
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFuture perspectives in green hydrogen production by catalyzed sono-photolysis of water
Piergiovanni
Domenighini
 *a,
Ferdinando
Costantino
b,
Pier Luigi
Gentili
b,
Anna
Donnadio
c,
Morena
Nocchetti
c,
Alceo
Macchioni
b,
Federico
Rossi
a and
Franco
Cotana
ad
*a,
Ferdinando
Costantino
b,
Pier Luigi
Gentili
b,
Anna
Donnadio
c,
Morena
Nocchetti
c,
Alceo
Macchioni
b,
Federico
Rossi
a and
Franco
Cotana
ad
aDipartimento di Ingegneria, Università degli Studi di Perugia, Via Goffredo Duranti, 93, 06125 Perugia PG, Italy. E-mail: piergiovanni.domenighini@unipg.it
bDipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, Via dell'Elce di Sotto, 8, 06123 Perugia PG, Italy
cDipartimento di Scienze Farmaceutiche, Università degli Studi di Perugia, Via del Liceo, 1, 06123 Perugia PG, Italy
dRicerca sul Sistema Energetico RSE spa, Via R. Rubattino 5420134, Milano MI, Italy
First published on 13th May 2024
Abstract
The quest for sustainable energy solutions has led to a growing interest in green hydrogen production, with catalyzed sono-photolysis of water emerging as a promising avenue. This perspective highlights the innovative combination of photocatalysis and acoustic cavitation to enhance the generation of green hydrogen from water splitting. By harnessing the power of semiconductor-based catalysts, the sono-photolysis approach capitalizes on solar radiation to initiate water dissociation. Simultaneously, high-intensity ultrasound waves trigger cavitation, creating reactive microbubbles and localized hotspots that further promote hydrogen evolution. Through systematic experimentation and optimization, researchers are investigating the influence of catalyst type, concentration, and ultrasonic parameters on hydrogen production. Excitingly, early results demonstrated a promising synergistic effect between photolysis and sonolysis. These findings traced a new path that is worth being pursued to open the door to scalable, cost-effective, and environmentally friendly green hydrogen production. In this perspective, catalyzed sono-photolysis holds tremendous potential for meeting the world's energy demands sustainably. Its innovative blend of light and sound-driven water splitting paves the way towards a greener future, offering a viable solution for the large-scale production of clean and renewable hydrogen.
Introduction
The global pursuit of sustainable and carbon-neutral energy sources has intensified in recent years, driven by the urgent need to mitigate climate change and reduce dependence on fossil fuels. Among various renewable energy alternatives, hydrogen has emerged as a promising candidate due to its high mass energy density and versatility (especially, non-toxicity and zero-carbon combustion). However, conventional methods of hydrogen production, such as steam methane reforming, require fossil fuels and generate significant carbon emissions. To overcome these limitations, the focus has shifted towards green hydrogen production, which utilizes renewable resources and produces no greenhouse gas emissions.Among the “green” hydrogen production technologies that utilize energy from renewable systems, the following are worth mentioning:
• Biomass gasification and pyrolysis: This process involves partial oxidation of biomass derived from animal waste, municipal or industrial waste, and agricultural waste. Hydrogen production yields typically range from 14.3 wt% to significant peaks of 17 wt% in the case of lignocellulosic biomass;1
• Biochemical processes: These include direct and indirect bio-photolysis, photo-fermentation, or fermentation in the absence of light;2
• Water electrolysis: This method involves the redox splitting of H2O into H2 and O2 induced by an electric current being passed through two electrodes in an aqueous solution.3 It currently represents 4% of the global hydrogen production,6 with production efficiencies ranging from 60% (alkaline cells) to 80% (molten carbonate cells);4,5
• Photoelectrolysis: This process utilizes photo-catalyzed electrolysis. Depending on the solar radiation intensity and the electrodes made of semiconductors that also act as photocatalysts, water splitting can be achieved with current densities ranging from 10 mA cm−2 to 30 mA cm−2 and an electrical potential difference of +1.35 V6.
Catalyzed sono-photolysis of water has emerged as an innovative and promising approach for green hydrogen production. This technique combines the principles of heterogeneous photocatalysis and acoustic cavitation to enhance the efficiency of hydrogen evolution. Heterogeneous photocatalysis involves the use of semiconductor materials to absorb solar radiation and induce the formation of electron–hole pairs that initiate the water-splitting reaction. Concurrently, acoustic cavitation induces the formation and implosive collapse of microbubbles in the liquid, creating localized hotspots and other extreme physicochemical conditions (such as extraordinarily high pressures) favorable for the dissociation of water molecules into their constitutive elements, which are H2 and O2. The integration of photocatalysis and acoustic cavitation in catalyzed sono-photolysis offers several distinct advantages. First, the use of sunlight as the primary energy source enables a renewable and abundant input for hydrogen generation. Furthermore, the incorporation of ultrasonic waves promotes the cavitation phenomenon, leading to the generation of additional reactive species and accelerating the overall hydrogen evolution process. Advantages over the other popular multifield synergistic catalysis such us photo-electrolysis, or photo-thermal, are mainly due to the extremely high energy contribution of the cavitation field. The cavitation phenomenon allows local temperatures and pressures to be reached which are otherwise inaccessible, which trigger the water-splitting reaction and improve the activity of the heterogeneous catalyst. Great opportunities may be unveiled by optimizing cavitation control, and, for this reason, this method of green hydrogen production has recently gained attention in research planning and financial support from both public–private partnerships in various nations and transnational bodies such as the European Union. Governments and organizations worldwide recognize the potential of green hydrogen as a key component in achieving climate and energy transition goals. As a result, they are actively promoting and investing in research and development efforts related to hydrogen production technologies, including catalyzed sono-photolysis.7,8 Partnerships between public and private entities are crucial in advancing the development and deployment of green hydrogen technologies. These collaborations bring together expertise, resources, and funding to support research, innovation, and commercialization efforts. They also foster knowledge sharing, standardization, and policy frameworks to create an enabling environment for the widespread adoption of green hydrogen. At the transnational level, the European Union has identified hydrogen as a strategic priority in its efforts to decarbonize the economy. Through initiatives such as the European Clean Hydrogen Alliance and the European Green Deal,9 the EU aims to drive research, innovation, and investment in green hydrogen technologies. This includes funding programs, regulatory support, and the establishment of hydrogen partnerships across member states. The involvement of research planning and financial support from diverse nations and transnational bodies reflects the global recognition of the potential of green hydrogen and its role in the transition to a sustainable and low-carbon future.10 These initiatives demonstrate a collective commitment to driving the development and adoption of green hydrogen technologies on a global scale.
The preliminary findings from sono-photolysis studies are promising, demonstrating significantly enhanced rates of hydrogen evolution and improved conversion efficiencies compared to conventional water-splitting methods. Moreover, the catalyzed sono-photolysis approach offers scalability, as it can be applied to large-scale systems with relatively simple reactor configurations. The cost-effectiveness and environmental friendliness of the technique further solidify its potential as a viable solution for green hydrogen production.
In this perspective paper, we aim to provide a comprehensive overview of the current state of heterogeneously catalyzed sono-photolysis of water for green hydrogen production starting from the photolysis and sonolysis processes separately. We will explore the fundamental principles, catalyst design strategies, and optimization approaches employed in this field. Additionally, we will discuss the challenges and opportunities associated with scaling up this technology and its potential integration into existing energy systems. Ultimately, this perspective seeks to shed light on the immense potential of catalyzed sono-photolysis in advancing the transition towards a sustainable, carbon-neutral future powered by green hydrogen.
Water splitting
Water splitting (1) is an endergonic redox reaction: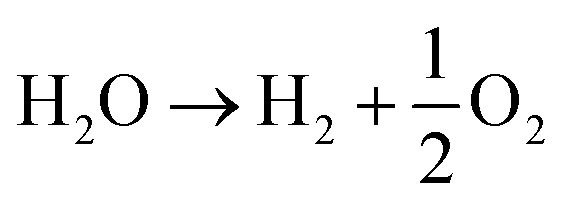 | (1) |
The protons of water are reduced to H2, whereas its dianionic oxygen is oxidized to O2. A noticeable amount of free energy is required to perform the reaction: under standard conditions, ΔG0 = 237.4 kJ mol−1. The real energy needed is, nevertheless, much higher than that predicted by thermodynamics, owing to the relevant kinetic barriers present on both sides of the redox process, especially in the oxidative one. The needed free energy to split water into molecular hydrogen and oxygen can be given in the form of UV-visible electromagnetic radiation emitted by the sun and reaching the surface of the earth (photolysis). Alternatively, it can be released by ultrasound (sonolysis) or both UV-visible electromagnetic radiation and ultrasounds (sonophotolysis). Active and robust co-catalysts, capable of offering alternative reaction pathways to water splitting, must be developed, and interfaced with the photocatalytic systems, in order to lower down the unfavorable kinetic barriers and thus maximize the energy conversion efficiency.11
The catalyzed photolysis
The composition of the solar spectrum corresponds to the thermal radiation emitted by a black body at 5800 K, which is the temperature of the solar surface. The solar radiation is incoherent and polychromatic: its spectrum12 ranges from UV-C (120 nm) to near-IR (100 μm). The chemical compounds of the atmosphere exert a filter effect and they screen the terrestrial biosphere from UV-C and some bands of the IR. The UV and visible radiations that reach the surface of the earth are not absorbed directly by water. Water molecules absorb some bands of the IR, but IR radiation does not have enough energy to split the chemical bonds of H2O unless solar concentrators are used to reach very high temperatures.13 Therefore, a photocatalyst is needed, i.e., a chemical compound that absorbs some radiation of the solar spectrum with the appropriate energy and transferring it to water, leading to its splitting into H2 and O2, thus catalyzing the process. From the point of view of large-scale hydrogen production, the use of heterogeneous photocatalysts in the form of particulate semiconductor materials is more practical than other solutions.14,15 A semiconductor absorbs photons that have energy equal to or larger than its band gap (BG) (see Fig. 1). Any photon absorbed promotes the jump of an electron from the valence (VB) to the conduction (CB) band of the semiconductor (see eqn (2)): a physically separated electron (e−) – hole (h+) pair is generated in the solar radiation harvesting photocatalyst.| (eVB− − hVB+)s + hν → (eCB−)s + (hVB+)s | (2) |
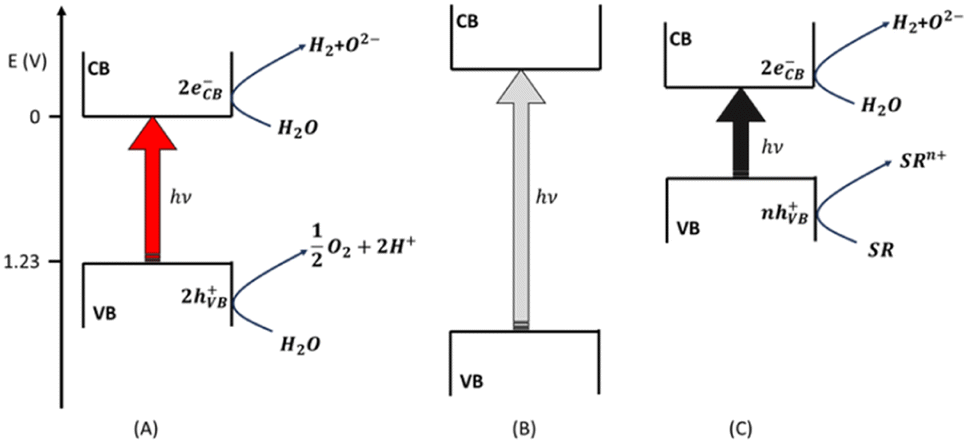 | ||
| Fig. 1 Water splitting photo-catalyzed by a semiconductor having appropriate electric potential values for the conduction (CB) and valence (VB) bands (A). The semiconductor in (B) has a highly negative CB electric potential and a very positive VB electric potential, guaranteeing the reduction of the over-potentials of the two redox semi-reactions, but generating a broad BG. Graph (C) shows the case of a semiconductor that (1) has a narrow BG, (2) oxidizes a sacrificial reagent (SR), and (3) has a negative CB electric potential assuring the production of H2 from water. | ||
These two opposite charges must migrate to the surface of the solid particles without recombining with each other. Then, as soon as they reach the surface, they must participate in the redox reactions required to split water molecules. The electron is supposed to reduce hydrogen whereas the hole converts the oxide to oxygen according to the following two redox semi-reactions (see Fig. 1A):
| 2(H+)aq + 2(eCB−)s → (H2)g | (3) |
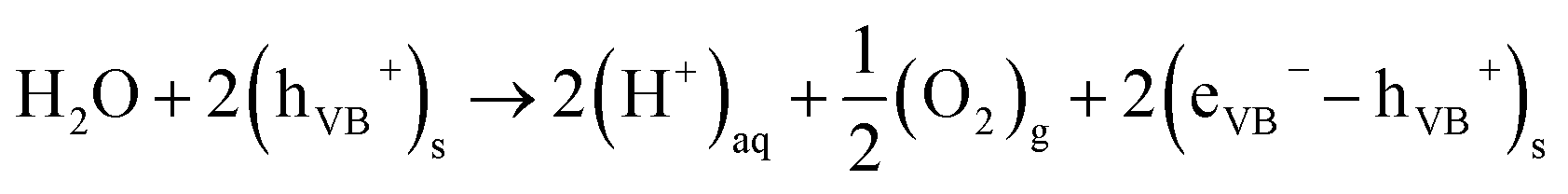 | (4) |
Reaction (3) is thermodynamically spontaneous if the electric potential associated with the electrons in the CB is more negative than the electric potential of H+/H2, which, under standard conditions, is equal to 0 V. On the other hand, eqn (4) requires that the electric potential of the holes in the VB is more positive than the potential of O2/H2O, which, under standard conditions, is equal to 1.23 V.
The sum of eqn (3) and (4) to eqn (2) multiplied by 2 gives the overall photo-induced water splitting:
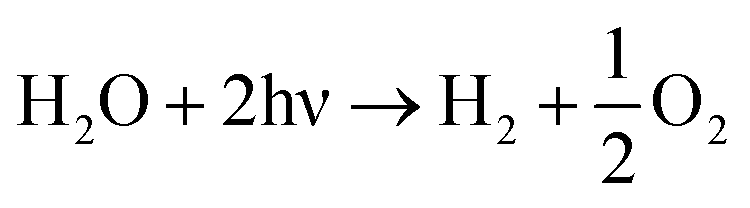 | (5) |
Based on these considerations, it might appear that two photons having an energy of 1.23 eV (i.e., a wavelength of λ = 1011 nm) are enough to split one water molecule into H2 and oxygen.16 Actually, the energy of an electron–hole pair after photon absorption is internal energy and not free energy. The contribution of entropy must be taken into account. The entropic factor reduces the range of solar wavelengths that can be effective in promoting water splitting to values above 610 nm.17 Other kinetic factors affect the efficiency of the solar-induced water splitting performed with a heterogeneous photocatalyst.18 For instance, activation barriers might hinder the migration of the photo-generated charges toward the surface of the solid particles. These barriers can be lowered by synthesizing the semiconductor photocatalysts in the form of nanomaterials having high crystallinity (reducing the internal defects that can act as traps of the photogenerated charges). Moreover, the superficial redox reactions might show over-potentials. This latter issue can be circumvented by loading the photocatalyst with co-catalysts for both the reductive and oxidative steps.19–21 Alternatively, the over-potentials can be overtaken if the electric potentials associated with the CB and VB are highly negative and positive, respectively (see Fig. 1B). This choice imposes large BGs on the semiconductors, which become capable of absorbing limited portions of the solar spectrum, almost exclusively restricted to the UV region.22–24 A strategy to avoid this blind alley relies upon the use of sacrificial reagents25 when the only water-splitting product that is of interest is the energy vector H2 (and O2 is just a by-product). A sacrificial reagent (SR) in the photo-induced hydrogen production from water is any species that can be oxidized more easily than the oxygen of water. Usual SRs are amines, alcohols, carboxylic acids, sulphide and sulphite salts. A farsighted choice consists of using an easily available biomass compound as a sacrificial reducing agent, guaranteeing the possibility of implementing a large-scale sustainable production of H2.16 Whenever a SR is used, the BG of the semiconductor can be shrunk, allowing the capture of a broad portion of the solar spectrum: the only strong thermodynamic requirement is regarding the CB electric potential which must be more negative than the couple H+/H2 (see Fig. 1C).
Strategies to optimize the photocatalytic process of H2 production through water splitting
1. Doping: Doping of metal oxides is usually carried out with sulphur or nitrogen atoms or suitable metals. The doping metals must have partially filled d-orbitals (such as Ni2+, Co2+, Fe3+, Mn2+, and Cr3+) or partially filled sp orbitals (such as Sn2+, Bi2+, Pb2+). The dopants introduce electron states in between the VB and the CB, making the material capable of absorbing visible light. However, the dopants are introduced in small percentages. Therefore, the number of visible photons that can be harvested is always limited.
2. Extension of the VB: The extension of the metal oxide VB is feasible by synthesizing metal oxy-nitrides and oxy-sulphides. The introduction of nitrogen or sulphur atoms in amounts comparable to that of oxygen enlarges the top of the VB towards less positive electric potential values. The same effect can be achieved by stoichiometric addition of metal ions having partially filled sp orbitals, such as Sn2+, Bi2+, or Pb2+.
3. Solid solutions: The mixture of white and colored oxides produces colored solid solutions that are good photocatalysts.26 Their synthesis is feasible when acidic and basic oxides are intermixed. Acidic and basic oxides are acceptors and donors of oxide anions, respectively. The acid–base character of a metal oxide depends on the electronegativity of the metal: the more electronegative the metal, the more acidic the metal oxide. The mixture of different metal oxides can give rise to new crystal structures that can be more active as photocatalysts than their parent phases.
Transition metal-based compounds, supported on a semiconductor surface, have proven to provide good results for the efficiency of the system.33–35 Due to their coloured features, transition metals can help in improving the light-harvesting properties of the system by adding a larger portion of the visible spectrum absorbed and helping the photo-induced charge separation. In addition, the formation of heterojunctions with the photocatalyst surface effectively promotes the charge separation lifetimes by means of two known processes: the formation of Schottky barriers which are electric fields generated by the co-catalyst and placed under the CB of the photocatalyst, which are able to inhibit the backflow of photogenerated electrons, resulting in more electrons to participate in the reaction of H2 evolution. The second process concerns the semiconducting properties of the co-catalyst, resulting in a band alignment of the CB at lower energy with respect to that of the photocatalysts, thus promoting electron migration upon light excitation. For the oxygen evolution side, the same process can occur with co-catalysts having the VB placed under the VB of the co-catalysts, promoting the hole-migration from photo-to the co-catalyst through the junction interface. In the literature, the most employed co-catalysts for the water splitting reaction based on transition metals belong to several families of compounds such as elemental metals, metal sulphides, nitrides, carbides, borides, phosphides, oxides, hydroxides and oxo-hydroxides.36–45 The pure solar-to-hydrogen process, which is solely based on the use of solar radiation without any external current supply or mediated by electrolyte solutions, is limited by the efficiency, being around 3%. As an example, a recent study reported a target value of about 9% by using concentrated solar irradiation and Ga/In nitride nanoparticles as a co-catalyst.46
The efficiency of co-catalysts in assisting the photocatalytic process is also related to other relevant factors:
• The microstructural features such as the crystal size distribution, the shape of the crystals and the active facets exposed, the presence of microstructural strain and local defects which can affect the charge separation process;
• The deposition methods such as photoassisted deposition, chemical vapour deposition/sputtering or impregnation with a solution containing the catalyst precursors;
• The dispersibility of the co-catalysts in low boiling solvents able to make homogeneous layers of nanometric size. Some recent papers reported that the use of nanometric-sized crystals, homogeneously distributed and deposited by physical methods such as vapor deposition or photo-deposition, are more effective in inducing stable and durable charge separation and achieving highly homogenous deposition of photo/co-catalysts.47,48
The sonolysis
Sonolysis is a process that involves the splitting of molecules within a medium due to the mechanical stimulation caused by the presence of acoustic waves, typically at ultrasonic frequencies (f > 20 kHz).56–58 Generally, sonolysis is strictly related to cavitation phenomena in the literature,59–61 with bubble generation in the fluid, and it is associated with emission of ultrasonic frequencies from a specific range, known as power ultrasound, with frequencies ranging from 20 kHz to 1 MHz, and a minimum power of 10 W, that can contribute to sonochemical effects.56 When ultrasound propagates through a liquid solution, it gives rise to various phenomena:• Heating of the solution occurs because of the dissipation of mechanical energy and atomization effects.59 This can lead to the formation of acoustic fountains at the gas–liquid interface, resulting in localized temperatures of up to 250 °C;62
• Small bubbles are generated, expanding during the pressure rarefaction phases caused by the oscillation of the ultrasound waves. Subsequently, these bubbles collapse during compression, giving rise to cavitation phenomena.60 Cavitation events can reach extremely high temperatures of up to 5000 °C and pressures of 2000 atm.63
In the case of water, the sonolysis process involves a series of reactions:64
| H2O → H˙ + ˙OH | (6) |
| H˙ + H˙ → H2 | (7) |
| ˙OH + ˙OH → H2O2 | (8) |
| H2O2 → H2O + ½O2 | (9) |
| H˙ + ˙OH → H2O | (10) |
Eqn (6) shows that water sonolysis produces two reactive radical species: H˙ and OH˙. Then, two H˙ can combine to produce molecular hydrogen (eqn (7)), and two OH˙ can produce hydrogen peroxide (eqn (8)). H2O2 is unstable and spontaneously decomposes into water and O2 (eqn (9)). Finally, H˙ and OH˙ can recombine to form the reagent water molecule (eqn (10)).
Cavitation and sonolysis processes for hydrogen production61 became popular in recent times because they represent an innovative way for green-hydrogen production (the so-called son-hydro-gen process65). Ultrasound power is reported as a conversion process with an overall efficiency of 80–90%, considering the electrical power input at the transducer stage, and a maximum potential loss of 10% for heat at the generator and transducer power conversion respectively.65
Combining the state of the art about sonolysis for sono-hydro-gen processes and more general sono-chemical applications, it is possible to list the main parameters which may influence the hydrogen production by sonolysis in water-filled sono-rectors (Fig. 2).
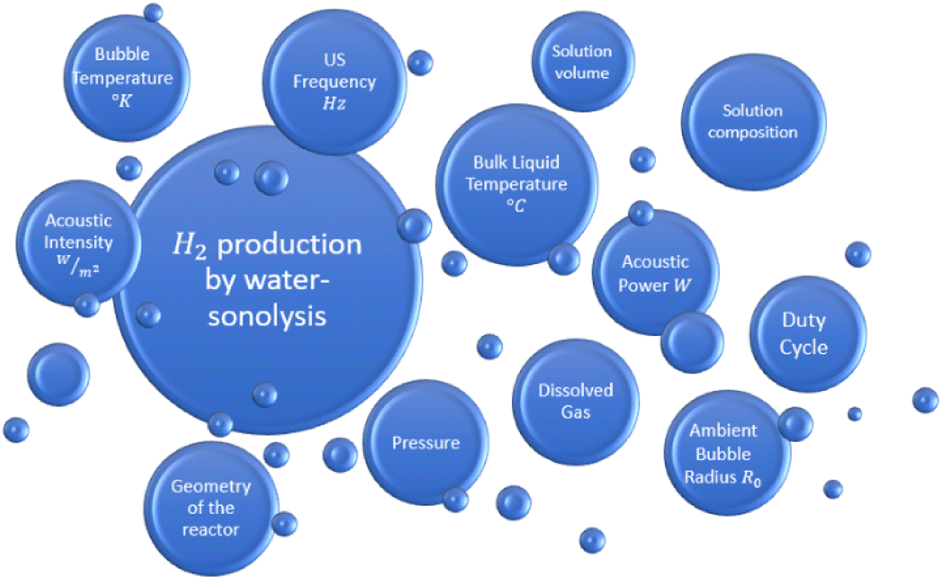 | ||
| Fig. 2 Factors affecting hydrogen production by water-sonolysis. | ||
Recent studies provided useful equations to calculate the cavitation energy Ecav in terms of input power Winput to a sonotrode (type-A sono-reactor, more information below):66,67
| Ecav = −8 × 10−4 × Winput2 + 0.4699 × Winput | (11) |
| ηSono-hydro-gen = 3 × 10−4 × Ecav2 + 0.014 × Ecav | (12) |
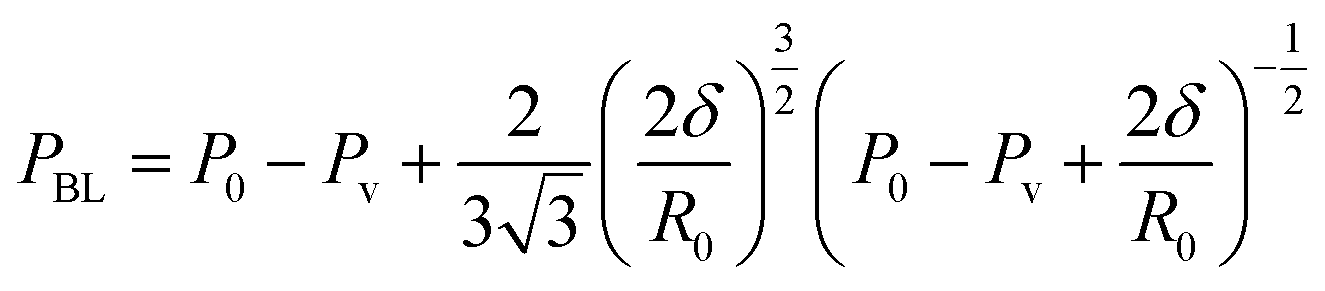 | (13) |
Furthermore, no sonochemical products are detected in a pure CO2 atmosphere.72,74
However, Ar should be considered as a promising dissolved gas in reactors for sono-hydro-gen processes and numerical work by Merouani et al.69,70 in dynamics for hydrogen production may prove it: applying the Keller–Miksas equation for cavitation and considering pressure and temperature by the ideal-gas law or van der Waals equation, the authors reported that argon gas shows a hydrogen production rate higher than natural air because of its greater polytropic ratio (γAr = 1.66 vs. γAir = 1.41) and lower thermal conductivity (λAr = 0.018 W m−2 K−1vs. λAir = 0.024 W m−2 K−1), causing bigger bubble radius, higher bubble temperature (6000 K with the van der Waals model71), and more violent explosion with a higher production of radicals from water. They reported a production of H2 61 times higher than that obtained in the air atmosphere (13.6 μmol min−1vs. 0.22 μmol min−1) in a type-A sonoreactor (Fig. 3) at 1 MHz.70
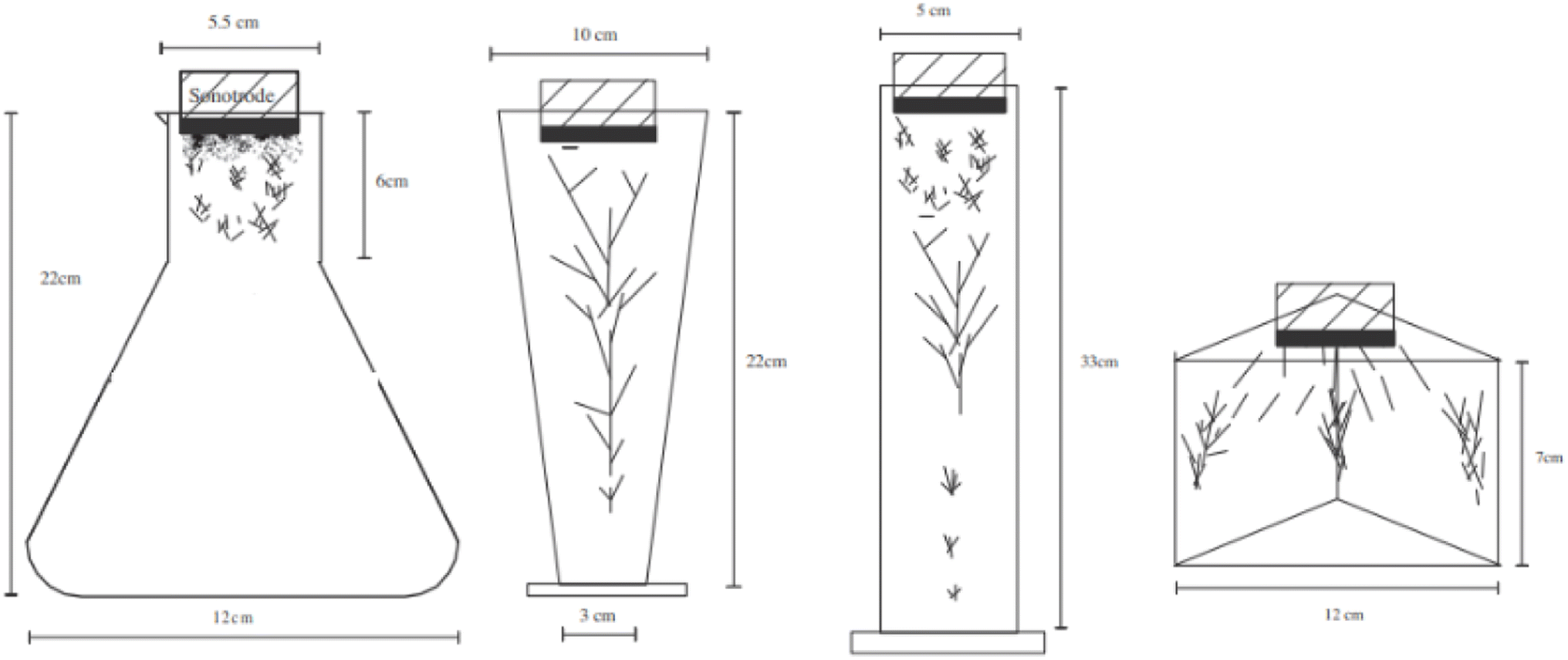 | ||
| Fig. 3 ALF structures in different reactors (P < 180 kPa).57 | ||
The yield depends on the geometry of the sonoreactor, acoustic power, and the number of sonotrodes. The geometry of the sonoreactor and its influence on the sonochemical process is a widely investigated topic. Starting from the three main sonoreactor schemes (Fig. 4), researchers performed numerical studies and experimental measurements on a wide range of cylindrical or rectangular base reactors. For a type-A sonoreactor,62 considerable differences in the acoustic pressure distribution are reported with an apparent advantage to the bottom-curved shape geometry at 20 kHz by numerical simulations in COMSOL Multiphysics.66 At the same frequency comparable results were obtained by Wei77 by simulations, but highlighting the impossibility to implement the bi-phase liquid section represented by the bubbles and the acoustic scattering provided by the bubble clusters as the main limitations to obtain reliable simulated acoustic pressure distributions. Consistently, Wang80 confirmed the potential of the sound wave-based propagation simulation approach but highlights lacking in the prediction of the influence of acoustic scattering on pressure distribution, and due to the lack of proof about the cavitation structure, high pressure zones cannot provide data to forecast cavitation activity. Cavitation structures are multiple, quite different from each other, and strictly related to the acoustic pressure P compared to a reference pressure P0 ≡ 190 kPa.73 If P > P0, because of primary Bjerknes forces, the cavitation evolves along the pressure lines arranging in different shapes as reported in Table 2, conversely, if P < P0, cavitation evolves along the pressure antinodes. Even the presence of a metal net in the fluid exposed to ultrasound influences the sound propagation by scattering, moving the CBS after the net (artificial conical bubble structure, ACBS).
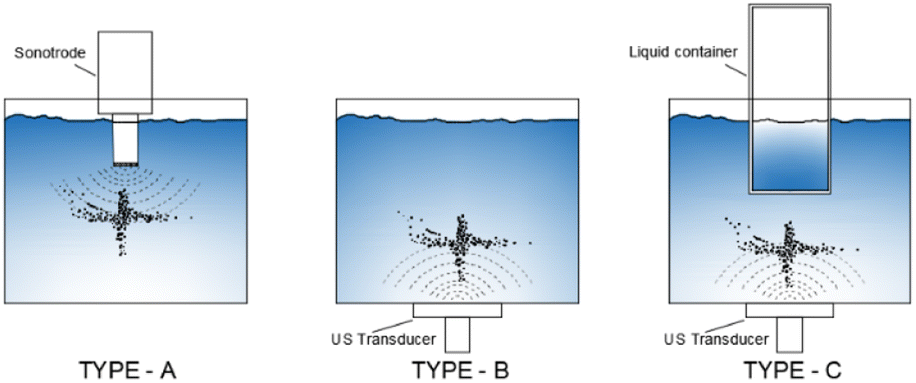 | ||
| Fig. 4 Common configuration of a sonoreactor. | ||
| CBS | ACBS | ALF | Smokers | Web | TBS | JBS | |
|---|---|---|---|---|---|---|---|
| P > P0 | X | X | X | X | |||
| P < P0 | X | X | X |
Correlation between the reactor geometry and cavitation distribution (or structure) is crucial for choosing the reactors and prediction of results by simulation. In multiple studies,65,66,73,77–80 there is a reference to the resonance of the acoustic field in the structure and its influence on the cavitation field. Nonetheless, many doubts arise about the real influence of eigenvalues and eigenvectors of geometries: ALF cavitation structure observation as a function of the shape of the reactor (Fig. 3) suggests that the influence of the geometry on the cavitation field is more related to the inclination of reflective surfaces rather than to the eigenvalues of the reactor geometry. Type-B reactors are less investigated. In rectangular shaped reactors, Koch81 found a correlation between the double layer cavitation structure and the acoustic power of ultrasound but there were no geometrical correlations, and comparing simulations and pressure measurements, eigenvalue influence was not proved, and symmetrical distribution of the sound pressure was discussed.82
The number of sonotrodes and the acoustic intensity applied by the transducer are key aspects investigated in the literature. Cavitation power is logarithmically related to the input power of the transducer,76,83 and increasing the electrical input power from 23 to 82 W, high values of the cavitation yield (∼3 × 10−7 mol W−1 min−1) can be achieved with larger liquid volume (to 0.3 L).84 Input power allowed determination of the cavitation threshold in a rectangular type-B reactor. At 45 kHz, with an ELMA 90 W transducer, cavitation structures appear at 14.8 W,81 with a clear smoking structure at 30.1 W, and a complete double layer from 58.5 W, while in the type-A reactor at 20 kHz the acoustic intensity threshold for cavitation is reported at ∼0.74 W cm−2.77 Numerically, Merouani et al.69 showed that with acoustic intensity increasing from 0.5 to 1 W cm−2, at different US frequencies, the maximum bubble temperature increases reporting an higher hydrogen production rate especially at higher US frequencies (over 1 MHz, on doubling the acoustic intensity, the production rate is 1010 higher, while at 20 kHz it is 102 higher).
Impedance matching tests were performed to enhance the cavitation energy conversion by Rooze et al.,73 measuring in air at 20 °C the US horn using a Hioki 3532-50 impedance analyzer and selecting three frequencies (20–41–62 kHz) as the frequencies with a lower impedance reporting a US-electric yield of 0.46 ± 0.03, 0.17 ± 0.01, and 0.21 ± 0.02 respectively, and consistently with the impedance values being measured. Regarding the number of sonotrodes in a type-A reactor,62 the cavitation percentage over the sonoreactor volume based on Blake cavitation analysis68 is higher for a 3 sonotrode configuration irradiant 20 kHz US in a rectangular reactor with 61.1% of cavitation.66 A higher number of sonotrodes for unit volume in a type-A sonoreactor does not provide a higher cavitation volume. The calculated conversion for one sonotrode logarithmically decreases while input power increases with an extreme 44% of energy conversion at 36 W input power to 1% at 580 W. Impedance matching test were performed to enhance the cavitation energy conversion by Rooze et al.,73 measuring in air at 20 °C the US horn using a Hioki 3532-50 impedance analyzer and selecting three frequencies (20–41–62 kHz) as the frequencies with a lower impedance reporting a US-electric yield of 0.46 ± 0.03, 0.17 ± 0.01, and 0.21 ± 0.02 respectively, and consistently with the impedance values measured. Consistently, the energy conversion efficiency decreases, but a flatter trend is reported with a decrease of only ∼10%, from ∼43% to 33% from 1 to 5 sonotrodes with each of power 36 W.
| Duty cycle (%) | On time (s) | Off time (s) | Iodine liberated (ppm) |
|---|---|---|---|
| 20 | 10 | 40 | 78.75 |
| 40 | 20 | 30 | 108.25 |
| 50 | 25 | 25 | 141.25 |
| 60 | 30 | 20 | 171.00 |
| 80 | 40 | 10 | 200.25 |
These are promising results that may be applied to hydrogen production as well.
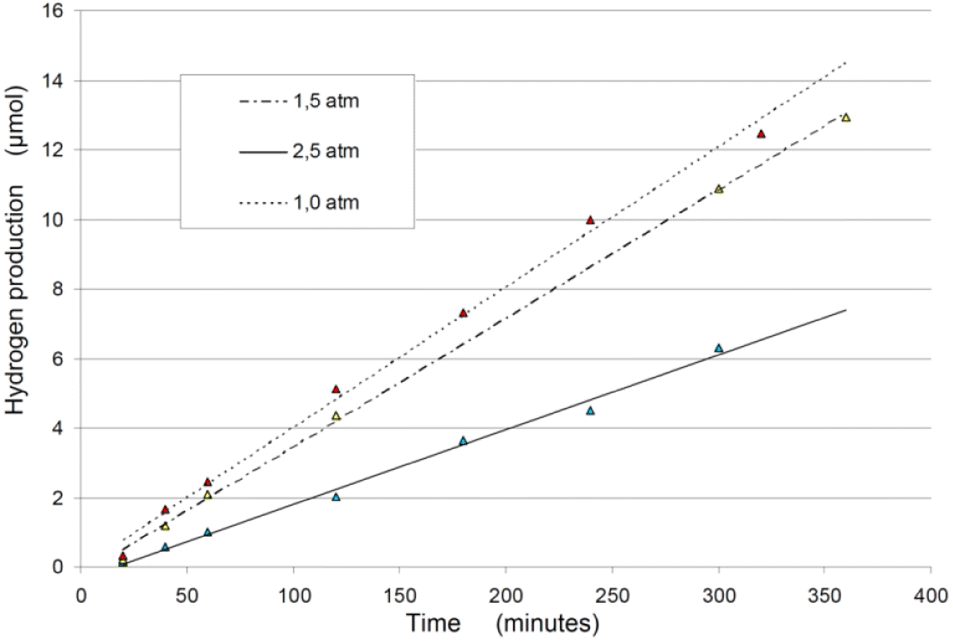 | ||
| Fig. 5 Hydrogen production rates during exposition time, changing the rector inner pressure.87 | ||
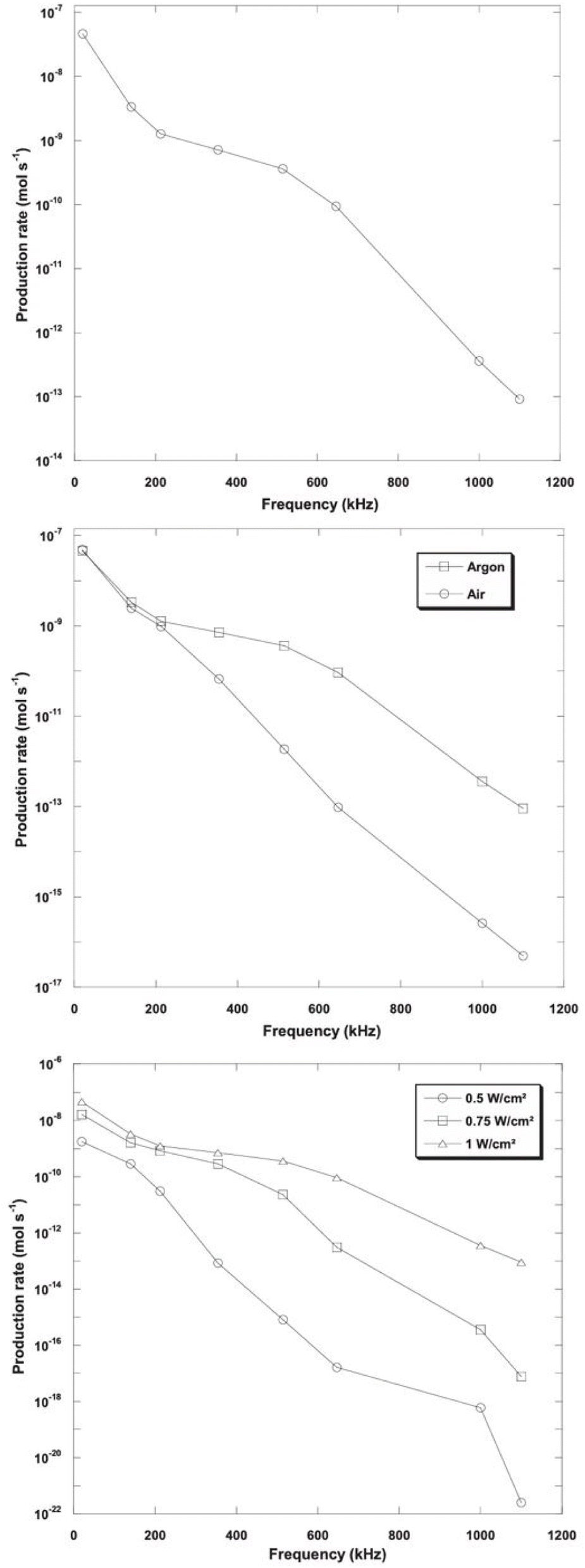 | ||
| Fig. 6 Hydrogen production as a function of frequency, atmosphere and intensity.70 | ||
The composition of the solution exposed to ultrasound has been explored as a possible way to enhance the sonolysis by introducing a sacrificial reagent, a chemical species that can be oxidized more easily than oxygen of water, reducing the recombination tendency of electrons and holes and accelerating the rate of hydrogen generation, into the solution.88 Bioethanol, a sugar fermentation biomass compound, was tested as a sacrificial agent in 300 mL of distilled water with a 20% volumetric concentration, and the results were compared with sheer water and sheer bioethanol tests.89 The presence of bioethanol in water solution resulted in a 40% improvement of the hydrogen production rate compared to sheer water solution (112 ± 3 vs. 80 ± 2 μmol h−1). Sheer bioethanol solution reported a lower production rate (5.5 ± 0.2 μmol h−1). The concentration of methanol in the solution for enhancing the sono-hydrogen production has been investigated in the literature.90 Henglein and Schultz reported that iodide oxidation by ultrasonic cavitation proportionally decreases with the increase of the methanol concentration in the solution due to sono activity inhibition above a certain concentration (5 mol in 30 mL solution) upon increasing temperature of the solution between 5 and 20 °C. But at 40 °C the presence of methanol did not influence the oxidation anymore. Büttner et al.91 confirmed the effect of methanol 83 increments of all species production in a 10% methanol–water solution. Theoretical studies reported the optimal methanol concentration for hydrogen production in water solution as 20%79 with an 80% argon concentration in the atmosphere, with an increase of hydrogen production of 4.7 times compared to a sheer water solution.90
The sono-photolysis and its synergistic effect
The combined action of solar radiation and ultrasound can exert a synergistic effect on some chemical reactions carried out in the liquid phase in the presence of a heterogeneous photocatalyst. Sono-photolysis was first employed for the degradation of water pollutants, for instance, phenols as reported by Wu and co-authors in 2001 in the absence or in the presence of a catalyst (FeSO4) using UV irradiation assisted by 30 kHz ultrasonication. The percentage of degraded phenols was slightly higher than that obtained by simple UV irradiation both in the presence or in the absence of dissolved Fe3+ ions.92 In 2010 a more pronounced synergistic degradation effect of UV/US irradiation (25 kHz, 1 kW) of phenolic solution in the presence of TiO2 as photocatalyst and H2O2 was also reported.93 Other important applications of catalysed sono-photolysis deal with the degradation of pollutants. In 2011 Cui et al. investigated a continuous process involving US (28 kHz, 300 W electric power)/UV (254 nm, 30 W, TiO2) combination, for the degradation of low concentration of methyl orange in water (25 mg L−1).94 In a recent paper the degradation of carmoisine was carried out by employing ˙OH radicals generated by photolysis and transient cavitation.95 A similar process was also reported in 2023 for the sono-photodegradation of carbaryl, a pollutant pesticide, by using a Ln@MOF as photocatalyst.96 Other organic pollutants as dyes were efficiently degraded by the synergic effect of photo-sonolysis by using CuO–TiO2/rGO (reduced graphene oxide rGO) as photocatalysts in which the coupled action of light and ultrasound enhanced the degradation kinetics by over 3 times.96Other examples of the use of sono-photolysis as an advanced oxidation process (AOP) for dye degradation, wastewater treatment and for biomass valorization have been also reported in a 2017 concept paper by Chatel and co-authors.97
The synergistic effect of sonophotolysis for the water-splitting reaction is a promising field of research although it is still scarcely investigated.
The synergistic effect of solar light and ultrasound in the production of H2 from water is usually quantified through the equation below:16,89,98–100
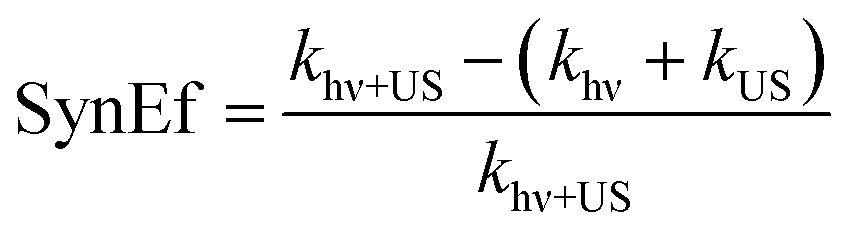 | (14) |
In (14), khν, kUS, khν+US are the kinetic constants of H2 production when simply electromagnetic radiation (hν) or simply ultrasound (US), or both light and ultrasound are applied, respectively. Whenever SynEf ≠ 0, there is a synergistic effect.
A recent paper reported on the synergistic and highly beneficial effect of ultrasound irradiation in visible light water photolysis using a Co-phtalocynine dye, namely Therephtal, as a homogeneous photocatalyst. The effect of ultrasound irradiation in the presence of light resulted in a large increment of oxygen evolution from about 35 (in the absence of ultrasound) to over 1400 μmol after 3 h of US/light irradiation. The authors proposed as a mechanism the US enhanced formation of ROS species such as (˙OH, ˙OOH, O2˙−) able to easily oxidize Co2+ to Co3+ which was mainly responsible for the oxygen evolution reaction.101 In 2017 Singh and Sinha reported the catalyzed sono-photolysis of water for the HER using CdS as a photocatalyst. The higher activity of H2 production of horn US connected to a piezoelectric transducer working at 20 kHz and 40 W, with respect to the pure UV irradiation was attributed to a faster removal of bubbles of hydrogen from the surface of photocatalyst due to the mechanical energy associated with ultrasound able to overcome surface tension.102 The different aspects of the synergy between light and ultrasound still need to be completely understood. It can be due to different factors. Some of them were recently discussed in a review by Wang and co-authors.101
It is evident that the two energy sources play a reciprocal positive feedback effect (see Fig. 7).
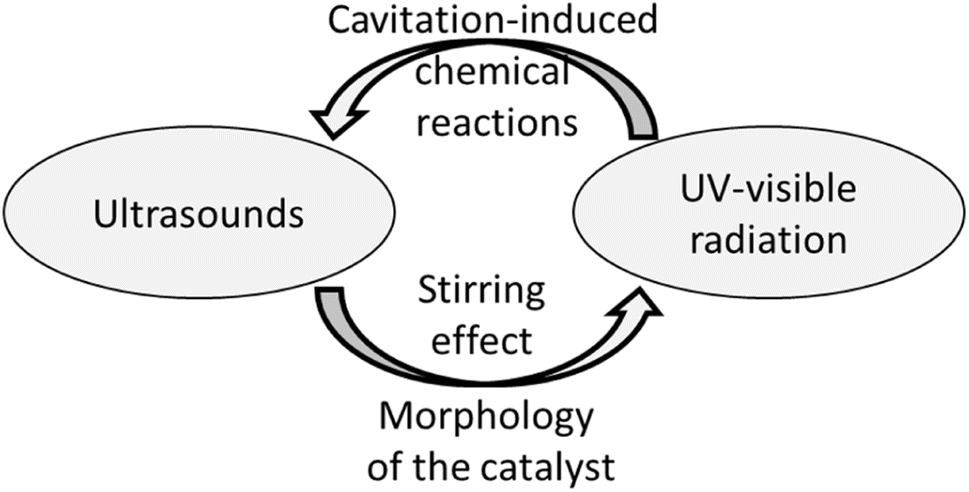 | ||
| Fig. 7 The reciprocal positive feedback action between the two energy sources. The ultrasound affects the morphology of the heterogeneous catalyst and exerts a stirring effect that favours water splitting. On the other hand, the UV-visible radiation produces chemical species that provide extra nuclei for bubble formation, promoting the cavitation phenomenon. | ||
It is well-known that acoustic cavitation induces the miniaturization of the catalyst's solid particles and an overall increase of the superficial area of the material. Hence, the semiconductor becomes a better photocatalyst.
Furthermore, the ultrasound waves exert a stirring effect in the reaction medium, favouring the physical contact between the liquid and solid phases. Moreover, the stirring effect lifts the catalytic solid particles from the bottom of the reactor up to the top part of the solution, facilitating the capture of light by the catalyst. On the other hand, the chemical species generated by photo-induced water splitting might provide extra nuclei for bubble formation, promoting the cavitation phenomenon. These concerted mechanisms may also affect the electronic structure of the catalyst. In particular, the collapse of cavitation bubbles could produce acoustic luminescence, thus promoting the migration of electrons from the valence band (VB) to the conduction band (CB). In contrast, the holes (h+) remain in the VB. Consequently, the photogenerated electron–hole pairs react with the system's oxidant to generate reactive oxygen species (ROS), similar to those created by the photocatalytic process.
An additional synergistic effect by ultrasound and photocatalysis combination is reported in the presence of piezoelectric photocatalysts. Besides the photocatalyst properties previously explored, these photocatalysts generate periodic electrical potential into the solution by interaction with ultrasound, resulting in piezoelectric material surface charging.103 Those potentials induce flexures of the energy band, enhancing the photogenerated charge separation.101 Furthermore, electronic jumps occur in the VB and CB of these materials providing additional electron–hole sources for the flow separation and transport.104 Meng et al.105 showed 1.32-fold increase of methyl orange photodegradation by Pt/BaTiO3 heterojunction fabrication for piezoelectric enhanced photocatalysis, and Liu et al.106 demonstrated 2.44 times higher photocatalysis activity due to the presence of the BaTiO3@ReS2 Schottky heterostructure. Hydrogen production by piezoelectric-assisted sono-photocatalysis was investigated with the g-C3N4 photocatalyst with a 50% H2 production rate improvement from water sonolysis (12.16 vs. 8 μmol (g−1 h−1)) and SynEf ∼ 0.32. Catalytic activity and the piezoelectric field of deformation are positively correlated as well as ultrasound intensity and the catalyst deformation degree.
The presence of photocatalyst powder dispersed into the solution may also affect the ultrasonic field behavior. Indeed, in the literature it has been reported that the cinematic viscosity of the solution varies with a calculable change of the acoustic absorption of the medium.100 Capillary viscosimeter measurements provided cinematic viscosity data of the solution reporting a variation of 1.7% (Table 4) when the photocatalyst was sulfur-doped. Applying the corrected Stokes formula for the volume viscosity, 3% variations of the acoustic property absorption of the solution were calculated (Table 4). This may be explained because of the decrease in the cinematic viscosity due to the increase in the density of the solution. Those parameters are proved to be inversely proportional to each other.107 Although this is not a proper declination of the synergistic effect of the photo-sonolysis, and the production improvement is sensible only for a short time period (up to 2 h of US exposition), further investigations are needed to evaluate any beneficial effect of the acoustic absorption variations of the solution on sono-photolysis.
| Photocatalyst | Cinematic viscosity![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ηc ηc |
Acoustic absorptionα |
Molecular mass g mol−1 | Synergistic H2 production improvement after 2 h μmol |
|---|---|---|---|---|
| LaGa0.5In0.5O3 | 0.0127 | 0.179 | 276.15 | 0.14 ± 0.01 |
| La0.8Ga0.5InO3 | 0.0117 | 0.166 | 329.69 | 0.14 ± 0.01 |
| S:La0.8Ga0.5InO3 | 0.0115 | 0.161 | 361.75 | 0.20 ± 0.01 |
Finally, our experiments108 have demonstrated that sonophotocatalysis for hydrogen production benefits from using metal oxide solid solutions, which are high-entropy materials.109 High-entropy materials are expected to be good catalysts because they guarantee the possibility of activating many parallel reactive paths. For this reason, high-entropy materials are enticing interests in energy conversion and storage.110–112
Conclusions/future perspectives
Sono-photolysis is a phenomenon which involves two different processes for hydrogen production by water splitting using ultrasound and solar radiation exposure of a water-based solution. This process may be the focus of a sustainable green hydrogen production reactor and a possible synergic alternative to electrolysers. The scientific literature has highlighted the following points:• The transparent covering of the photo-sono reactor should be made of quartz glass to allow for a broader transmission of light, particularly in the UV spectrum.
• The presence of dispersed photocatalysts in the solution and the use of sacrificial agents are necessary for the proper functioning of the photolysis process. The sulfur doping of catalysts significantly affects the photolysis yields, with up to 17-fold increase. Semiconductor catalysts, particularly S:Y0.8Ga0.2InO3, have experimentally shown the highest rates of hydrogen production (0.14 ± 0.05 μ moles (H2) per h).
• The use of a bioproduct as a sacrificial reagent allows the production of H2 and avoids the production of O2, which can partially deplete the desired hydrogen through combustion reactions, and instead produce by-products (such as aldehydes or caraboxylic acids) which remain in solution and more easily separated from H2.
• Introduce capsules of up-converting materials to reduce the waste of low-energy photons.
• The hydrogen production through ultrasound (US) is sensitive to the chemical composition of the solution. Solutions containing 20% vol of water and ethanol exhibit production rates around 40% faster than solutions composed solely of distilled water.
• The volume of the solution and its geometry within the reactor, determined by the peak resonance points of the water volume exposed to the US frequency, significantly enhances the efficiency (up to 112 μ moles (H2) per h 89).
• Atmospheric pressure conditions for the solution yield the best results in terms of efficiency for sono- and photo-sonolysis.87
• Type A or B sonoreactors can achieve the best performances for low-frequency range ultrasounds, with a duty cycle transducer activity of 60%, in an Ar atmosphere to achieve the largest bubble dimensions at the highest temperature.
• Catalysts that reduce the kinematic viscosity of the solution enhance sonolysis yields by decreasing the solution's absorption coefficient.
• Sono-photolysis exhibits synergistic coupling between sonolysis and photolysis, resulting in efficiencies higher than the sum of the individual phenomena. This synergy is approximately 13%, and sulfur doping of catalysts has experimentally shown advantages in efficiency during the initial 3 hours of combined US and solar light exposure.
In envisioning the potential future advancements for sono-photolysis and reactor design, several key areas stand out when compared to the current state of the art. First, there is a growing focus on the environmental sustainability of sacrificial agents in the photolysis process,89 with discussions in the literature highlighting bioethanol as a viable option. However, challenges arise as the catalysts explored often involve a 24-hour calcination process at 1373 K or the use of rare earth metals, both of which pose energy-intensive hurdles for large-scale photo-sonolysis applications. Another avenue for progress lies in enhancing photolysis efficiencies through the development of next-generation catalysts. Transition metal hydroxides and coordination polymers, particularly those based on Fe, Co, and Ni, are being explored. Simultaneously, there is an emphasis on extending the photolysis range within the solution by leveraging luminescent materials.113 Facilitating the electron promotion into the CB by US-activated intermittent electrical fields into the solution by dispersion of photo-catalysts with piezoelectric properties is another promising application that must be further explored. Refinement of the reactor geometry is also a key consideration, with an emphasis on maximizing solution volume based on resonance peaks.89 This approach aligns with existing studies in the literature, aiming for optimal conditions for sono-photolysis. However, more investigations are required to predict the contribution of the vessel geometry compared to the modal excitations of the solution volume exposed to ultrasound. The literature77 highlighted that the current impossibility of simulating the cavitation-bubbling evolution and spatial distribution precisely may compromise the usefulness of finite volume software approaches due to the absence of acoustic scattering of ultrasound waves by the bubbles. This is a factor which affects the spatial distribution of acoustic pressure into the vessel, rising differences between measurements and mechanical modal analysis.82 Furthermore, efforts are underway to investigate the energy efficiency between the mechanical energy induced by ultrasonication in the solution and the subsequent extraction of hydrogen. Although an energy cavitation efficiency in the solution up to 44% has been reported in the literature,66 direct measurements of acoustic amplitude in the solution and the optimization of impedance matching between the reactor and the ultrasonic transducer are required. The potential expressed in contemporary literature114 in the production of oxygen through sono-photolysis (500 μmol h−1) can only be welcomed as an extremely important point to be further investigated. It represents an additional synergistic effect of the described process, with clear application opportunities. However, the energy conversion of the whole hydrogen production system must be measured to further prove the potentiality of the method, and to evaluate its margins for improvement. Lastly, efforts are focused on studying the synergistic interaction between photolysis and sonolysis to optimize and maximize hydrogen production efficiency. Researchers aim to uncover how these processes complement each other, seeking to identify conditions that enhance overall efficiency and hold promise for advancing hydrogen production technologies.
The sono-photolysis can play a promising role in the energy transition because of its transversal application potentiality, it can be implemented in existing processes such as water purification by ultrasonic cavitation,115 as well as it can be integrated into existing energy systems such as photovoltaic plants. Differentiating the energy production into electricity and hydrogen for energy storage purposes, sono-photolysis combination with existing energy production technologies systems may be a ready-to-use competitive hydrogen production application. Furthermore, combined with electrical production systems, the water purification process as a by-product of sono-photolysis can play a key role compared with existing hydrogen production technologies such as electrolysers (Fig. 8), and it may be a synergistic effect extremely promising for applications in areas where there is a high solar radiation intensity magnitude and the necessity to provide off-grid energy and purified water for domestic use.
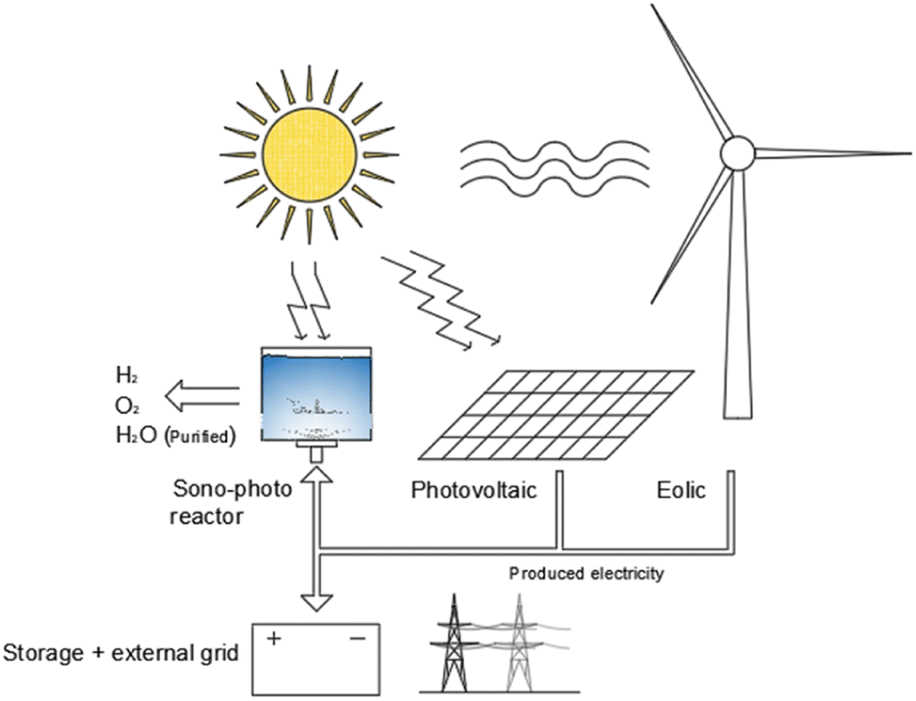 | ||
| Fig. 8 Sono-photolysis technology integration in existing renewable production systems. The synergistic production of hydrogen, oxygen and purified water is highlighted as a key parameter for the application of this technology. | ||
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- J. Turner, G. Sverdrup, M. K. Mann, P. C. Maness, B. Kroposki, M. Ghirardi, R. J. Evans and D. Blake, Int. J. Energy Res., 2008, 32, 379–407 CrossRef CAS.
- D. B. Levin, L. Pitt and M. Love, Int. J. Hydrogen Energy, 2004, 29, 173–185 CrossRef CAS.
- C. M. Kalamaras and A. M. Efstathiou, Conference Papers in Energy, 2013, 2013, 1–9 CrossRef.
- J. Ivy, Summary of Electrolytic Hydrogen Production: Milestone Completion Report, Golden, CO (United States), 2004 Search PubMed.
- A. Hauch, S. D. Ebbesen, S. H. Jensen and M. Mogensen, J. Mater. Chem., 2008, 18, 2331–2340 RSC.
- S. E. Lindquist and C. Fell, Encyclopedia of Electrochemical Power Sources, 2009, pp. 369–383 Search PubMed.
- Ministero dello Sviluppo Economico MISE, Strategia Nazionale Idrogeno – Linee Guida Preliminari, Italy, 2020 Search PubMed.
- Gruppo di Lavoro Idrogeno, Prime Indicazioni Per Una Strategia Italiana Ricerca Idrogeno (S I R I), 2020 Search PubMed.
- European Commission, Communication From the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions a Hydrogen Strategy for a Climate-Neutral Europe, 2020 Search PubMed.
- Council Regulation (EU Euratom) 2020/2093, Laying Down the Multiannual Financial Framework for the Years 2021 to 2027, 2020 Search PubMed.
- L. Baldinelli, G. Menendez Rodriguez, I. D. ’ Ambrosio, A. M. Grigoras, R. Vivani, L. Latterini, A. Macchioni, F. De, A. Acde and G. Bistoni, Chem. Sci., 2024, 15, 1348–1363 RSC.
- K. N. Liou, Int. Geophys., 2002, 84, 37–64 CrossRef.
- J. E. Funk, Int. J. Hydrogen Energy, 2001, 26, 185–190 CrossRef CAS.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2008, 38, 253–278 RSC.
- M. A. Basile, C. Carfagna, L. Dipasquale, A. Fontana, A. Gambacorta, G. G. d'Ayala, M. Malinconico and P. Cerruti, Hydrogen Production: Prospects and Processes, 2015, pp. 411–420 Search PubMed.
- J. R. Bolton, S. J. Strickler and J. S. Connolly, Nature, 1985, 316, 495–500 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2008, 38, 253–278 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- S. Wang, A. Lu and C. J. Zhong, Nano Convergence, 2021, 8, 1–23 CrossRef CAS PubMed.
- Z. Chen, W. Wei and B. J. Ni, Curr. Opin. Green Sustainable Chem., 2021, 27, 100398 CrossRef CAS.
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237–268 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2008, 38, 253–278 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- V. Kumaravel, M. D. Imam, A. Badreldin, R. K. Chava, J. Y. Do, M. Kang and A. Abdel-Wahab, Catalysts, 2019, 9, 276 CrossRef CAS.
- P. L. Gentili, M. Penconi, F. Costantino, P. Sassi, F. Ortica, F. Rossi and F. Elisei, Sol. Energy Mater. Sol. Cells, 2010, 94, 2265–2274 CrossRef CAS.
- Z. Chen, W. Wei, L. Song and B. J. Ni, Sustainable Horizons, 2022, 1, 100002 CrossRef.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- A. Macchioni, Eur. J. Inorg. Chem., 2019, 2019, 7–17 CrossRef CAS.
- F. Zaccaria, G. Menendez Rodriguez, L. Rocchigiani and A. Macchioni, Front. Catal., 2022, 2, 892183 CrossRef.
- C. Trotta, G. Menendez Rodriguez, L. Tensi, A. V. Yakimov, L. Rocchigiani, A. Donnadio, C. Zuccaccia, C. Copéret and A. Macchioni, Eur. J. Inorg. Chem., 2023, 26, e202300211 CrossRef CAS.
- L. Fagiolari, F. Zaccaria, F. Costantino, R. Vivani, C. K. Mavrokefalos, G. R. Patzke and A. Macchioni, Dalton Trans., 2020, 49, 2468–2476 RSC.
- B. Yang, D. Luo, S. Wu, N. Zhang and J. Ye, Sci. Technol. Adv. Mater., 2022, 23, 587–616 CrossRef CAS PubMed.
- S. Nishioka, F. E. Osterloh, X. Wang, T. E. Mallouk and K. Maeda, Nat. Rev. Methods Primers, 2023, 3, 42 CrossRef CAS.
- H. Zhao, L. Jian, M. Gong, M. Jing, H. Li, Q. Mao, T. Lu, Y. Guo, R. Ji, W. Chi, Y. Dong and Y. Zhu, Small Struct., 2022, 3, 2100229 CrossRef CAS.
- N. N. Rosman, R. M. Yunus, N. R. A. M. Shah, R. M. Shah, K. Arifin, L. J. Minggu and N. A. Ludin, Int. J. Energy Res., 2022, 46, 11596–11619 CrossRef CAS.
- L. Chen, J. T. Ren and Z. Y. Yuan, Green Chem., 2022, 24, 713–747 RSC.
- L. C. Goveas, S. Nayak, R. Vinayagam, R. Selvaraj and A. Pugazhendhi, Fuel, 2023, 348, 128460 CrossRef CAS.
- E. Heo, S. Lee and H. Yoon, Mater. Chem. Front., 2023, 7, 6154–6187 RSC.
- A. Sherryna, M. Tahir and W. Nabgan, Int. J. Hydrogen Energy, 2022, 47, 862–901 CrossRef CAS.
- T. Simon, N. Bouchonville, M. J. Berr, A. Vaneski, A. Adrovic, D. Volbers, R. Wyrwich, M. Döblinger, A. S. Susha, A. L. Rogach, F. Jäckel, J. K. Stolarczyk and J. Feldmann, Nat. Mater., 2014, 13, 1013–1018 CrossRef CAS PubMed.
- H. Lin, B. Sun, H. Wang, Q. Ruan, Y. Geng, Y. Li, J. Wu, W. Wang, J. Liu and X. Wang, Small, 2019, 15, 1804115 CrossRef PubMed.
- G. Ge, M. Liu, C. Liu, W. Zhou, D. Wang, L. Liu and J. Ye, J. Mater. Chem. A, 2019, 7, 9222–9229 RSC.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- Z. Sun, H. Zheng, J. Li and P. Du, Energy Environ. Sci., 2015, 8, 2668–2676 RSC.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS.
- X. Shi, M. Fujitsuka, S. Kim and T. Majima, Small, 2018, 14, 1703277 CrossRef PubMed.
- L. Zhang, X. Fu, S. Meng, X. Jiang, J. Wang and S. Chen, J. Mater. Chem. A, 2015, 3, 23732–23742 RSC.
- A. Shalav, B. S. Richards and M. A. Green, Sol. Energy Mater. Sol. Cells, 2007, 91, 829–842 CrossRef CAS.
- Y. Y. Cheng, B. Fückel, T. Khoury, R. G. C. R. Clady, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, J. Phys. Chem. A, 2011, 115, 1047–1053 CrossRef CAS PubMed.
- M. Penconi, F. Ortica, F. Elisei and P. L. Gentili, J. Lumin., 2013, 135, 265–270 CrossRef CAS.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- O. S. Kwon, H. S. Song, J. Conde, H. Il Kim, N. Artzi and J. H. Kim, ACS Nano, 2016, 10, 1512–1521 CrossRef CAS PubMed.
- G. Quaglia, B. Bartolomei, P. L. Gentili and L. Latterini, J. Mater. Chem. C, 2022, 10, 9073–9080 RSC.
- H. Islam, O. S. Burheim and B. G. Pollet, Ultrason. Sonochem., 2019, 51, 533–555 CrossRef PubMed.
- T. Leong and S. Kentish, The fundamentals of power ultrasound-A review, 2011, vol. 39 Search PubMed.
- N. Merabet and K. Kerboua, Int. J. Hydrogen Energy, 2022, 47, 17879–17893 CrossRef CAS.
- J. L. Laborde, A. Hita, J. P. Caltagirone and A. Gerard, Ultrasonics, 2000, 38, 297–300 CrossRef CAS PubMed.
- K. Yasui, Acoustic Cavitation and Bubble Dynamics, Springer International Publishing, Cham, 2018 Search PubMed.
- P. Bruno, Power Ultrasound in Electrochemistry : from Versatile Laboratory Tool to Engineering Solution Sonoelectrochemistry, Wiley, 2012 Search PubMed.
- M. Legay, N. Gondrexon, S. Le Person, P. Boldo and A. Bontemps, Int. J. Chem. Eng., 2011, 2011, 1–17 CrossRef.
- K. S. Suslick, Science (1979), 1990, 247, 1439–1445 CAS.
- M. D. Archer and J. R. Bolton, Biopolymers, 1990, 94, 975 Search PubMed.
- S. S. Rashwan, I. Dincer, A. Mohany and B. G. Pollet, Int. J. Hydrogen Energy, 2019, 44, 14500–14526 CrossRef CAS.
- S. S. Rashwan, A. Mohany and I. Dincer, Int. J. Hydrogen Energy, 2021, 46, 15219–15240 CrossRef CAS.
- Y. Son, M. Lim, M. Ashokkumar and J. Khim, J. Phys. Chem. C, 2011, 115, 4096–4103 CrossRef CAS.
- Z. Zhang, T. Gao, X. Liu, D. Li, J. Zhao, Y. Lei and Y. Wang, Ultrason. Sonochem., 2018, 42, 787–794 CrossRef CAS PubMed.
- S. Merouani, O. Hamdaoui, Y. Rezgui and M. Guemini, Int. J. Hydrogen Energy, 2015, 40, 4056–4064 CrossRef CAS.
- S. Merouani, O. Hamdaoui, Y. Rezgui and M. Guemini, Int. J. Hydrogen Energy, 2016, 41, 832–844 CrossRef CAS.
- K. Kerboua and O. Hamdaoui, Ultrason. Sonochem., 2018, 40, 194–200 CrossRef CAS PubMed.
- K. Kerboua, S. Merouani, O. Hamdaoui, A. Alghyamah, M. H. Islam, H. E. Hansen and B. G. Pollet, Ultrason. Sonochem., 2021, 72, 105422 CrossRef CAS PubMed.
- J. Rooze, E. V. Rebrov, J. C. Schouten and J. T. F. Keurentjes, Ultrason. Sonochem., 2011, 18, 209–215 CrossRef CAS PubMed.
- H. Harada, C. Hosoki and M. Ishikane, J. Photochem. Photobiol., A, 2003, 160, 11–17 CrossRef CAS.
- T. J. Mason, J. P. Lorimer and D. M. Bates, Ultrasonics, 1992, 30, 40–42 CrossRef CAS.
- P. R. Gogate, S. Shaha and L. Csoka, Chem. Eng. J., 2014, 239, 364–372 CrossRef CAS.
- Z. Wei and L. K. Weavers, Ultrason. Sonochem., 2016, 31, 490–498 CrossRef CAS PubMed.
- Y. Son, M. Lim, J. Khim and M. Ashokkumar, Ultrason. Sonochem., 2012, 19, 16–21 CrossRef CAS PubMed.
- K. Kerboua and O. Hamdaoui, Chem. Phys., 2019, 519, 27–37 CrossRef CAS.
- Y. C. Wang and M. C. Yao, Ultrason. Sonochem., 2013, 20, 565–570 CrossRef CAS PubMed.
- C. Koch, Ultrason. Sonochem., 2016, 29, 439–446 CrossRef CAS PubMed.
- P. Domenighini, A. M. Gambelli, F. Rossi and F. Cotana, J. Phys.: Conf. Ser., 2023, 2648, 012059 CrossRef.
- Y. Son, M. Lim and J. Khim, Ultrason. Sonochem., 2009, 16, 552–556 CrossRef CAS PubMed.
- M. Lim, M. Ashokkumar and Y. Son, Ultrason. Sonochem., 2014, 21, 1988–1993 CrossRef CAS PubMed.
- L. Bai, W. Xu, J. Deng, C. Li, D. Xu and Y. Gao, Ultrason. Sonochem., 2014, 21, 1696–1706 CrossRef CAS.
- J. Rooze, E. V. Rebrov, J. C. Schouten and J. T. F. Keurentjes, Ultrason. Sonochem., 2011, 18, 209–215 CrossRef PubMed.
- F. Cotana and F. Rossi, in 3rd Int. green energy Conf., 2007, pp. 3–8 Search PubMed.
- Q. Li, X. Li and J. Yu, Interface Sci. Technol., 2020, 31, 313–348 CrossRef CAS PubMed.
- M. Penconi, F. Rossi, F. Ortica, F. Elisei and P. L. Gentili, Sustainability, 2015, 7, 9310–9325 CrossRef.
- A. Dehane, L. Nemdili, S. Merouani and M. Ashokkumar, Top. Curr. Chem., 2023, 381, 1–25 CrossRef.
- J. Büttner, M. Gutiérrez and A. Henglein, J. Phys. Chem., 1991, 95, 1528–1530 CrossRef.
- H. Rahmani, M. Gholami, A. H. Mahvi, M. Ali-Mohammadi and K. Rahmani, Journal of Water Chemistry and Technology, 2014, 36, 317–324 CrossRef.
- I. M. Khokhawala and P. R. Gogate, Ultrason. Sonochem., 2010, 17, 833–838 CrossRef CAS PubMed.
- P. Cui, Y. Chen and G. Chen, Ind. Eng. Chem. Res., 2011, 50, 3947–3954 CrossRef CAS.
- S. Chakma and V. S. Moholkar, J. Ind. Eng. Chem., 2016, 33, 276–287 CrossRef CAS.
- S. G. Babu, P. Karthik, M. C. John, S. K. Lakhera, M. Ashokkumar, J. Khim and B. Neppolian, Ultrason. Sonochem., 2019, 50, 218–223 CrossRef CAS PubMed.
- G. Chatel, S. Valange, R. Behling and J. C. Colmenares, ChemCatChem, 2017, 9, 2615–2621 CrossRef CAS.
- M. Mrowetz, C. Pirola and E. Selli, Ultrason. Sonochem., 2003, 10, 247–254 CrossRef CAS PubMed.
- R. A. Torres, J. I. Nieto, E. Combet, C. Pétrier and C. Pulgarin, Appl. Catal., B, 2008, 80, 168–175 CrossRef CAS.
- P. L. Gentili, M. Penconi, F. Ortica, F. Cotana, F. Rossi and F. Elisei, Int. J. Hydrogen Energy, 2009, 34, 9042–9049 CrossRef CAS.
- C. Li, X. Wang, J. Wu, J. Gao, R. Zhao, S. Xia, H. Yang, Z. Chen, L. Li and W. Wang, Ultrason. Sonochem., 2023, 99, 106584 CrossRef CAS.
- A. Singh and A. S. K. Sinha, J. Energy Chem., 2018, 27, 1183–1188 CrossRef.
- S. Tu, Y. Guo, Y. Zhang, C. Hu, T. Zhang, T. Ma, H. Huang, S. C. Tu, Y. H. Zhang, C. Hu, H. W. Huang, Y. X. Guo, T. R. Zhang and T. Y. Ma, Adv. Funct. Mater., 2020, 30, 2005158 CrossRef CAS.
- M. B. Starr and X. Wang, Nano Energy, 2015, 14, 296–311 CrossRef CAS.
- H. Meng, Z. Chen, Z. Lu and X. Wang, J. Mol. Liq., 2023, 369, 120846 CrossRef CAS.
- W. Liu, P. Wang, Y. Ao, J. Chen, X. Gao, B. Jia, T. Ma, W. Liu, P. F. Wang, Y. H. Ao, J. Chen, X. Gao, B. Jia and T. Ma, Adv. Mater., 2022, 34, 2202508 CrossRef CAS PubMed.
- A. J. Zuckerwar and R. L. Ash, Phys. Fluids, 2006, 18, 47101 CrossRef.
- P. L. Gentili, F. Rossi, M. Penconi, F. Ortica and F. Elisei, in Hydrogen Production: Prospects and Processes, ed. D. Honnery and P. Moriarty, Nova Science Publishers, New York, 2011, pp. 413–422 Search PubMed.
- M. Penconi, A. Cesaretti, F. Ortica, F. Elisei and P. L. Gentili, J. Lumin., 2016, 177, 314–324 CrossRef CAS.
- C. Oses, C. Toher and S. Curtarolo, Nat. Rev. Mater., 2020, 5, 295–309 CrossRef CAS.
- Y. Sun and S. Dai, Sci. Adv., 2021, 7, eabg1600 CrossRef CAS PubMed.
- Y. Ma, Y. Ma, Q. Wang, S. Schweidler, M. Botros, T. Fu, H. Hahn, T. Brezesinski and B. Breitung, Energy Environ. Sci., 2021, 14, 2883–2905 RSC.
- M. Ashokkumar and F. Grieser, Chem. Commun., 1998, 561–562 RSC.
- O. V. Kharissova, L. M. Torres Martínez, E. Luevano Hipólito, L. F. Garay-Rodríguez, M. R. Alfaro Cruz and B. I. Kharissov, J. Photochem. Photobiol., A, 2023, 437, 114463 CrossRef CAS.
- R. Singh and S. Dutta, Sustainable Fuel Technologies Handbook, 2021, pp. 541–549 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |