
DFT study of CH4 activation by d0 Cl2LnZ (Z = H, CH3) complexes
Abstract
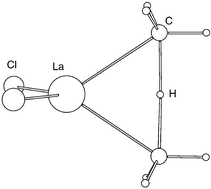
DFT(B3PW91) calculations of the activation of CH4 by models (Cl2LnZ) of Cp*2LnZ (Z = H, Me) have been carried out for the entire lanthanide series. Cl2LnZ appears to be a good model for Cp*2LnZ. It reproduces well the coordination around the lanthanide. The energetics of the transformation X2LnH + CH4 → X2LnCH3 + H2 are fairly close for X = Cl and Cp and the difference in behavior can be attributed to the stronger electron donating ability of Cp. Formation of the lanthanide hydride complex is calculated to be exothermic in agreement with experimental evidence. The energy profiles of the reactions Cl2LnH + CH4 → Cl2LnCH3 + H2; Cl2LnH* + CH4 → Cl2LnH + H*CH3; Cl2LnCH*3 + CH4 → Cl2LnCH3 + H–CH*3 have been calculated. The transition states for the first and third transformations are energetically accessible, in good agreement with the known experimental data. The second reaction has a transition state of very high energy indicating an unfeasible reaction. The geometry of the transition stuctures are suggestive of a proton transfer between two anionic species (Z and CH3−; Z = H− and CH3−) in the field of the lanthanide fragment.
 Please wait while we load your content...
Please wait while we load your content...