
All-inorganic copper-halide perovskites for large-Stokes shift and ten-nanosecond-emission scintillators†
Tobias
Haposan
 a,
Arramel
Arramel
*b,
Pramitha Yuniar Diah
Maulida
b,
Sri
Hartati
b,
Afif Akmal
Afkauni
b,
Muhammad Haris
Mahyuddin
cd,
Lei
Zhang
e,
Dominik
Kowal
f,
Marcin Eugeniusz
Witkowski
g,
Konrad Jacek
Drozdowski
g,
Michal
Makowski
f,
Winicjusz
Drozdowski
g,
Lina Jaya
Diguna
*a and
Muhammad Danang
Birowosuto
*f
a,
Arramel
Arramel
*b,
Pramitha Yuniar Diah
Maulida
b,
Sri
Hartati
b,
Afif Akmal
Afkauni
b,
Muhammad Haris
Mahyuddin
cd,
Lei
Zhang
e,
Dominik
Kowal
f,
Marcin Eugeniusz
Witkowski
g,
Konrad Jacek
Drozdowski
g,
Michal
Makowski
f,
Winicjusz
Drozdowski
g,
Lina Jaya
Diguna
*a and
Muhammad Danang
Birowosuto
*f
aDepartment of Renewable Energy Engineering, Universitas Prasetiya Mulya, Kavling Edutown I.1, Jl. BSD Raya Utama, Tangerang 15339, Indonesia. E-mail: lina.diguna@prasetiyamulya.ac.id
bNano Center Indonesia, Jl. PUSPIPTEK, Tangerang Selatan 15314, Indonesia. E-mail: arramel@nano.or.id
cQuantum and Nano Technology Research Group, Institut Teknologi Bandung, Jl. Ganesha 10, Bandung 40132, Indonesia
dResearch Center for Nanoscience and Nanotechnology, Institut Teknologi Bandung, Jl. Ganesha 10, Bandung 40132, Indonesia
eDepartment of Physics, National University of Singapore, 2 Science Drive 3, Singapore 117542, Singapore
fŁukasiewicz Research Network-PORT Polish Center for Technology Development, Stabłowicka 147, 54-066 Wrocław, Poland. E-mail: muhammad.birowosuto@port.lukasiewicz.gov.pl
gInstitute of Physics, Faculty of Physics, Astronomy, and Informatics, Nicolaus Copernicus University in Torun, ul. Grudziadzka 5, 87-100 Torun, Poland
First published on 12th January 2024
Abstract
The recent surge of interest in low-dimensional lead-free copper halide perovskites (CHPs) has driven significant progress in optoelectronics and scintillating materials. However, the development of green-based synthetic routes for CHPs, aimed at creating fast-decaying scintillators with ultrasensitive X-ray detection, remains elusive. In this study, we utilize a mechanochemical method to obtain 1D CHP (CsCu2I3) and 0D CHPs (Cs3Cu2X5 (X = I, Br)) focusing on the mixing of I and Br anions with different molar ratios (I![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Br = 4:1 and 3:2). CsCu2I3 and Cs3Cu2I5 exhibit a substantial large Stokes shift (SS) of 1.75 ± 0.02 eV and 1.57 ± 0.05 eV with the former displaying the absence of afterglow, whereas the latter has a deep trap of ∼500 meV, complicating the scintillation mechanism and resulting in a slower decay component. The CsCu2I3 scintillation decay time is primarily characterized by a fast component (τ1) of 9.30 ± 0.01 ns, accounting for contribution (C1) of 43% from the total emission. This fast decay component of ∼10 ns has not been previously reported in the family of CHPs. Similarly, τ1 of 10.9 ± 0.6 ns is obtained in Cs3Cu2I5, but when compared to its counterpart, C1 is only 3%. Upon increasing the Br substitution in Cs3Cu2I5, we observe that the traps become shallower, with energies ranging from 208 ± 21 to 121 ± 18 meV, along with an appreciable trap concentration of ∼104. The C1 of τ1 also increases with higher Br concentration, reaching a maximum value of 29%. Unfortunately, this increased contribution in decay times is accompanied by a decrease in light yields (Cs3Cu2I5 has 16.5 ph per keV at room temperature (RT)) as thermal quenching processes predominate throughout the entire series of CHPs at RT. Our work provides valuable insights into the tunable structure–property relationship through the I:Br composition ratio of CHPs, hence advancing scintillation performance by rational design towards timing applications.
:Br = 4:1 and 3:2). CsCu2I3 and Cs3Cu2I5 exhibit a substantial large Stokes shift (SS) of 1.75 ± 0.02 eV and 1.57 ± 0.05 eV with the former displaying the absence of afterglow, whereas the latter has a deep trap of ∼500 meV, complicating the scintillation mechanism and resulting in a slower decay component. The CsCu2I3 scintillation decay time is primarily characterized by a fast component (τ1) of 9.30 ± 0.01 ns, accounting for contribution (C1) of 43% from the total emission. This fast decay component of ∼10 ns has not been previously reported in the family of CHPs. Similarly, τ1 of 10.9 ± 0.6 ns is obtained in Cs3Cu2I5, but when compared to its counterpart, C1 is only 3%. Upon increasing the Br substitution in Cs3Cu2I5, we observe that the traps become shallower, with energies ranging from 208 ± 21 to 121 ± 18 meV, along with an appreciable trap concentration of ∼104. The C1 of τ1 also increases with higher Br concentration, reaching a maximum value of 29%. Unfortunately, this increased contribution in decay times is accompanied by a decrease in light yields (Cs3Cu2I5 has 16.5 ph per keV at room temperature (RT)) as thermal quenching processes predominate throughout the entire series of CHPs at RT. Our work provides valuable insights into the tunable structure–property relationship through the I:Br composition ratio of CHPs, hence advancing scintillation performance by rational design towards timing applications.
1 Introduction
Lead halide perovskites have been attractive for scintillation detection due to their high quantum yield and their potential for X-ray imaging applications.1–3 A light yield (LY) of 24 ± 2 ph per keV and a decay time of 5.97 ns have been achieved for CsPbBr3 nanocrystals.4 Moreover, CsPbBr3 nanocrystals embedded into zero-dimensional (0D) Cs4PbBr6 microstructure materials have shown a high LY of 64 ± 2 ph per keV and a fast scintillation time of 1.3 ns (88%) and 6.7 ns (12%).5 Unfortunately, the commonly used heavy metal lead in perovskites is still a major concern due to its toxicity.6 Furthermore, the relatively poor stability7 and strong light self-absorption8 are also the major challenges for the introduction of lead-based materials into the marketplace. To address these issues, research efforts have been devoted to developing lead-free halide perovskites.9,10 In this regard, copper halide perovskites (CHPs) have been considered as alternatives due to their stability and bright emission,11–15 especially in the stable form of Cu1+.16 Substituting Pb with Cu has been previously reported to lead to a larger Stokes shift (SS), which may be beneficial to enhance LY by suppressing the reabsorption loss.10,17 Cheng et al. have reported the LYs of one-dimensional (1D) CsCu2I3 and zero-dimensional (0D) Cs3Cu2I5 single crystals to be 16 ph per keV and 32 ph per keV, respectively.17,18 Some larger LYs were also observed for Tl-doped and nanocrystal-formed Cs3Cu2I5.13,19 However, the slow scintillation decay remains a major concern, given that the previously reported fast component of scintillation decay is 39 ns (82%) for Cs3Cu2I5 single-crystals.20 This value is still far from competing with lead-based perovskites, and thus faster scintillation decay is still necessary to be pursued for practical timing application.3 To note, several strategies have been adopted to regulate the scintillation and luminescence properties such as phase control, doping, the pressure effect, polarization dependence, and chemical composition.21–26 The last approach is quite feasible and attractive since it allows access to bandgap control and emission tuning via halogen substitution.27,28 For a comparison, Roccanova et al. reported a powdered form of CsCu2Br1.5I1.5 with the PL peak and SS at 584 nm and 249 nm, respectively.12 In addition, the powdered forms of Cs3Cu2Br1.25I3.75, Cs3Cu2Br2.5I2.5 and Cs3Cu2Br3.75I1.25 have been reported with tunable PL peaks (456–443 nm) and SSs (164–135 nm); however, the corresponding LYs are not investigated.29 Thus, the scintillation properties at the different I:Br ratios remain elusive in these intriguing mixed-halide CHP systems.
The widely adopted solution processing methods for perovskites use organic solvents such as N,N-dimethylformamide and dimethyl sulfoxide solutions which are not green.30,31 On the other hand, mechanochemistry offers a simpler and faster synthesis method without the employment of complex organic solvents and heat treatment.32,33 Through this method, all inorganic CHPs have been successfully synthesized with bright emission.32,34 Here, we investigate the potential of the mechanochemical synthesis method for CHPs and study their scintillation properties. First, we vary the molar ratio of CsI and CuI precursors and characterise their scintillation properties. Both Cs3Cu2I5 and CsCu2I3 have similar LY (13 and 16.5 ph per keV, respectively) and 10 ns scintillation decay fast component. This is faster than those reported before for Cs3Cu2I5 and CsCu2I3.13,17–19 In different dimensionalities, Cs3Cu2I5 has a smaller SS and higher LY, but slower average decay time than those of CsCu2I3. The average slow decay times are attributed to the presence of dominating slow components between 100 and 1000 ns. In this study, we add the CsBr precursor for Br doping in Cs3Cu2I5 to increase the contribution of 10 ns fast components to the emission. However, the LYs of Cs3Cu2I3Br2 decrease maximally down to one of the third from that of Cs3Cu2I5. Therefore, from this study, we expect that the optimum values of LYs and the contribution of the 10 ns decay component of CHP scintillators can be estimated. Such control of 10 ns scintillation decay components paves the way for scalable, low-cost, and high-throughput production of high-quality fast lead-free perovskite scintillators for applications of photon counting computed tomography (PCCT) and positron emission tomography (PET).3
2 Experimental
2.1 Synthesis
Caesium iodide (CsI, anhydrous, 99.9999% trace metal basis), copper iodide (CuI, anhydrous, 99.9999% trace metal basis), and caesium bromide (CsBr, anhydrous, 99.9999% trace metal basis) were obtained from Sigma-Aldrich. Acetone (ACS Reag., for analysis) was acquired from Millipore. All chemicals were used as received without undergoing any purification steps. All samples of CHPs were synthesized mechanochemically, CsI and CuI, in 1:2 and 3:2 molar ratios as carried out in a previous report,33 and were each ground for 10 minutes with 300 μL of acetone, hereinafter referred to as CsCu2I3 and Cs3Cu2I5, respectively. The same method was used for CsI, CuI, and CsBr in the molar ratios of 2:2:1 and 1:2:2, hereinafter referred to as Cs3Cu2I4 Br and Cs3Cu2I3Br2, respectively. The grinding process immediately produced strongly blue-emitting and yellow-emitting powders, as shown in Fig. S1a–d (ESI†), indicating the formation of the desired perovskites.33
2.2 Scanning electron and transmission electron microscopy
Sample micrographs were taken at 5 kV accelerating voltage in the secondary electron mode using a ZEISS EVO MA10 scanning electron microscope (SEM) system with 1.00 K times magnification. Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) analyses were performed utilizing a double-Cs-corrected Titan G2 60–300 (S)TEM microscope. Before HRTEM analysis, the electron beam conditions were adjusted based on the sample material. In this study, an acceleration voltage of 300 kV was employed. Samples were prepared by dissolving 4 mg of the compound in 4 mL of ethanol, followed by 1 hour sonication. The samples were transferred to Cu holey carbon grids (∼5 μL droplet) and allowed to dry. Both TEM and STEM modes were employed, also at very high magnifications allowing high-resolution imaging. For image acquisition, the Ultrascan US1000 camera was used for TEM and the HAADF detector for STEM. The STEM-EDS maps were also acquired with a Super-X EDS detector. Due to sample sensitivity to the beam, the data were collected with the smallest possible dose at the expense of the signal-to-noise ratio, which is particularly seen on EDS maps.2.3 Fourier-transform infrared and Raman spectroscopies
Fourier-transform infrared (FTIR) spectra were obtained using an ATR-FTIR Thermo Scientific Nicolet iS10 spectrometer (ThermoFisher Scientific, USA), covering the spectral range of 800–400 cm−1. The as-synthesised CHPs were directly mounted and compacted into the sample holder without any further preparation. Raman spectra were acquired with a ThermoFisher Scientific DXR instrument (ThermoFisher Scientific, USA) utilizing a 532 nm excitation laser source. The measurements were conducted at room temperature (RT), employing laser powers ranging from 0.2 to 3.0 mW.2.4 X-ray diffractometry
The structure of the CHPs was inspected with a powder X-ray diffractometer (XRD) (PANalytical AERIS, Malvern Panalytical, the Netherlands) with Kα of 1.54 Å. Data acquisition was conducted at RT with a scanning speed of 0.12 s per step and a step size of 0.0217°. Further Rietveld refinement was carried out using Profex,35 while visualization system for electronic and structural analysis (VESTA) was used for crystallography visualisations.362.5 X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy (XPS) analyzed 1 mm diameter samples using a magnesium Kα X-ray source (excitation energy at 1254 eV). For calibration, the Ag 3d5/2 peak (centered at 368.10 eV) from argon-ion-sputtered silver foil was utilised. CasaXPS software package was used for deconvolution analysis.372.6 Density functional theory calculations
Density functional theory (DFT) calculations were performed under the Kohn–Sham formulation38,39 as implemented in the Vienna ab initio simulation package (VASP).40,41 The projector augmented wave (PAW) method42,43 was used to describe the interaction between ion cores and electrons. The electron exchange–correlation was treated using spin polarization by the generalized gradient approximation (GGA) method based on the Perdew–Burke–Ernzerhof (PBE) functional.44 The rotationally invariant GGA+U approach introduced by Dudarev et al.45 was used with an effective Hubbard parameter Ueff being 11.0 eV for the Cu d orbital. The plane wave basis sets with a cut-off energy of 500 eV were used for all calculations. The Brillouin zone was sampled according to the Monkhorst–Pack scheme46 with a k-point grid of 3 × 3 × 5. The zero-damping D3 method47 was adopted to account for the dispersion correction. During calculations, all atoms were allowed to fully relax. The conjugate gradient method was employed for cell optimizations, and the calculations were considered to converge when the maximum forces on each atom were less than 0.01 eV Å−1.2.7 Absorption and photoluminescence
The samples were excited for photoluminescence (PL) measurement using a picosecond laser diode (Master Oscillator Fibre Amplifier, Picoquant GmbH, Berlin, Germany) with a repetition rate of 31.25 kHz and a wavelength of 266 nm. The laser diode had a pulse duration of 50 ps and an average power of 2 mW at RT. Excitation focusing and signal collection were carried out using a microscope objective (Nikon Corporation, Tokyo, Japan) with a magnification of 20× and a numerical aperture of 0.4. The PL signal was filtered and acquired using a high-sensitivity visible light spectrometer (Ocean Optics, Florida, USA). Absorption spectra were measured with a homemade setup with the same commercial spectrometer in the transmission mode. Since the samples were put inside the quartz tubes, the spectra were corrected for the absorption of the tubes.2.8 Temperature-dependent radioluminescence
For radioluminescence (RL) measurement, X-ray excitation was performed using an Inel XRG3500 X-ray generator with a Cu-anode tube operating at 45 kV/10 mA. The RL signal was recorded using an Acton Research Corporation SpectraPro-500i monochromator equipped with a 500 nm blazed grating and a Hamamatsu R928 photomultiplier. The temperature of the sample was controlled using an APD Cryogenics Inc. closed-cycle helium cooler, along with a Lake Shore 330 programmable temperature controller, in the range of 10 to 350 K. To prevent any thermal release of charge carriers, measurements were carried out in reverse, starting at 350 K and ending at 10 K.2.9 Thermoluminescence
Thermoluminescence (TL) measurement involved exposing the samples to X-ray excitation for 600 s at 10 K, followed by observing the afterglow upon X-ray cessation. After a waiting period of at least 2500 s to ensure decay of the afterglow, TL glow peaks were recorded while raising the temperature from 10 K to 350 K at a rate of 0.14 K s−1.2.10 Pulse height and scintillation decay
We used a 137Cs (662 keV) radioisotope for the γ-ray source and a Hamamatsu R878 photomultiplier (PMT) for detecting the converted photons. To operate the PMT, we applied a voltage of 1.25 kV. The corresponding output signal from the PMT is integrated with a charge-sensitive pre-amplifier. The output then feeds a spectroscopic amplifier with a shaping time of 2 μs and an analog-to-digital converter (Ortec series). The photoelectron yield was obtained by comparing the position of the photopeak to the position of the mean value of the single electron response in pulse high spectra measurements. The actual LY for the radiation conversion in photons per MeV was obtained after the photoelectron yield was divided by the quantum efficiencies of the PMT. Scintillation decay measurements were performed by the delayed coincidence single photon counting method.48 A 137Cs radioactive source, two Hamamatsu photomultiplier tubes (R1104 and R928 for “starts” and “stops”, respectively), a Canberra 2145 time-to-amplitude converter, and a TUKAN-8K-USB multichannel analyzer were used.3 Results and discussion
In Fig. 1a, the attenuation length of the CHPs is compared with the attenuation length of CsPbX3 (where X = Br or I).4 In the range of 10 to 33 keV, the attenuation length of CsPbX3 is shorter than that of the CHPs. In the range of 33 to 42 keV however, the attenuation length of the CHPs outperforms that of CsPbBr3, and is still shorter than that of CsPbI3 of up to 46 keV. In the case of the CHPs, the doping of bromine results in a longer, less effective attenuation length compared to the undoped CHPs within this energy range. The CHPs hold promise for medical or security imaging applications within the X- or γ-ray energy spectrum.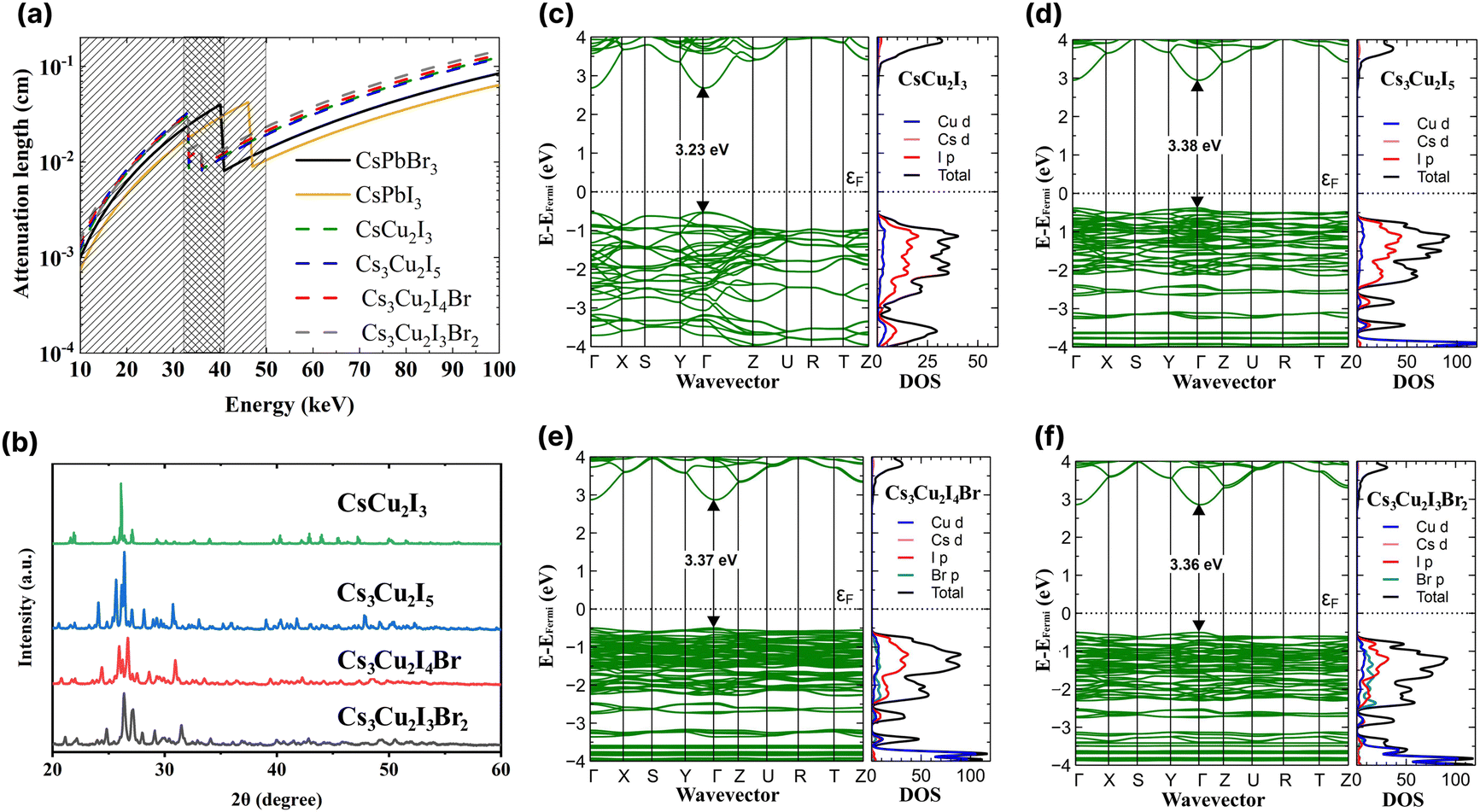 | ||
| Fig. 1 (a) X-ray attenuation lengths for the investigated caesium copper halide perovskites (CHPs) with CsPbI3 and CsPbBr3 used as in ref. 4. (b) XRD spectra of the CHPs. Band structures and density of states of (c) CsCu2I3, (d) Cs3Cu2I5, (e) Cs3Cu2I4 Br, and (f) Cs3Cu2I3Br2. | ||
Subsequent to grinding, the morphology of CHPs was inspected with SEM, as displayed in Fig. S2a–d (ESI†). The CsCu2I3 powder has a rod-like shape, while Cs3Cu2I5, Cs3Cu2I4 Br, and Cs3Cu2I3Br2 have particle-like shape, suggesting that a different crystal system is present between the two configurations. The particle size distribution observed from SEM is displayed in Fig. S3a–d (ESI†). To investigate the CHPs in smaller dimensions, TEM, HRTEM, and EDS mapping are displayed in Fig. S4 (ESI†). Major peaks of XRD patterns depicted in Fig. 1b indicate that a structural difference is present between CsCu2I3 and Cs3Cu2X5 compounds. A shift to a higher angle is observed with increasing Br doping concentration in Cs3Cu2I5, Cs3Cu2I4 Br, and Cs3Cu2I3Br2, which can be attributed to the decrease of lattice distance. Rietveld refinement was further carried out with calculated CIF, as displayed in Fig. S5 (ESI†), confirming the orthorhombic structure shared by all of the CHPs. The 1D CsCu2I3 XRD pattern was best fitted with the Cmcm space group, while the 0D Cs3Cu2I5, Cs3Cu2I4 Br, and Cs3Cu2I3Br2 were best fitted with the Pnma space group. Furthermore, the refinement parameters shown in Table S1 (ESI†) reveal that the increased concentration of Br doping reduces the unit cell volume, from 1698.750 ± 0.046, to 1679.527 ± 0.047, and to 1637.956 ± 0.617 Å3 for Cs3Cu2I5, Cs3Cu2I4 Br, and Cs3Cu2I3Br2, respectively. This trend is consistent with CHPs synthesised by solution-processing and mechanochemistry.33,49,50 In the case of CHPs, its fundamental structure is typically arranged in a tetrahedral form. This is quite distinct from the octahedral configuration commonly found in Pb-based,4 PbBi halide double perovskite,51 and various lead-free halide perovskites such as 2D Cu-,52 Sn-53,54 Mn-based,55 and AgBi halide double perovskites.56 This structural divergence leads to two different views in the community, with some not recognising CHP as a perovskite. Nevertheless, a part of the community perceives CHP as a variant of perovskites,57–60 a stance which is adopted in this study. FTIR and Raman spectra of the CHPs are presented in Fig. S6 and S7 (ESI†), respectively. Additionally, XPS analysis confirms the presence of metal cations (Cs and Cu) and anions (I and Br) accordingly, as shown in Fig. S8 (ESI†).
Rational design carried out by modifying the I:Br molar ratio of 0D CHPs not only affects the crystallographic structure of the CHPs, but can also be used to modify band gaps and optoelectronic properties. We proceeded to determine the density of states (DOS) utilising DFT and the optical bandgap. Fig. 1c–f displays the band structure, inclusive of the total (black) and individual (coloured) DOSs for CsCu2I3, Cs3Cu2I5, Cs3Cu2I4 Br, and Cs3Cu2I3Br2. Within the DOS, Cu d is symbolised by the blue line, Cs d by pink, I p by red, and Br p by green. The calculated band gaps are 3.23 ± 0.20, 3.38 ± 0.20, 3.37 ± 0.20, and 3.36 ± 0.20 eV for CsCu2I3, Cs3Cu2I5, Cs3Cu2I4 Br, and Cs3Cu2I3Br2, respectively. All CHPs exhibit a direct band gap at the Γ point. Given that Cu states majorly influence both the conduction and valence bands, the band gaps for Cs3Cu2I5, Cs3Cu2I4 Br, and Cs3Cu2I3Br2 are strikingly similar to each other. The calculated band gaps for CsCu2I3 and Cs3Cu2I5 concur with prior findings from the Kubelka–Munk plot12 and the hybrid HSE06 functional.61
The optical properties of the CHPs were investigated by collecting their absorption and PL emission spectra, as shown in Fig. 2. The maximum absorbance of the CHPs is 3.83, 4.08, 4.11, and 4.16 eV for CsCu2I3, Cs3Cu2I5, Cs3Cu2I4 Br, and Cs3Cu2I3Br2, respectively. From the absorption spectra, we obtain the band gaps through Elliott fitting (see eqn (S1) and S2 (ESI†))62 as depicted in Fig. S9 (ESI†). As summarised in Table 1, the experimental band gaps (Eexpg) are 3.72 ± 0.02, 3.96± 0.05, 3.99 ± 0.03, and 4.08 ± 0.02 eV for CsCu2I3, Cs3Cu2I5, Cs3Cu2I4 Br, and Cs3Cu2I3Br2, respectively. From these data, we further calculated the SS, determined as the difference between Eexpg and maximum PL emission.63 All CHPs exhibit large SS with negligible self-absorption, as the characteristics of the CHPs. For example, the 1D yellow-emitting CsCu2I3 has a vast PL spectrum, showcasing a peak PL intensity at 2.07 eV. This leads to an SS of 265.23 ± 1.78 nm (1.65 ± 0.02 eV). Lowering the dimensionality causes the PL maximum to blue-shift, with the PL peak for Cs3Cu2I5 standing at 2.57 eV, correlating with an SS of 168.73 ± 3.91 nm (1.39 ± 0.05 eV). The addition of a Br dopant to Cs3Cu2I5 induces a red shift in the PL peaks, registering values of 2.55 eV for Cs3Cu2I4 Br and 2.54 eV for Cs3Cu2I3Br2. The incorporation of Br also results in a SS of 176.39 ± 2.35 nm (1.45 ± 0.03 eV) for Cs3Cu2I4 Br and 183.25 ± 1.48 nm (1.53 ± 0.02 eV) for Cs3Cu2I3Br2, respectively. An increased concentration of Br doping is known to produce a larger SS, paired with a red shift in the PL peak emission trend.29 For CsCu2I3, the SS is higher and comparable to its single-crystal (257 nm),64 nanocrystals (240 nm),65 and mechanochemically synthesised powder (244 nm)33 form. For Cs3Cu2I5, the SS is also comparable to and larger than those of its single-crystal (141 nm),49 nanocrystals (161 nm),13 thin film (156 nm)66 and mechanochemically synthesised powder (130 nm)33 form. The SS of the CHPs herein is much larger than those of Pb-based ones, as summarised in Table S3 (ESI†), along with their other comparable optical properties. All the CHPs exhibited a relatively broad PL emission spectrum compared to other forms, with full-width-at-half-maxima (FWHM) ranging from 0.37 to 0.67 eV. As a case in point, the FWHM of CsCu2I3 for single-crystals is 75 nm9 as reported by Lin et al., while that for a mechanochemically synthesised powder is 120 nm33 as reported by Grandhi et al. We attribute the relatively broad PL emission spectra and their variation to the irregularities that may be caused by the hand-grinding approach. We further comment on the reproducibility of the experimental approach with PL reliability study as depicted in Fig. S10 (ESI†).
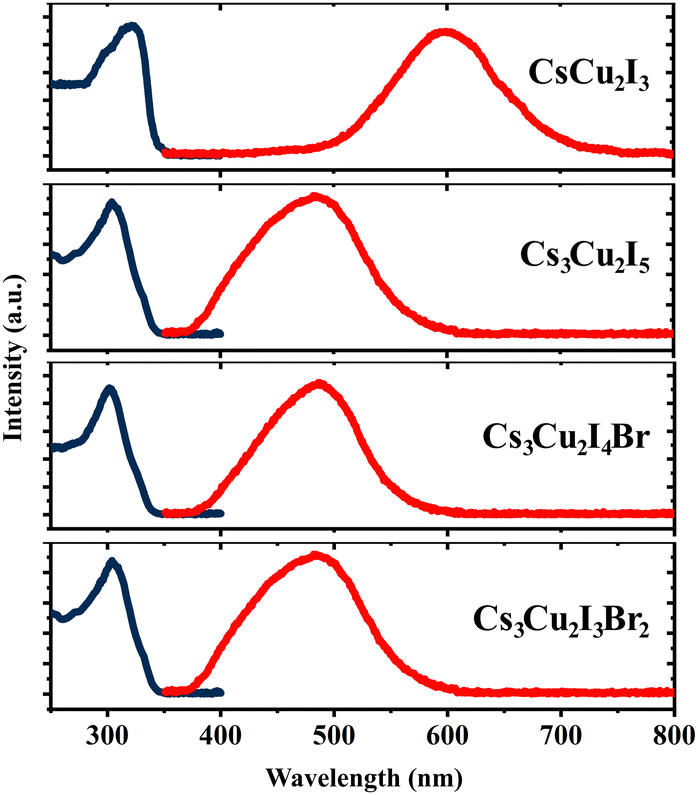 | ||
| Fig. 2 Absorption (dark blue lines) and photoluminescence (red lines) spectra of the CHPs measured at RT. | ||
| CHP | E calcg (eV) | E expg (eV) | Abs. maximum (eV) | PL maximum (eV) | Stokes shift (SS) (nm) | Stokes shift (SS) (eV) | FWHM (nm) | FWHM (eV) |
|---|---|---|---|---|---|---|---|---|
| CsCu2I3 | 3.23 ± 0.20 | 3.72 ± 0.02 | 3.83 | 1.97 ± 0.01 | 294.49 ± 2.81 | 1.75 ± 0.02 | 127.31 ± 6.05 | 0.20 ± 0.01 |
| Cs3Cu2I5 | 3.38 ± 0.20 | 3.96 ± 0.05 | 4.08 | 2.39 ± 0.01 | 203.54 ± 4.35 | 1.57 ± 0.05 | 102.26 ± 3.01 | 0.19 ± 0.01 |
| Cs3Cu2I4 Br | 3.37 ± 0.20 | 3.99 ± 0.03 | 4.11 | 2.54 ± 0.01 | 119.31 ± 21.92 | 0.83 ± 0.20 | 87.18 ± 2.66 | 0.18 ± 0.01 |
| Cs3Cu2I3Br2 | 3.36 ± 0.20 | 4.08 ± 0.02 | 4.16 | 2.55 ± 0.01 | 176.08 ± 3.00 | 1.44 ± 0.03 | 83.26 ± 3.00 | 0.17 ± 0.01 |
RL study was conducted to examine emission characteristics of the CHPs in the cryogenic state and at RT. RL spectra of the CHPs measured in the temperature range of 10 to 350 K are presented in Fig. 3a–d. The alteration of dimensionality shows a significant change in their RL properties. Upon excitation by an X-ray source, the 1D CsCu2I3 exhibits an increase in the RL intensity from the temperature of 10 up to ∼230 K, showing that CsCu2I3 is suitable for RL within this range. The RL intensity above 270 K is weakened before reaching RT, indicating thermal quenching (TQ) behaviour. It is also notable that the RL peak is blue-shifted as the temperature increases, from ∼579 nm at ∼60 K to ∼562 nm at ∼173 K. We discuss that the physical origin of such peak-shifting behaviour is mainly related to the temperature effect on the luminescent centre of the 1D CHP system. In contrast to CsCu2I3, the 0D Cs3Cu2I5 exhibits an RL peak at ∼445 nm, commencing at a very low temperature and demonstrating a slight red shift in RL maxima as the temperature rises. Upon approaching RT, the RL intensity diminishes, showing TQ. This finding aligns similarly to solution-grown Cs3Cu2I5 single crystals.67,68 The increased doping of Br, however, results in TQ occurring earlier. Br doping onto the conventional 0D CHP has a non-negligible impact on contour mapping, which broadens the RL intense peak wavelength. TQ behaviour can also be seen with the broadening of RL FWHM as the temperature increase. For example, the FWHM of CsCu2I3 at 150 K is 81.93 nm, while at 200 K it broadens to 90.46 nm. In Cs3Cu2I5, Cs3Cu2I4 Br, and Cs3Cu2I3Br2, the FWHM also broadens from 54.56, 61.40, and 73.09 nm at 150 K to 61.85, 69.42, and 79.43 nm at 200 K. The RL emission peak aligns closely with the PL peaks, suggesting a shared radiative recombination path.13 The variety of emissive transitions of CHPs in different dimensionality shows the possibility of RL property tuning by rational design.
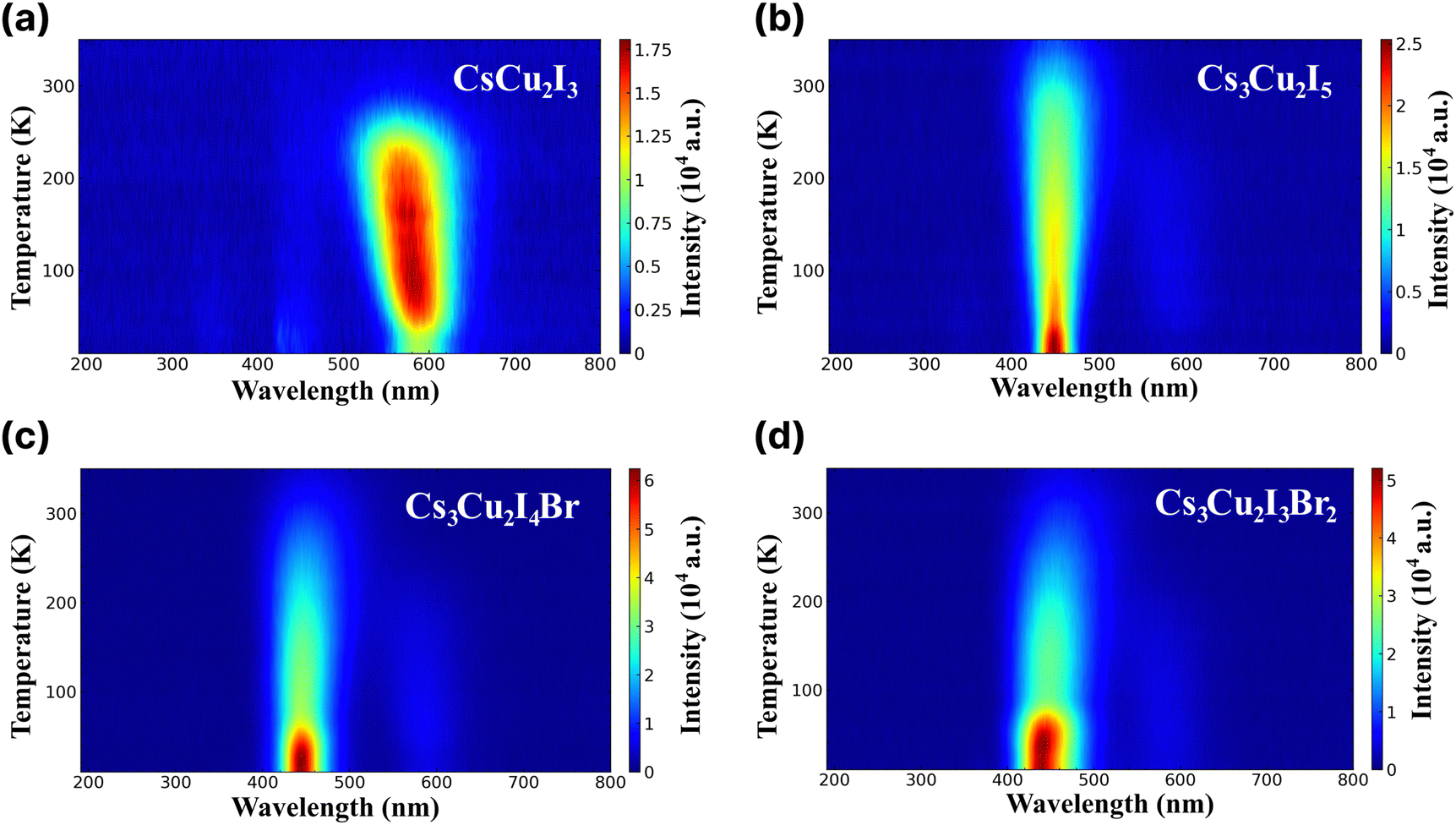 | ||
| Fig. 3 Radioluminescence spectra across different wavelengths at different temperatures ranging from 10 to 350 K of (a) CsCu2I3, (b) Cs3Cu2I5, (c) Cs3Cu2I4 Br, and (d) Cs3Cu2I3Br2. | ||
In the scintillation mechanism, the depth and concentration of traps are key determinants of the scintillation characteristics of the CHPs. Herein, the trap state properties of the CHPs are investigated via TL characterisation, as presented in Fig. 4. Steady-state TL measurements at 10 K depict the absence of decay after the X-ray source is turned off. The absence of afterglow in all CHPs shows the absence of super shallow traps. Typically, this afterglow phenomenon is commonly observed in all-inorganic lead halide and hybrid organic–inorganic tin-based perovskites, as reported in our prior research.10,69 During the TL experiments, we waited for 2500 seconds after we switched off the X-ray source and subsequently raised the temperature of the respective CHPs sample from 10 to 350 K. The purpose of such sequence is to eliminate the afterglow contribution to the background intensities. This temperature increase revealed observable TL peak intensities for all CHPs except the 1D CsCu2I3. For CsCu2I3, the lack of an emission signal with increasing temperature indicates that both shallow and deep traps are absent. To reveal the depth and concentration of traps, a glow curve fitting with the Randall-Wilkins equation as depicted in Fig. S11 (ESI†) was carried out towards the 0D CHPs.1,70 The fitting parameters summarised in Table S4 (ESI†) reveal that the depth of traps for 0D CHPs (all of which are observable below RT) is suppressed with an increase in trap concentration as Br doping is introduced. Trap states in Cs3Cu2I5 are revealed at depths of 271, 512, and 521 meV, while Cs3Cu2I4 Br exhibits trap depths at 151 and 208 meV, and Cs3Cu2I3Br2 at 121 and 184 meV. These traps can be attributed to the implication of internal scattering and structural defects, with the increment of Br doping.71,72 When compared to other metals or organic components in our previous works, the overall relatively low concentration of traps in the order between 103 and 104 may be partially attributed to the incorporation of Cs+ cations.73 These low trap concentration characteristics are also observed in Cs3Cu2I5 single crystals.20
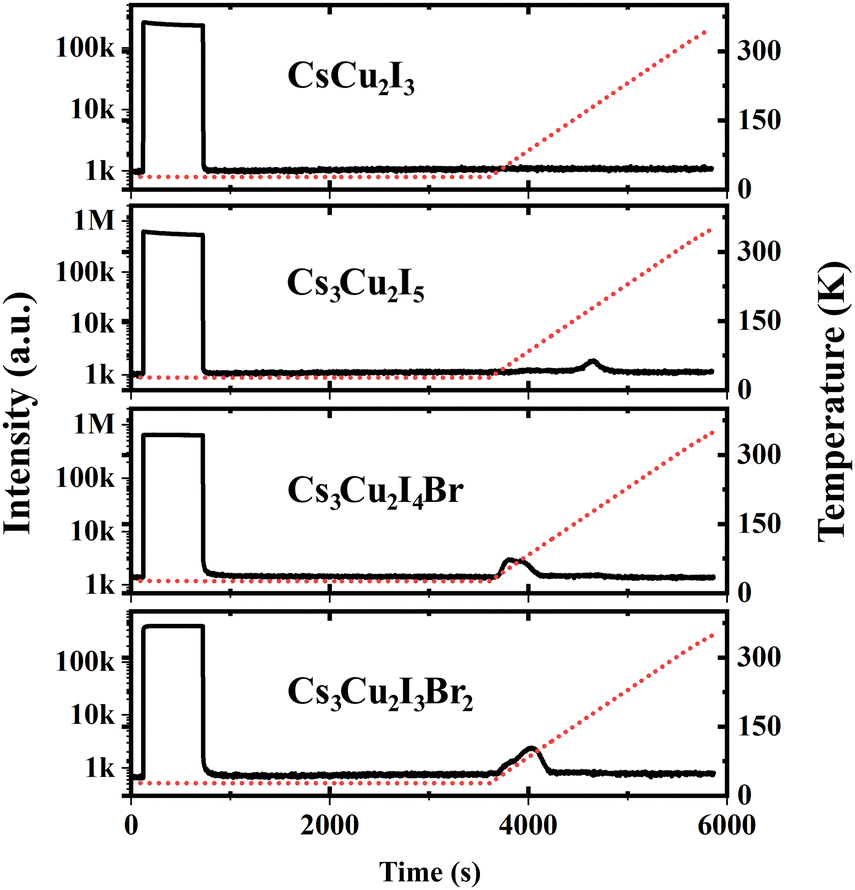 | ||
| Fig. 4 Afterglow and thermoluminescence of CHPs. The red dashed lines indicate the temperature changes over time. | ||
A considerable property in scintillators is a fast scintillation decay time, which is beneficial for timing applications, e.g. PET and PCCT. Upon γ-irradiation exposure, the decay times of CHPs were measured at RT in which three-component decay fittings are performed as shown in Fig. 5 (with fitting parameters shown in Table 2). We note that the first component (τ1) is associated with the direct recombination of excitons, the second component (τ2) is attributed to the self-trapped excitation (STE), and the third component (τ3) corresponds to the recombination of trapped electrons and holes as Frenkel defects.74 We found that CHPs exhibit a fast value of τ1 with varied photon decay contribution. 1D CsCu2I3 demonstrated a value of 9.30 ± 0.01 ns (τ1 contribution; 43%). Upon lowering the dimensionality, 0D Cs3Cu2I5 demonstrated a τ1 value of 10.90 ± 0.55 ns (τ1 contribution; 3%). Herein, the overall value of τ1 of the CHP powder which was synthesized by mechanochemistry is relatively faster than that of the single-crystal of CsCu2I3 and Cs3Cu2I5,17,18 surpassing a notable fast component of 39 ns (82%) observed on a Cs3Cu2I5 single-crystal synthesised using the aqueous solution method.20 This fast component of ∼10 ns has also not been previously reported in the family of CHPs and is faster than that of the existing commercial inorganic scintillators, Ce-doped LaBr3.75 Perovskite scintillators in powder form exhibit faster X-ray or scintillation decay times compared to their single crystal or thin film counterparts primarily due to their increased surface area and defect levels.76,77 The powder form allows for more efficient energy transfer and quicker electron–hole recombination.78,79 Furthermore, we observed incremental changes in the decay time for the 0D system, where the contribution of τ3 varied from 387.15 ± 6.05 ns (27%) for CsCu2I3 to 924.85 ± 4.05 (90%) for Cs3Cu2I5. It turns out that by doping Br into the Cs3Cu2I5 lattice (Cs3Cu2I4 Br and Cs3Cu2I3Br2), τ1 of ∼10 ns is still retained while simultaneously increasing the contribution of τ1 (10.40 ± 0.10 ns (12%) for Cs3Cu2I4 Br and 11.55 ± 0.55 ns (29%) for Cs3Cu2I3Br2). It is also worth noting that the contribution of τ2 is significant in 1D CsCu2I3 (54.5 ± 0.01 ns at 30%) and less dominant in Cs3Cu2I5 (116.15 ± 0.35 ns at 7%), and it becomes more dominant towards the increased Br doping (116.15 ± 0.35 ns at 7%, 92.85 ± 6.95 ns at 11%, and 110.05 ± 32.85 ns at 15% for Cs3Cu2I5, Cs3Cu2I4 Br, and Cs3Cu2I3Br2 respectively).
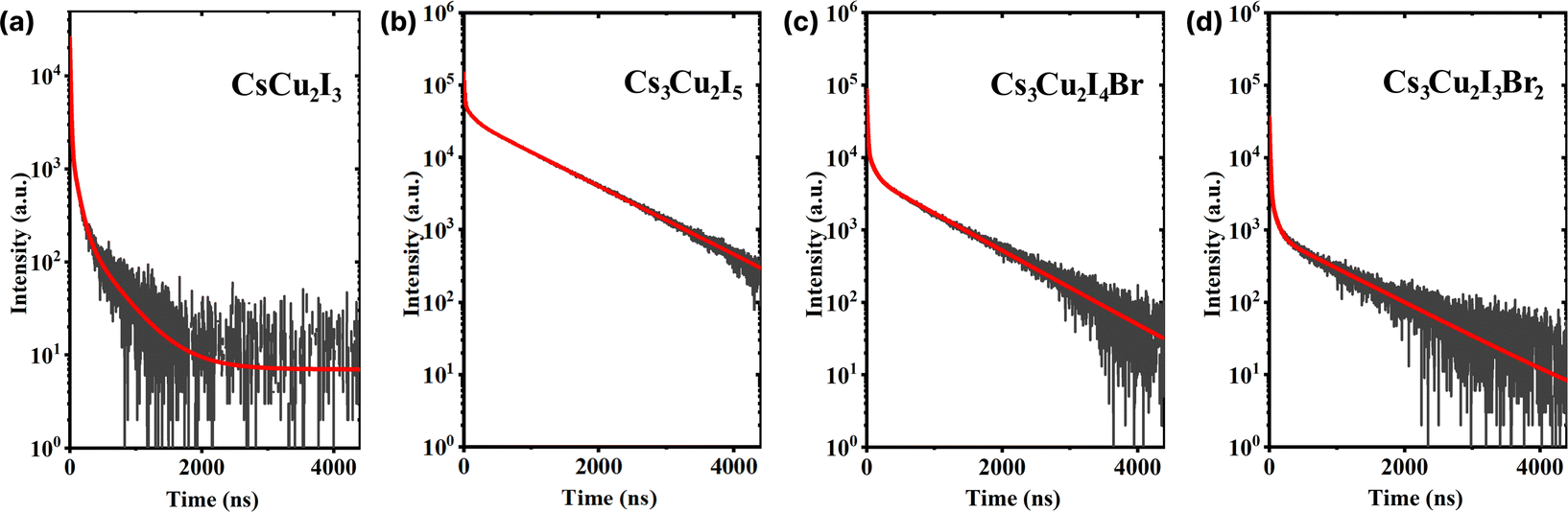 | ||
| Fig. 5 Scintillation decay curves of (a) CsCu2I3, (b) Cs3Cu2I5, (c) Cs3Cu2I4 Br, and (d) Cs3Cu2I3Br2. The red lines indicate the respective three-exponential decay fitting. | ||
| CHP | τ 1 (ns) | C 1 (%) | τ 2 (ns) | C 2 (%) | τ 3 (ns) | C 3 (%) | τ avg (ns) |
|---|---|---|---|---|---|---|---|
| CsCu2I3 | 9.30 ± 0.01 | 43 | 54.50 ± 0.01 | 30 | 387.15 ± 6.05 | 27 | 124.88 ± 1.63 |
| Cs3Cu2I5 | 10.90 ± 0.55 | 3 | 116.15 ± 0.35 | 7 | 924.85 ± 4.05 | 90 | 840.82 ± 3.65 |
| Cs3Cu2I4 Br | 10.40 ± 0.10 | 12 | 92.85 ± 6.95 | 11 | 873.60 ± 28.50 | 77 | 684.13 ± 22.72 |
| Cs3Cu2I3Br2 | 11.55 ± 0.55 | 29 | 110.05 ± 32.85 | 15 | 1050.80 ± 129.00 | 56 | 604.45 ± 46.15 |
We found that the quantum yield (QY) is enhanced by lowering the CHP dimensionality from 1D CsCu2I3 (13.59 ± 2.85%) to 0D Cs3Cu2I5 (47.19 ± 8.83%), as summarised in Table 3 (CsPbBr3 quantum dots were used as a reference, see Fig. S12 and eqn (S4) (ESI†)). In particular, we note that the QY of mechanically ground CsCu2I3 is fairly comparable to those of its single-crystal (18%)80 and nanocrystals (11%)61 and differs in contrast with those of mechanochemically-synthesised powder (6%)33 and thin films (20.6%).81 In the case of Cs3Cu2I5, the corresponding QY is fairly competitive compared to those reported for single-crystals (93.5%),80 microcrystals (60%),48 thin films (up to 91%),60,82,83 nanocrystals (87%),61 and mechanochemically-synthesised powder (62%).33 In terms of the QY value in mechanochemically-synthesised CHPs, we consider this to be related to the irregularities of the hand-grinding method. As we introduced the Br dopant onto the perovskite lattices, a lower QY (23.74 ± 4.15 and 7.37 ± 1.24% for Cs3Cu2I4 Br and Cs3Cu2I3Br2, respectively) resulted.
| CHP | QY (%) | LYcalc(S = 1) (ph per keV) | LYexp relative to Cs3Cu2I5 at 300 K (× 16.5 ph per keV) | S exp (%) |
|---|---|---|---|---|
| CsCu2I3 | 13.59 ± 2.85 | 15.88 ± 0.08 | 0.76 ± 0.03 | 78.70 ± 16.99 |
| Cs3Cu2I5 | 47.19 ± 8.83 | 51.82 ± 0.23 | 1 ± 0.21 | 31.84 ± 6.50 |
| Cs3Cu2I4Br | 23.74 ± 4.15 | 25.87 ± 0.11 | 0.67 ± 0.06 | 42.52 ± 7.94 |
| Cs3Cu2I3Br2 | 7.37 ± 1.24 | 7.85 ± 0.03 | 0.33 ± 0.03 | 70.06 ± 12.40 |
Furthermore, the calculated and experimental LYs at RT (LYcalc and LYexp, respectively) are also presented in Table 3. Utilising the QYs, the highest LYcalc value in ph per keV is ascertained using eqn (S5) (ESI†). LYexp values of the CHPs were determined by the pulse height spectrum method using a 137Cs source at 661.7 keV, as shown in Fig. S13 (ESI†). Here, we found that the LYexp is lower than its calculated value and this finding is commonly observed in the other scintillation studies. Moreover, we measured pulse height spectra to obtain the LYexp as a function of temperature (10–350 K) as presented in Fig. 6. The values of LYexp at RT relative to Cs3Cu2I5 (16.5 ph per keV) are 0.76 ± 0.03, 0.67 ± 0.06, and 0.33 ± 0.03 for CsCu2I3, Cs3Cu2I4 Br, and Cs3Cu2I3Br2, respectively. Considering its powder form, the LY of 1D CsCu2I3 and 0D Cs3Cu2I5 is comparable to that of the single-crystal CHPs reported by Cheng et al., at 16 ph per keV17 and 29 ph per keV18 for CsCu2I3 and 0D Cs3Cu2I5 single-crystals, respectively. The peak of CsCu2I3 was observed to be in the temperature range of 100–200 K, showing its potential for use in this temperature range. The temperature-dependent LY spectra also show that the LY intensity is diminished by TQ upon approaching RT. As a side note, the LYs and scintillation decay times of Pb-based perovskites are depicted in Table S5 (ESI†).
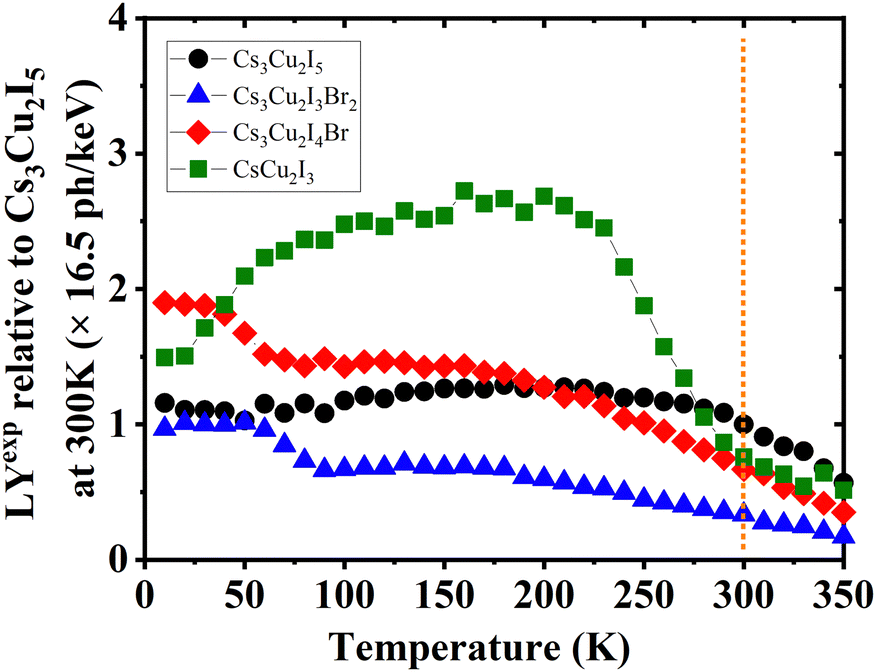 | ||
| Fig. 6 Temperature-dependent experimental LY (LYexp) spectra of Cs3Cu2I5, Cs3Cu2I4 Br, Cs3Cu2I3Br2, and CsCu2I3, normalised to the LY of Cs3Cu2I5 at 300 K derived using integrated RL intensities. The vertical dashed line indicates where the value of LYexp is taken using pulse height spectra at RT, see Fig. S13 (ESI†). | ||
In general, we are looking for an optimised composition of CHPs. First, we investigate the 1D CsCu2I3. CsCu2I3 possesses large SS, a notable contribution from its rapid scintillation decay component (9.30 ± 0.01 ns for 43%), and its attenuation length is shorter than that of Pb-based perovskites for energies between 33 and 42 keV (see Fig. 1a). In our pursuit to shorten attenuation length and to enhance LY and QY, we transitioned from the structure of CsCu2I3 to Cs3Cu2I5 through rational design by adjusting the molar ratio of its constituents. This transition to Cs3Cu2I5 resulted in a reduced attenuation length, a boost in QY (13.59 ± 2.85 to 47.19 ± 8.83%), and an elevated LY (0.76 ± 0.03 to 1 ± 0.21 × 16.5 ph per keV). Yet, a trade-off was observed, which is an extended scintillation decay due to the augmented τ3 component. We also observe deep traps within the Cs3Cu2I5 system. In an attempt to tune the properties, we introduced Br doping into the 0D Cs3Cu2I5 through rational design. Br doping proves to produce 0D CHPs with shallower and more concentrated traps (see Table S4 (ESI†)), an increase in scintillation decay fast component (see Table 2), but as a consequence of longer attenuation length for energies between 33 and 43 keV, and lower LY (see Table 3). The trend of less efficient QY in contrast with a more efficient S indicates that defects are necessary to induce transport.20,68,84 The localisation of excitons achieved by rational design is attributed to the tunable optical and scintillation properties of the CHPs.
Herein, we evaluate the presence of multiple STEs in CHPs from the theoretical61,85–88 perspectives and experimental findings49 that are mainly governed by the direct consequence of exciton localisation and excited-state structural reorganization.89 As the latter, the energetic positions can be modulated by altering the halogen composition of spatially isolated Cu-X polyhedra via rational design. According to the absorption and PL results (see Table 2), we schematically propose the configuration coordinate of the mixed halide CHP with I:Br molar ratios of 4:1 and 3:2 in Fig. 7a and b, respectively. It turns out that the energetic difference between STEs to the ground states of the two ratios is almost negligible (∼10 meV), which is lower than thermal activation energy (∼25 meV). Thus, we believe that the STE emission efficiency (radiative recombination profile) would not differ substantially from the perspective of the energetic landscape approach. On the other hand, previous reports21,85,90 have shown that the STE emissions of either 1D or 0D are marginally substantiated due to the lattice deformation mode.91
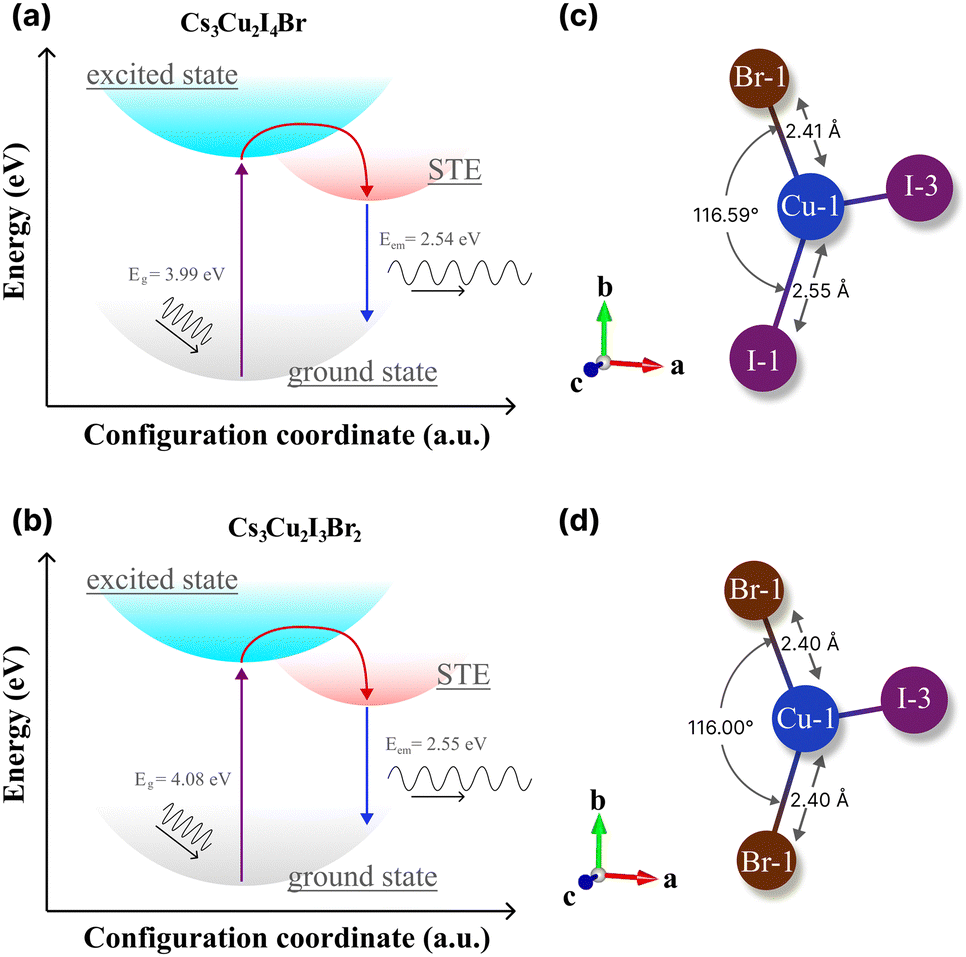 | ||
| Fig. 7 The configuration coordinate diagrams of the STE process of (a) Cs3Cu2I4 Br and (b) Cs3Cu2I3Br2. The theoretical bond length and angle of (c) Cs3Cu2I4 Br and (d) Cs3Cu2I3Br2, extracted from theoretical structural files by the VESTA software package.36 | ||
In CHPs, the geometric configuration of the crystals plays a critical role in influencing the self-trapped excitation mechanism, which in turn significantly affects the absorption and luminescence processes, as well as the optical and scintillation properties. Hence, we proceed to extend our discussion in these mixed halide CHP systems by interpreting the calculated geometric structure acquired from DFT calculations, as exemplified in Fig. 7c (Cs3Cu2I4 Br) and 7d (Cs3Cu2I3Br2). The Cu1–X bond lengths of Cs3Cu2I5, Cs3Cu2I4 Br, and Cs3Cu2I4 Br obtained from the structural CIFs notably display a bond length reduction. For example, as we lowered the CHP dimension from 1D CsCu2I3 (2.61 Å), the bond length is shortened to 2.56 Å for 0D Cs3Cu2I5 (see Fig. S14 and Table S6 (ESI†)). This is in agreement with the previous finding by Ma et al. in thin films.91 When the I atom is substituted by a single Br atom (Fig. 7c), the Cu1–Br1 bonds are measured at ∼2.41 Å in the case of Cs3Cu2I4 Br. Upon the substitution of Br atoms, the bond length is found to be 2.40 Å for Cs3Cu2I3Br2. We also fairly compare CHP structural changes in terms of their bond angles as depicted in Fig. 7c and d. I1–Cu1–I1 angles are determined at ∼116.42° (CsCu2I3) and 117.07° (Cs3Cu2I5). Upon Br doping, we find that the Br1–Cu1–I1 angle is 116.59° (Cs3Cu2I4 Br) and slightly reduced to 116.00° (Cs3Cu2I3Br2) (see Table S6 (ESI†)). Such bond length and angle reduction in 0D CHPs signify the fact that our attempt to incorporate Br atoms onto the CHP host lattice affects the proportion ratio between the well-known three STE configurations.92,93 Since the first decay component of lifetimes (τ1) is associated with the direct recombination of excitons,74,93,94 we argue that the [Cu2X5]3− species in the mixed halide system (I:Br 4:1 and 3:2) powders have a slightly distorted structure compared to their conventional 0D Cs3Cu2I5. This eventually decreased the perovskite volume, mitigating the depth of traps that are largely found in bromine-rich CHPs.94 In addition, we postulate that the Jahn–Teller distortion in the d orbital of Cu centres is strongly enhanced when the unequal occupation of the other degenerate t2g 3d orbitals is indirectly affected by their interaction due to Br incorporation.49 This, in turn, manifests in the large SS and multiple STE emissions as outlined by previous works.29,34,90,94 We believe that such STE modulation not only improves τ1 but simultaneously reduced the slowest τ3 proportion significantly (see Table 2). This rationalizes the QY results shown in Table 3 of the reported CHPs by a mechanochemical approach. The indirect interplay between bond lengths and angles due to Br doping is pivotal in shaping the exciton localisation hence STE formation, where such luminescent properties can be used to optimise the desirable scintillation properties with fast decay time with higher τ1 contribution.
4 Conclusions
In summary, we have investigated the optoelectronics and scintillation properties of 1D (CsCu2I3) and 0D (Cs3Cu2I5, Cs3Cu2I4 Br, and Cs3Cu2I3Br2) CHP powders synthesised by mechanochemistry. 1D CsCu2I3 exhibits an impressive SS of 1.65 ± 0.04 eV accompanied by a short attenuation length (33 to 42 keV), a fast decay component of 9.30 ± 0.01 ns, and competitive experimental LY (state value) compared to different CHP morphologies such as thin films or single crystals at RT. However, 0D Cs3Cu2I5 exhibits deep traps (∼500 meV) that gradually converted to shallow traps upon Br doping. In addition, temperature-dependent RL measurements across the entire samples reveal that RL emission diminishes due to TQ. Introducing Br doping in 0D CHP, the observed reduction of QYs in 0D CHP is proposed due to the contribution of surface trap states induced by the Br content within these materials, highlighting the importance of geometric consideration. By carefully adjusting the I:Br ratio, the lattice distortions can impart a minute change in the Jahn–Teller distortion across Cu tetrahedral networks, leading to a localised exciton and hence STE tunability. Thus, dimensionality control and halide compositional engineering shed some light on the future of scintillator-based CHPs.
Author contributions
Conceptualisation: A., L. J. D., and M. D. B.; data curation: P. Y. D. M., S. H., L. Z., D. K., M. E. W., K. J. D., M. M., W. D., and M. D. B.; formal analysis: T. H., A., S. H., A. A. A., M. H. M., L. J. D., and M. D. B.; investigation: T. H., A., S. H., A. A. A., M. H. M., L. J. D., and M. D. B.; methodology: T. H. and L. J. D.; project administration: A., L. J. D., and M. D. B.; resources: A., P. Y. D. M., S. H., M. H. M., L. Z., M. E. W., K. J. D., M. M., W. D., L. J. D., and M. D. B.; supervision: A., L. J. D., and M. D. B.; visualisation: T. H., M. H. M., and M. D. B.; writing – original draft: T. H., A., and L. J. D. and M. D. B.; writing – review and editing: T. H., A., L. J. D., and M. D. B. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the facilities and scientific and technical support from Advanced Characterization Laboratories Serpong, National Research and Innovation Agency through E-Layanan Sains, Badan Riset dan Inovasi Nasional (BRIN). T. H. and L. J. D. acknowledge the support from Universitas Prasetiya Mulya. Authors from Nano Center Indonesia express their gratitude to PT Nanotech Indonesia Global Tbk for the start-up research grant. M. H. M. acknowledges research fund from Institut Teknologi Bandung under the “Riset ITB 2024” scheme (grant no. 308/IT1.B07.1/TA.01/2024). M. D. B. acknowledges research funds from the National Science Center, Poland, under grant OPUS-24 no. 2022/47/B/ST5/01966. RL and TL measurements were performed at the National Laboratory for Quantum Technologies, Nicolaus Copernicus University and supported by the European Regional Development Fund.References
- M. D. Birowosuto, D. Cortecchia, W. Drozdowski, K. Brylew, W. Lachmanski, A. Bruno and C. Soci, Sci. Rep., 2016, 6, 37254 CrossRef CAS PubMed.
- H. Yu, T. Chen, Z. Han, J. Fan and Q. Pei, ACS Appl. Nano Mater., 2022, 5, 14572–14581 CrossRef CAS.
- A. Wibowo, M. A. K. Sheikh, L. J. Diguna, M. B. Ananda, M. A. Marsudi, A. Arramel, S. Zeng, L. J. Wong and M. D. Birowosuto, Commun. Mater., 2023, 4, 21 CrossRef CAS.
- F. Maddalena, A. Xie, X. Y. Chin, R. Begum, M. E. Witkowski, M. Makowski, B. Mahler, W. Drozdowski, S. V. Springham, R. S. Rawat, N. Mathews, C. Dujardin, M. D. Birowosuto and C. Dang, J. Phys. Chem. C, 2021, 125, 14082–14088 CrossRef CAS.
- Q. Xu, J. Wang, W. Shao, X. Ouyang, X. Wang, X. Zhang, Y. Guo and X. Ouyang, Nanoscale, 2020, 12, 9727–9732 RSC.
- J. Li, H.-L. Cao, W.-B. Jiao, Q. Wang, M. Wei, I. Cantone, J. Lü and A. Abate, Nat. Commun., 2020, 11, 310 CrossRef CAS PubMed.
- A. H. Slavney, R. W. Smaha, I. C. Smith, A. Jaffe, D. Umeyama and H. I. Karunadasa, Inorg. Chem., 2017, 56, 46–55 CrossRef CAS PubMed.
- M. C. Brennan, J. Zinna and M. Kuno, ACS Energy Lett., 2017, 2, 1487–1488 CrossRef CAS.
- R. Lin, Q. Guo, Q. Zhu, Y. Zhu, W. Zheng and F. Huang, Adv. Mater., 2019, 31, 1905079 CrossRef CAS PubMed.
- L. J. Diguna, L. Jonathan, M. H. Mahyuddin, Arramel, F. Maddalena, I. Mulyani, D. Onggo, A. Bachiri, M. E. Witkowski, M. Makowski, D. Kowal, W. Drozdowski and M. D. Birowosuto, Mater. Adv, 2022, 3, 5087–5095 RSC.
- Z.-X. Zhang, C. Li, Y. Lu, X.-W. Tong, F.-X. Liang, X.-Y. Zhao, D. Wu, C. Xie and L.-B. Luo, J. Phys. Chem. Lett., 2019, 10, 5343–5350 CrossRef CAS PubMed.
- R. Roccanova, A. Yangui, G. Seo, T. D. Creason, Y. Wu, D. Y. Kim, M.-H. Du and B. Saparov, ACS Mater. Lett., 2019, 1, 459–465 CrossRef CAS.
- L. Lian, M. Zheng, W. Zhang, L. Yin, X. Du, P. Zhang, X. Zhang, J. Gao, D. Zhang, L. Gao, G. Niu, H. Song, R. Chen, X. Lan, J. Tang and J. Zhang, Adv. Sci., 2020, 7, 2000195 CrossRef CAS PubMed.
- X. Bin, J. Liu, R. Zeng, H. Liu, J. Zhao and T. Lin, J. Mater. Chem. C, 2024, 12, 922–929 RSC.
- J. J. Van Blaaderen, L. A. Van Den Brekel, K. W. Krämer and P. Dorenbos, Chem. Mater., 2023, 35, 9623–9631 CrossRef CAS PubMed.
- P. Yang, G. Liu, B. Liu, X. Liu, Y. Lou, J. Chen and Y. Zhao, Chem. Commun., 2018, 54, 11638–11641 RSC.
- S. Cheng, A. Beitlerova, R. Kucerkova, E. Mihokova, M. Nikl, Z. Zhou, G. Ren and Y. Wu, ACS Appl. Mater. Interfaces, 2021, 13, 12198–12202 CrossRef CAS PubMed.
- S. Cheng, A. Beitlerova, R. Kucerkova, M. Nikl, G. Ren and Y. Wu, Phys. Status Solidi RRL, 2020, 14, 2000374 CrossRef CAS.
- D. Yuan, ACS Appl. Mater. Interfaces, 2020, 12, 38333–38340 CrossRef CAS PubMed.
- Q. Yao, J. Li, X. Li, X. Zheng, Z. Wang and X. Tao, Adv. Opt. Mater., 2022, 10, 2201161 CrossRef CAS.
- Z. Ma, X. Ji, S. Lin, X. Chen, D. Wu, X. Li, Y. Zhang, C. Shan, Z. Shi and X. Fang, Adv. Mater., 2023, 2300731 CrossRef CAS PubMed.
- S. Li, Z. Shi, F. Zhang, L. Wang, Z. Ma, D. Yang, Z. Yao, D. Wu, T.-T. Xu, Y. Tian, Y. Zhang, C. Shan and X. J. Li, Chem. Mater., 2019, 31, 3917–3928 CrossRef CAS.
- X. Huang, Q. Sun and B. Devakumar, Mater. Today Chem., 2020, 17, 100288 CrossRef CAS.
- Y. Liu, M. S. Molokeev and Z. Xia, Energy Mater. Adv., 2021, 2021, 2585274 Search PubMed.
- J.-M. Yang, Y.-K. Jung, J.-H. Lee, Y. C. Kim, S.-Y. Kim, S. Seo, D.-A. Park, J.-H. Kim, S.-Y. Jeong, I.-T. Han, J.-H. Park, A. Walsh and N.-G. Park, Nanoscale Horiz., 2021, 6, 987–997 RSC.
- L. Manna, O. M. Bakr, S. Brovelli and H. Li, Energy Mater. Adv., 2022, 2022, 9865891 Search PubMed.
- Z. Luo, Q. Li, L. Zhang, X. Wu, L. Tan, C. Zou, Y. Liu and Z. Quan, Small, 2020, 16, 1905226 CrossRef CAS PubMed.
- Y. Li, P. Vashishtha, Z. Zhou, Z. Li, S. B. Shivarudraiah, C. Ma, J. Liu, K. S. Wong, H. Su and J. E. Halpert, Chem. Mater., 2020, 32, 5515–5524 CrossRef CAS.
- R. Roccanova, A. Yangui, H. Nhalil, H. Shi, M.-H. Du and B. Saparov, ACS Appl. Electron. Mater., 2019, 1, 269–274 CrossRef CAS.
- V. Scailteur and R. R. Lauwerys, Toxicology, 1987, 43, 231–238 CrossRef CAS PubMed.
- E. G. Worthley and C. D. Schott, Toxicol. Appl. Pharmacol., 1969, 15, 275–281 CrossRef CAS PubMed.
- S. Fang, Y. Wang, H. Li, F. Fang, K. Jiang, Z. Liu, H. Li and Y. Shi, J. Mater. Chem. C, 2020, 8, 4895–4901 RSC.
- G. K. Grandhi, N. S. M. Viswanath, H. B. Cho, J. H. Han, S. M. Kim, S. Choi and W. B. Im, J. Phys. Chem. Lett., 2020, 11, 7723–7729 CrossRef CAS PubMed.
- P. Sebastia-Luna, J. Navarro-Alapont, M. Sessolo, F. Palazon and H. J. Bolink, Chem. Mater., 2019, 31, 10205–10210 CrossRef CAS.
- N. Doebelin and R. Kleeberg, J. Appl. Cryst., 2015, 48, 1573–1580 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Cryst., 2011, 44, 1272–1276 CrossRef CAS.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci., 2021, 5, 100112 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- L. Xie, B. Chen, F. Zhang, Z. Zhao, X. Wang, L. Shi, Y. Liu, L. Huang, R. Liu, B. Zou and Y. Wang, Photon. Res., 2020, 8, 768 CrossRef CAS.
- T. Jun, K. Sim, S. Iimura, M. Sasase, H. Kamioka, J. Kim and H. Hosono, Adv. Mater., 2018, 30, 1804547 CrossRef PubMed.
- F. Zhang, Z. Zhao, B. Chen, H. Zheng, L. Huang, Y. Liu, Y. Wang and A. L. Rogach, Adv. Opt. Mater., 2020, 8, 1901723 CrossRef CAS.
- F. Maddalena, M. E. Witkowski, M. Makowski, A. Bachiri, Arramel, T. Yang, M. H. Mahyuddin, M. Baravaglio, M. Boutchich, W. Drozdowski, C. Dujardin, M. D. Birowosuto and C. Dang, J. Mater. Chem. C, 2022, 10, 11266–11275 RSC.
- Arramel, A. Xie, X. Yin, C. S. Tang, M. H. Mahyuddin, C. Hettiarachchi, M. F. Sahdan, K. Yoshizawa, C. Dang, M. D. Birowosuto, A. T. S. Wee and A. Rusydi, ACS Appl. Energy Mater., 2020, 3, 7500–7511 CrossRef CAS.
- S. Hartati, P. Y. D. Maulida, T. Zakly, I. Mulyani, D. Onggo, M. H. Mahyuddin, A. Noviyanto, A. Arramel and N. T. Rochman, NHC, 2023, 40, 1–6 Search PubMed.
- L. J. Diguna, S. Kaffah, M. H. Mahyuddin, Arramel, F. Maddalena, S. A. Bakar, M. Aminah, D. Onggo, M. E. Witkowski, M. Makowski, W. Drozdowski and M. D. Birowosuto, RSC Adv., 2021, 11, 20635–20640 RSC.
- P. Y. D. Maulida, D. Wang, F. Maddalena, C. S. Tang, X. Yin, C. Diao, I. Mulyani, D. Onggo, A. Noviyanto, M. D. Birowosuto, Arramel, A. T. S. Wee and A. Rusydi, J. Phys. Chem. C, 2022, 126, 15801–15808 CrossRef CAS.
- H. Lei, D. Hardy and F. Gao, Adv. Funct. Mater., 2021, 31, 2105898 CrossRef CAS.
- G. S. Long, M. Wei and R. D. Willett, Inorg. Chem., 1997, 36, 3102–3107 CrossRef CAS PubMed.
- A. M. Elseman, A. E. Shalan, S. Sajid, M. M. Rashad, A. M. Hassan and M. Li, ACS Appl. Mater. Interfaces, 2018, 10, 11699–11707 CrossRef CAS PubMed.
- Y. Zhou, X. Wang, T. He, H. Yang, C. Yang, B. Shao, L. Gutiérrez-Arzaluz, O. M. Bakr, Y. Zhang and O. F. Mohammed, ACS Energy Lett., 2022, 7, 844–846 CrossRef CAS.
- J. Qu, S. Xu, H. Shao, P. Xia, C. Lu, C. Wang and D. Ban, J. Mater. Chem. C, 2023, 11, 6260–6275 RSC.
- L. Lian, M. Zheng, P. Zhang, Z. Zheng, K. Du, W. Lei, J. Gao, G. Niu, D. Zhang, T. Zhai, S. Jin, J. Tang, X. Zhang and J. Zhang, Chem. Mater., 2020, 32, 3462–3468 CrossRef CAS.
- R. J. Elliott, Phys. Rev., 1957, 108, 1384–1389 CrossRef CAS.
- B. Ullrich, A. K. Singh, P. Barik, H. Xi and M. Bhowmick, Opt. Lett., 2015, 40, 2580 CrossRef CAS PubMed.
- Q. Li, Z. Chen, B. Yang, L. Tan, B. Xu, J. Han, Y. Zhao, J. Tang and Z. Quan, J. Am. Chem. Soc., 2020, 142, 1786–1791 CrossRef CAS PubMed.
- P. Vashishtha, G. V. Nutan, B. E. Griffith, Y. Fang, D. Giovanni, M. Jagadeeswararao, T. C. Sum, N. Mathews, S. G. Mhaisalkar, J. V. Hanna and T. White, Chem. Mater., 2019, 31, 9003–9011 CrossRef CAS.
- X. Liu, Y. Yu, F. Yuan, C. Zhao, H. Dong, B. Jiao and Z. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 52967–52975 CrossRef CAS PubMed.
- Y. Tratsiak, L. Stand, R. Lalk, M. Zhuravleva and C. L. Melcher, J. Lumin., 2023, 263, 119952 CrossRef CAS.
- B. Liu, Y. Sun, L. Chen, B. Liao, J. Ruan, L. Zhou, Y. Li and X. Ouyang, Inorg. Chem., 2022, 61, 16141–16147 CrossRef CAS PubMed.
- F. Maddalena, A. Xie, Arramel, M. E. Witkowski, M. Makowski, B. Mahler, W. Drozdowski, T. Mariyappan, S. V. Springham, P. Coquet, C. Dujardin, M. D. Birowosuto and C. Dang, J. Mater. Chem. C, 2021, 9, 2504–2512 RSC.
- J. T. Randall and M. H. F. W. Wilkins, Proc. R. Soc. London, Ser. A, 1945, 184, 365–389 CAS.
- P. Schulz, ACS Energy Lett., 2018, 3, 1287–1293 CrossRef CAS.
- C. Giansante and I. Infante, J. Phys. Chem. Lett., 2017, 8, 5209–5215 CrossRef CAS PubMed.
- Y. Hu, E. M. Hutter, P. Rieder, I. Grill, J. Hanisch, M. F. Aygüler, A. G. Hufnagel, M. Handloser, T. Bein, A. Hartschuh, K. Tvingstedt, V. Dyakonov, A. Baumann, T. J. Savenije, M. L. Petrus and P. Docampo, Adv. Energy Mater., 2018, 8, 1703057 CrossRef.
- F. Maddalena, L. Tjahjana, A. Xie, Arramel, S. Zeng, H. Wang, P. Coquet, W. Drozdowski, C. Dujardin, C. Dang and M. Birowosuto, Crystals, 2019, 9, 88 CrossRef.
- E. V. D. van Loef, P. Dorenbos, C. W. E. van Eijk, K. W. Krämer and H. U. Güdel, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 045108 CrossRef.
- Y. Zhou, Z. Deng, B. Wang, P. Li, L. Li, W. Han, J. Huang, W. Jia, X. Ouyang, Q. Xu and K. K. Ostrikov, Chem. Eng. J., 2023, 471, 144431 CrossRef CAS.
- V. Naresh, S. Singh, H. Soh, J. Lee and N. Lee, Mater. Today Nano, 2023, 100364 CrossRef CAS.
- M. S. J. Marshall, J. Wang, S. Miller, B. Singh and V. Nagarkar, 2020 IEEE Nuclear Science Symposium and Medical Imaging Conference (NSS/MIC), Boston, MA, USA, 2020, pp. 1–2 Search PubMed.
- J. Liu, W. Shao, Q. Xu, Y. Liu and X. Ouyang, IEEE Photon. Technol. Lett., 2020, 32, 635–638 CAS.
- Q. Xu, J. Wang, Q. Zhang, X. Ouyang, M. Ye, W. Xie, X. Yan, D. Li, X. Ouyang, X. Tang and X. Zhang, Photon. Res., 2021, 9, 351 CrossRef.
- Z. Ma, Z. Shi, C. Qin, M. Cui, D. Yang, X. Wang, L. Wang, X. Ji, X. Chen, J. Sun, D. Wu, Y. Zhang, X. J. Li, L. Zhang and C. Shan, ACS Nano, 2020, 14, 4475–4486 CrossRef CAS PubMed.
- N. Li, Z. Xu, Y. Xiao, Y. Liu, Z. Yang and S. F. Liu, Adv. Opt. Mater., 2022, 10, 2102232 CrossRef CAS.
- T. Jiang, J. Wang, L. Xie, C. Bai, M. Wang, Y. Wu, F. Zhang, Y. Zhao, B. Chen and Y. Wang, ACS Appl. Nano Mater., 2022, 5, 508–516 CrossRef CAS.
- M. D. Birowosuto and P. Dorenbos, Phys. Status Solidi A, 2009, 206, 9–20 CrossRef CAS.
- M.-H. Du, ACS Energy Lett., 2020, 5, 464–469 CrossRef CAS.
- R. Lin, Q. Zhu, Q. Guo, Y. Zhu, W. Zheng and F. Huang, J. Phys. Chem. C, 2020, 124, 20469–20476 CrossRef CAS.
- Y.-K. Jung, S. Kim, Y. C. Kim and A. Walsh, J. Phys. Chem. Lett., 2021, 12, 8447–8452 CrossRef CAS PubMed.
- Y. Hu, X. Yan, L. Zhou, P. Chen, Q. Pang and Y. Chen, J. Phys. Chem. Lett., 2022, 13, 10786–10792 CrossRef CAS PubMed.
- M. A. Halcrow, Chem. Soc. Rev., 2013, 42, 1784–1795 RSC.
- H. Chen, J. M. Pina, F. Yuan, A. Johnston, D. Ma, B. Chen, Z. Li, A. Dumont, X. Li, Y. Liu, S. Hoogland, Z. Zajacz, Z. Lu and E. H. Sargent, J. Phys. Chem. Lett., 2020, 11, 4326–4330 CrossRef CAS PubMed.
- Z. Ma, Z. Shi, D. Yang, Y. Li, F. Zhang, L. Wang, X. Chen, D. Wu, Y. Tian, Y. Zhang, L. Zhang, X. Li and C. Shan, Adv. Mater., 2021, 33, 2001367 CrossRef CAS PubMed.
- R. Kentsch, M. Morgenroth, M. Scholz, K. Xu, J. Schmedt Auf Der Günne, T. Lenzer and K. Oum, J. Phys. Chem. Lett., 2020, 11, 4286–4291 CrossRef CAS PubMed.
- C.-Y. Chen, Y.-I. Lin, P.-T. Lai, H.-C. Lin, G.-H. Tan, H.-W. Lin and R. D. Schaller, Adv. Opt. Mater., 2022, 10, 2200005 CrossRef CAS.
- G. F. Samu, T. S. Zsigmond, C. Hajdu, M. Hunyadi, L. Csige, A. Csík, J. Kopniczky, B. Hopp and C. Janáky, Adv. Opt. Mater., 2023, 2300825 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc03977c |
| This journal is © The Royal Society of Chemistry 2024 |