DOI:
10.1039/C0SC00240B
(Edge Article)
Chem. Sci., 2010,
1, 461-468
Substituent effects in ditetrel alkyne analogues: multiple vs. single bonded isomers†
Received
29th March 2010
, Accepted 10th May 2010
First published on
18th June 2010
Abstract
The synthesis and characterization of a series of digermynes and distannynes stabilized by terphenyl ligands are described. The ligands are based on the Ar′ (Ar′ = C6H3-2,6(C6H3-2,6-iPr2)2) or Ar* (Ar* = C6H3-2,6(C6H2-2,4,6-iPr3)2) platforms which were modified at the meta or para positions of their central aryl rings to yield 4-X-Ar′ (4-X-Ar′ = 4-X-C6H2-2,6(C6H3-2,6-iPr2)2, X = H, F, Cl, OMe, tBu, SiMe3, GeMe3) and 3,5-iPr2-Ar′ or Ar* and 3,5-iPr2-Ar*. The compounds were synthesized by reduction of the terphenyl germanium(II) or tin(II) halide precursors with a variety of reducing agents. The precursors were obtained by the reaction of one equivalent of the lithium terphenyl with GeCl2 dioxane or SnCl2. For germanium, their X-ray crystal structures showed them to be either Ge–Ge bonded dimers with trans-pyramidal geometries or V-shaped monomers. In contrast, the terphenyl tin halides had no tin–tin bonding but existed either as halide bridged dimers or V-shaped monomers. Reduction with a variety of reducing agents afforded the digermynes ArGeGeAr (Ar = 4-Cl-Ar′, 4-SiMe3-Ar′ or 3,5-iPr2-Ar*) or the distannynes ArSnSnAr (Ar = 4-F-Ar′, 4-Cl-Ar′, 4-MeO-Ar′, 4-tBu-Ar′, 4-SiMe3-Ar′, 4-GeMe3-Ar′, 3,5-iPr2-Ar′, 3,5-iPr2-Ar*), which were characterized structurally and spectroscopically. The digermynes display planar trans-bent core geometries with Ge–Ge distances near 2.26 Å and bending angles near 128° consistent with Ge–Ge multiple bonding. In contrast, the distannynes had either multiple bonded geometries with Sn–Sn distances that averaged 2.65 Å and an average bending angle near 123.8°, or single bonded geometries with a Sn–Sn bond length near 3.06 Å and a bending angle near 98°. The 3,5-iPr2-Ar*SnSnAr*-3,5-iPr2 species had an intermediate structure with a longer multiple bond near 2.73 Å and a variable torsion angle (14–28°) between the tin coordination planes. Mössbauer data for the multiple and single bonded species displayed similar isomer shifts but had different quadrupole splittings.
Introduction
The isolation of heavier group 14 element homologues of alkynes has been a major synthetic challenge owing to their tendency to oligomerize and their inherently high reactivity.1,2 The first stable example, the lead derivative Ar*PbPbAr* Ar* (Ar* = C6H3-2,6-(C6H2-2,4,6-iPr3)2), was synthesized in 2000.3 In sharp contrast to linear acetylene, it had a strongly bent (Pb–Pb–C = 94.26(4)°) core structure, (A in Scheme 1) and a long Pb–Pb bond (3.1881(1) Å) consistent with single bonding. In the ensuing period several examples of stable or quasi-stable alkyne analogues REER (E = Si,4–7 Ge,8–10 Sn11), all of which were stabilized by bulky substituents, have been isolated and characterized. X-Ray crystallography showed that they all had a trans-bent, planar or almost planar geometry with E–E distances that were shortened with respect to single bonds which is indicative of multiple bond character (B in Scheme 1). In addition, the ditetrel alkyne species [{tBuC(NC6H3-2,6-iPr2)2}Ge]2,12 [{PhC(NtBu)2}Ge]213 and [{2,3-(Me2NCH2)C6H3}Sn]214 stabilized by bulky multidentate ligands have been synthesized. However, these feature long E–E single bonds exclusively.
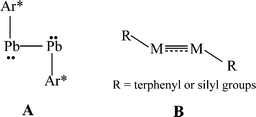 |
| | Scheme 1 Single bonded or multiple bonded trans-bent geometry of RMMR (M = Si, Ge, Sn, Pb; R = terphenyl or silyl substituents) | |
The bonding in the heavier group 14 element alkyne analogues has been the subject of considerable discussion.15,16 Some maintain that with the exception of the lead species multiple bonding is present in the heavier Si, Ge and Sn alkyne analogues even though the amount of bond shortening relative to a single bond diminishes as the group is descended.17,18 The silicon alkyne analogues display short Si–Si bond lengths near 2.06 Å and spectroscopic properties6,7,19 that are consistent with bond orders near three although this has been disputed.20,21 The germanium species have Ge–Ge bond lengths consistent with an approximate double bond22 and the distannyne has a Sn–Sn distance that is consistent with significant multiple bond character23 (Table 1). Computational work by Nagase and Takagi predicted that Ar*SnSnAr* would have a more trans-bent structure with a longer Sn–Sn bond distance (2.90 Å) and a narrower bending angle (Sn–Sn–C(ipso) 111.0°) than those experimentally measured for Ar′SnSnAr′ (Ar′ = C6H3-2,6-(C6H3-2,6-iPr2)2; Sn–Sn = 2.6675(4) Å, Sn–Sn–C = 125.24(7)°)23 (Scheme 2). However, by altering the Ar′ ligand through replacement of the para-hydrogen of the central aryl ring with a SiMe3 group, (Scheme 3) we showed that a single bonded structure could be obtained experimentally.24 Nagase and Takagi calculated that the energy difference between the single bonded and multiple bonded isomers of Ar′SnSnAr′ or 4-Me3Si-Ar′SnSnAr′-4-SiMe3 was only ca. 5 kcal mol−1, which indicated that a relatively small modification of the ligand could cause a large structural change,25 a view corroborated by calculations on simple MeEEMe (E = Si, Ge, Sn or Pb) model species.26 In addition, Frenking et al. had shown earlier that the strongly bent geometry of Ar*PbPbAr* was partly a result of packing effects and an isomer with a less bent geometry was close in energy.27 Calculations by Nagase and Takagi for Ar*PbPbAr* showed that in the gas phase a less strongly bent structure is slightly more stable.28 We wished to explore how changes in the steric and electronic properties of the terphenyl ligand affected the metal–metal bonding across a range of substituents. As a result, we developed a series of ligands related to Ar′ 29 or Ar* 30 (Scheme 3) with different electronic and steric properties to explore the occurrence of different isomeric structures. We now report our findings for the digermyne and distannyne series using these modified terphenyl ligands.
Table 1 Selected structural data for the first structurally characterized heavier element “ditetrelynes” with comparison data for acetylene
R–E![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) E–R E–R |
E–E–C/° |
EE/Å |
E–E/Å, single bond |
Ref. |
|
R′ = SiPri{CH(SiMe3)2}2 or C6H2-2,6-(CH(SiMe3)2)2-4-C(SiMe3)3.
|
| R–CC–R |
180 |
1.21 |
1.54 |
— |
| R′SiSiR′ a |
137.44(4) |
2.0622(9) |
2.34 |
6, 7
|
| Ar′GeGeAr′ |
128.67(8) |
2.285(6) |
2.44 |
8
|
| Ar′SnSnAr′ |
125.24(7) |
2.6675(4) |
2.81 |
11
|
| Ar*PbPbAr* |
94.26(14) |
3.1881(1) |
2.9 |
3
|
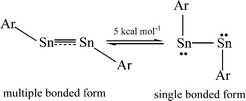 |
| | Scheme 2 Small energy difference between multiple bonded and single bonded ArSnSnAr. | |
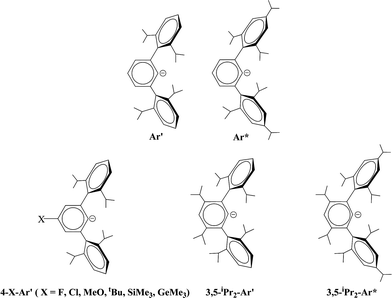 |
| | Scheme 3 Schematic drawings of Ar′, Ar* and modified ligands based on Ar′ and Ar*. | |
Results and discussion
The precursors (ArMCl)n (M = Ge or Sn; n = 1, 2)
Synthesis and spectroscopy.
Full details of the experimental work are given in the ESI.† The aryl Ge(II) and aryl Sn(II) halides were prepared by a route (route 1 in Scheme 4) similar to that reported for the preparation of (Ar′MCl)2 and Ar*MCl (M = Ge, Sn) by the straightforward reaction of 1 equivalent of ArLi with GeCl2(dioxane) or SnCl2.9,31–34 The compound [4-Me3Ge-Ar′SnCl]2 was prepared by a slightly different procedure (route 2 in Scheme 4) because of the cleavage of aromatic carbon–germanium bonds upon quenching with I2.35 They were isolated as either orange or yellow crystals. The UV-Vis spectra and 119Sn NMR spectroscopic data for aryl Sn(II) halide species are summarized in Table 2. The UV-Vis spectra of Ge(II) aryl halide and Sn(II) aryl halide which have absorptions near 350 and 400 nm respectively are assignable to the transition from the non-bonded pair to the respective 4p(Ge) or 5p(Sn) orbitals. 119Sn spectra for Sn(II) halide species in hydrocarbon solvent also displayed a single resonance in the range ca. 800 ± 400 ppm.
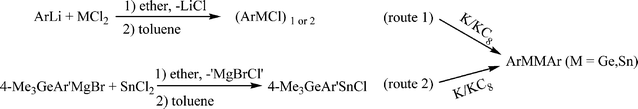 |
| | Scheme 4 Synthetic route for (ArMCl)1, 2 and ArMMAr (Ar = terphenyl ligand; M = Ge, Sn). | |
Table 2 UV-Vis (λmax, nm (ε, mol L−1 cm−1)) and 119Sn spectroscopic data for aryl M(II) halides in hexane
| |
Ar*GeCl |
(4-Cl-Ar′GeCl)2 |
(4-SiMe3-Ar′GeCl)2 |
3,5-iPr2-Ar*GeI |
|
λ
max (ε) |
393 (950) |
358 (2550) |
379 (2700) |
388 (800) |
| |
| |
(Ar*SnCl)1, 2 |
Ar*SnI |
(4-F-Ar′SnCl)2 |
(4-Cl-Ar′SnCl)2 |
(4-MeO-Ar′SnCl)2 |
(4-tBu-Ar′SnCl)2 |
(4-SiMe3-Ar′SnCl)2 |
(4-GeMe3-Ar′SnCl)2 |
(3,5-iPr2-Ar′SnCl)2 |
3,5-iPr2-Ar*SnCl |
|
λ
max (ε) |
395 (1040) |
428 (3000) |
395 (2200) |
394 (2600) |
402 (730) |
402 (1240) |
406 (2400) |
408 (740) |
399 (2200) |
416 (2700) |
| δ 119Sn NMR |
793.4 |
1140 |
719.5 |
443.3 |
1115 |
1001 |
904 |
|
749 |
751 |
The chemical shifts are affected by the substituents at the para or meta positions on the central ring of the ligand. The downfield shifts indicate that the aryl Sn(II) halide species are dissociated to monomers in solution. The 119Sn NMR shifts of compounds [4-F-Ar′SnCl]2 and [4-Cl-Ar′SnCl]2 are upfield of those of other aryl Sn(II) halides, which is opposite to what is expected on the basis of σ-inductive effects of the electronegative ring substituents. This observation may be explained on the basis that the paramagnetic shielding is decreased by more electronegative substituents, leading to upfield shifts.36,37
X-Ray crystal structures.
The structures of the ArGeCl species, where Ar = C6H3-2,6-Mes2 (Mes = C6H2-2,4,6-Me3), Ar′, Ar* were reported earlier. The data showed that {2,6-Mes2-H3C6GeCl}2,38 {Ar′GeCl}29 and {Ar*GeCl}234 had Ge–Ge bonded dimeric structures with pyramidal geometry at the germaniums and Ge–Ge distances in the range ca. 2.36–2.46 Å. Although no new X-ray structures of an ArGeCl derivative of a modified Ar′ ligand were obtained in the current work, it is reasonable to assume that the 4-X-Ar′GeCl, (X = Cl, Me3Si) structure would be similar to that of {Ar′GeCl}2. In contrast, a monomeric structure was observed for the extremely hindered iodo derivative 3,5-iPr2-Ar*GeI, which was obtained as a result of halide exchange upon reaction of GeCl2 dioxane with LiAr*-3,5-iPr2 generated from its iodo precursor. Its structure resembles that of the monomer Ar*GeCl which could be obtained in preference to its Ge–Ge bonded dimeric form (Ar*GeCl)2 by recrystallization under different conditions.38 The observation of monomeric structures for these very bulky derivatives is a result of steric effects. With the less bulky C6H3-2,6-Mes2 and Ar′, weak association occurs to afford the above mentioned Ge–Ge bonded dimers. Oddly, the Ge–Ge bond length in {Ar*GeCl}2 is the shortest of the three compounds (Ge–Ge = 2.4624(4) Å for {Ar′GeCl}2; 2.363(2) Å for Ar*GeCl; 2.443(2) Å for (2,6-Mes2H3C6GeCl)2). The longer Ge–Ge bonds in the less crowded C6H3-2,6-Mes2 or Ar′ derivatives is rationalized on the basis of secondary interactions between the germanium and one of the o-mesityl or o-isopropyl rings, which effectively increases the coordination number and weakens the Ge–Ge interactions. In the bulkier Ar* or 3,5-iPr2-Ar* derivative, this interaction does not occur for steric reasons, which permits a shorter Ge–Ge bond despite the larger size of the ligand. The only other monomeric aryl germanium(II) halide is Ge(Cl)C6H2-2,4,6-tBu3 whose monomeric character was also thought to be due to the large size of the organic substituent.39
With the exception of the monomeric 3,5-iPr2-Ar*SnCl, the terphenyl tin(II) halide species are dimerized by halide bridging (Fig. 1). The different dimerization modes observed for the aryl germanium and tin halides reflect the weaker metal–metal bonding and greater non-bonding pair character of tin. Selected Sn–C bond and Sn–Cl bond distances, which resemble those reported for Sn(II)–Cl bridged dimers,40–42 for these derivatives are listed in Table 3. As in the case of 3,5-iPr2-Ar*GeI, the 3,5-iPr2-Ar* ligand also stabilizes the monomeric structure for 3,5-iPr2-Ar*SnCl. In this case two metaiPr groups on the central phenyl rings push the flanking aryl rings forward to afford greater steric protection at the metal center, which is sufficient to prevent dimerization and stabilize the monomeric structure (Fig. 2). In the compound 3,5-iPr2-Ar*SnCl,33 the angle at tin, 98.77(9)°, is narrower than the 102.6(3)° in Ar*SnCl and 102.31(15)° in Ar*SnI due to the higher steric effect of the ligand.32
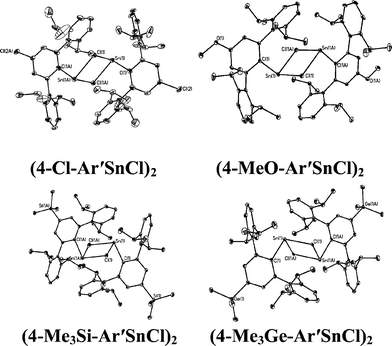 |
| | Fig. 1 Thermal ellipsoid (30%) drawings of (4-Cl-Ar′SnCl)2, (4-MeO-Ar′SnCl)2, (4-Me3Si-Ar′SnCl)2 and (4-Me3Ge-Ar′SnCl)2. Hydrogen atoms are not shown. Selected bond lengths (Å) and angles (°): (4-Cl-Ar′SnCl)2: Sn1–C1 2.230(5), Sn1–Cl1 2.540(2), Sn1–Cl1A 2.7099(18), C1–Sn1–Cl1 91.90(16), C1–Sn1–Cl1A 106.23(13), Cl1–Sn1–Cl1A 82.88(6); (4-MeO-Ar′SnCl)2: Sn1–C1 2.2225(2), Sn1–Cl1 2.5892(8), Sn1–Cl1A 2.6867(7), Cl–Sn1–Cl1 93.54(6), C1–Sn1–Cl1A 105.70(6), Cl1–Sn1–Cl1A 82.30(2); (4-Me3Si-Ar′SnCl)2: Sn1–C1 2.247(5), Sn1–Cl1 2.6679(11), Sn1–Cl1A 2.7193(11), C1–Sn1–Cl1 92.85(12), C1–Sn1–Cl1A 100.86(12), Cl1–Sn1–Cl1A 87.75(3); (4-Me3Ge-Ar′SnCl)2: Sn1–C1 2.2463(17), Sn1–Cl1 2.6157(5), Sn1–Cl1A 2.6376(5), C1–Sn1–Cl1 92.89(4), C1–Sn1–Cl1A 100.55(5), Cl1–Sn1–Cl1A 85.629(15). | |
Table 3 Selected Sn–C and Sn–Cl distances (Å) for (4-Cl-Ar′SnCl)2, (4-MeO-Ar′SnCl)2, (4-SiMe3-Ar′SnCl)2, (4-GeMe3-Ar′SnCl)2 and 3,5-iPr2-Ar*SnCl
| |
(4-Cl-Ar′SnCl)2 |
(4-MeO-Ar′SnCl)2 |
(4-MeSi3-Ar′SnCl)2 |
(4-MeGe3-Ar′SnCl)2 |
3,5-iPr2-Ar*SnCl |
| Sn–C |
2.230(5) |
2.225(2) |
2.2463(17) |
2.247(5) |
2.176(6) |
| 2.395(5) |
|
|
|
|
| Sn–Cl |
2.540(2) |
2.5892(8) |
2.6157(5) |
2.7193(11) |
2.371(3) |
| 2.668(4) |
2.6867(7) |
2.6376(5) |
2.6679(11) |
|
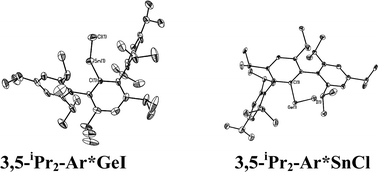 |
| | Fig. 2 Thermal ellipsoid (30%) drawings of 3,5-iPr2-Ar*GeI and 3,5-iPr2-Ar*SnCl. H atoms are not shown. Selected bond lengths (Å) and angles (°): 3,5-iPr2-Ar*GeI: Ge1–C1 2.015(4), Ge1–I1 2.6221(6), C1–Ge1–I1 108.99(6); 3,5-iPr2-Ar*SnCl: Sn1–C1 2.176(6), Sn1–Cl1 2.371(3), C1–Sn1–Cl1 133.68(14). | |
Digermynes and distannynes
Synthesis and spectroscopy.
The digermynes and distannynes were prepared by the reduction of Ge(II) aryl halide or Sn(II) aryl halide, usually with stoichiometric amounts of K or KC8 in hydrocarbon solvent. (Scheme 4) The germanium species are red in color, whereas the tin analogues form dark blue-green (teal) solutions. Their color undergoes no apparent change when they are dissolved in hydrocarbons. The UV-Vis spectra of the germanium species feature two absorbances in the range 320–380 and 430–510 nm, whereas the corresponding wavelength ranges for the tin species are 402–430 and 582–612 nm, respectively. The shifts to longer wavelength absorptions on descending a group are common in the electronic spectra of many element–element multiple bonded main group compounds.43 The two well separated absorptions in each class of compounds were calculated to be the symmetry allowed n− → n+ and π → π* transitions25 (Table 4). Despite numerous attempts to record solution 119Sn NMR spectra, with use of a variety of parameters, no signals have been detected so far. This may be a result of the unsymmetric electronic environment at tin causing rapid relaxation through anisotropy of the chemical shift tensor, thereby broadening the signal into the baseline. However the solid state 119Sn NMR spectra for Ar′SnSnAr′ (δiso = +335.3 ppm) and Ar*SnSnAr* (δiso = −36.2 ppm) showed that they had substantially different spectroscopic parameters, which indicated that they had different solid-state structures.44
Table 4 UV-Vis data λmax, nm (ε, mol L−1 cm−1) for neutral digermynes and distannynes in hexane
| Ar′GeGeAr′ |
Ar*GeGeAr* |
(4-Cl-Ar′Ge)2 |
(4-SiMe3-Ar′Ge)2 |
(3,5-iPr2-Ar′Ge)2 |
| 371 (34000) |
365 (31000) |
324 (5100) |
380 (5600) |
378 (3033) |
| 501 (7500) |
490 (5500) |
438 (1800) |
510 (1050) |
496 (407) |
| |
| Ar′SnSnAr′ |
Ar*SnSnAr* |
(4-F-Ar′Sn)2 |
(4-Cl-Ar′Sn)2 |
(4-MeO-Ar′Sn)2 |
(4-tBu-Ar′Sn)2 |
(4-SiMe3-Ar′Sn)2 |
(4-GeMe3-Ar′Sn)2 |
(3,5-iPr2-Ar′Sn)2 |
(3,5-iPr2-Ar*Sn)2 |
| 410 (2000) |
409 (1800) |
425 (4200) |
406 (4300) |
402 (3960) |
406 (4600) |
416 (4700) |
430 (4450) |
414 (4600) |
424 (4900) |
| 597 (1700) |
593 (1400) |
592 (3100) |
592 (1550) |
582 (1050) |
592 (1100) |
608 (1200) |
602 (2200) |
608 (1200) |
612 (550) |
X-Ray crystal structures.
The structures of the digermynes are illustrated in Fig. 3 and the corresponding tin species are illustrated in Fig. 4 and 5. Selected structural data are summarized in Table 5. In contrast to the usual linear structures of alkynes, they all possess trans-bent CGeGeC or CSnSnC core arrays. The most important structural parameters are the trans-bending angles and the M–M distances. The Ge–Ge bonds for Ar′GeGeAr′ (2.2850(8) Å) and 4-Cl-Ar′GeGeAr′-4-Cl (2.3071(3) Å) are slightly longer than the Ge–Ge distances (2.2060(7) and 2.2260(7) Å) in BbtGeGeBbt (Bbt = 2,6-bis[bis(trimethylsilyl)methyl]-4-[tris(trimethylsilyl)methyl]phenyl), whereas the Ge–Ge bond lengths for 4-Me3Si-Ar′GeGeAr′-4-SiMe3 (2.2438(8) Å) and 3,5-iPr2-Ar*GeGe-3,5-iPr2Ar* (2.2125(13) Å) are almost as short as they are in BbtGeGeBbt. They lie in the lower half of the known Ge–Ge bond length range (2.21–2.46 Å) for digermenes45,46 and are considerably shorter than the Ge–Ge single bond distance (2.44 Å) in elemental germanium,47 which suggests considerable Ge–Ge multiple bond character. Based on the structural data, it can be argued that the bond order in these digermyne species is at least two, which is in agreement with the order calculated for the model species MeGeGeMe.18,22 The different Ge–Ge bond distances can be rationalized, at least in part, on the basis of the electronic properties of the substituents. Thus BbtGeGeBbt, 4-Me3Si-Ar′GeGeAr′-4-SiMe3 and 3,5-iPr2-Ar*GeGe-3,5-iPr2Ar*, which have the shortest Ge–Ge bonds, all possess electron releasing alkyl or silyl substituents on the central aryl rings, whereas Ar′GeGeAr′, which does not have this feature, and 4-Cl-Ar′GeGeAr′-4-Cl, which contains an electron withdrawing Cl substituent, have longer Ge–Ge bonds. Employing arguments similar to those used to explain inversion barriers, electron releasing groups tend to decrease bending, whereas electron withdrawing groups have the opposite effect. A lower degree of bending leads to a higher bond order and a shorter Ge–Ge bond. Thus 4-Me3Si-Ar′GeGeAr′-4-SiMe3 and 3,5-iPr2-Ar*GeGe-3,5-iPr2Ar* have relatively wide Ge–Ge–C angles. The 136.13(17)° observed in the very crowded 3,5-iPr2-Ar*GeGe-3,5-iPr2Ar* approaches the 137.44(4)° in Sekiguchi's disilyne {SiiPr(CH(SiMe3)2)2}SiSi{SiiPr(CH(SiMe3)2)2} which has a very short Si–Si bond (2.0622(9) Å) that is triple in character.6 The angular correlation is not exact (cf. the 128.67(8)° in Ar′GeGeAr) but it should be borne in mind that packing forces (see below) can also influence the structural parameters. Except for the very sterically crowded 3,5-iPr2-Ar*GeGe-3,5-iPr2Ar* in which the angle between central ligand ring and C(ipso)GeGeC(ipso) array is ca. 47.2(2)°, the central rings of the digermyne ligands are essentially coplanar with the C(ipso)GeGeC(ipso) array.
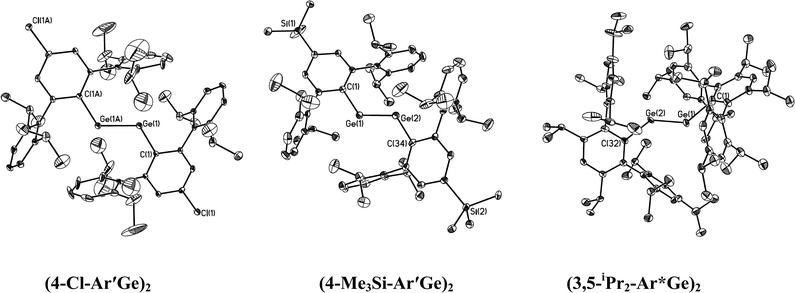 |
| | Fig. 3 Thermal ellipsoid (30%) drawings (without H atoms) of three digermynes. | |
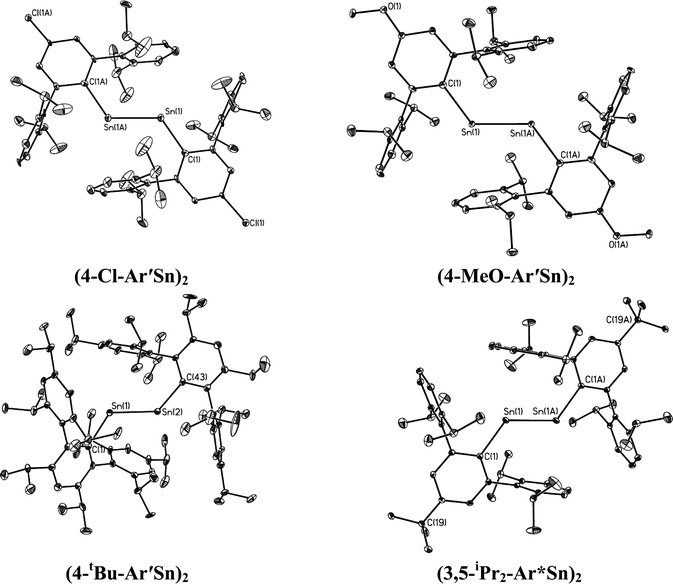 |
| | Fig. 4 Thermal ellipsoid (30%) drawings of (4-Cl-Ar′Sn)2, (4-MeO-Ar′Sn)2, (4-tBu-Ar′Sn)2 and (3,5-iPr2-Ar*Sn)2. Hydrogen atoms are not shown. | |
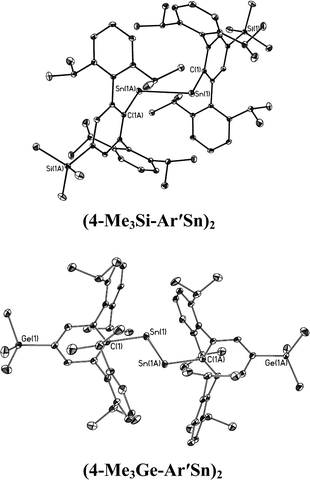 |
| | Fig. 5 Thermal ellipsoid (30%) drawings of single bonded (4-Me3Si-Ar′Sn)2 and (4-Me3Ge-Ar′Sn)2. Hydrogen atoms are not shown. | |
Table 5 Selected structural data for digermynes ((4-Cl-Ar′Ge)2, (4-Me3Si-Ar′Ge)2and (3,5-iPr2-Ar*Ge)2) and distannynes ((4-Cl-Ar′Sn)2, (4-MeO-Ar′Sn)2, (4-tBu-Ar′Sn)2, (4-Me3Si-Ar′Sn)2, (4-Me3Ge-Ar′Sn)2 and (3,5-iPr2-Ar*Sn)2)
|
|
Ar′GeGeAr′ |
(4-Cl-Ar′Ge)2 |
(4-Me3Si-Ar′Ge)2 |
(3,5-iPr2-Ar*Ge)2 |
| Ge–Ge/Å |
2.2850(8) |
2.3071(3) |
2.2438(8) |
2.2125(13) |
| Ge–C/Å |
1.996(3) |
1.966(5) |
1.973(5) |
2.021(6) |
| C–Ge–Ge/° |
128.67(8) |
124.19(16) |
128.44(16) |
136.13(17) |
| C–Ge–Ge–C/° |
180 |
180 |
180 |
165.8(2) |
| |
| |
Ar′SnSnAr |
(4-Cl-Ar′Sn)2 |
(4-MeO-Ar′Sn)2 |
(4-tBu-Ar′Sn)2 |
(4-Me3Si-Ar′Sn)2 |
(4-Me3Ge-Ar′Sn)2 |
(3,5-iPr2-Ar*Sn)2 |
| Sn–Sn/Å |
2.6675(4) |
2.672(2) |
2.6480(12) |
2.6461(3) |
3.0577(2) |
3.077(12) |
2.7205(12)–2.7360(14) |
| Sn–C/Å |
2.191(3) |
2.159(12) |
2.165(7) |
2.173(2) |
2.2084(11) |
2.196(7) |
2.193(1)–2.200(8) |
| C–Sn–Sn/° |
125.24(7) |
121.8(4) |
124.2(2) |
123.98(5) |
99.07(3) |
97.79(17) |
125.1(2)–127.6(2) |
| C–Sn–Sn–C/° |
180 |
180 |
180 |
180 |
180 |
180 |
151.97–166.22 |
In contrast to the digermynes, two dramatically different bonding modes are observed in their tin analogues (Fig. 4 and 5). A multiple bonded isomeric form is found in Ar′SnSnAr′, 4-Cl-Ar′SnSnAr′-4-Cl, 4-MeO-Ar′SnSnAr′-4-MeO and 4-tBu-Ar′SnSnAr′-4-tBu (Fig. 4) which have short Sn–Sn bond distances in the range 2.6461(3) Å to 2.6675(4) Å (cf. Sn–Sn single bond 2.81 Å for element tin)47 and relatively moderate bending angles in the range 121.8(4)–125.24(7)°. In contrast, the introduction of p-SiMe3 and GeMe3 substituents at the para position of the central aryl ring of the Ar′ ligand results in single bonded isomers (Fig. 5) with Sn–Sn distances of 3.0577(2) and 3.077(12) Å and sharp Sn–Sn–C bending angles of 99.07(3) and 97.79(17)°. The Sn–Sn distances are slightly shorter than the 2.68(1) Å found in the tristannaallene Sn{Sn(SitBu3)2}2 but are longer than the Sn–Sn double bond (2.59(1) Å) in the cyclotristannene (tBu3Si)2SnSn(SitBu3)Sn(SitBu3)48 which resemble the 2.60 Å predicted for the sum of the double bond radii for tin.49 On the basis of these comparisons, a bond order between 1.5 and 2.0 can be estimated for the Sn–Sn bond in compounds Ar′SnSnAr′, 4-Cl-Ar′SnSnAr′-4-Cl, 4-MeO-Ar′SnSnAr′-4-OMe and 4-tBu-Ar′SnSnAr′-4-tBu. For the previously reported single bonded isomer 4-SiMe3-Ar′SnSnAr′-4-SiMe3, the calculations of Nagase and Takagi,28 and electronic spectroscopy, showed that this structure relaxes to the multiple bonded form in solution. We wished to explore these unusual structural differences between the solid state and solution phases. We expected that the replacement of the para-SiMe3 substituent with a similar tBu group would also afford the single bonded isomer. To our surprise a multiple bonded isomer was obtained in the solid state which had a short Sn–Sn bond (2.6461(3) Å) and a wide Sn–Sn–C angle (123.98(5)°). We then decided to synthesize corresponding GeMe3 and SnMe3 substituted structures to further investigate this phenomenon. Unfortunately, due to problems with Sn–C bond cleavage, a para-SnMe3 substituted ligand has not yet been obtained. However, the use of the –GeMe3 ligand led to a single bonded distannyne 4-Me3Ge-Ar′SnSnAr′-4-GeMe3 whose structure was very similar to its SiMe3 substituted analogue 4-Me3Si-Ar′SnSnAr′-4-SiMe3 (Table 5). These experimental data confirmed the earlier reported structural results for 4-Me3Si-Ar′SnSnAr′-4-SiMe3 and show that seemingly minor alterations to the ligand produce very large changes in the structure of the distannyne. The data are also in harmony with computational work of Nagase and Head-Gordon, which indicated only small (ca. 5 kcal mol−1) energy differences between the multiple bonded and single bonded isomers.25,26 This energy difference is sufficiently small to be influenced by packing effects. On the other hand, Nagase and Takagi25 calculated that the multiple bonded Ar′GeGeAr′ is more stable than its single bonded form by 14.7 kcal mol−1, which is apparently too high an energy difference to allow the structure to be changed by packing forces. As a result only the multiple bonded isomer is observed for the digermynes. A new data set for 4-Me3Si-Ar′SnSnAr′-4-SiMe3 was also obtained and a comparison of its structure and spectroscopic properties with those of the hydride derivative 4-Me3Si-Ar′Sn(μ-H)2SnAr′-4-SiMe3 ruled out the remote possibility that 4-Me3Si-Ar′SnSnAr′-4-SiMe3 and 4-Me3Ge-Ar′SnSnAr′-4-GeMe3 contained bridging hydride.50 Inspection of the packing diagram of 4-Me3Si-Ar′SnSnAr′-4-SiMe3 and 4-tBu-Ar′SnSnAr′-4-tBu does not reveal an obvious explanation for the packing differences (Fig. 6). Finally we note that in the multiple bonded structure the central aryl rings of the ligands are coplanar with the C(ipso)SnSnC(ipso) array, whereas in the case of the single bonded structures these rings lie essentially perpendicular to the core (ca. 87.7(4)°) (Fig. 7). The complex 3,5-iPr2-Ar*SnSn-3,5-iPr2Ar* has a structure intermediate between the multiple and single bonded extremes, with Sn–Sn distances near 2.73 Å and C–Sn–Sn–C torsion angles in the range 151.97–160.22°. The angles between the coordination planes at Sn1 and Sn2 and the central aryl rings of the terphenyl ligand are in the range 38.64–70.07°. In related diaryldiphosphenes, ArP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PAr, calculations have shown that the twist angles between the plane of the aryl ring and that of the core affects the stability of lowest energy excited states and all such angles are accessible for under 5 kcal mol−1.51,52 This quantity is similar to the energy difference between the multiple and single bonded distannyne isomers so that the twist angles likely also play a role in stabilizing the isomeric forms.
PAr, calculations have shown that the twist angles between the plane of the aryl ring and that of the core affects the stability of lowest energy excited states and all such angles are accessible for under 5 kcal mol−1.51,52 This quantity is similar to the energy difference between the multiple and single bonded distannyne isomers so that the twist angles likely also play a role in stabilizing the isomeric forms.
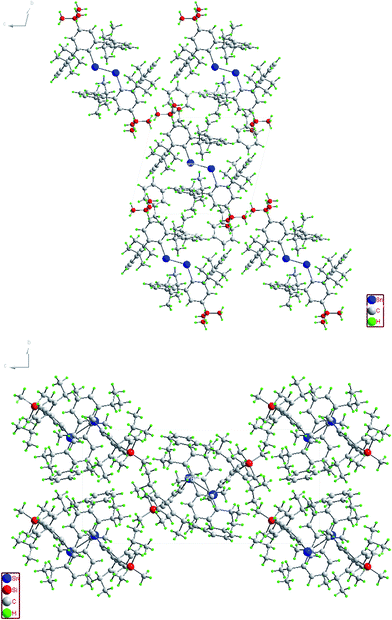 |
| | Fig. 6 Packing diagram of (4-tBu-Ar′Sn)2 (top) and (4-Me3Si-Ar′Sn)2 (bottom) along the a-axis. | |
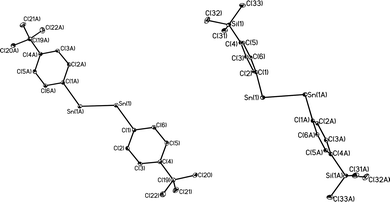 |
| | Fig. 7 Comparison of core geometries for (4-tBu-Ar′Sn)2 (left) and (4-Me3Si-Ar′Sn)2 (right); flanking aryl groups are not shown. | |
Mössbauer spectroscopy of compounds Ar′SnSnAr′, Ar*SnSnAr*, 4-tBu-Ar′SnSnAr′-4-tBu and 4-Me3Si-Ar′SnSnAr′-4-SiMe3
Mössbauer spectral data were reported earlier for Ar′SnSnAr′ and Ar*SnSnAr*. These indicated that they had different structures in the solid state.44 However, this could not be verified in detail due to the poor diffraction characteristics of Ar*SnSnAr*. To obtain more information about the two structural modes of distannyne, the multiple bonded 4-tBu-Ar′SnSnAr′-4-tBu and single-bonded 4-SiMe3-Ar′SnSnAr′-4-SiMe3 were selected for Mössbauer study. The Mössbauer data are given in Table 6. It can be seen that all compounds possess a single tin(II) environment with a doublet absorption at shifts of 2.658(2) mm s−1 for Ar′SnSnAr′,44 2.60(2) mm s−1 for 4-tBu-Ar′SnSnAr′-4-tBu, 2.69 mm s−1 for Ar*SnSnAr*44 and 2.908(4) mm s−1 for 4-Me3Si-Ar′SnSnAr′-4-SiMe3. The quadrupolar splitting (QS) was very similar for the multiple-bonded compounds Ar′SnSnAr′ (2.995(2) mm s−1) and 4-tBu-Ar′SnSnAr′-4-tBu (3.01(2) mm s−1). For 4-Me3Si-Ar′SnSnAr′-4-SiMe3, however, the quadrupolar splitting (4.736(4) mm s−1) is much larger, which is consistent with the more strongly bent geometry at tin observed in its crystal structure. The splitting for Ar*SnSnAr* (3.730(3) mm s−1) lies between the single and multiple bonded extremes and is indicative of an intermediate structure. In the absence of X-ray data it is not possible to correlate this intermediate isomer shift with the structural details. One possibility is that the Sn–Sn distance and bending angle for Ar*SnSnAr* lie between those of the single and multiple bond isomers. However, it is also possible that the structure of Ar*SnSnAr* resembles that of 3,5-iPr2-Ar*SnSn-3,5-iPr2Ar* in which torsion angles 14–28° exist between the tin coordination planes as well as a slightly longer Sn–Sn distance and marginally wider Sn–Sn–C bending angles. These structural differences may be sufficient to produce the observed changes in the Mössbauer spectrum.
Table 6 Mössbauer data for Ar′SnSnAr′, Ar*SnSnAr*, (4-tBu-Ar′Sn)2 and (4-Me3Si-Ar′Sn)2
| |
Ar′SnSnAr′ |
(4-tBu-Ar′Sn)2 |
Ar*SnSnAr* |
(4-Me3Si-Ar′Sn)2 |
| IS/mm s−1 |
2.658(2) |
2.60(2) |
2.69(3) |
2.908(4) |
| QS/mm s−1 |
2.995(2) |
3.01(2) |
3.730(3) |
4.736(4) |
Conclusions
A variety of germanium and tin analogues of alkyne derivatives of modified Ar′ and Ar* ligands were prepared by reducing their aryl Ge(II) halide or aryl Sn(II) halide species using K or KC8. X-Ray crystallographic data showed that aryl Ge(II) halides and aryl Sn(II) halides stabilized by Ar′ and modified Ar′ ligands have Ge–Ge bonded dimeric structures, however, the corresponding tin species are all dimerized through bridging halogen atoms with no Sn–Sn bonding. More sterically demanding ligands such as Ar* and 3,5-iPr2-Ar* can stabilize monomeric aryl M(II) halide structures. Reduction of these aryl metal halides afforded germanium and tin analogues of alkynes. The digermynes displayed essentially planar trans-bent structures with short Ge–Ge bonds and an approximate bond order of at least two. However, their tin congeners displayed two distinct solid-state structures that either had a multiple bonded structure similar to the germanium species or had a more strongly bent, single-bonded structure. All the germanium and tin alkyne analogues have multiple bonded structures in hydrocarbon solution. These data are corroborated by computational studies, which show that there is a very small energy difference between the multiple and single bonded tin structures. The above findings are consistent with the increasing preference for non-bonding pair character in the heavier main group p-block elements.
Acknowledgements
We are grateful to the National Science Foundation (CHE-0948417) for financial support.
Notes and references
- P. Jutzi, Angew. Chem., Int. Ed., 2000, 39, 3797 CrossRef CAS.
- M. Weidenbruch, J. Organomet. Chem., 2002, 646, 39 CrossRef CAS.
- L. Pu, B. Twamley and P. P. Power, J. Am. Chem. Soc., 2000, 122, 3524 CrossRef CAS.
- N. Wiberg, M. Niedermayer, G. Fischer, H. Nöth and M. Suter, Eur. J. Inorg. Chem., 2002, 1066 CrossRef CAS.
- N. Wiberg, S. K. Vasisht, G. Fischer and P. Z. Mayer, Z. Anorg. Allg. Chem., 2004, 630, 1823 CrossRef CAS.
- A. Sekiguchi, R. Kingo and M. Ichinohe, Science, 2004, 305, 1755 CrossRef CAS.
- T. Sasamori, K. Hironaka, Y. Sugiyama, N. Takagi, S. Nagase, Y. Hosoi, Y. Furukawa and N. Tokitoh, J. Am. Chem. Soc., 2008, 130, 13856 CrossRef CAS.
- M. Stender, A. D. Phillips, R. J. Wright and P. P. Power, Angew. Chem., Int. Ed., 2002, 41, 1785 CrossRef CAS.
- L. Pu, A. D. Phillips, A. F. Richards, M. Stender, R. S. Simons, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2003, 125, 11626 CrossRef CAS.
- Y. Sugiyama, T. Sasamori, Y. Hosoi, Y. Furukama, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 1023 CrossRef.
- A. D. Phillips, R. J. Wright, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2002, 124, 5930 CrossRef CAS.
- S. P. Green, C. Jones, P. C. Junk, K. A. Lippert and A. Stasch, Chem. Commun., 2006, 3978 RSC.
- S. Nagendran, S. S. Sen, H. W. Roesky, D. Koley, H. Grubmuller, A. Pal and R. Herbst-Irmer, Organometallics, 2008, 27, 5459 CrossRef CAS.
- R. Jambor, B. Kašná, K. N. Kirschner, M. Schürmann and K. Jurkschat, Angew. Chem., Int. Ed., 2008, 47, 1650 CrossRef CAS.
- P. P. Power, Chem. Commun., 2003, 2091 RSC.
- P. P. Power, Organometallics, 2007, 26, 4362 CrossRef CAS.
- M. Lein, A. Krapp and G. Frenking, J. Am. Chem. Soc., 2005, 127, 6290 CrossRef CAS.
- C. R. Landis and F. Weinhold, J. Am. Chem. Soc., 2006, 128, 7335 CrossRef CAS.
- V. Kravchenko, R. Kinjo, A. Sekiguchi, M. Ichinohe, R. West, Y. S. Balazs, A. Schmidt, M. Karni and Y. Apeloig, J. Am. Chem. Soc., 2006, 128, 14472 CrossRef CAS.
- G. Frenking, A. Krapp, S. Nagase, N. Takagi and A. Sekiguchi, ChemPhysChem, 2006, 7, 799 CrossRef CAS.
- C. A. Pignedoli, A. Curioni and W. Andreoni, ChemPhysChem, 2006, 7, 801 CrossRef CAS.
- T. L. Allen, W. H. Fink and P. P. Power, J. Chem. Soc., Dalton Trans., 2000, 407 RSC.
- N. Takagi and S. Nagase, Organometallics, 2001, 20, 5498 CrossRef CAS.
- R. C. Fischer, L. Pu, J. C. Fettinger, M. A. Brynda and P. P. Power, J. Am. Chem. Soc., 2006, 128, 11366 CrossRef CAS.
- N. Takagi and S. Nagase, Organometallics, 2007, 26, 469 CrossRef CAS.
- Y. Jung, M. Brynda, P. P. Power and M. Head-Gordon, J. Am. Chem. Soc., 2006, 128, 7185 CrossRef CAS.
- Y. Chen, M. Hartmann, M. Diedenhofen and G. Frenking, Angew. Chem., Int. Ed., 2001, 40, 2051 CrossRef.
- N. Takagi and S. Nagase, Organometallics, 2007, 26, 3627 CrossRef CAS.
- B. Schiemenz and P. P. Power, Angew. Chem., Int. Ed. Engl., 1996, 35, 2150 CrossRef CAS.
- B. Schiemenz and P. P. Power, Organometallics, 1996, 15, 958 CrossRef CAS.
- T. Fjeldberg, A. Haaland, B. E. R. Schilling, M. F. Lappert and A. J. Thorne, J. Chem. Soc., Dalton Trans., 1986, 1551 RSC.
- L. Pu, M. M. Olmstead, P. P. Power and B. Schiemenz, Organometallics, 1998, 17, 5602 CrossRef CAS.
- B. E. Eichler, L. Pu, M. Stender and P. P. Power, Polyhedron, 2001, 20, 551 CrossRef CAS.
- M. Stender, L. Pu and P. P. Power, Organometallics, 2001, 20, 1820 CAS.
- S. M. Moerlein, J. Org. Chem., 1987, 52, 664 CrossRef CAS.
- B. Wrackmeyer, Annu. Rep. NMR Spectrosc., 1999, 38, 203 CrossRef CAS.
- B. E. Eichler, B. L. Phillips, P. P. Power and M. P. Augustine, Inorg. Chem., 2000, 39, 5450 CrossRef.
- R. S. Simons, L. Pu, M. M. Olmstead and P. P. Power, Organometallics, 1997, 16, 1920 CrossRef CAS.
- P. Jutzi and C. Leue, Organometallics, 1994, 13, 2898 CrossRef CAS.
- M. Brynda, R. Herber, P. B. Hitchcock, M. F. Lappert, I. Nowik, P. P. Power, A. V. Protchenko, A. Ruzicka and J. Steiner, Angew. Chem., Int. Ed., 2006, 45, 4333 CrossRef CAS.
- R. W. Chorley, P. B. Hitchcock, B. S. Jolly, M. F. Lappert and G. A. Lawless, J. Chem. Soc., Chem. Commun., 1991, 1302 RSC.
- C. Eaborn, P. B. Hitchcock, J. D. Smith and S. E. Sozerli, Organometallics, 1997, 16, 5653 CrossRef CAS.
- N. Tokitoh, T. Arai, T. Sasamori, R. Okazaki, S. Nagase, H. Uekusa and Y. Okashi, J. Am. Chem. Soc., 1998, 120, 433 CrossRef CAS.
- G. H. Spikes, J. R. Giuliani, M. P. Augustine, I. Nowik, R. H. Herber and P. P. Power, Inorg. Chem., 2006, 45, 9132 CrossRef CAS.
- J. Escudie and H. Ranaivonjatovo, Adv. Organomet. Chem., 1999, 44, 113 CAS.
- P. P. Power, Chem. Rev., 1999, 99, 3463 CrossRef CAS.
-
A. F. Wells, Structural Inorganic Chemistry, 5th edn, Clarendon Press, UK, 1986, p. 1279 Search PubMed.
- N. Wiberg, H. W. Lerner, S. K. Vasisht, S. Wagner, K. Karaghiosoff, H. Nöth and W. Ponikwar, Eur. J. Inorg. Chem., 1999, 1211 CrossRef CAS.
- P. Pyykko and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770 CrossRef.
- E. Rivard, R. C. Fischer, R. Wolf, Y. Peng, W. A. Merrill, N. D. Schley, Z. Zhu, L. Pu, J. C. Fettinger, S. J. Teat, I. Nowik, R. H. Herber, N. Takagi, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2007, 129, 16197 CrossRef CAS.
- H.-L. Peng, J. L. Payton, J. D. Protasiewicz and M. C. Simpson, J. Phys. Chem. A, 2009, 113, 7054 CrossRef CAS.
- Y. Amatatsu, J. Phys. Chem. A, 2009, 113, 9667 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and spectroscopic data. CCDC reference numbers 771266–771280. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00240b |
| ‡ Current address: Department of Chemistry, Institute of Inorganic Chemistry, Graz University of Technology, Stremayrgasse 16/IV, A-8010 Graz, Austria. |
| § Current address: California State University, Dominguez Hills, 1000 East Victoria Street, Carson, California, 90747, USA. |
| ¶ Current address: Department of Chemistry, Acadia University, Wolfville, Nova Scotia, Canada B4P 2R6. |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.