
Supramolecular assembly of multifunctional protein gels via an N-glycosylation consensus sequence fusion domain†
Eric D.
Hill
 ,
Stephen
Michel
,
Natasha R.
Sequeira
,
Benjamin G.
Keselowsky
* and
Gregory A.
Hudalla
*
,
Stephen
Michel
,
Natasha R.
Sequeira
,
Benjamin G.
Keselowsky
* and
Gregory A.
Hudalla
*
J. Crayton Pruitt Family Department of Biomedical Engineering, University of Florida, Biomedical Sciences J293, PO BOX 116131, 1275 Center Drive, Gainesville, FL, USA. E-mail: ghudalla@bme.ufl.edu
First published on 24th May 2024
Abstract
Polypeptide fusion tags that can direct the assembly of folded proteins into supramolecular networks are attractive for creating functional biomaterials. A practical challenge is identifying polypeptide sequences that form supramolecular networks in response to specific user-controlled stimuli, which is advantageous for producing polypeptide–protein fusions using cell-based expression hosts. Here, we report an N-glycosylation tag, (GGGSGGGSGGNWTT)10 or “NGT,” that assembles into a supramolecular network at reduced temperatures when fused to a folded protein. For example, NGT fused to superfolder green fluorescent protein (NGTsfGFP) formed materials that emitted green fluorescence in blue light, while NGT fused to NanoLuc luciferase (NGTnL) formed materials that emitted blue light in the presence of the chemical substrate furimazine. Oscillatory rheology established the materials as weak viscoelastic gels that can undergo shear-thinning and self-healing. Gel formation could be disrupted by mutating the asparagines in NGT to glutamines, introducing a chaotropic agent, or modifying the asparagines in NGT with glucose, suggesting a role for hydrogen bonds involving asparagine in supramolecular network formation. A mixture of soluble NGTsfGFP and NGTnL formed a multifunctional gel at reduced temperature that demonstrated bioluminescence resonance energy transfer between the nL and sfGFP domains in the presence of furimazine. Collectively, these data establish NGT as a temperature-responsive polypeptide tag that can be used to create functional biomaterials from soluble fusion proteins synthesized by cell-based hosts.
Design, System, ApplicationCertain amino acid sequences provide polypeptides that can form supramolecular networks in response to an external stimulus through specific, repeated interactions. Fusing these polypeptides onto proteins is an established method to create functional supramolecular materials. However, a practical challenge exists in synthesizing the protein–polypeptide fusion molecule. Merging the polypeptide and the functional protein at the genetic level can allow for expression in living hosts, such as microbes; but, the polypeptide should ideally remain in the unassembled (i.e., soluble) state to facilitate recovery and purification. Here, we identified an N-glycosylation consensus sequence that, when fused to either a fluorescent protein or luciferase enzyme, formed supramolecular gels in response to reduced temperatures. These gels exhibited self-healing after mechanical disruption, demonstrating potential as injectable drug carriers. As repeated asparagine residues in the consensus sequence contributed to gel formation, the fluorescent protein construct and luciferase enzyme construct could co-assemble into a two-component gel that simultaneously emitted bioluminescence and fluorescence. The modular design of this platform suggests a wide range of potential protein fusion partners, leading to biomaterials with a rich assortment of functional capabilities. |
1 Introduction
Modification of proteins with natural or synthetic polymers can impart unique properties and functionalities to the protein. Of particular interest is modifying proteins such that they can assemble into supramolecular biomaterials that demonstrate activity of the protein domain.1,2 For example, catalytic micellar rods can be created from polystyrene-modified proteins,3 enzymatically active inclusion bodies can be created by fusing leucine-rich surfactant-like peptides onto proteins,4 cell-penetrating disks can be created from polyarginine,5 biocatalytic films can be created from poly(N-isopropylacrylamide),6 calcium-responsive hydrogels can be created from polyethylene glycol tetraacrylate conjugated to calmodulin,7 and a variety of protein–polymer conjugates have been used to create nanoparticles.8Modifying a functional protein with a peptide-based assembly domain via genetic fusion affords precise control of the amino acid sequence of the assembly domain. The sequence, in turn, can be tailored to limit the extent of assembly or control the conditions under which assembly occurs, which allows for manufacturing of soluble products in cell-based hosts. For example, fusions of a functional protein and a sequence that undergoes slow secondary structure transition from an α-helix to a β-sheet can be recovered from microbial hosts in the soluble fraction and then incorporated into fibrillar biomaterials.9 Sequences encoding α-helical coiled-coils with different strand numbers can be used to create bifunctional assemblies with tunable numbers of protein domains,10 while sequences encoding heterogeneous coiled-coils can be used to form assemblies with modular protein domain composition.11 Charge-complementary sequences that selectively co-assemble to form β-sheets can be used to create functional microscopic and macroscopic biomaterials.12,13 Amino acid sequences can also be tailored to endow responsiveness to external stimuli.14 For instance, thermally responsive elastin like polypeptides (ELPs)15,16 have been engineered to form temperature sensitive micelles with a multivalent display of fibronectin type III domain or thioredoxin through precise control of the amino acid sequence.17,18 Similar approaches have also been used to engineer ELP micelles for anti-tumor activity19,20 and wound healing21via recombinant fusion of different bioactive cargo.
Protein–peptide and protein–polymer conjugates are often used to create mechanically robust gels through self-assembly22–24 or chemical crosslinking.25–29 However, examples of bioactive gels fabricated from protein–polypeptide fusion are uncommon. Existing strategies have fused a self-assembling α-helical polypeptide22 or calcium-dependent β-roll directly to functional cargo.30,31 Fusing the α-helical polypeptide and alcohol dehydrogenase or organophosphate hydrolase provided catalytically active hydrogels,32,33 while fusing the polypeptide onto fluorescent proteins could be used to create multifunctional hydrogels.34 Likewise, the calcium-dependent β-roll formed hydrogels when fused to maltose binding protein.35 These examples, which are based on ligand-responsive, pH-responsive, and temperature responsive supramolecular assembly domains, show that user-controlled changes in the chemical or physical properties of the system can be harnessed to create bioactive gels from protein–polypeptide fusions. Informed by these examples, opportunities exist to establish whether other stimuli-responsive polypeptide fusion sequences can also be used to create bioactive gels.
Here, we show that an N-glycosylation fusion tag, (GGGSGGGSGGNWTT)10, which we refer to as “NGT,” can drive the temperature-dependent formation of functional supramolecular protein gels. NGT is a repeat of a peptide sequence that was recently identified through a high-throughput screen as a consensus glycosylation motif for the Actinobacillus pleuropneumoniae N-glycosyltransferase;36 NGT has no known supramolecular assembly propensity. Recombinant fusions of NGT and either superfolder green fluorescent protein (sfGFP) or NanoLuc luciferase (nL) formed bioactive viscoelastic gels at reduced temperature. NGT protein gels softened with increased temperature. Studies using sodium thiocyanate or an asparagine-to-glutamine NGT mutant (“QGT”) suggested that non-covalent intermolecular interactions mediate gel formation. Likewise, enzymatic glycosylation of NGT also disrupted gel formation. Binary mixtures of NGTsfGFP and NGTnL yielded gels that demonstrated bioluminescence resonance energy transfer (BRET) in the presence of the nL substrate, furimazine. This suggested that different NGT-fusion proteins can co-assemble in close spatial proximity to afford multifunctional supramolecular biomaterials demonstrating synergistic activity of the different protein domains.
2 Materials and methods
2.1 Recombinant protein expression and purification
Genes encoding for Actinobacillus pleuropneumoniae N-glycosyltransferase (ApNGT), NGTnL, NGTsfGFP, and QGTsfGFP were inserted into pET-21d(+) plasmid vectors between NcoI and XhoI restriction sites (Genscript). Plasmids were transformed into Origami™ B(DE3) E. coli (Novagen) and cultured on LB/agar plates with ampicillin (50 μg mL−1) and kanamycin B (15 μg mL−1) overnight at 37 °C. Cultures were selected from the agar plate and grown overnight at 37 °C and 225 rpm on an orbital shaker in 5 mL of LB media containing ampicillin (50 μg mL−1), kanamycin B (15 μg mL−1), and 1% glucose for catabolite repression.NGTnL was sub-cultured into 1 L of LB media (10 g tryptone, 5 g yeast extract, 5 g NaCl) with additives for autoinduction (5 g glycerol, 2 g α-lactose, 0.5 g glucose, 0.493 g MgSO4, trace elements) and grown for 24 hours at 37 °C, 185 rpm.
NGTsfGFP, QGTsfGFP, and ApNGT were sub-cultured into 1 L of LB media and grown at 37 °C, 185 rpm until reaching an optical density of 0.6–0.8 at a wavelength of 600 nm. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was then supplemented into the culture at 0.5 mM concentration, and 1 L cultures were incubated for 18 hours at 18 °C, 185 rpm.
Bacteria were pelleted via centrifugation (8800 × g at 4 °C for 10 minutes) with a Sorvall™ RC 6 Plus Superspeed Centrifuge (ThermoFisher) and washed with phosphate buffered saline (PBS, pH 7.4). Bacterial pellets were then mechanically disrupted in a solution of B-PER™ bacterial protein extraction reagent (Thermo Scientific) (4 mL per 1 g of bacteria mass), 2400 units per mL DNAse I (ThermoFisher), 50 mg mL−1 lysozyme (ThermoFisher), and pierce protease inhibitor tablet (Thermo Scientific), and rocked for 15 minutes at room temperature. The bacterial lysate was then centrifuged (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g at 4 °C for 25 minutes) to separate the soluble protein fraction from bacterial debris.
000 × g at 4 °C for 25 minutes) to separate the soluble protein fraction from bacterial debris.
Recombinant proteins of interest were recovered from the soluble protein fraction using immobilized metal affinity chromatography (IMAC). Briefly, high density cobalt agarose beads (GoldBio) were packed between two porous polyethylene filters in a polypropylene column. 6-Histidine-tagged recombinant proteins were loaded onto the cobalt agarose beads and washed with 1× PBS. A stepwise gradient of 0–200 mM imidazole was used to elute the recombinant proteins of interest. Purity of proteins collected from IMAC was analyzed using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and coomassie brilliant blue G-250 staining. Molar concentration of proteins was determined using a NanoDrop spectrophotometer (ThermoFisher).
2.2 Gel electrophoresis
Protein molecular weight was determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), staining by coomassie brilliant blue, and comparison to a protein ladder (PageRuler™ Plus Prestained Protein Ladder, ThermoFisher Scientific). Following IMAC purification, 5 μg of protein were diluted in water and loaded into 2× Laemmli sample buffer (Bio-Rad) with 5% β-mercaptoethanol at a 1:1 ratio. This mixture was heated to 95 °C for 10 minutes to ensure denaturation of sample proteins. Denatured samples were loaded into wells of Any kD™ Mini-PROTEAN® TGX™ Precast Protein Gels (Bio-Rad). Electrophoresis was performed in ice cold SDS-PAGE running buffer (192 mM glycine, 25 mM Tris base, 3.4 mM SDS) at 100 V for 1 h and 45 min. After electrophoresis, gels were stained with coomassie brilliant blue for 30 minutes, followed by >1 hour of destaining in a solution of 50% water, 40% methanol, and 10% glacial acetic acid. Photographic images were taken after destaining.
2.3 MALDI-TOF mass spectrometry
Proteins were desalted and concentrated to >80 μM using Amicon® Ultra 0.5 mL centrifugal filters (Millipore Sigma). Sinapinic acid matrix was prepared by dissolving 10 mg of matrix in 50% water, 50% acetonitrile, and 0.1% TFA. Proteins were mixed into sinapinic acid matrix at a 1:4 v/v ratio and vortexed for 30 seconds. 2 μL of sinapinic acid protein mixture was spotted onto a MTP 384 ground steel target plate (Bruker), dried, and overspotted with an additional 2 μL. After drying, the target plate was inserted into a Bruker Autoflex LRF MALDI TOF and set to linear mode to analyze proteins larger than 20 kDa. For good signal to noise ratio, laser intensity was set to 95% and gain to 68×, and proteins embedded in the matrix were desorbed and ionized using a nitrogen laser (335 nm).
2.4 Recombinant protein supramolecular assembly and gel preparation
Post-IMAC purification, purified proteins were concentrated to 30 μM using Amicon® Ultra 15 mL centrifugal filters (Millipore Sigma) by spinning at 4100 × g. Protein concentrate was moved into 10 K MWCO SnakeSkin™ Dialysis Tubing (ThermoFisher Scientific) and dialyzed overnight at 4 °C into PBS (pH 7.4). Dialyzed protein concentrate was moved to centrifuge tubes and frozen at −80 °C for storage. Proteins were thawed at 4 °C and protein assemblies were centrifuged at 14100 × g for 7 min to form protein gels.
2.5 Recombinant protein gel concentration measurement
NGTsfGFP gel was heated to 55 °C to reduce the viscosity of the gel, and subsequently well mixed. A measured amount of NGTsfGFP gel material was pipetted into diluent water, heated to 55 °C, and vortexed vigorously to resuspend the NGTsfGFP. The diluted NGTsfGFP was measured via a NanoDrop spectrophotometer (ThermoFisher) and multiplied by the dilution factor to determine the concentration of the gel.NGTnL gel concentration was measured using a NanoDrop spectrophotometer (ThermoFisher) to measure the concentration before and after protein assembly. The mass of NGTnL protein in the gel was then determined and divided by the volume of the gel to determine the concentration. NGTnL could not be heated to 55 °C for resuspension in diluent, as this would cause the NGTnL gel to form an insoluble aggregate.
2.6 Turbidity measurements
Turbidity measurements of NGTsfGFP and QGTsfGFP were performed on a SpectraMax® M3 Multi-Mode microplate reader (Molecular Devices) at an absorbance of 600 nm wavelength. For sodium thiocyanate (NaSCN) measurements, NaSCN was added to 100 μM NGTsfGFP to a final concentration of 1 M or 0.1 M NaSCN. 100 μL of each sample was added to clear plastic bottom 96-well microplate. The microplate was incubated at 4 °C for 30 minutes to induce aggregation of NGTsfGFP. Absorbance readings were taken at 4 °C. The spectrophotometer temperature was set to 55 °C and absorbance was read every minute until the sample reached 55 °C.Turbidity measurements comparing NGTsfGFP to glucose modified NGT were performed using identical methods, except the plate reader was heated to 55 °C prior to the 96-well microplate being inserted.
2.7 Glycosylation of NGTsfGFP
Glycosylation methods were adapted from previous reports.36 The enzymatic reaction catalyzed by Actinobacillus pleuropneumoniae N-glycosyltransferase, in which a single glucose is coupled to asparagine, requires a consensus sequence of amino acids flanking the asparagine residue of N-X-S/T, where X is any amino acid except proline. This consensus sequence exists in the NGT repeat as N-W-T. Briefly, NGTsfGFP was buffer exchanged into 100 mM HEPES (pH 8) and 500 mM NaCl. 50 μM NGTsfGFP was incubated with 0.1 μM Actinobacillus pleuropneumoniae N-glycosyltransferase and 25 mM uracil-diphosphate glucose at 30 °C for 4 hours. Glycosylation efficiency was confirmed with MALDI-TOF MS.2.8 Vial inversion test
Vial inversion tests were performed in 2 mL scintillation vials with an outer diameter of 12 mm. 250 μL of 500 μM protein was placed into the vial, incubated at the indicated temperature for study, and inverted. We used the vial inversion test as a preliminary assessment of gel formation for all proteins reported in this manuscript. For any protein preparation that passed the vial inversion test, that is it did not flow due to gravity, we also acquired viscosity and storage/loss modulus data using a rheometer as described in the methods. Likewise, for any protein preparation that failed the vial inversion test, that is it flowed due to gravity, we did not analyze that sample using a rheometer.2.9 Oscillatory rheology
Oscillatory rheology methods were adapted from previous work.37 For all rheological measurements, NGTsfGFP or NGTnL gels were loaded into 0.5 mL 27 G allergy syringes (BD). 80 μL of gel was extruded into cylindrical gel molds and kept at 4 °C until loaded onto the rheometer plate. All rheology measurements were made on an Anton Paar MCR302 with 8 mm diameter parallel plates separated by 1.3 mm.2.10 Co-assembly and BRET assay
BRET assay was performed according to previously described methods.11 All measurements were taken in a SpectraMax® M3 Multi-Mode microplate reader (Molecular Devices). After IMAC purification, NGTnL and NGTsfGFP concentrated solutions were put into 10 K MWCO SnakeSkin™ Dialysis Tubing (ThermoFisher Scientific) in equimolar amounts (30 μM NGTsfGFP + 30 μM NGTnL) and dialyzed overnight at 4 °C against PBS. For luminescence and BRET measurements, 10 μL NGTnL or 10 μL NGTsfGFP + NGTnL co-assembled gels were extruded into black opaque bottom 96-well microplates, and 90 μL of PBS pipetted on top of each gel. Immediately prior to reading, 100 μL of diluted furimazine solution (1 μL stock furimazine + 49 μL Nano-Glo™ buffer) (Promega) was added to the well. Emission of NGTnL or NGTsfGFP + NGTnL was measured over a 400–700 nm wavelength sweep at 1 nm increments and 500 ms integration time. For fluorescent measurements of NGTsfGFP, 10 μL NGTsfGFP gels were extruded into a white opaque 96-well microplate, and 90 μL of PBS pipetted on top of each gel. Wells were excited at 485 nm with a cut-off set at 495 nm. Fluorescence emission was read over a 400–700 nm wavelength sweep at 1 nm increments and 500 ms integration time.3 Results and discussion
N-glycotag (NGT) was recombinantly fused onto the N-terminus of either superfolder green fluorescent protein (sfGFP) or NanoLuc luciferase (nL), a bioluminescent protein, via a flexible glycine–serine (GS) linker (Fig. 1a). These proteins were chosen as model effector proteins due to their ease of detection and characterization using spectroscopic methods. SDS-PAGE and MALDI-TOF MS confirmed that full-length NGTnL and NGTsfGFP could be expressed in E. coli and recovered in the soluble phase in good purity at approximately 4 mg per liter of expression media (Fig. 1b and c). Assemblies that were visible to the naked eye formed when purified NGT-fusion proteins were subjected to a reduced temperature. When centrifuged at 14100 × g, these protein assemblies packed into a self-supporting material that did not flow under gravity at room temperature (Fig. S1†). No change in emission wavelength, relative luminescence units (RLU), or relative fluorescence units (RFU) were seen when NGT-fusion proteins were compared to the unmodified proteins (Fig. 1d and e). NGTsfGFP and NGTnL concentrated to 500 μM and 2 mM, respectively, and processed by freezing at −80 °C and thawing at 4 °C formed a self-supporting material that did not flow due to gravity, whereas unmodified sfGFP and nL did flow in the same conditions (Fig. 1f and S2†). When diluted in excess 1× PBS at 37 °C, sfGFP dispersed until equilibrium was reached in the vial, whereas 93% of the NGTsfGFP remained at the bottom of the vial after 66 hours as a self-supporting material phase (Fig. 1g).
 | ||
| Fig. 1 NGT-fusion proteins form self-supporting, bioactive materials. (a) Design of NGT-fusion proteins. (b) SDS-PAGE analysis of purified NGT-fusion proteins; (i) NGTnL; (ii) molecular weight ladder; (iii) NGTsfGFP. (c) MALDI-TOF MS spectra of NGTnL (blue) and NGTsfGFP (green). (d) Offset emission spectra and relative luminescence units (RLU) of nL and NGTnL in the presence of chemical substrate furimazine, n = 3 technical replicates, significance determined by Student's t-test (pairwise comparison). (e) Offset emission spectra and relative fluorescence units (RFU) of sfGFP and NGTsfGFP in the presence of λ = 465 nm wavelength light, n = 3 technical replicates, significance determined by Student's t-test (pairwise comparison). (f) 500 μM sfGFP (−) and NGTsfGFP (+) inverted after being frozen at −80 °C and thawed at 4 °C. (g) Blue-light transillumination of 1 mM sfGFP (−) NGTsfGFP (+) before and 66 hours after dilution in excess 1× PBS at 37 °C. | ||
Oscillatory rheology measurements showed that NGTsfGFP and NGTnL formed viscoelastic gels (Fig. 2). The storage (G′) modulus was greater than the loss (G′′) modulus for 1 mM preparations of NGTsfGFP or 3 mM preparations of NGTnL at 0.3% strain and angular frequencies of 0.1–10 rad s−1 (Fig. 2a). The damping factor for 1 mM NGTsfGFP and 3 mM NGTnL gels ranged from 0.1–1 (Fig. 2b), indicating that the self-supporting materials were weak viscoelastic gels under these conditions.38 1 mM NGTsfGFP gels showed an average G′ of 0.62 ± 0.31 kPa, while 3 mM NGTnL gels showed an average G′ of 0.45 ± 0.15 kPa, suggesting that the functional protein domains (e.g. sfGFP or nL) did not appreciably alter material mechanics despite differing in size and charge. NGTsfGFP concentrated to 500 μM and incubated at 4 °C did not flow due to gravity in a vial inversion test, while 500 μM NGTsfGFP incubated at 37 °C failed to form a self-supporting material and flowed when inverted (Fig. S3†). This suggested a minimum temperature required for gel formation. NGTsfGFP that was kept at 4 °C without a freeze–thaw step formed gels that were softer than gels formed via freeze–thaw processing (Fig. S4†), demonstrating that NGT gel mechanics can be augmented by reduced temperatures.
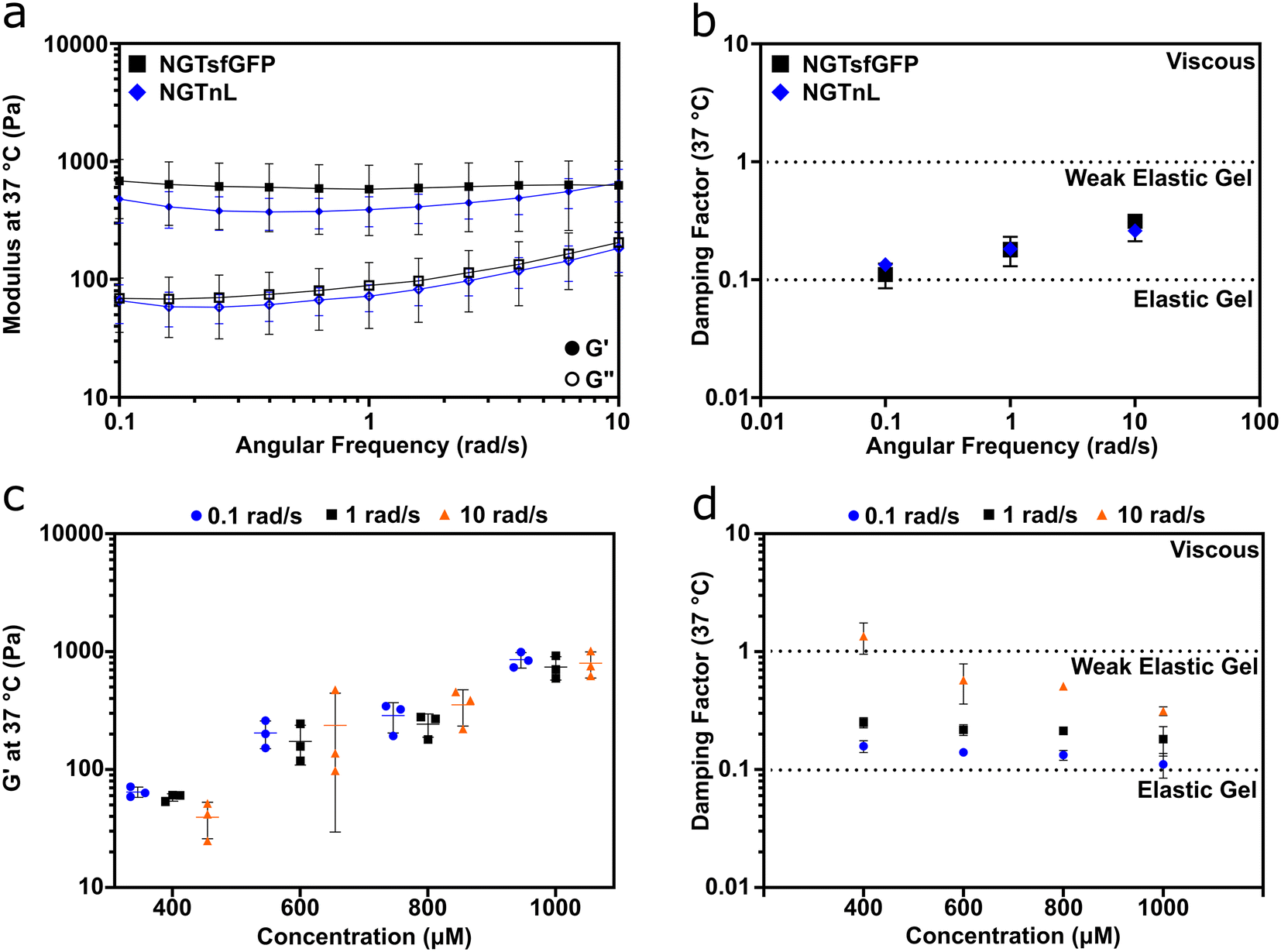 | ||
| Fig. 2 NGT-fusion proteins form concentration dependent, weak elastic gels. (a) G′ (closed circles) and G′′ (open circles) of NGTsfGFP and NGTnL gels at 37 °C from 10 to 0.1 rad s−1. (b) Damping factor (G′′/G′) of NGTsfGFP and NGTnL. (c) G′ of NGTsfGFP gels at varying concentrations at angular frequencies of 0.1, 1, and 10 rad s−1. (d) Damping factor (G′′/G′) of NGTsfGFP gels at varying concentrations and angular frequencies of 0.1, 1, and 10 rad s−1. Measurements taken at a strain of 0.3%. | ||
Informed by these data, all gels used in subsequent studies were processed with a freeze–thaw cycle. The mechanical properties of NGTsfGFP materials were concentration-dependent, with stiffness increasing over the range of 400 μM to 1000 μM (Fig. 2c). The damping factor indicated that NGTsfGFP gels approached the elastic regime with increasing concentration, whereas at 400 μM and 10 rad s−1 NGTsfGFP entered the viscous regime, suggesting that the gel point is in the low 0.1 mM range (Fig. 2d).37 Both NGTsfGFP and NGTnL had increased viscous character with increased angular frequency, displayed by G′′ approaching G′, suggesting dissociation of the gel network. This is in contrast with covalently crosslinked gels39,40 or gels formed from physically entangled nanofibers,37,41,42 which are typically frequency insensitive in the range of 0.1–10 rad s−1. This suggested that the NGT-protein gels may form via weak intermolecular bonds rather than physical entanglement or strong intermolecular contacts.
Temperature-dependent rheology and turbidity measurements demonstrated that NGT-fusion proteins formed gels via non-covalent interactions involving the asparagine residue (Fig. 3). 1000 μM NGTsfGFP gels were a viscoelastic solid at 4 °C, 37 °C, and 55 °C, as indicated by G′ > G′′ in each condition (Fig. 3a–c). However, the average G′ and G′′ decreased with increasing temperature, indicating that heat softened the gels. Likewise, G′′ approached G′ at increasing angular frequency at all temperatures, as also reported in Fig. 2a. NGTsfGFP gels could also be recycled via heating to 55 °C, freezing at −80 °C, and thawing at 4 °C without appreciable loss of stiffness (Fig. S5†). Together, these rheological characteristics suggested that NGT fusion proteins existed in a supramolecular network of intermolecular interactions following cold-temperature processing and centrifugation.
 | ||
| Fig. 3 NGTsfGFP gels form via intermolecular bonds involving asparagine. Oscillatory rheology of NGTsfGFP gels (1000 μM) at (a) 4 °C, (b) 37 °C, and (c) 55 °C. (d) Transmittance at λ = 600 nm of 100 μM NGTsfGFP heated from 4 °C to 55 °C in the presence of 0 M, 0.1 M, or 1 M NaSCN. (e) Background subtracted absorbance at λ = 600 nm of 500 μM NGTsfGFP and glutamine mutant QGTsfGFP at 4 °C. (f) Absorbance at λ = 600 nm of 500 μM NGTsfGFP and glucose modified NGTsfGFP (“Glc-NGTsfGFP”) heated from 4 °C to 55 °C. | ||
Informed by these data, we probed the role of hydrogen bonds in the formation of the supramolecular network. At 100 μM, NGTsfGFP formed a turbid solution at 4 °C, which became more transparent upon heating to 55 °C. The chaotropic agent sodium thiocyanate (NaSCN) increased the transparency of NGTsfGFP solutions at both 4 °C and 55 °C in a NaSCN concentration-dependent manner (Fig. 3d). This behavior persisted over several cycles of heating and cooling (Fig. S6†). NaSCN has been used in prior reports to decrease the phase transition temperature of upper critical solution temperature polymers by disrupting intermolecular hydrogen bonding between amide groups.43–46 Informed by these studies, our results suggested that the repeated asparagine in the NGT domain is important for gel formation.
To probe the role of asparagine in gel formation by NGTsfGFP, we first studied the assembly behavior of the mutant QGTsfGFP, wherein all asparagine residues in the NGT domain were mutated to glutamine residues (i.e. (GGGSGGGSGGQWTT)10). SDS-PAGE and MALDI-TOF MS confirmed that full length QGTsfGFP was recombinantly expressed from E. coli and recovered from the soluble phase (Fig. S7†). QGTsfGFP showed no change in emission spectra or RFU when compared to unmodified sfGFP (Fig. S8†). QGTsfGFP failed to form a gel at 500 μM (Fig. S9†), while turbidity measurements suggested little to no aggregation of QGTsfGFP after freezing at −80 °C and thawing 4 °C (Fig. 3e).
Next, we studied the assembly behavior of an NGTsfGFP variant, Glc-NGTsfGFP, in which all asparagine residues in the NGT consensus sequence were modified with glucose via treatment with the N-glycosyltransferase from Actinobacillus pleuropneumoniae and UDP-glucose (Fig. S10†).47 Glc-NGTsfGFP showed no change in emission spectra or RFU when compared to unmodified sfGFP (Fig. S11†). Based on sequence alignment, sfGFP lacks the glycosylation consensus sequence, N-X-S/T, and so asparagine residues in sfGFP cannot be modified by Actinobacillus pleuropneumoniae N-glycosyltransferase. Indeed, incubation of sfGFP with N-glycosyltransferase and UDP-sugar did not result in a mass shift (Fig. S12†). Solutions of Glc-NGTsfGFP were transparent at both 4 °C and 55 °C, whereas NGTsfGFP formed a gel with temperature-dependent transparency (Fig. 3f), demonstrating that glycosylation of the asparagine residues prevented assembly. Collectively, these data show that asparagine contributes to the supramolecular assembly of NGT fusion proteins into viscoelastic materials via cold-temperature processing. However, the QGTsfGFP control showed that increasing the side-chain length by one methylene unit disrupted gel formation. Thus, simply the presence of a side-chain amide is insufficient to drive gel formation; rather, that amide must exist in an appropriate conformational arrangement. Similar relationships have recently been reported for aspartate and glutamate in β-sheet peptide supramolecular assemblies as well.48
NGTsfGFP and NGTnL gels recovered from shear thinning and high strain disruption, as measured by oscillatory rheology (Fig. 4). Both NGTsfGFP and NGTnL at 37 °C experienced considerable loss of viscosity when exposed to high shear rate (100 Hz), but partially recovered after transition from high to low shear rate (Fig. 4a and b). NGTsfGFP and NGTnL gels experienced shear thinning at low shear rate (0.5 Hz) on the first cycle, but not subsequent cycles. 1 mM NGTsfGFP gels did not flow during vial inversion directly after extrusion from a 25 gauge syringe, displaying immediate recovery to a solid material (Fig. S13, Movie S1, ESI†). NGTnL and NGTsfGFP gels subjected to high-strain disruption (1000% strain) recovered to a G′/G′′ ratio of >1 within 30 seconds after cessation of high strain, showing recovery to a weak viscoelastic gel. NGTsfGFP recovered 81% of its initial stiffness after 10 minutes, and NGTnL recovered 94% of its initial stiffness in the same amount of time. Rheology measurements at plate temperatures of 4 °C or 37 °C showed that percent recovery of stiffness of 1 mM NGTsfGFP was not dependent on temperature in this range (Fig. S14†). The recovery from high shear and strain is indicative of a self-healing network,49 which may be amenable to minimally invasive delivery via injection through a needle.
 | ||
| Fig. 4 NGTsfGFP and NGTnL networks recover after high shear and high strain disruption. High shear recovery of (a) NGTsfGFP and (b) NGTnL gels. Viscosity was measured at 0.5 Hz shear rate before and after the dashed lines, and 100 Hz shear rate between the dashed lines. Viscoelastic restoration of (c) NGTsfGFP and (d) NGTnL gels after high-strain disruption. G′ (closed circles) and G′′ (open circles) measured at 0.3% strain (before and after dashed lines) and 1000% strain (in-between dashed lines). | ||
NGTsfGFP and NGTnL were co-assembled into a multifunctional gel. An equimolar solution of NGTsfGFP and NGTnL was dialyzed against PBS, frozen at −80 °C, thawed at 4 °C, and centrifuged at 14100 × g to generate a gel containing both NGT-fusion proteins. Bioluminescence resonance energy transfer (BRET) was used to probe NGTsfGFP and NGTnL co-assembly into a heterogeneous gel (Fig. 5a), as NGTnL emission in the presence of furimazine will induce emission from NGTsfGFP if the protein pairs are within 100 Å from one another.50 NGTnL gel alone displayed a strong emission peak at λ = 440 nm in the presence of furimazine (Fig. 5b, blue trace), and NGTsfGFP alone displayed a strong emission peak at λ = 515 nm when excited with λ = 485 nm light (Fig. 5b, green trace) or blue light from a transilluminator (Fig. 5b, (ii) inset). In the presence of furimazine, co-assembled NGTnL and NGTsfGFP displayed emission peaks at λ = 440 nm and λ = 515 nm (Fig. 5b, black trace), consistent with the single-component gel peaks. The perceived color of the co-assembled gel in the presence of furimazine was predicted to be a combination of nL bioluminescence and sfGFP fluorescence, which was observed (Fig. 5b, (iii) inset). These data suggested that NGTnL and NGTsfGFP were within 100 Å of one another when co-assembled in the gel state. The stronger intensity of the acceptor emission peak of NGTsfGFP relative to the donor emission peak of NGTnL indicated efficient transfer between co-assembled NGTnL and NGTsfGFP, given that energy transfer rate of BRET is dependent on the inverse sixth power of intermolecular separation.51
 | ||
| Fig. 5 Bioluminescence resonance energy transfer (BRET) occurs in co-assembled gels of NGTsfGFP and NGTnL. (a) Schematic representation of BRET between NGTnL and NGTsfGFP in the gel. BRET is initiated by the enzymatic reaction between NanoLuc luciferase and the substrate furimazine. (b) Emission spectra of (i) NGTnL gel in the presence of furimazine, (ii) NGTsfGFP gel exposed to λ = 485 nm light, (iii) and NGTnL co-assembled with NGTsfGFP. Inset: digital images of gels corresponding to plate reader conditions. | ||
4 Conclusion
NGT is a polypeptide fusion tag that can mediate the assembly of folded proteins into bioactive supramolecular gels. NGT fusion proteins assembled into gels at reduced temperatures, whereas they remained soluble at elevated temperatures. This temperature responsiveness allowed for NGT fusion proteins to be expressed in microbial hosts and recovered from the soluble fraction. NGT gels demonstrated recovery from high shear and high strain, which suggests they may find use as minimally-invasive delivery vehicles. Disruption of gelation via glycosylation sheds light on an unexpected relationship between designer post-translational modification of proteins and their supramolecular behavior, establishing that glycosylation can disrupt intermolecular interactions even in non-natural molecules. As assembly is mediated by interactions involving the asparagine residues of the NGT tag, different proteins could be incorporated into NGT gels alone and in combination, suggesting broad potential versatility of the NGT fusion tag to create modular and multifunctional biomaterials.Author contributions
E. D. H. designed experiments, conducted experiments, analysed data, and wrote the paper. S. M. conducted experiments and analysed data. N. R. S. conducted experiments. B. G. K. conceived of the project, analysed data, and wrote the paper. G. A. H. conceived of the project, designed experiments, analysed data, and wrote the paper.Conflicts of interest
Patents have been applied for by the University of Florida that name E. D. H., B. G. K., and G. A. H. as co-inventors.Acknowledgements
This research was supported by the National Institute of Health. Support to B. G. K. is acknowledged from NIDDK R01DK129690 and NIAID R01AI171045. Support to G. A. H. is acknowledged from NIGMS R35GM133697. We would like to acknowledge the University of Florida Mass Spectrometry Research and Education Center for technical assistance and training with MALDI-TOF MS, which is funded by NIH S10OD021758 and S10OD030250.References
- C. A. Stevens, K. Kaur and H.-A. Klok, Adv. Drug Delivery Rev., 2021, 174, 447–460 CrossRef CAS.
- C. J. Wilson, A. S. Bommarius, J. A. Champion, Y. O. Chernoff, D. G. Lynn, A. K. Paravastu, C. Liang, M.-C. Hsieh and J. M. Heemstra, Chem. Rev., 2018, 118, 11519–11574 CrossRef CAS PubMed.
- K. Velonia, A. E. Rowan and R. J. M. Nolte, J. Am. Chem. Soc., 2002, 124, 4224–4225 CrossRef CAS PubMed.
- B. Zhou, L. Xing, W. Wu, X.-E. Zhang and Z. Lin, Microb. Cell Fact., 2012, 11, 10 CrossRef CAS PubMed.
- E. Vazquez, M. Roldán, C. Diez-Gil, U. Unzueta, J. Domingo-Espín, J. Cedano, O. Conchillo, I. Ratera, J. Veciana, X. Daura, N. Ferrer-Miralles and A. Villaverde, Nanomedicine, 2010, 5, 259–268 CrossRef CAS PubMed.
- A. Huang, G. Qin and B. D. Olsen, ACS Appl. Mater. Interfaces, 2015, 7, 14660–14669 CrossRef CAS.
- W. L. Murphy, W. S. Dillmore, J. Modica and M. Mrksich, Angew. Chem., Int. Ed., 2007, 46, 3066–3069 CrossRef CAS PubMed.
- L. P. Herrera Estrada and J. A. Champion, Biomater. Sci., 2015, 3, 787–799 RSC.
- G. A. Hudalla, T. Sun, J. Z. Gasiorowski, H. Han, Y. F. Tian, A. S. Chong and J. H. Collier, Nat. Mater., 2014, 13, 829–836 CrossRef CAS PubMed.
- S. A. Farhadi, R. Liu, M. W. Becker, E. A. Phelps and G. A. Hudalla, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2024117118 CrossRef CAS PubMed.
- S. A. Farhadi, A. Restuccia, A. Sorrentino, A. Cruz-Sánchez and G. A. Hudalla, Mol. Syst. Des. Eng., 2022, 7, 44–57 RSC.
- D. T. Seroski, A. Restuccia, A. D. Sorrentino, K. R. Knox, S. J. Hagen and G. A. Hudalla, Cell. Mol. Bioeng., 2016, 9, 335–350 CrossRef CAS.
- D. T. Seroski, X. Dong, K. M. Wong, R. Liu, Q. Shao, A. K. Paravastu, C. K. Hall and G. A. Hudalla, Commun. Chem., 2020, 3, 172 CrossRef CAS PubMed.
- R. L. DiMarco and S. C. Heilshorn, Adv. Mater., 2012, 24, 3923–3940 CrossRef CAS PubMed.
- D. T. McPherson, J. Xu and D. W. Urry, Protein Expression Purif., 1996, 7, 51–57 CrossRef CAS PubMed.
- D. E. Meyer and A. Chilkoti, Nat. Biotechnol., 1999, 17, 1112–1115 CrossRef CAS PubMed.
- W. Hassouneh, K. Fischer, S. R. MacEwan, R. Branscheid, C. L. Fu, R. Liu, M. Schmidt and A. Chilkoti, Biomacromolecules, 2012, 13, 1598–1605 CrossRef CAS PubMed.
- M. R. Dreher, A. J. Simnick, K. Fischer, R. J. Smith, A. Patel, M. Schmidt and A. Chilkoti, J. Am. Chem. Soc., 2008, 130, 687–694 CrossRef CAS PubMed.
- P. Zhao, D. Atanackovic, S. Dong, H. Yagita, X. He and M. Chen, Mol. Pharmaceutics, 2017, 14, 1494–1500 CrossRef CAS PubMed.
- N. Liu, M. Cui, N. Hu, F. Yang, Y. Mu, C. Guo, X. Guan and Z. Xie, Colloids Surf., B, 2022, 220, 112976 CrossRef CAS.
- B. Bulutoglu, A. Acun, S. L. Deng, S. Mert, E. Lupon, A. G. Lellouch, C. L. Cetrulo, B. E. Uygun and M. L. Yarmush, Adv. Healthcare Mater., 2022, 11, 2102795 CrossRef CAS PubMed.
- W. A. Petka, J. L. Harden, K. P. McGrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389–392 CrossRef CAS PubMed.
- J. S. Lee, M. J. Kang, J. H. Lee and D. W. Lim, Biomacromolecules, 2022, 23, 2051–2063 CrossRef CAS PubMed.
- A. A. Dinerman, J. Cappello, H. Ghandehari and S. W. Hoag, Biomaterials, 2002, 23, 4203–4210 CrossRef CAS PubMed.
- A. Araújo, B. D. Olsen and A. V. Machado, Biomacromolecules, 2018, 19, 329–339 CrossRef PubMed.
- H. Wang, L. Cai, A. Paul, A. Enejder and S. C. Heilshorn, Biomacromolecules, 2014, 15, 3421–3428 CrossRef CAS PubMed.
- D. Asai, D. Xu, W. Liu, F. Garcia Quiroz, D. J. Callahan, M. R. Zalutsky, S. L. Craig and A. Chilkoti, Biomaterials, 2012, 33, 5451–5458 CrossRef CAS PubMed.
- D. W. Lim, D. L. Nettles, L. A. Setton and A. Chilkoti, Biomacromolecules, 2007, 8, 1463–1470 CrossRef CAS PubMed.
- Z. Sui, W. J. King and W. L. Murphy, Adv. Mater., 2007, 19, 3377–3380 CrossRef CAS.
- K. Dooley, Y. H. Kim, H. D. Lu, R. Tu and S. Banta, Biomacromolecules, 2012, 13, 1758–1764 CrossRef CAS PubMed.
- K. Dooley, B. Bulutoglu and S. Banta, Biomacromolecules, 2014, 15, 3617–3624 CrossRef CAS PubMed.
- I. R. Wheeldon, E. Campbell and S. Banta, J. Mol. Biol., 2009, 392, 129–142 CrossRef CAS PubMed.
- H. D. Lu, I. R. Wheeldon and S. Banta, Protein Eng., Des. Sel., 2010, 23, 559–566 CrossRef CAS PubMed.
- I. R. Wheeldon, S. C. Barton and S. Banta, Biomacromolecules, 2007, 8, 2990–2994 CrossRef CAS PubMed.
- B. Bulutoglu and S. Banta, Gels, 2019, 5, 10 CrossRef CAS PubMed.
- W. Kightlinger, L. Lin, M. Rosztoczy, W. Li, M. P. DeLisa, M. Mrksich and M. C. Jewett, Nat. Chem. Biol., 2018, 14, 627–635 CrossRef CAS PubMed.
- B. S. Morales, R. Liu, J. Olguin, A. M. Ziegler, S. M. Herrera, K. L. Backer-Kelley, K. L. Kelley and G. A. Hudalla, Biomater. Sci., 2021, 9, 2494–2507 RSC.
- Z. A. Syahariza and H. Y. Yong, Food Measure, 2017, 11, 1586–1591 CrossRef.
- A. Upadhyay, R. Kandi and C. P. Rao, ACS Sustainable Chem. Eng., 2018, 6, 3321–3330 CrossRef CAS.
- X. Liu, X. Yang, Z. Yang, J. Luo, X. Tian, K. Liu, S. Kou and F. Sun, ACS Appl. Nano Mater., 2018, 1, 1579–1585 CrossRef CAS.
- Y. Sun, X. Li, M. Zhao, Y. Chen, Y. Xu, K. Wang, S. Bian, Q. Jiang, Y. Fan and X. Zhang, Bioact. Mater., 2022, 8, 396–408 CAS.
- M. A. Greenfield, J. R. Hoffman, M. O. De La Cruz and S. I. Stupp, Langmuir, 2010, 26, 3641–3647 CrossRef CAS PubMed.
- J. Seuring, F. M. Bayer, K. Huber and S. Agarwal, Macromolecules, 2012, 45, 374–384 CrossRef CAS.
- A. Augé, D. Fortin, X. Tong and Y. Zhao, Polym. Chem., 2018, 9, 4660–4673 RSC.
- H. C. Haas, R. D. Moreau and N. W. Schuler, J. Polym. Sci., Polym. Phys. Ed., 1967, 5, 915–927 CAS.
- L. Hua, M. Xie, Y. Jian, B. Wu, C. Chen and C. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 43641–43648 CrossRef CAS PubMed.
- F. Schwarz, Y.-Y. Fan, M. Schubert and M. Aebi, J. Biol. Chem., 2011, 286, 35267–35274 CrossRef CAS PubMed.
- R. Liu, X. Dong, D. T. Seroski, B. Soto Morales, K. M. Wong, A. S. Robang, L. Melgar, T. E. Angelini, A. K. Paravastu, C. K. Hall and G. A. Hudalla, Angew. Chem., 2023, 62, e202314531 CrossRef CAS PubMed.
- J. L. Mann, A. C. Yu, G. Agmon and E. A. Appel, Biomater. Sci., 2018, 6, 10–37 RSC.
- R. B. Sekar and A. Periasamy, J. Cell Biol., 2003, 160, 629–633 CrossRef CAS PubMed.
- H. Schneckenburger, Methods Appl. Fluoresc., 2019, 8, 013001 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4me00029c |
| This journal is © The Royal Society of Chemistry 2024 |