Urea electrolysis: direct hydrogen production from urine†
Bryan K.
Boggs
,
Rebecca L.
King
and
Gerardine G.
Botte
*
Department of Chemical and Biomolecular Engineering, Ohio University, Athens, OH, USA. E-mail: botte@ohio.edu; Fax: +1 740 593 0873; Tel: +1 740 593 9670
First published on 1st July 2009
Abstract
A new technology has been developed that accomplishes the direct conversion of urine and urea to pure hydrogenvia electrochemical oxidation with an inexpensive nickel catalyst.
The utilization of wastewater for useful fuel has been gathering recent attention due to society’s need for alternative energy sources. The electrooxidation of urea found at high concentrations in wastewater simultaneously accomplishes fuel production and remediation of harmful nitrogen compounds that currently make their way into the atmosphere and groundwater. Pure hydrogen was collected in the cathode compartment at 1.4 V cell potential, where water electrolysis does not occur appreciably. It was determined that an inexpensive nickel catalyst is the most active and stable for the process.
Urine is the most abundant waste on Earth. The largest constituent of urine is urea, which is a significant organic source of H, C, O, and N. Despite the numerous benefits of using urea/urine for hydrogen production,1 there is not a single technology that directly converts urea to hydrogen.1,2 In addition to sustaining hydrogen resources, such a process could denitrificate urea-rich water that is commonly purged into rivers, creeks, and tributaries from municipal wastewater treatment plants. Currently, nitrate concentration in these waters is regulated at 10 mg L−1, but available denitrification technologies are expensive and inefficient.3 Converting urea to valuable products before it naturally hydrolyzes to ammonia, which generates gas-phase ammonia emissions and contributes to ammonium sulfate and nitrate formation in the atmosphere, will save billions of dollars spent each year on health costs.4 Here we demonstrate a technology for improving hydrogen resources for energy sustainability by recycling waste materials such as human excreta. We have developed an electrochemical process that produces H2 from urine/urea as shown in Fig. 1.5

Our results demonstrate that human urine, with an average concentration of 0.33 M urea,6 can be electrochemically oxidized with an inexpensive transition metal, nickel, according to eqn (1–4).
| CO(NH2)2(aq) + 6OH− → N2(g) + 5H2O(l) + CO2(g) + 6e− | (1) |
| Ni(OH)2(s) + OH− → NiOOH(s) + H2O(l) + e− | (2) |
| 6H2O(l) + 6e− → 3H2(g) + 6OH− | (3) |
| CO(NH2)2(aq) + H2O(l) → N2(g) + 3H2(g) + CO2(g) | (4) |
Nitrogen is generated from the anode demonstrating nitrate remediation of wastewater while water is reduced at the cathode producing valuable hydrogen for the impending hydrogen economy.
Fig. 2a shows the cyclic voltammogram (CV) comparison of different electrocatalysts (Pt, Pt–Ir, Rh, and Ni) for the electrooxidation of urea in alkaline media. Polarization curves between the various metals in the presence and absence of 0.33 M urea and 5 M KOH at a scan rate of 10 mV s−1 from −0.1 to 0.8 V versus Hg/HgO reference supported by a Luggin capillary at 25 °C shows that Ni is the most active catalyst in terms of current density. Scanning the potential more negative than −0.1 V revealed no oxidation current. The electrodes were 4 cm2 based on geometric area of Ti foil (inert) deposited with an average 10.0 ± 0.1 mg of respective metal. The counter electrode was a 25 cm2 Pt foil. All electrochemical experiments were performed in a conventional three-electrode cell powered by a Solartron 1281 Multiplexer potentiostat. Fig. 2b, constant voltage analysis at 1.4 V in 5 M KOH–0.33 M urea at 25 °C, further shows Ni is the most stable and active electrocatalyst for the electrooxidation of urea in alkaline media. This potential was chosen from the fact that waterreduction is kinetically friendly at −0.83 V vs. SHE7–9 (standard hydrogen electrode), and the electrooxidation of urea is occurring at 0.55 V vs. Hg/HgO according to Fig. 2c. Nickel in basic media is rapidly converted to Ni(OH)2 (Ni2+) which is further oxidized to NiOOH (Ni3+). This transition from Ni2+ to Ni3+ enhances catalytic electrooxidation behavior of small organic compounds.9–11 The oxidation of Ni(OH)2to NiOOH is represented by anodic peak a1. Fig. 2c shows that urea electrolysis begins at the same potential where NiOOH is formed, suggesting that Ni3+ is the active form for ureaoxidation. This is seen as an increase of current density at a1 in the presence of urea. Furthermore, a change in slope due to the onset of water electrolysis can be seen at more positive potentials.
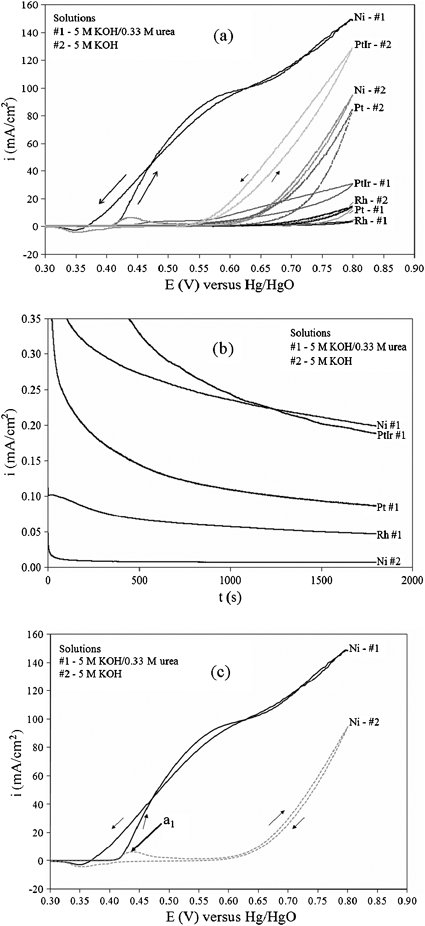 | ||
| Fig. 2 Anode catalyst analysis at 25 °C: (a) cyclic voltammograms obtained in 5 M KOH with and without the presence of urea on Ti-foil supported electrodes with a 10 mV s−1 scan; (b) constant voltage test with 1.4 V potential step with 5 M KOH–0.33 M urea; (c) cyclic voltammogram of Ni/Ti electrode in the absence (grey) and presence (black) of 0.33 M KOH in 5 M KOH solution. | ||
We found that nickel oxyhydroxide modified nickel electrodes (NOMN) for urea electrooxidation on different metallic substrates (Ni foil, Ni gauze, Ti foil, and Ti gauze) that have been electroplated with 10.0 ± 0.1 mg of Ni using a Watts bath then activated following the procedure developed by Vaze et al.10 yield higher current densities than those of M/Ni, where M represents the metallic substrate.
NOMN electrodes were used for the remaining electrochemical behavior analyses. Fig. 3 demonstrates that there is an influence of scan rate on the cyclic voltammetry behavior of NOMN electrodes. The electrooxidation of urea in this system was characterized with CVs from 0.0 to 0.6 V versus Hg/HgO at scan rates of 5 to 95 mV s−1. Fig. 3a shows that the cathodic peak does not shift in potential as the scan rate increases in the presence of urea. The curves are shown from 0.0 to 0.5 V for scaling purposes. Fig. 3b indicates that cyclic voltammetry peak cathodic currents (Ipc) followed a linear correlation with the square root of the scan rate (R2 = 0.976). Together, these criteria confirm that the production of NiOOH from Ni(OH)2 is a reversible diffusion-controlled process. The increase in cathodic currents as a function of scan rate indicates that the electrooxidation of urea is slower than the electrooxidation of nickel species to a higher valence state. Therefore, we hypothesize that the rate-limiting step is the reaction between NiOOH and urea absorbed on the surface.
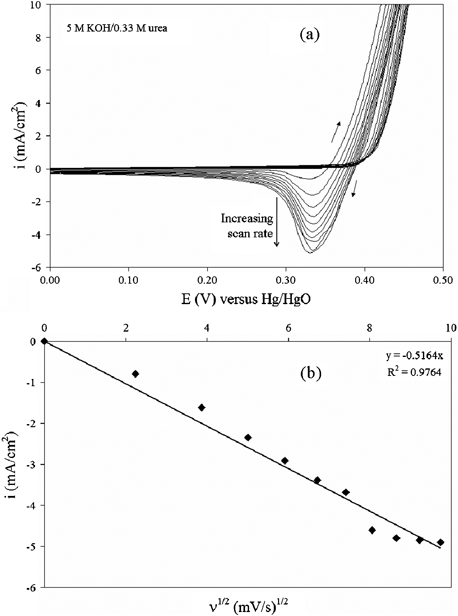 | ||
| Fig. 3 (a) Cyclic voltammograms obtained in 5 M KOH + 0.33 M urea for the NOMN electrode with various scan rates (ν) from 5 mV s−1 to 95 mV s−1. (b) The plot of cathodic current density variation with ν1/2. | ||
The electrocatalytic behavior of the NOMN electrode towards ureaoxidation in basic media was further studied with cyclic voltammetry and constant voltage analyses at varying operating conditions. It was found that the current density increases with temperature. Also, higher concentrations of KOH favor the reaction rate. As the concentration of KOH exceeded 5 M, the NOMN electrode lost activity as seen by a decrease in current density during constant voltage analyses. This could be due to faster disappearance of the oxide layer, which was visibly evident.
Energy dispersive X-ray (EDX) microanalyses of a Ti foil (99.99% pure) electrode (deposited with 10.0 ± 0.1 mg of Ni and then activated into a NOMN electrode) before and after urea electrolysis at 1.4 V for 30 minutes in 5 M KOH–0.33 M urea shows that the amount of atomic carbon and oxygen on the electrode surface increases during electrolysis. This may be contributed to adsorption of products onto the surface. As a result, the surface atomic composition of Ni decreases leading to decay in the current density during the constant voltage study.
Anode and cathode gases were collected separately in a Hoffman apparatus filled with a solution of 5 M KOH in the presence and absence of 0.33 M urea and analyzed viagas chromatography. The electrolyses were performed at a constant voltage of 1.5 V and 25 °C for 22 hours. Currents observed were 20 mA and less than 1 mA in the presence and absence of urea, respectively. This verifies that water electrolysis is not occurring to an appreciable extent. Pure H2 was observed at the cathode while N2 (96.1%) with trace amounts of O2 (1.9%) and H2 (2.0%) were detected at the anode for urea electrolysis. A small amount of hydrogen (0.28%) was detected at the anode in the absence of urea as well, which suggests this hydrogen is not a product of urea electrolysis. Instead, it is likely due to the nickel transition reaction Ni(OH)2 → NiOOH. Carbon dioxide was not detected as part of the gas phase for urea electrolysis, but is believed to have formed potassium carbonate in the liquid phase. After 22 electrolysis hours, 13% of the urea was converted into hydrogen, nitrogen, and potassium carbonate, as determined using a heat treatment method for urea determination.
We have demonstrated that urea at the concentrations found in urine can be used for the production of H2 through this new technology utilizing inexpensive Ni. The electrolysis of urine was also demonstrated viacyclic voltammetry (see ESI† , Fig. S4). Theoretically, hydrogen can be produced at $0.69 kg−1 based on an electricity cost of $0.07 kWh−1 and the proposed electrochemical reactions (eqn (1–4)) that have been developed from electrochemical data and gas analyses.
Table 1 shows energy consumption (Wh per gram of hydrogen) and cost of hydrogen comparison between urea and water electrolysis at experimental conditions with Ni anodes. Utilizing the same cell configuration as the GC analysis mentioned in the ESI† , voltages for both urea (1.4 V) and water electrolysis (2.0 V) were found that resulted in cell currents of 20 mA. Using these voltages for comparison, we found that 30% less energy is required for urea electrolysis, which generated 36% cheaper hydrogen compared to water electrolysis.
In the past, research pertaining to urea electrolysis exclusively involved the possibility of developing artificial kidneys for portable dialysis devices utilizing platinum electrodes in acidic buffers.12–15 There is great interest in the scientific community for finding non-platinized catalyst alternatives such as Ni for hydrogen production. We have demonstrated that the technology is effective for both urea and urine.
Notes and references
- Research on Applied Bioelectrochemistry, Quarterly Progress Report No. 2, Magna Corporation Contract NASw-623, 1963.
-
H. B. H. Cooper, I. Spencer and W. Herbert, Methods for the production of ammonia from urea and/or biuret, and uses for NOx and/or particulate matter removal, U.S. Pat., 6
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 077491, 2000 Search PubMed.
077491, 2000 Search PubMed. - D. C. Bouchard, M. K. Williams and R. Y. Surampalli, J. Am. Water Works Assoc., 1992, 84, 85–90 CAS.
- D. R. McCubbin, B. J. Apelberg, S. Roe and F. Divita, Environ. Sci. Technol., 2002, 36, 1141–1146 CrossRef CAS.
-
G. G. Botte, Electrolysis of urea/urine to produce ammonia and hydrogen, electrolysis of urea/urine to ammonia, and methods, uses, and fuel cells related thereto, U.S. Pat., 60/980056, 2007 Search PubMed.
- Urine, Britannica Online Encyclopedia, http://www.britannica.com/EBchecked/topic/619857/urine, 2007.
- F. Vitse, M. Cooper and G. G. Botte, J. Power Sources, 2005, 142, 18–26 CrossRef CAS.
-
G. G. Botte, F. Vitse and M. Cooper, Electrocatalysts for the oxidation of ammonia and their application to hydrogen production, fuel cells, sensors, and purification processes, U.S. Pat., 7485211 2003 Search PubMed.
- Q. Yi, W. Huang, J. Zhang, X. Liu and L. Li, J. Electroanal. Chem., 2007, 610, 163–170 CrossRef CAS.
- A. S. Vaze, S. B. Sawant and V. G. Pangarkar, J. Appl. Electrochem., 1997, 27, 584–588 CrossRef CAS.
- H. J. Schafer, Top. Curr. Chem., 1987, 142, 101–129.
- A. E. Bolzan and T. Iwasita, Electrochim. Acta, 1988, 33, 109–112 CrossRef CAS.
- V. Climent, A. Rodes, J. M. Ort, A. Aldaz and J. M. Feliu, J. Electroanal. Chem., 1999, 461, 65–75 CrossRef CAS.
- F. C. Nart and T. Iwasita, Electrochim. Acta, 1992, 37, 385–391 CrossRef CAS.
- S. J. Yao, K. S. Walfson, B. K. Ahn and C. C. Liu, Nature, 1973, 241, 471–472 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, cyclic voltammograms, constant voltage analyses, and gas chromatographs. See DOI: 10.1039/b905974a |
| This journal is © The Royal Society of Chemistry 2009 |