 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On-surface Ullmann polymerization via intermediate organometallic networks on Ag(111)†
Johanna
Eichhorn
abc,
Thomas
Strunskus
d,
Atena
Rastgoo-Lahrood
abc,
Debabrata
Samanta
e,
Michael
Schmittel
e and
Markus
Lackinger
*abf
aDepartment of Physics, Technische Universität München, James-Franck-Str. 1, 85748 Garching, Germany
bCenter for NanoScience, Schellingstrasse 4, 80799 Munich, Germany
cTUM School of Education, Technische Universität München, Marsstraße 12, 80335 München, Germany
dInstitute for Materials Science – Multicomponent Materials, Christian-Albrechts-Universität zu Kiel, Kaiserstr. 2, 24143 Kiel, Germany
eCenter of Micro- & Nanochemistry & Engineering, Organische Chemie I, Universität Siegen, Adolf-Reichwein-Str. 2, 57068 Siegen, Germany
fDeutsches Museum, Museumsinsel 1, 80538, Munich, Germany. E-mail: markus@lackinger.org; Tel: +49-89-2179-605
First published on 21st May 2014
Abstract
The role of organometallic intermediates during on-surface polymerization via Ullmann coupling was studied on Ag(111). The polymerization progress was monitored by X-ray photoelectron spectroscopy (XPS). Scanning Tunneling Microscopy (STM) was used to characterize organometallic and covalent networks and to identify the temperature regimes for organometallic and covalent bond formation.
In recent years, low-dimensional organic covalent materials have gained significant research interest due to unprecedented chemical and mechanical stability. In combination with their adjustable electronic properties, 2D polymers are perfectly suited for applications in (opto)electronic devices.1,2 On-surface Ullmann polymerization has become an established route for the tailored fabrication of covalent nanostructures.3–6 The observed low structural quality of 2D networks originates from the irreversibility of the newly formed C–C cross-links, inhibiting error correction processes. On the other hand, non-covalent self-assembly is well known to yield highly ordered structures. In this respect, the formation of metastable organometallic chains7,8 and even 2D networks9 as established intermediates in on-surface Ullmann couplings may be used for improving the structural quality of 2D covalent networks. A necessity therefore is the reversibility of the organometallic bonds. Since C–Cu bonds are relatively strong, silver appears to be the most promising metal due to its intermediate reactivity. In this context, detailed knowledge about the temperature regimes where organometallic C–Ag–C bonds may become reversible and where they are irreversibly converted into covalent C–C bonds is required.
Herein, we use finely-tuned tempering protocols to demonstrate a stepwise on-surface Ullmann polymerization leading to 2D porous covalent networks on Ag(111) via intermediate organometallic networks. On-surface Ullmann coupling relies on the catalytic properties of the metal surface for cleavage of weakly bound halogen substituents. Therefore, the halogen side groups play a decisive role: at room temperature, deiodination takes place spontaneously on Cu, Ag, and Au.4,10 On the other hand, bromine is fully split off on copper,9,11 partially on silver,3 and not at all on gold.12 On copper and silver surfaces, the surface-bound radicals directly form metastable organometallic structures based on carbon–metal–carbon bonds. On gold, however, organometallic bonds are only occasionally observed.13 An important difference is the formation of disordered organometallic networks on copper as opposed to partly ordered structures on silver.14 These observations suggest the irreversibility of C–Cu bonds that are normally stronger than C–Ag bonds. On the other hand, the relatively low binding energy of the C–Ag bond promotes its reversibility. This enables the possibility of structural rearrangements during the polymerization of the organometallic intermediate. Accordingly, self-assembly of organometallic precursor networks may be used to improve the structural quality of subsequently formed covalent networks. One requirement, however, is that the organometallic network can undergo equilibration and error correction prior to the formation of irreversible covalent bonds by releasing the interlinking silver atom. Interestingly, on copper this conversion is not always possible, because the required temperature for breaking C–Cu bonds can already exceed the thermal stability of the monomer.15
To study the potential differences between I and Br functionalization, 1,3-bis(p-bromophenyl)-5-(p-iodophenyl)benzene (BIB, cf. inset Fig. 1(c)) was chosen as the monomer. BIB was deposited in UHV onto Ag(111) either held at room temperature (RT) or heated above 125 °C. Additionally, X-ray photoelectron spectroscopy (XPS) was conducted for detailed insights into the temperature dependence of debromination. The resulting structures and the influence of different preparation protocols – RT deposition and heating vs. high temperature deposition – were studied by STM.
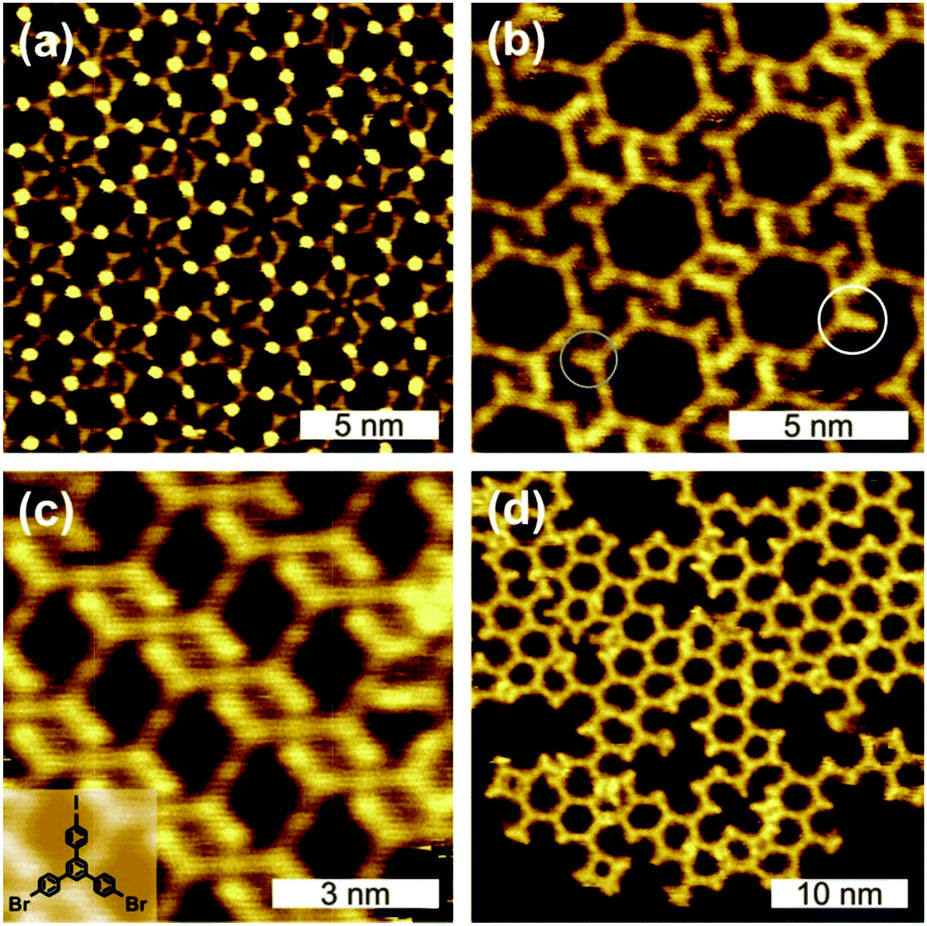 | ||
| Fig. 1 STM images of self-assembled structures directly obtained after RT deposition of BIB onto Ag(111) (cf. ESI† for tunnelling parameter). In (b) intact as well as dehalogenated side groups can be distinguished (white/grey circle). The inset in (c) depicts the chemical structure of BIB. | ||
RT deposition of BIB onto Ag(111) results in a great variety of coexisting partly ordered arrangements that range from closely packed to porous structures (Fig. 1). The three-fold structure of BIB is clearly recognizable in the STM images. The STM data feature two different contrasts, whereby the molecules are either interconnected by clearly discernible bright (Fig. 1(a)) or by barely visible faint dots (Fig. 1(b)–(d)). In both cases, the center-to-center distance of adjacent molecules is 1.62 ± 0.10 nm. In accordance with DFT calculations, this length is characteristic of organometallic complexes with straight intermolecular C–Ag–C bonds (cf. ESI,† Fig. S3). The formation of these complexes at RT indicates the availability of a sufficient amount of Ag adatoms. For the unconnected molecular lobes of BIB, STM reveals clear length differences, suggesting the coexistence of intact brominated and even debrominated molecular side groups (Fig. 1(b), highlighted by circles). Deiodinated and debrominated side groups, however, are indistinguishable in the organometallic complexes. For RT deposition C–I cleavage is well documented on Ag(111),10 whereas the temperature progression of the debromination is less clear. To obtain detailed insights, XP spectra of Br 3d core levels were acquired after each step of successive heating (cf.Fig. 2(a)). Directly after RT deposition, XPS reveals the coexistence of two chemically distinct Br species (each giving rise to a spin–orbit doublet), confirming the coexistence of carbon-bound (Br 3d5/2 BE 69.8 eV) and chemisorbed bromine (Br 3d5/2 BE 68.2 eV).16 Accordingly, after RT deposition ∼38% of the bromine is split off, and the debromination is completed to ∼92% after annealing at 260 °C. A similar behavior, yet with a substantially higher amount of initial RT debromination, was already reported for brominated tetrathienoanthracene on Ag(111).3
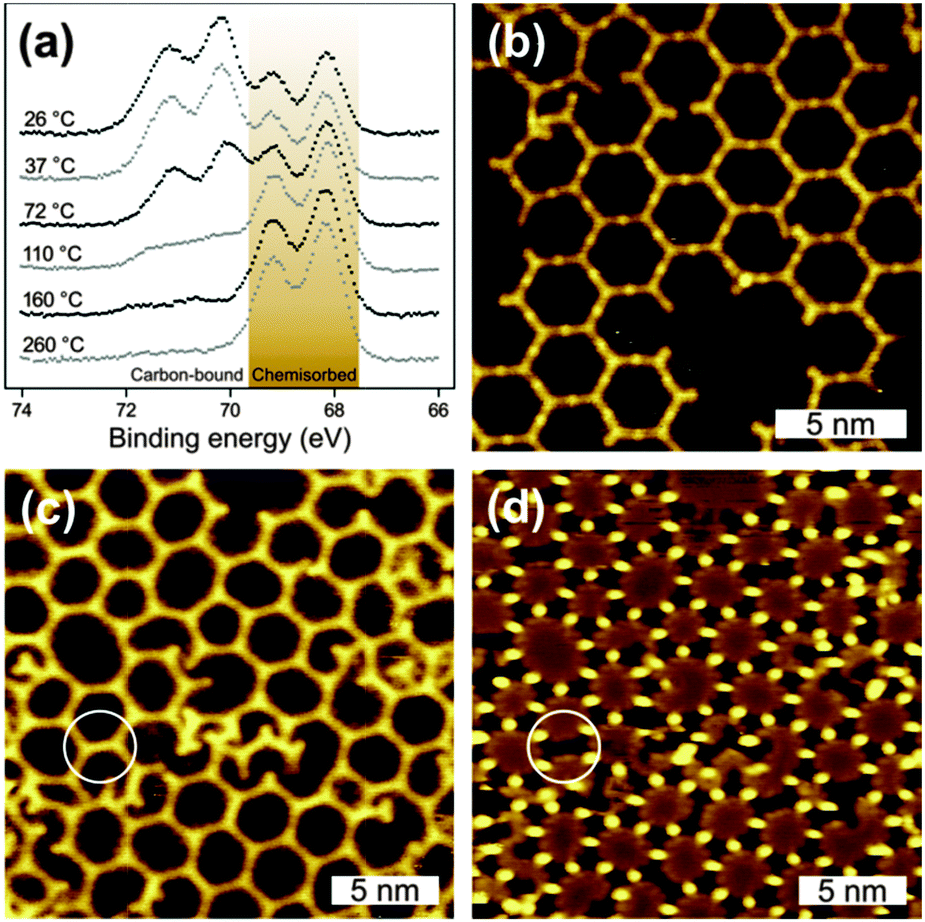 | ||
| Fig. 2 (a) XP spectra of Br 3d on Ag(111) after RT deposition and successive tempering steps up to 260 °C. STM images of BIB deposited on Ag(111) (b) at RT with subsequent heating to 125 °C (11 pA, +0.80 V) and (c) + (d) preheated to 170 °C (7.8 pA, (c) +0.90 V, (d) +0.20 V). The white circles highlight individual already formed covalent bonds. | ||
Guided by the XPS data, the temperature dependent formation of fully cross-linked organometallic structures was studied by tempering at 125 °C to activate almost complete debromination. The corresponding STM image in Fig. 2(b) shows rather regular hexagonal porous networks based on straight intermolecular bonds as commonly observed for Ag+ complexes. Both the bond lengths and the faint dots between the molecules indicate organometallic networks. Upon heating to 125 °C, further debromination initiates the reorganization of the partly ordered RT arrangements and results in the formation of regular porous organometallic networks. In contrast to RT deposition, the obtained networks largely feature fully debrominated monomers. Some defects, however, can be attributed to still intact brominated molecular lobes (cf.Fig. 2(b)). The rearrangement processes inevitably require breaking and reorganization of organometallic bonds, confirming the dynamic properties of C–Ag bonds on Ag(111) below ∼125 °C. Newly formed covalent bonds have never been observed in this temperature range.
To investigate the effect of the preparation protocol samples were also prepared by depositing BIB directly onto preheated substrates. The resulting organometallic networks exhibit comparable structural quality (cf.Fig. 2(c) and (d)). However, now monomers can also become trapped within the pores, i.e. even at the elevated deposition temperature monomers cannot escape the pores by diffusing below or above the organic framework.
A further intention of these experiments was to determine the temperature threshold for conversion of organometallic into covalent bonds. In this context, the question arises whether the organometallic networks can be equilibrated before the irreversible formation of covalent networks sets in. Therefore, a series of experiments with increasing substrate temperatures was performed, the corresponding STM data of BIB deposition onto preheated Ag(111) at 170 °C are shown in Fig. 2(c) and (d). Both images depict the same sample area, however, with different STM contrasts, originating from different bias voltages. In Fig. 2(c) the silver atoms are barely visible, whereas in (d) they are clearly resolved as protrusions while the organic backbones of the network appear as depressions. Most intermolecular bonds are mediated by silver atoms and can thus be assigned to organometallic bonds. On the other hand, few interconnections do not exhibit any internal STM contrast and are ∼0.26 ± 0.1 nm shorter in length, indicating the formation of covalent bonds. In conclusion, the conversion from organometallic to covalent bonds becomes noticeable at ∼170 °C, while individual covalent bonds can form even at ∼155 °C (cf. ESI†). The coexistence of both types of bonds suggests a gradual conversion, whereby the molecules retain their original arrangement. Remarkably, the temperature regimes for the reversible equilibration of the C–Ag bonds and the final irreversible C–C bond formation are well separated. This opens up the possibility of employing self-assembly and error correction of organometallic networks prior to conversion into covalent networks. The potential of this approach was further explored by tempering samples at extremely slow heating rates (0.07 or 0.31 °C min−1) with the intention of establishing and retaining organometallic equilibrium structures before covalent bond formation occurs. Again, room vs. high temperature deposition was compared. In both approaches, polymerization was initiated by slow tempering to 250 °C. The resulting networks shown in Fig. 3 exhibit decreased intermolecular distances of 1.3 nm and a lattice parameter of 2.3 nm, indicating covalent interlinks. RT deposition with subsequent tempering results in branched networks with predominantly hexagonal pores and only a few irregular pentagonal pores (cf.Fig. 3(a)).
 | ||
| Fig. 3 STM images of covalent networks on Ag(111). BIB was deposited at RT ((a) −1.9 V, 24.7 pA) or deposited onto a preheated sample at 150 °C (b) − (d). Subsequently, both samples were tempered up to 250 °C ((b) + (c) 4.6 pA, +3.9 V, (d) 5.5 pA, +2.6 V). The circle in (d) marks a still present organometallic bond in a disordered domain. | ||
Samples prepared by deposition on a preheated surface (150 °C) and subsequent tempering up to 250 °C, instead, feature more compact networks (Fig. 3(b)–(d)), whereby highly ordered and more disordered domains can be distinguished. The close-up image in Fig. 3(d) provides a detailed view of a more irregular domain. Although the relative amount of regular hexagonal pores is still relatively high, irregular single pentagonal pores can be recognized and molecules trapped within the pores are frequently observed. The bright dots within the pores are assigned to split off halogens (ESI,† S2), yet a distinction between Br and I was not possible. Surprisingly, few organometallic bonds are still present, but were only observed in disordered regions (cf.Fig. 3(d)).
In summary, we studied the formation of 2D covalent networks by on-surface Ullmann coupling on Ag(111) with the focus on intermediate organometallic networks. In contrast to the polymerization of the comparable monomer 1,3,5-tris(4-bromophenyl)benzene (TBB) on Au(111)12 and Cu(111),9 the covalent networks on Ag(111) exhibit a higher degree of regularity with structurally perfect domains extending up to 10 nm (cf.Fig. 3(c)). Fasel et al. account for differences in monomer mobility as an important surface influence on the structural quality of 2D polymers.10 Additionally, we propose a decisive role of organometallic intermediates, in particular, on Ag(111). By moderate tempering at ∼125 °C highly ordered organometallic networks could be stabilized, while a sizable conversion to covalent networks sets in at markedly higher temperatures of ∼170 °C. Future experiments focus on a better understanding of error correction in organometallic networks.
Financial support from DFG (LA1842/4-1) and Nanosystems Initiative Munich is gratefully acknowledged. We thank the Helmholtz-Zentrum Berlin for the allocation of synchrotron radiation beamtime and financial support. We are grateful to Dr. Alexei Nevedov and Prof. Christoph Wöll (Karlsruhe Institute of Technology) for making their UHV system available for XPS measurements.
Notes and references
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453 CrossRef CAS PubMed.
- D. F. Perepichka and F. Rosei, Science, 2009, 323, 216 CrossRef CAS PubMed.
- R. Gutzler, L. Cardenas, J. Lipton-Duffin, M. El Garah, L. E. Dinca, C. E. Szakacs, C. Fu, M. Gallagher, M. Vondracek, M. Rybachuk, D. F. Perepichka and F. Rosei, Nanoscale, 2014, 6, 2660 RSC.
- S. Schlögl, W. M. Heckl and M. Lackinger, Surf. Sci., 2012, 606, 999 CrossRef PubMed.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687 CrossRef CAS PubMed.
- G. Eder, E. F. Smith, I. Cebula, W. M. Heckl, P. H. Beton and M. Lackinger, ACS Nano, 2013, 7, 3014 CrossRef CAS PubMed.
- M. Di Giovannantonio, M. El Garah, J. Lipton-Duffin, V. Meunier, L. Cardenas, Y. Fagot Revurat, A. Cossaro, A. Verdini, D. F. Perepichka, F. Rosei and G. Contini, ACS Nano, 2013, 7, 8190 CrossRef CAS PubMed.
- W. Wang, X. Shi, S. Wang, M. A. Van Hove and N. Lin, J. Am. Chem. Soc., 2011, 133, 13264 CrossRef CAS PubMed.
- R. Gutzler, H. Walch, G. Eder, S. Kloft, W. M. Heckl and M. Lackinger, Chem. Commun., 2009, 4456 RSC.
- M. Bieri, M.-T. Nguyen, O. Gröning, J. Cai, M. Treier, K. Aït-Mansour, P. Ruffieux, C. A. Pignedoli, D. Passerone, M. Kastler, K. Müllen and R. Fasel, J. Am. Chem. Soc., 2010, 132, 16669 CrossRef CAS PubMed.
- H. Walch, R. Gutzler, T. Sirtl, G. Eder and M. Lackinger, J. Phys. Chem. C, 2010, 114, 12604 CAS.
- M. O. Blunt, J. C. Russell, N. R. Champness and P. H. Beton, Chem. Commun., 2010, 46, 7157 RSC.
- A. Saywell, W. Greń, G. Franc, A. Gourdon, X. Bouju and L. Grill, J. Phys. Chem. C, 2013, 118, 1719 Search PubMed.
- M. Bieri, S. Blankenburg, M. Kivala, C. A. Pignedoli, P. Ruffieux, K. Müllen and R. Fasel, Chem. Commun., 2011, 47, 10239 RSC.
- C. J. Villagómez, T. Sasaki, J. M. Tour and L. Grill, J. Am. Chem. Soc., 2010, 132, 16848 CrossRef PubMed.
- Y.-J. Wu, W.-H. Wang and C.-M. Chiang, Langmuir, 2002, 18, 1449 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional STM data and DFT results. See DOI: 10.1039/c4cc02757d |
| This journal is © The Royal Society of Chemistry 2014 |