 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and reactivity of molybdenum–dinitrogen complexes bearing an arsenic-containing ANA-type pincer ligand†
Yoshiaki Tanabe, Shogo Kuriyama, Kazuya Arashiba, Yoshihiro Miyake, Kazunari Nakajima and Yoshiaki Nishibayashi*
Institute of Engineering Innovation, School of Engineering, The University of Tokyo, Yayoi, Bunkyo-ku, Tokyo, 113-8656, Japan. E-mail: ynishiba@sogo.t.u-tokyo.ac.jp; Fax: +81-3-5841-1175; Tel: +81-3-5841-1175
First published on 12th August 2013
Abstract
Novel mono- and dimolybdenum–dinitrogen complexes bearing an arsenic-containing ANA-type pincer ligand are prepared and characterized by X-ray analyses. These complexes afford a stoichiometric amount of ammonia by treatment with sulfuric acid at room temperature.
The preparation and reactivity of transition metal–dinitrogen complexes have been intensively explored in order to develop a new nitrogen fixation system under ambient conditions.1 The choice and design of the ligands to compromise the coordination sphere are shown to be some of the most important points for the efficient coordination and activation of molecular dinitrogen. In fact, we have recently succeeded in constructing another catalytic system2 to convert molecular dinitrogen directly into ammonia using a dinitrogen-bridged dimolybdenum complex trans,trans-[{Mo(N2)2(tBuPNP)}2(μ-N2)] (1a), where up to 23 equiv. of ammonia have been produced based on the catalyst (12 equiv. of ammonia based on the molybdenum atom).3
As an extension of our study, we have now focused on the use of an arsenic-containing ANA-type pincer ligand (Chart 1), where phosphines in the PNP-type pincer ligand are substituted for arsines.4 Arsines are known to have similar coordination modes to phosphines, but are also recognized to be more reluctant to oxidation, sterically bulkier, and poorer σ-donors and π-acceptors compared to phosphines. As a result, the use of arsines in the pincer ligand leads to contrasting steric and electronic effects on coordination spheres different from that of phosphines.5 There have been some examples of transition metal–dinitrogen complexes with arsines as auxiliary ligands, however, their reactivities are not well investigated except for [Mo(N2)(triphos)(diars)] (triphos = PhP(CH2CH2PPh2)2, diars = 1,2-bis(dimethylarsino)benzene) which contains both triphosphine and diarsine ligands.6 Herein, we describe the preparation of molybdenum–dinitrogen complexes bearing an arsenic-containing ANA-type pincer ligand tBuANA (2b, 2,6-bis[(di-tert-butylarsino)methyl]pyridine) and their reactivity toward protonolysis to produce ammonia from the coordinated molecular dinitrogen.
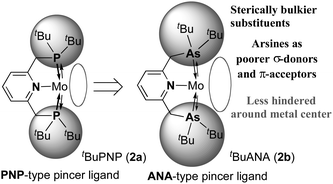 | ||
| Chart 1 ANA-type pincer ligand: introduction of arsine moieties into the pincer ligand. | ||
The arsenic-containing tridentate pincer ligand tBuANA (2b) can be newly prepared in a similar way to the preparation of tBuPNP (2a).7 Treatment of 2,6-lutidine with 2.5 equiv. of nBuLi in diethyl ether at reflux for 15 h gave a solution of dilithiated 2,6-lutidine, where 2.5 equiv. of tBu2AsCl in diethyl ether were slowly added at −78 °C. The reaction mixture was then gradually warmed up to room temperature to afford 2b in 91% yield (Scheme 1). To the best of our knowledge, 2b has been the first ANA-type pincer ligand reported so far.8
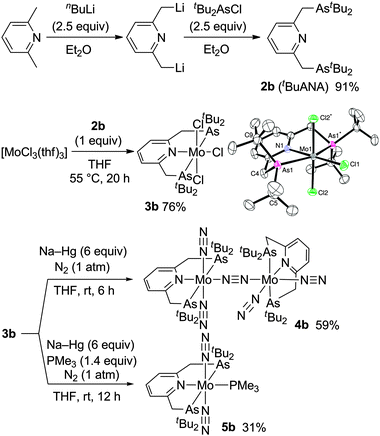 | ||
| Scheme 1 Preparation of molybdenum–dinitrogen complexes bearing ANA-type pincer ligands. | ||
Next, we examined the preparation of molybdenum–dinitrogen complexes ligated by the ANA-type pincer ligand. Chelation of 2b onto the molybdenum atom was successful when 2b was treated with [MoCl3(thf)3] in THF at 55 °C for 20 h to afford the paramagnetic molybdenum(III) ANA-type pincer complex [MoCl3(tBuANA)] (3b) in 76% yield (Scheme 1). The molecular structure of 3b was unambiguously determined by the X-ray analysis (Scheme 1 and Fig. S1, ESI†), which demonstrated that 3b has a distorted octahedral geometry around the molybdenum center, as has been observed for the analogous PNP-type pincer complex [MoCl3(tBuPNP)] (3a).3 In addition, 3b appears to be slightly bulkier than 3a, with the interatomic distances between molybdenum and arsenic atoms (2.65 Å, mean) and between arsenic and carbon atoms (2.00 Å, mean) in 3b slightly longer than those between molybdenum and phosphorus atoms (2.61 Å, mean) and between phosphorus and carbon atoms (1.88 Å, mean) in 3a,3 due to the difference in covalent radii of arsenic and phosphorus atoms.9 On the other hand, a cyclic voltammetric study of 3b revealed the oxidation potential of the couples Mo(III)/Mo(IV) to be +0.15 V vs. Fc/Fc+ comparable to the value for 3a (+0.14 V vs. Fc/Fc+), suggesting that the change in the electronic effect on the molybdenum center by the substitution of arsenic for phosphorus is rather small compared to the steric effect.
The reduction of 3b with an excess amount of Na–Hg (6 equiv.) in THF at room temperature for 6 h under an atmospheric pressure of dinitrogen gave the dinitrogen-bridged dimolybdenum complex trans,cis-[{Mo(N2)2(tBuPNP)}2(μ-N2)] (4b) in 59% yield (Scheme 1). A strong Raman band attributable to the bridging N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch is observed at 1904 cm−1 for 4b, which is not observed in the IR spectrum and is comparable to the value (1890 cm−1) observed for 1a.5 On the other hand, the IR spectrum of 4b exhibits two strong broadened absorptions at 1955 and 1870 cm−1 attributable to NN stretching frequencies in contrast to the single absorption at 1936 cm−1 observed for 1a.5 The 1H NMR resonances due to protons in the tBuANA ligand also appear to be unsymmetrical, with the methylene proton signals at δ 3.48, 3.43, and 3.39–3.28 in an intensity ratio of 2
N stretch is observed at 1904 cm−1 for 4b, which is not observed in the IR spectrum and is comparable to the value (1890 cm−1) observed for 1a.5 On the other hand, the IR spectrum of 4b exhibits two strong broadened absorptions at 1955 and 1870 cm−1 attributable to NN stretching frequencies in contrast to the single absorption at 1936 cm−1 observed for 1a.5 The 1H NMR resonances due to protons in the tBuANA ligand also appear to be unsymmetrical, with the methylene proton signals at δ 3.48, 3.43, and 3.39–3.28 in an intensity ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:4 and the tert-butyl proton signals at δ 1.32–1.23 and 1.17 in an intensity ratio of 54:18. These spectroscopic features indicate that the molecular structure of 4b is different from that of 1a.
:2:4 and the tert-butyl proton signals at δ 1.32–1.23 and 1.17 in an intensity ratio of 54:18. These spectroscopic features indicate that the molecular structure of 4b is different from that of 1a.
The detailed structure of 4b has been determined by an X-ray analysis of 4b·C6H14, which clearly demonstrates that 4b has an unsymmetrical structure where a trans-[Mo(N2)2(tBuANA)] unit and a cis-[Mo(N2)2(tBuANA)] unit are bridged by one dinitrogen ligand in an end-on fashion (Fig. 1). The difference in the coordination geometry around each molybdenum center is in good accord with the spectroscopic data, suggesting that the dimolybdenum structure bridged by a dinitrogen ligand with both cis- and trans-bis(dinitrogen) molybdenum units is maintained even in solution. It must be noted that we have recently isolated similar dinitrogen-bridged dimolybdenum complexes with unsymmetric PNP-type pincer ligands 2-R2PCH2-6-tBu2PCH2-pyridine (tBuRPNP; R = Ad, Ph, iPr, Cy), where trans,trans-[{Mo(N2)2(tBuRPNP)}2(μ-N2)] (1c: R = Ad, 1d: R = Ph) and trans,cis-[{Mo(N2)2(tBuRPNP)}2(μ-N2)] (4e: R = iPr, 4f: R = Cy) were obtained selectively depending on the varieties of PNP-type pincer ligands (Chart 2).10 The reason why trans,cis isomer 4b instead of the trans,trans isomer was obtained may be due to the less hindered environment around molybdenum atoms compared to the complex 1a (Chart 1).
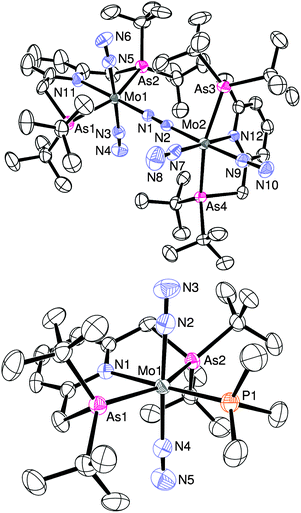 | ||
| Fig. 1 ORTEP drawing of 4b (upper) and 5b (lower). Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
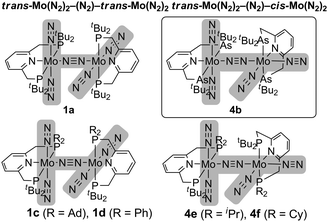 | ||
| Chart 2 Comparison of structures of dinitrogen-bridged dimolybdenum–dinitrogen complexes. | ||
When the reduction of 3b with an excess amount of Na–Hg (6 equiv.) in THF at room temperature for 12 h under an atmospheric pressure of dinitrogen was carried out in the presence of 1.4 equiv. of trimethylphosphine, the corresponding mononuclear molybdenum–dinitrogen complex bearing both the ANA-type pincer and phosphine ligands trans-[Mo(N2)2(PMe3)(tBuANA)] (5b) was obtained in 31% yield (Scheme 1). The IR spectrum exhibits a strong absorption assignable to the NN asymmetric stretching at 1915 cm−1, which is almost the same as that in the corresponding PNP-type pincer complex trans-[Mo(N2)2(PMe3)(tBuPNP)] (5a) (νNN = 1915 cm−1), which has been prepared recently by our group.11 The molecular structure of 5b was confirmed by X-ray crystallography, where the trimethylphosphine ligand occupies the position trans to the pyridine group of an ANA ligand as shown in Fig. 1. The N–N (1.07 Å, mean) bond distances in 5b are slightly shorter than those in 5a (1.13 Å, mean), which may suggest that the dinitrogen ligands in 5b may be less activated than 5a owing to weaker σ-donation from arsines.
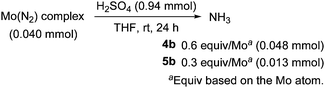
We next investigated the reactivity of these molybdenum–dinitrogen complexes bearing ANA-type pincer ligands toward protonolysis. When dimolybdenum complex 4b was treated with an excess amount of sulfuric acid in THF at room temperature for 24 h, 0.6 equiv. of ammonia was obtained based on the molybdenum atom (Scheme 2), which is almost the same as those obtained from the protonolysis of 1a (0.6 equiv. of ammonia and 0.06 equiv. of hydrazine).11 On the other hand, the amount of ammonia obtained from the protonolysis of 5b is rather small (0.3 equiv. of ammonia), compared to that obtained from the protonolysis of 5a (0.9 equiv. of ammonia and 0.08 equiv. of hydrazine).11 No formation of hydrazine was observed in the protonolysis of 4b and 5b.
 | ||
| Scheme 2 Protonation of molybdenum–dinitrogen complexes. | ||
The catalytic reduction of molecular dinitrogen to ammonia by using 4b as a catalyst was investigated according to the previous procedure.10 Unfortunately, we observed only 2 equiv. of ammonia based on the catalyst (1 equiv. of ammonia based on the Mo atom in 4b) together with 16 equiv. of molecular dihydrogen based on the catalyst. This result indicates that only a stoichiometric amount of ammonia was formed, and most of the cobaltocene was consumed to form molecular dihydrogen from the proton source. This is in sharp contrast to the catalytic formation of ammonia using 1a as a catalyst, where 12 equiv. of ammonia were produced based on the catalyst (6 equiv. of ammonia based on the Mo atom in 1a).3 We have not yet obtained the exact reason why the dinitrogen-bridged dimolybdenum complex with ANA-type pincer ligands 4b did not work as a catalyst toward the formation of ammonia under the same reaction conditions, but we previously confirmed that similar dinitrogen-bridged dimolybdenum complexes with PNP-type pincer ligands 4e and 4f, which have trans,cis structures, also did not work as catalysts.10
In summary, we have synthesized a series of molybdenum–dinitrogen complexes bearing an arsenic-containing ANA-type pincer ligand. Protonation of the dinitrogen-bridged dimolybdenum–dinitrogen complex with an excess amount of sulfuric acid in THF afforded ammonia in good yields. Further studies12 on the preparation and reactivity of other transition metal complexes bearing ANA-type pincer ligands are currently underway.
Notes and references
- For recent reviews, see: (a) S. Hinrichsen, H. Broda, C. Gradert, L. Söncksen and F. Tuczek, Annu. Rep. Prog. Chem., Sect. A, 2012, 108, 17 RSC; (b) Y. Nishibayashi, Dalton Trans., 2012, 41, 7447 RSC; (c) K. C. MacLeod and P. L. Holland, Nat. Chem., 2013, 5, 559 CrossRef CAS PubMed; (d) Y. Tanabe and Y. Nishibayashi, Coord. Chem. Rev., 2013, 257, 2551 CrossRef CAS PubMed.
- (a) D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76 CrossRef CAS PubMed; (b) R. R. Schrock, Acc. Chem. Res., 2005, 38, 955 CrossRef CAS PubMed; (c) R. R. Schrock, Angew. Chem., Int. Ed., 2008, 47, 5512 CrossRef CAS PubMed.
- K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120 CrossRef CAS PubMed.
- For recent reviews, see: (a) The Chemistry of Pincer Compounds, ed. D. Morales-Morales and C. G. M. Jensen, Elsevier, Amsterdam, 2007 Search PubMed; (b) J. I. van der Vlugt and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 8832 CrossRef CAS PubMed; (c) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588 CrossRef CAS PubMed; (d) Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer-Verlag, Berlin, 2013 Search PubMed.
- (a) W. Levason and G. Reid, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier Science, Amsterdam, vol. 1, ch. 1.16, 2004 Search PubMed; (b) F. Mohr, S. H. Privér, S. K. Bhargava and M. A. Bennett, Coord. Chem. Rev., 2006, 250, 1851 CrossRef CAS PubMed; (c) D. Lu and G. Salem, Coord. Chem. Rev., 2013, 257, 1026 CrossRef CAS PubMed.
- The reaction of [Mo(N2)(triphos)(diars)] with an excess amount of hydrobromic acid at room temperature produced both ammonia and hydrazine, whose amounts were less than those obtained from the reaction of [Mo(N2)(triphos)(PMe2Ph)2] with hydrobromic acid, suggesting that phosphines rather than the diarsine contribute more to the activation of the molecular dinitrogen coordinated to the molybdenum atom; (a) T. A. George and R. C. Tisdale, J. Am. Chem. Soc., 1985, 107, 5157 CrossRef CAS; (b) T. A. George and R. C. Tisdale, Inorg. Chem., 1988, 27, 2909 CrossRef CAS.
- M. Kawatsura and J. F. Hartwig, Organometallics, 2001, 20, 1960 CrossRef CAS.
- Preparation of ACA-type pincer ligand 1,3-bis[(di-tert-butylarsino)methyl]benzene has been reported: C. M. Jensen, D. Sun, B. Lewandowski, K. K. Kumashiro, W. P. Niemczura, D. Morales-Morales and Z. Wang, Proceedings of the 2001 U.S. DOE Hydrogen Program Review, National Renewable Energy Laboratory, Golden, CO, 2001, p. 500 Search PubMed.
- (a) B. Cordezo, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC; (b) P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186 CrossRef PubMed.
- E. Kinoshita, K. Arashiba, S. Kuriyama, Y. Miyake, R. Shimazaki, H. Nakanishi and Y. Nishibayashi, Organometallics, 2012, 31, 8437 CrossRef CAS.
- K. Arashiba, K. Sasaki, S. Kuriyama, Y. Miyake, H. Nakanishi and Y. Nishibayashi, Organometallics, 2012, 31, 2035 CrossRef CAS.
- We have quite recently found the first successful example of iron-catalyzed reduction of molecular dinitrogen under ambient reaction conditions; see: M. Yuki, H. Tanaka, K. Sasaki, Y. Miyake, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2012, 3, 1254 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 949577–949579. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc45228j |
| This journal is © The Royal Society of Chemistry 2013 |