 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and reactivity of a dinitrogen-bridged dimolybdenum-tetrachloride complex†
Kazuya
Arashiba
,
Shogo
Kuriyama
,
Kazunari
Nakajima
and
Yoshiaki
Nishibayashi
*
Institute of Engineering Innovation, School of Engineering, The University of Tokyo, Yayoi, Bunkyo-ku, Tokyo, 113-8656, Japan. E-mail: ynishiba@sogo.t.u-tokyo.ac.jp; Fax: +81-3-5841-1175
First published on 23rd October 2013
Abstract
A dinitrogen-bridged dimolybdenum-tetrachloride complex is prepared and reduced with Super-Hydride (LiBHEt3) to afford the corresponding dimolybdenum-dinitrogen complex together with the formation of molecular dihydrogen. This reaction proceeds via the ligand exchange of the coordinated dihydrogen generated in situ with molecular dinitrogen.
The development of a nitrogen fixation system under mild reaction conditions is one of the most important subjects in chemistry.1 Quite recently, we have found another successful example2 of the catalytic formation of ammonia from molecular dinitrogen under ambient conditions, where dinitrogen-bridged dimolybdenum-dinitrogen complexes work as an effective catalyst.3 In this reaction system, a sequential process of protonation and reduction is necessary to transform the coordinated molecular dinitrogen into ammonia at the molybdenum atom of the catalyst. Interestingly, a similar process may occur at the active site of nitrogenase to convert molecular dinitrogen into ammonia under ambient conditions.4
As the next stage of the previous work, we have focused on the development of the catalytic formation of ammonia from molecular dinitrogen and dihydrogen at ambient temperature and pressure.5,6 Previously, we have found the stoichiometric formation of ammonia from the reaction of a tungsten–dinitrogen complex with an excess amount of ruthenium–dihydrogen complex under mild reaction conditions.6 After the reaction, a ruthenium–hydride complex was observed together with high oxidative tungsten species without nitrogenous ligands. To achieve the catalytic formation of ammonia as the next nitrogen fixation, in place of the Haber–Bosch process,7 the ruthenium–hydride species should reduce the high oxidative tungsten species to regenerate the corresponding tungsten–dinitrogen complex. However, unfortunately, the tungsten species can not be reduced with the ruthenium–hydride species or with other hydride species such as LiBHEt3.6
As the first stage of the development of the catalytic formation of ammonia from molecular dinitrogen and dihydrogen under mild reaction conditions, we envisaged the reaction of the high oxidative molybdenum complexes with hydride species to regenerate the corresponding dinitrogen complexes as starting catalytic species. In this reaction, the high oxidative molybdenum complexes can be reduced into the corresponding dinitrogen complexes, where the ligand exchange of the coordinated dihydrogen with molecular dinitrogen may be involved as a key step to regenerate the corresponding dinitrogen complexes. Herein, we describe the preparation of the dinitrogen-bridged dimolybdenum-tetrachloride complex bearing a PNP-type pincer ligand and the reduction of the dimolybdenum-tetrachloride complex with Super-Hydride (LiBHEt3) to afford the corresponding molybdenum-dinitrogen complex together with the formation of molecular dihydrogen.
Treatment of [MoCl3(PNP)]3 (1:PNP = 2,6-bis(di-tert-butylphosphinomethyl)pyridine) with 1 equiv. of KC8 in THF at room temperature for 20 h under an atmospheric pressure of dinitrogen gave the dinitrogen-bridged dimolybdenum-tetrachloride complex [MoCl2(PNP)]2(μ-N2) (2) in 64% yield (Scheme 1). No informative data on the structure of 2 were obtained from its NMR spectra due to the paramagnetism. Complex 2 exhibits μeff = 3.2 μB in THF-d8 at 296 K (Evans NMR method), which is consistent with an S = 1 system (μeff = 2.83 μB) on each Mo atom. No Raman absorption attributable to the bridging N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch is observed at 1800–2100 cm−1 for 2. Additionally, no IR absorption attributable to the terminal NN stretch is also observed for 2, in contrast to the value (1936 cm−1) observed for [Mo(N2)2(PNP)]2(μ-N2) (3).3 A cyclic voltammetric study of 2 reveals two reversible cathodic waves at −1.98 and −1.02 V and two quasi-reversible anodic waves at −0.06 and +0.17 V vs. Fc/Fc+, respectively.
N stretch is observed at 1800–2100 cm−1 for 2. Additionally, no IR absorption attributable to the terminal NN stretch is also observed for 2, in contrast to the value (1936 cm−1) observed for [Mo(N2)2(PNP)]2(μ-N2) (3).3 A cyclic voltammetric study of 2 reveals two reversible cathodic waves at −1.98 and −1.02 V and two quasi-reversible anodic waves at −0.06 and +0.17 V vs. Fc/Fc+, respectively.
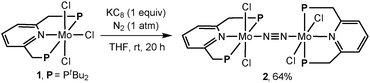 | ||
| Scheme 1 Preparation of a dinitrogen-bridged dimolybdenum-tetrachloride complex bearing PNP pincer ligand (2). | ||
A more detailed molecular structure of 2 is determined by X-ray crystallographic study (Fig. 1). The molecular structure of 2 contains two [MoCl2(PNP)] moieties bearing two chloride ligands in trans form. The two molybdenum atoms are bridged by one dinitrogen ligand in an end-on fashion with the almost linear Mo–N–N–Mo bonding. The two molybdenum fragments are twisted around the Mo–N–N–Mo axis with respect to each other away from the steric interaction between two PNP ligands: the torsion angle for Cl(1)–Mo(1)–Mo(2)–Cl(3) is 96.73(2)°. As shown in Table 1, the bond distance of bridging-dinitrogen (N(1)–N(2) 1.169(3) Å) is slightly longer than that of 3 (N–N 1.146(4) Å), while the Mo–N(bridging) distances in 2 (Mo(1)–N(1) 1.925(2) Å, Mo(2)–N(2) 1.927(2) Å) are shorter than that of 3 (Mo–N 2.024(3) Å). These results indicate that the N–N bond lengths of the dinitrogen-bridged dimolybdenum complexes 2 and 3 decrease as their Mo centers are reduced from Mo(II) to Mo(0). These phenomena can be explained by the enhancement of the N–N π-bonding between the two nitrogen atoms of the bridging dinitrogen ligand. The reduction of 2 into 3 increases the population of the LUMO, which is responsible for the N–N π-bonding character. Similar phenomena were observed in a series of dinitrogen-bridged dimolybdenum complexes bearing bulky amido ligands developed by Cummins and co-workers.8
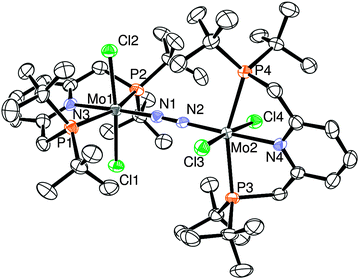 | ||
| Fig. 1 ORTEP drawing of 2. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms and solvent molecules are omitted for clarity. Selected bond lengths (Å) and angles (°): Mo1–N1 1.925(2), Mo2–N2 1.927(2), Mo1–N3 2.192(2), Mo2–N4 2.197(2), N1–N2 1.169(3), Mo1–N1–N2 177.1(2), Mo2–N2–N1 177.9(2), P1–Mo1–P2 157.59(3), P3–Mo2–P4 156.94(3), Cl1–Mo1–Cl2 173.41(3), Cl3–Mo2–Cl4 174.64(3). | ||
| 2 (L = Cl) | 3 (L = N2) | |
|---|---|---|

|
||
| Distances (Å) | ||
| N–N(bridging) | 1.169(3) | 1.146(4) |
| Mo–N(bridging) | 1.925(2) and 1.927(2) | 2.024(3) |
| Mo–N(pyridine) | 2.192(2) and 2.197(2) | 2.182(3) |
| Angles (°) | ||
| Mo–N–N | 177.1(2) and 177.9(2) | 178.9(2) |
| P–Mo–P | 157.59(3) and 156.94(3) | 157.07(4) |
| L–Mo–Mo–L | 96.73(2) | 61.41(9) |
Next, the novel dinitrogen-bridged dimolybdenum-tetrachloride complex 2 was applied as a catalyst toward the catalytic reaction of molecular dinitrogen to form ammonia under ambient conditions.3 Unfortunately, 2 did not work as an effective catalyst toward the catalytic reaction. This result prompts us to investigate the stoichiometric reactivity of the coordinated dinitrogen ligand toward the protonation. The reaction of 2 with an excess amount of sulfuric acid in THF at room temperature for 24 h did not result in the formation of ammonia and hydrazine at all (Scheme 2). This result suggests that the bridging-dinitrogen moiety in 2 may be less reactive toward the protonation than the terminal dinitrogen ligands in 3, where the formation of stoichiometric amounts of ammonia and hydrazine (0.6 equiv. of ammonia and 0.06 equiv. of hydrazine based on the Mo atom in 3, respectively) was observed under the same reaction conditions.3
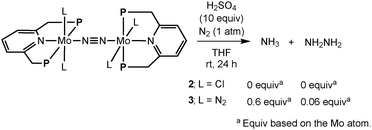 | ||
| Scheme 2 Protonation of dinitrogen-bridged dimolybdenum complexes (2 and 3). | ||
Next, we investigated the reduction of 2 with an excess amount (10 equiv.) of LiBHEt3 in THF at room temperature for 20 h under an atmospheric pressure of dinitrogen to afford 3 in 43% isolated yield together with the formation of molecular dihydrogen in 200% GLC yield (2 equiv. based on 2) (Scheme 3). Separately, we confirmed the almost complete consumption of 2 by ESI-TOF-MS. Based on the result of the formation of molecular dihydrogen, we consider the following proposed reaction pathway leading to the formation of 3 (Scheme 4). At first, a dinitrogen-bridged dimolybdenum-tetrahydride complex (A) may be formed from the reaction of dimolybdenum-tetrachloride complex 2 with LiBHEt3 and then the dinitrogen-bridged dimolybdenum-tetrahydride complex A is converted into the dinitrogen-bridged dimolybdenum-bis(dinitrogen) complex (C) via a dinitrogen-bridged dimolybdenum-bis(dihydrogen) complex (B) as a key intermediate. The ligand exchange of the coordinated molecular dihydrogen with molecular dinitrogen may be a key step to transform 2 into 3.9,10 In fact, unfortunately, we have not yet observed the formation of the dinitrogen-bridged dimolybdenum-bis(dihydride) and bis(dihydrogen) complexes such as A and B by ESI-TOF-MS. However, when the reduction of 2 with LiBHEt3 was carried out under an atmospheric pressure of argon, in place of dinitrogen, the formation of the dinitrogen-bridged dimolybdenum-dichloride-dihydride complex [Mo(H)Cl(PNP)]2(μ-N2) (4) was observed by ESI-TOF-MS from a reaction mixture after the reduction.11
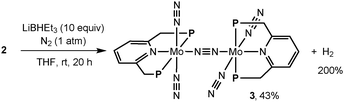 | ||
| Scheme 3 Reduction of 2 with Super-Hydride to form a dinitrogen-bridged dimolybdenum-dinitrogen complex (3). | ||
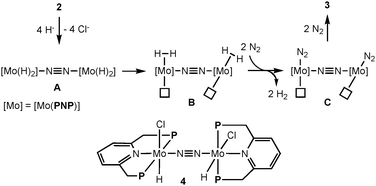 | ||
| Scheme 4 Proposed reaction pathway from 2 to 3via dimolybdenum-tetrahydride and -bis(dihydrogen) complexes. | ||
Separately, we confirmed that only a small amount of 3 was observed by 1H NMR in the direct reduction of 1 with an excess amount of LiBHEt3 in THF at room temperature for 20 h under an atmospheric pressure of dinitrogen. This result indicates that the stepwise reduction of 1via2 as a reactive intermediate into 3 provides a novel approach to the preparation of dinitrogen-bridged dimolybdenum-dinitrogen complexes. We believe that this process provides useful information to design the catalytic formation of ammonia from molecular dinitrogen and dihydrogen under mild reaction conditions.9,10
Interestingly, we confirmed the formation of 2 from the oxidation of 3 with 4 equiv. of AgCl in THF at room temperature for 20 h under an atmospheric pressure of dinitrogen (Scheme 5). This is the reverse of the reductive transformation of 2 into 3. These results indicate that the bridging-dinitrogen ligand may be tightly coordinated to the two molybdenum atoms in 2 and 3 during the oxidation process.
 | ||
| Scheme 5 Oxidation of 3 with AgCl to form 2. | ||
In summary, we have prepared a novel dinitrogen-bridged dimolybdenum-tetrachloride complex bearing PNP-type pincer ligands and reduced the dimolybdenum-tetrachloride complex with Super-Hydride to form the corresponding dinitrogen-bridged dimolybdenum-dinitrogen complex together with molecular dihydrogen. We believe that the reduction of high oxidative molybdenum complexes with hydride species to regenerate the corresponding dinitrogen complexes is one of the most important steps to realize the catalytic formation of ammonia from molecular dinitrogen and dihydrogen under mild reaction conditions.12 Further studies on the preparation and reactivity of molybdenum- and iron-complexes bearing other pincer ligands are currently underway.13–15
This work was supported by the Funding Program for Next Generation World-Leading Researchers (GR025). S.K. is a recipient of the JSPS Predoctoral Fellowships for Young Scientists.
Notes and references
- For recent reviews, see: (a) S. Hinrichsen, H. Broda, C. Gradert, L. Söncksen and F. Tuczek, Annu. Rep. Prog. Chem., Sect. A, 2012, 108, 17 RSC; (b) Y. Nishibayashi, Dalton Trans., 2012, 41, 7447 RSC; (c) K. C. MacLeod and P. L. Holland, Nat. Chem., 2013, 5, 559 CrossRef CAS PubMed; (d) M. D. Fryzuk, Chem. Commun., 2013, 49, 4866 RSC; (e) Y. Tanabe and Y. Nishibayashi, Coord. Chem. Rev., 2013, 257, 2551 CrossRef CAS PubMed.
- (a) D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76 CrossRef CAS PubMed; (b) R. R. Schrock, Acc. Chem. Res., 2005, 38, 955 CrossRef CAS PubMed; (c) R. R. Schrock, Angew. Chem., Int. Ed., 2008, 47, 5512 CrossRef CAS PubMed.
- (a) K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120 CrossRef CAS PubMed; (b) K. Arashiba, K. Sasaki, S. Kuriyama, Y. Miyake, H. Nakanishi and Y. Nishibayashi, Organometallics, 2012, 31, 2035 CrossRef CAS; (c) E. Kinoshita, K. Arashiba, S. Kuriyama, Y. Miyake, R. Shimazaki, H. Nakanishi and Y. Nishibayashi, Organometallics, 2012, 31, 8437 CrossRef CAS.
- Nitrogen Fixation, ed. M. W. Ribbe, Human Press, New York, 2011 Search PubMed.
- (a) J. A. Pool, E. Lobkovsky and P. J. Chirik, Nature, 2004, 427, 527 CrossRef CAS PubMed; (b) M. M. Rodriguez, E. Bill, W. W. Brennessel and P. L. Holland, Science, 2011, 334, 780 CrossRef CAS PubMed; (c) T. Shima, S. Hu, G. Luo, X. Kang, Y. Luo and Z. Hou, Science, 2013, 340, 1549 CrossRef CAS PubMed.
- (a) Y. Nishibayashi, S. Iwai and M. Hidai, Science, 1998, 279, 540 CrossRef CAS; (b) Y. Nishibayashi, S. Takemoto, S. Iwai and M. Hidai, Inorg. Chem., 2000, 39, 5946 CrossRef CAS; (c) Y. Nishibayashi, I. Wakiji, K. Hirata, M. R. DuBois and M. Hida, Inorg. Chem., 2001, 40, 578 CrossRef CAS.
- Ammonia Synthesis Catalysis: Innovation and Practice, ed. H. Liu, World Scientific, Beijing, 2013 Search PubMed.
- J. J. Curley, T. R. Cook, S. Y. Reece, P. Müller and C. C. Cummins, J. Am. Chem. Soc., 2008, 130, 9394 CrossRef CAS PubMed.
- For a review, see: J. Ballmann, R. F. Munhá and M. D. Fryzuk, Chem. Commun., 2010, 46, 1013 RSC.
- (a) M. Hidai, K. Tominari and Y. Uchida, J. Am. Chem. Soc., 1972, 94, 110 CrossRef CAS; (b) L. Archer and T. A. George, Inorg. Chem., 1979, 18, 2079 CrossRef CAS; (c) R. Pierantozzi and G. L. Geoffroy, Inorg. Chem., 1980, 19, 1821 CrossRef CAS.
- ESI-TOF-MS (THF) of 4 1084 (M).
- J. D. Gilberson, N. K. Szymczak and D. R. Tyler, J. Am. Chem. Soc., 2005, 127, 10184 CrossRef PubMed.
- We have recently found the first successful example of iron-catalyzed reduction of molecular dinitrogen under ambient reaction conditions; see: M. Yuki, H. Tanaka, K. Sasaki, Y. Miyake, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2012, 3, 1254 CrossRef PubMed.
- Quite recently, Peters and co-workers have reported the first successful example of the iron-catalyzed transformation of atmospheric pressure molecular dinitrogen into ammonia at −78 °C: J. A. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84 CrossRef CAS PubMed.
- For recent examples of molybdenum-dinitrogen complexes bearing other types of ligands, see: (a) T. Miyazaki, Y. Tanabe, M. Yuki, Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem.–Eur. J., 2013, 19, 11874 CrossRef CAS PubMed; (b) Y. Tanabe, S. Kuriyama, K. Arashiba, Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2013, 49, 9290 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 953144. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc46946h |
| This journal is © The Royal Society of Chemistry 2013 |