation of monofluorinated sulfones via selective fluorination of non-fluorinated sulfones are rare.8 In this communication, we wish to report a ZnCl2-mediated selective monofluorination of active methylene compounds using an electrophilic fluorination reagent at room temperature.
At the onset of our investigation, we focused on the monofluorination of bis(phenylsulfonyl)methane (1a), since selective monofluorination of 1a can lead to a highly useful monofluoromethylation reagent 2a.3b,9 Previously, efforts have been made by the Hu9a and Shibata9b groups through electrophilic fluorination to synthesize 2a; however, in both cases the formation of the difluorinated by-product 3a was observed.9a,10 After a brief screening, we found that the employment of a bulky base was beneficial to minimize difluorination. Therefore, we chose sodium tert-butoxide (1.0 equiv.) as a base to react with 1a in THF at room temperature, and then 2.0 equiv. of SelectFluor was added. It turned out that a good yield of the monofluorinated 2a was obtained, with only a small amount of the difluorinated by-product 3a being observed by 19F NMR (Table 1, entry 1). However, when we increased the equivalents of the base to improve the conversion of the starting material 1a, the tendency of overfluorination unfortunately increased (Table 1, entries 2–4).
|
|
||||
|---|---|---|---|---|
| Entry | t BuONa (equiv.)a | ZnCl2 (equiv.)a | 2a (%)b | 2a (%)b |
| a The equivalent is relative to that of 1a. b Determined by 19F NMR analysis of the crude reaction mixture using PhCF3 as an internal standard. | ||||
| 1 | 1.0 | — | 73 | 2 |
| 2 | 1.2 | — | 71 | 3 |
| 3 | 1.5 | — | 61 | 19 |
| 4 | 1.8 | — | 60 | 22 |
| 5 | 1.8 | 1.0 | 79 | 3 |
| 6 | 1.8 | 1.5 | 57 | — |
| 7 | 1.8 | 1.8 | 45 | — |
| 8 | 2.2 | 1.5 | 91 | 7 |
| 9 | 2.2 | 1.8 | 79 | — |
| 10 | 2.5 | 2.2 | 94 | 3 |
| 11 | 2.5 | 2.5 | 91 | 1 |
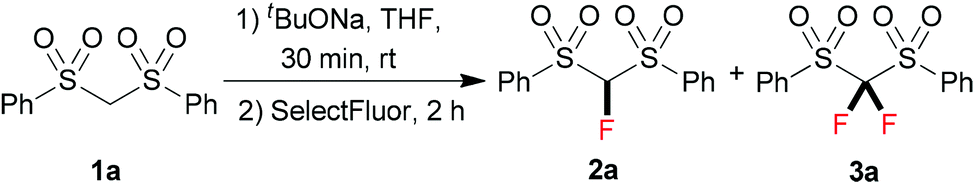
We presumed that the van der Waals radius of fluorine resembles that of hydrogen, leading to the difficulty in preventing overfluorination. Inspired by the monoiodination of methylene-containing sulfones reported by Imamoto,11 we supposed that it could be possible to prepare the corresponding organozinc reagent12 after deprotonation of active methylene compounds. Given the fact that the basicity of the organozinc reagent is relatively weak, over-deprotonation should be significantly inhibited. To test our hypothesis, 1.0 equiv. of anhydrous ZnCl2 was added to the reaction mixture. We were satisfied to find that the monofluorinated product 2a was formed in 79% yield, with the difluorinated by-product 3a being formed in 3% yield (Table 1, entry 5). After further tuning the molar ratio between tBuONa and ZnCl2 (entries 6–11), we found that an optimal yield (91%) of 2a was obtained when 2.5 equiv. of tBuONa and 2.5 equiv. of ZnCl2 were employed (Table 1, entry 11).
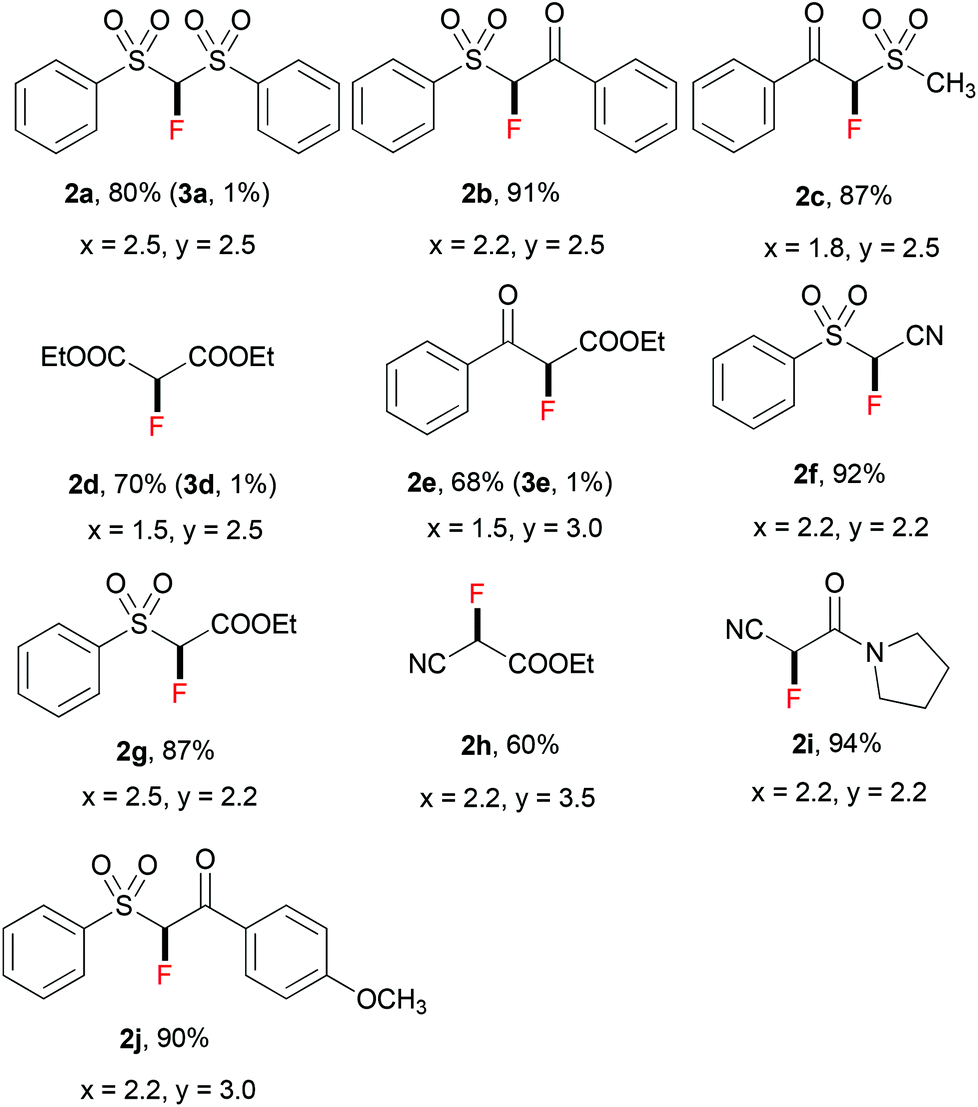
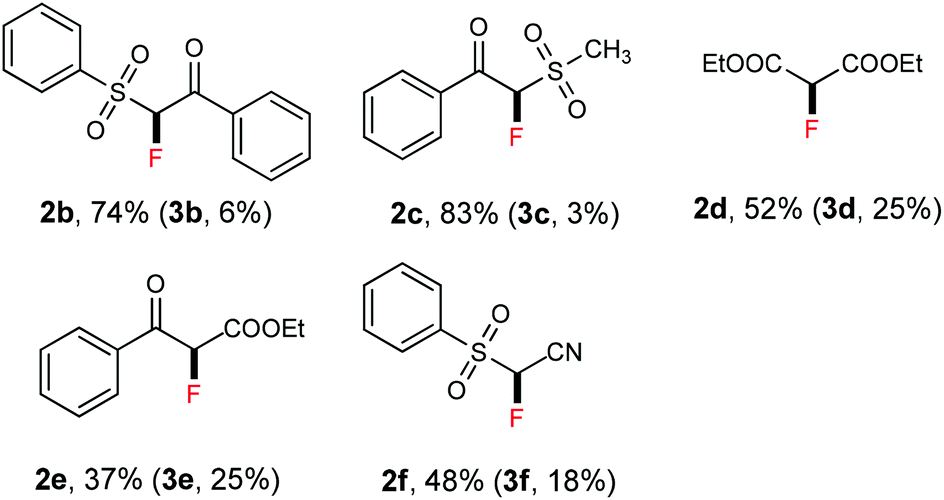
Thereafter, we continued to examine the substrate scope of this new protocol of ZnCl2-mediated monofluorination. Initially, we applied the optimized reaction conditions (as described in Table 1, entry 11) to other active methylene compounds such as 1b, 1c, and 1d; however, we quickly realized that the reaction is very sensitive to different substrates, and further optimizations are needed for different active methylene substrates.13 It is obvious that a significant change in the C–H acidity of 1 caused by different substituents results in a great influence in the degree of deprotonation of methylene compounds, and therefore, the alteration of the basicity of the reaction mixture could be crucial to the selectivity of deprotonation. We found that when substituents R1 and R2 on the methylene compound 1 are more electron-withdrawing, a decrease of the amount of tBuONa and/or an increase of that of ZnCl2 could improve the selectivity of monofluorination.14 It should be mentioned that, by tuning the ratio between tBuONa and ZnCl2, selective monofluorination was accomplished with a variety of structurally diverse active methylene compounds (Table 2). This ZnCl2-mediated new method was also found to be amenable to other active methylene compounds without sulfonyl groups (see 2d, 2e, 2h and 2i). To verify the important role of ZnCl2 in selective monofluorination of active methylene compounds, we chose some methylene-containing substrates (1b–1f) to be fluorinated in the absence of ZnCl2: 1.0 equiv. sodium tert-butoxide was added to react with 1 in THF at room temperature, and then 2.0 equiv. of SelectFluor was added. In all cases, difluorinated by-products were observed by 19F NMR analysis. Moreover, for substrates 1d, 1e and 1f, these reactions suffered from overfluorination severely (Table 3).
|
|
|---|
| a In all cases, the equivalents of tBuONa (x) and ZnCl2 (y) are based on that of the corresponding starting material 1. b The yield of 2 refers to the isolated yield. c Unless otherwise mentioned, difluorinated products 3 were not observed by 19F NMR; when 3 was formed, the yield of 3 was determined by 19F NMR. d The range of pKa values of C–H bonds among these methylene compounds 1 is between 11.4 and 17.2 in DMSO. e EWG = electron-withdrawing group. |
|
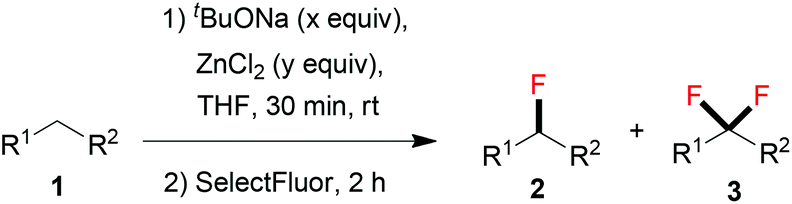
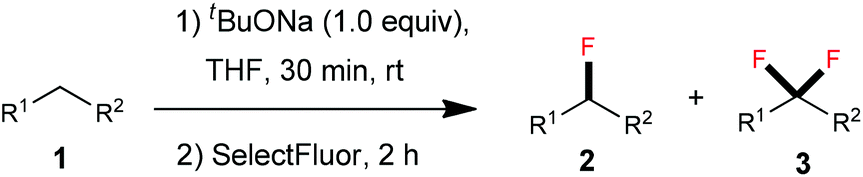
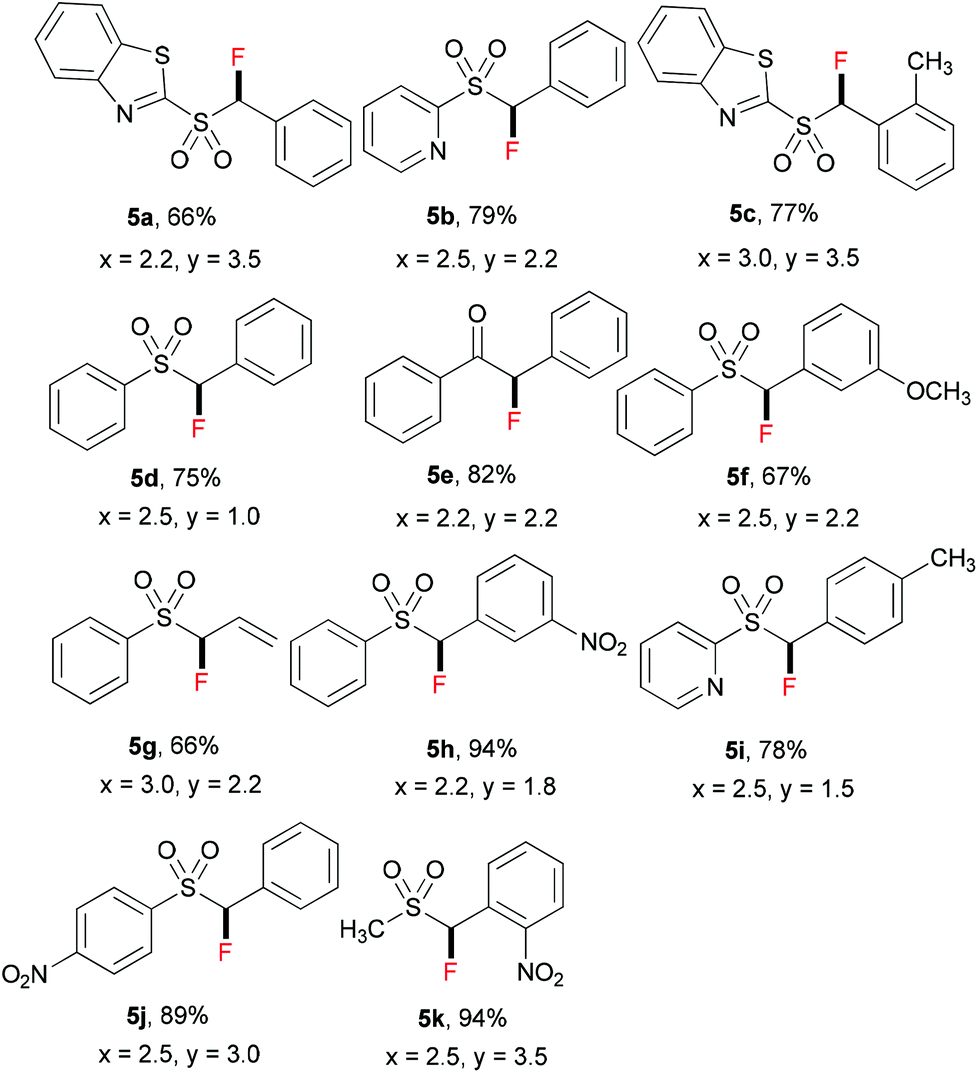
Inspired by the achievement of selective monofluorination with active methylene compounds 1, we extended the substrate scope of this reaction to relatively inactive methylene compounds 4 (see Table 4). Fluorinated benzothiazolyl sulfones have been reported as general synthons for the fluoro-Julia–Kocienski olefinations, and the resulting monofluoroalkene moiety can be used as a non-hydrolyzable mimetic of amide in the peptidomimetic unit of protease inhibitors.15 We envisaged that, for relatively inactive methylene compounds with the less acidic –CH2– unit, a stronger base than tBuONa should be used. Therefore, lithium hexamethyldisilazide (LiHMDS) was chosen as a base for the reaction, and the amounts of base and ZnCl2 were further optimized for each substrate 4 (for details of optimization, see ESI†). As shown in Table 4, a variety of relatively inactive methylene compounds can be selectively fluorinated to give the corresponding products 5; in all cases, no difluorinated by-products were observed.
|
|
|---|
| a In all cases, the equivalents of LiHMDS (x) and ZnCl2 (y) are based on that of starting materials 4. b The yield of 5 refers to the isolated yield. c In all cases, the difluorinated by-products are not observed. d The range of pKa values of C–H bonds among these methylene compounds is between 17.7 and 23.4 in DMSO. |
|
To gain more insight into this ZnCl2-mediated selective monofluorination, we carried out some experiments. First of all, we compared the tendency of deprotonation and fluorination between methylene compounds and the corresponding monofluorinated ones using (PhSO2)2CH2 and (PhSO2)2CHF as model compounds. Equal equivalents of (PhSO2)2CH2 and (PhSO2)2CHF were mixed in THF-d8 in the presence of a substoichiometric amount of LiHMDS, and the mixture was characterized by 1H NMR (1A, 1B, for details, see ESI†). It was found that (PhSO2)2CH2 was more easily deprotonated than (PhSO2)2CHF, which was consistent with the theoretical calculation reported by Prakash and Olah.16 Secondly, we mixed equal equivalents of (PhSO2)2CH2 and (PhSO2)2CHF in THF-d8 in the presence of an excess amount of NaH, and thereafter, the reaction was quenched with a substoichiometric amount of N-fluorobisphenylsulfonimide (NFSI) (2A–2F, for details, see ESI†). It was found that (PhSO2)2CHF showed higher tendency for fluorination. According to these experimental results, the key to acquire the most monofluorinated product and inhibit the difluorination is to maximize the deprotonation of the methylene compound (PhSO2)2CH2 and minimize the further deprotonation of the monofluorinated intermediate (PhSO2)2CHF. Thirdly, we added (PhSO2)2CH2 to THF-d8 under the optimized conditions (2.5 equiv. of tBuONa and 2.5 equiv. of ZnCl2) (3A, 3B, for details, see ESI†). Unexpectedly, (PhSO2)2CH2 was almost intact (i.e., the deprotonation hardly occurred), which was indicated by 1H NMR.
As reported previously,17 mixing tBuONa with ZnCl2 could generate the tert-butoxyzincate complex Na2[Zn(OtBu)3]2, whose basicity was too weak to deprotonate (PhSO2)2CH2. Indeed, we observed a new species by 1H NMR spectroscopy when tBuONa and ZnCl2 were mixed (Fig. 1, 4B).
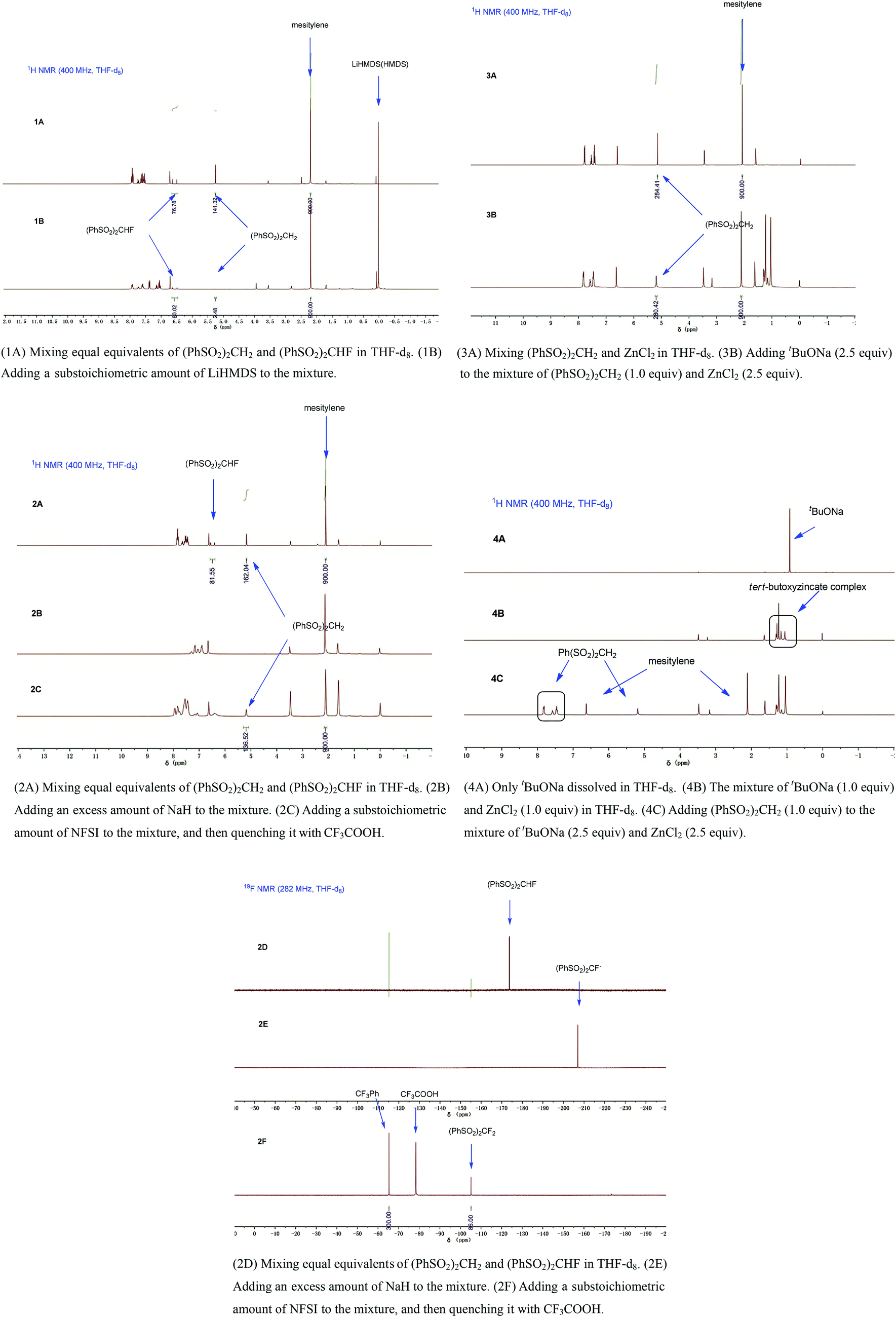 | ||
| Fig. 1 Mechanistic study through 1H NMR experiments. | ||
Based on the aforementioned results, the selective monofluorination reaction is explained as depicted in Scheme 1. A trace amount of “(PhSO2)2CHZn” is generated in an equilibrium when (PhSO2)2CH2 is added to the mixture of tBuONa and ZnCl2 (through in situ formation of Na2[Zn(OtBu)3]2). Subsequently, SelectFluor is added to this mixture. “(PhSO2)2CHZn” is therefore fluorinated, which shifts the equilibrium to the side of “(PhSO2)2CHZn” (Scheme 1). Furthermore, for the reactions with different methylene substrates, tBuONa and ZnCl2 should exist in proper ratios to prevent over-deprotonation and therefore overfluorination. It is obvious that the deprotonation of (PhSO2)2CHF by Na2[Zn(OtBu)3]2 is almost negligible. In the cases of relatively inactive methylene compounds (as shown in Table 4), we also presume that the reaction proceeds through a similar pathway in the presence of LiHMDS and ZnCl2.18
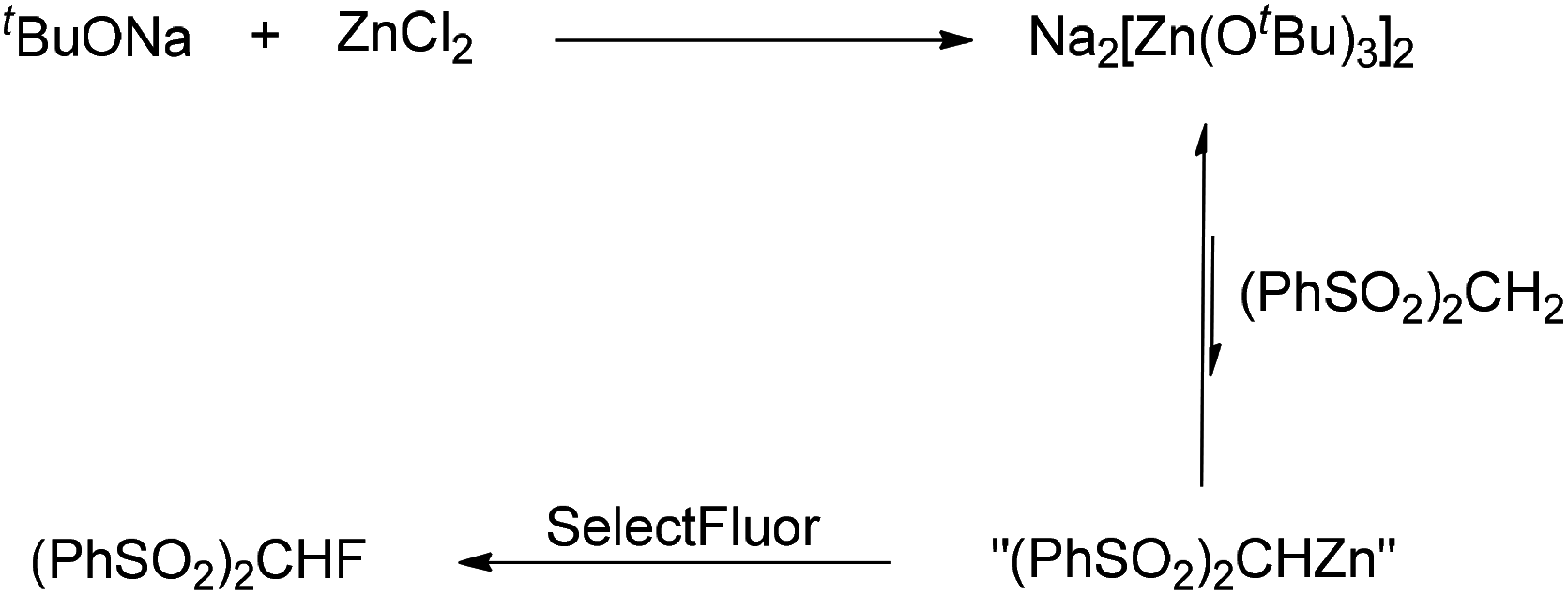 | ||
| Scheme 1 The equilibrium in the process of fluorination. | ||
Conclusions
In conclusion, we have developed a highly selective and efficient monofluorination of methylene compounds with inhibition of undesired difluorination products. ZnCl2 plays an important role in selective monofluorination through its interaction with a Brønsted base to control the deprotonation of the starting methylene compounds rather than the monofluorinated products. Given the mild reaction conditions and the ready availability of the reagents, this method has promising important applications in the synthesis of monofluoromethylene-containing compounds.Acknowledgements
Support of our work by the National Basic Research Program of China (2012CB215500 and 2012CB821600), the National Natural Science Foundation of China (21372246 and 20825209), the Shanghai QMX program (13QH1402400), and the Chinese Academy of Sciences is gratefully acknowledged.Notes and references
- (a) A. Massa, Curr. Org. Chem., 2012, 16, 2159 CrossRef CAS; (b) C. E. Inman, R. E. Oesterling and E. A. Tyczkowski, J. Am. Chem. Soc., 1958, 80, 6533 CrossRef CAS; (c) R. Connor, C. L. Fleming and T. Clayton, J. Am. Chem. Soc., 1936, 58, 1386 CrossRef CAS.
- (a) K. L. Kirk, J. Fluorine Chem., 2006, 127, 1013 CrossRef CAS; (b) G. K. S. Prakash, X. Zhao, S. Chacko, F. Wang, H. Vaghoo and G. A. Olah, Beilstein J. Org. Chem., 2008, 4 Search PubMed; (c) B. E. Smart, J. Fluorine Chem., 2001, 109, 3 CrossRef CAS.
- (a) R. D. Chambers and J. Hutchinson, J. Fluorine Chem., 1998, 92, 45 CrossRef CAS; (b) C. Ni, L. Zhang and J. Hu, J. Org. Chem., 2009, 74, 3767 CrossRef CAS PubMed; (c) H. Loghmani-Khouzani, M. R. Poorheravi, M. M. M. Sadeghi, L. Caggiano and R. F. W. Jackson, Tetrahedron, 2008, 64, 7419 CrossRef CAS; (d) W. M. Peng and J. M. Shreeve, Tetrahedron Lett., 2005, 46, 4905 CrossRef CAS; (e) K. Radwan-Olszewska, F. Palacios and P. Kafarski, J. Org. Chem., 2011, 76, 1170 CrossRef CAS PubMed; (f) F. A. Davis, W. Han and C. K. Murphy, J. Org. Chem., 1995, 60, 4730 CrossRef CAS.
- (a) G. Verniest, E. Van Hende, R. Surmont and N. De Kimpe, Org. Lett., 2006, 8, 4767 CrossRef CAS PubMed; (b) M. S. Marma, L. A. Khawli, V. Harutunian, B. A. Kashemirov and C. E. McKenna, J. Fluorine Chem., 2005, 126, 1467 CrossRef CAS.
- Selective examples are as follows: (a) F. A. Davis, W. Han and C. K. Murphy, J. Org. Chem., 1995, 60, 4730 CrossRef CAS; (b) E. Fuglseth, T. H. K. Thvedt, M. F. Moll and B. H. Hoff, Tetrahedron, 2008, 64, 7318 CrossRef CAS; (c) T. Kitamura, S. Kuriki, M. H. Morshed and Y. Hori, Org. Lett., 2011, 13, 2392 CrossRef CAS PubMed; (d) G. Stavber, M. Zupan and S. Stavber, Tetrahedron Lett., 2007, 48, 2671 CrossRef CAS; (e) J. C. Xiao and J. M. Shreeve, J. Fluorine Chem., 2005, 126, 473 CrossRef.
- (a) M. Nielsen, C. B. Jacobsen, N. Holub, M. W. Paixao and K. A. Jorgensen, Angew. Chem., Int. Ed., 2010, 49, 2668 CrossRef CAS PubMed; (b) D. C. Meadows and J. Gervay-Hague, Med. Res. Rev., 2006, 26, 793 CrossRef CAS PubMed; (c) A. El-Awa, M. N. Noshi, X. M. du Jourdin and P. L. Fuchs, Chem. Rev., 2009, 109, 2315 CrossRef CAS PubMed.
- (a) G. K. S. Prakash and J. Hu, Acc. Chem. Res., 2007, 40, 921 CrossRef CAS PubMed. Selective examples are as follows: (b) Y. Zhao, B. Gao and J. Hu, J. Am. Chem. Soc., 2012, 134, 5790 CrossRef CAS PubMed; (c) X. Shen, W. Zhang, C. Ni, Y. Gu and J. Hu, J. Am. Chem. Soc., 2012, 134, 16999 CrossRef CAS PubMed; (d) X. Shen, W. Zhang, L. Zhang, T. Luo, X. Wan, Y. Gu and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 6966 CrossRef CAS PubMed; (e) C. Ni, J. Liu, L. Zhang and J. Hu, Angew. Chem., Int. Ed., 2007, 46, 786 CrossRef CAS PubMed; (f) Y. Zhao, W. Huang, L. Zhu and J. Hu, Org. Lett., 2010, 12, 1444 CrossRef CAS PubMed.
- I. Vints, J. Gatenyo and S. Rozen, J. Fluorine Chem., 2013, 146, 66 CrossRef CAS.
- Selective examples are as follows: (a) C. Ni, Y. Li and J. Hu, J. Org. Chem., 2006, 71, 6829 CrossRef CAS PubMed; (b) T. Fukuzumi, N. Shibata, M. Sugiura, H. Yasui, S. Nakamura and T. Toru, Angew. Chem., Int. Ed., 2006, 45, 4973 CrossRef CAS PubMed; (c) S. Mizuta, N. Shibata, Y. Goto, T. Furukawa, S. Nakamura and T. Toru, J. Am. Chem. Soc., 2007, 129, 6394 CrossRef CAS PubMed; (d) X. Shen, L. Zhang, Y. Zhao, L. Zhu, G. Li and J. Hu, Angew. Chem., Int. Ed., 2011, 50, 2588 CrossRef CAS PubMed; (e) G. K. S. Prakash, S. Chacko, S. Alconcel, T. Stewart, T. Mathew and G. A. Olah, Angew. Chem., Int. Ed., 2007, 46, 4933 CrossRef CAS PubMed; (f) A.-N. Alba, X. Companyo, A. Moyano and R. Rios, Chem. – Eur. J., 2009, 15, 7035 CrossRef CAS PubMed.
- We repeated the experimental procedure of monofluorination of (PhSO2)2CH2 demonstrated by Shibata, and found that (PhSO2)2CHF (64%) and (PhSO2)2CF2 (14%) were formed (determined by 19F NMR analysis using PhCF3 as an internal standard).
- T. Imamoto and H. Koto, Synthesis, 1985, 982 CrossRef CAS.
- (a) P. Knochel and P. Jones, Organozinc reagents: a practical approach, Oxford University Press, Oxford, New York, 1999 Search PubMed; (b) P. Knochel and R. D. Singer, Chem. Rev., 1993, 93, 2117 CrossRef CAS; (c) P. Knochel, J. J. Almena Perea and P. Jones, Tetrahedron, 1998, 54, 8275 CrossRef CAS.
- When substrates 1b–d were subjected to the standard reaction conditions (as described in Table 1, entry 11), 87% of 2b (11% of 3b), 80% of 2c (20% of 3c), and 65% of 2d (23% of 3d) were formed, respectively. All the yields were determined by 19F NMR analysis of the crude reaction mixture using PhCF3 as an internal standard.
- The pKa of C–H bonds collected by D.A. Evans proves to be a good reference to give constructive guide in adjusting the amount of base and ZnCl2.
- (a) P. R. Blakemore, W. J. Cole, P. J. Kocieński and A. Morley, Synlett, 1998, 26 CrossRef CAS; (b) A. K. Ghosh and B. Zajc, Org. Lett., 2006, 8, 1553 CrossRef CAS PubMed.
- G. K. S. Prakash, F. Wang, N. Shao, T. Mathew, G. Rasul, R. Haiges, T. Stewart and G. A. Olah, Angew. Chem., Int. Ed., 2009, 48, 5358 CrossRef CAS PubMed.
- (a) R. M. Fabicon, M. Parvez and H. G. Richey, J. Am. Chem. Soc., 1991, 113, 1412 CrossRef CAS; (b) A. P. Purdy and C. F. George, Polyhedron, 1994, 13, 709 CrossRef CAS.
- Previously, the bis[bis(trimethylsilyl)amido] complex of zinc was reported. See: (a) H. Bürger, W. Sawodny and U. Wannagat, J. Organomet. Chem., 1965, 3, 113 CrossRef; (b) O. Just, D. A. Gaul and W. S. Rees Jr., Polyhedron, 2001, 20, 815 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and the characterization data of new compounds. See DOI: 10.1039/c4qo00090k |
| This journal is © the Partner Organisations 2014 |