 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Concluding remarks: Reflections on the Faraday Discussion on New Directions in Molecular Scattering
Mark
Brouard

Department of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OXON OX1 3TA, UK. E-mail: mark.brouard@chem.ox.ac.uk; Tel: +44 1865 275457
First published on 11th July 2024
Abstract
These concluding remarks summarize the Faraday Discussion on New Directions in Molecular Scattering. The discussion brought together scientists from a wide range of disciplines, from astrochemistry to coherent quantum control, and the submitted papers highlighted the need for innovation in experimental methods and computational tools to tackle more complex systems, relevant to chemistry in the real world. As recorded in the previous pages of this discussion, the meeting saw lively debate on numerous topical issues. This summary outlines some of the highlighted key developments in the field, and points towards future directions of molecular scattering research.
Introduction
Molecular collisions are ubiquitous, occurring in diverse environments all the way from cold traps to Titan. Their study is largely the province of chemists and physicists who wish to understand more about energy transfer and reactive processes involving atoms and molecules. One of the most powerful methods for studying molecular collisions involves directly interrogating the scattering event. This often involves using molecular beam techniques, which enable collisions to be studied under rigorously defined conditions, facilitating the most detailed comparisons to be made with dynamical theory. Papers presented in this Faraday Discussion illustrate the ways in which colliding molecules can be cooled, state selected, and manipulated, how scattered molecules can be probed in minute detail, and how theory can be used to enhance fundamental understanding of experimental measurements.Molecular scattering is a mature field,1–5 with the first crossed molecular beam experiments performed in the early 1960s by Dudley Herschbach and Yuan T. Lee and their coworkers, which, together with the pioneering work of John Polanyi on infrared chemiluminescence, led to the 1986 Nobel Prize in Chemistry for ‘contributions concerning the dynamics of chemical elementary processes’. To observers outside the field, it may seem surprising that after all this time molecular scattering is still a topic of such current scientific interest. That it is so is a reminder of the fundamental importance of collisional processes of one form or another in much of physical and biological science.
Given the maturity of the field, the Faraday Discussion focussed on new challenges in molecular scattering, in particular those of investigating larger, more complex systems (see Fig. 1). Such endeavours require enhanced experimental and theoretical capabilities, building on existing expertise in quantum-state preparation, stereochemical control of reactants, and the detailed characterisation of products typically used in studies of smaller systems. The discussion explored four broad themes: control of translational energy or stereochemistry; scattering of larger molecules; scattering in extreme environments; and scattering at condensed-phase surfaces. The importance of new theoretical developments required to shed light on more complex systems was recognized throughout, both in the submitted papers and the discussion. In the following summary, I have attempted to align the papers and associated discussion broadly according to the four themes originally outlined for the meeting, whilst recognizing that the boundaries between them are somewhat blurry.
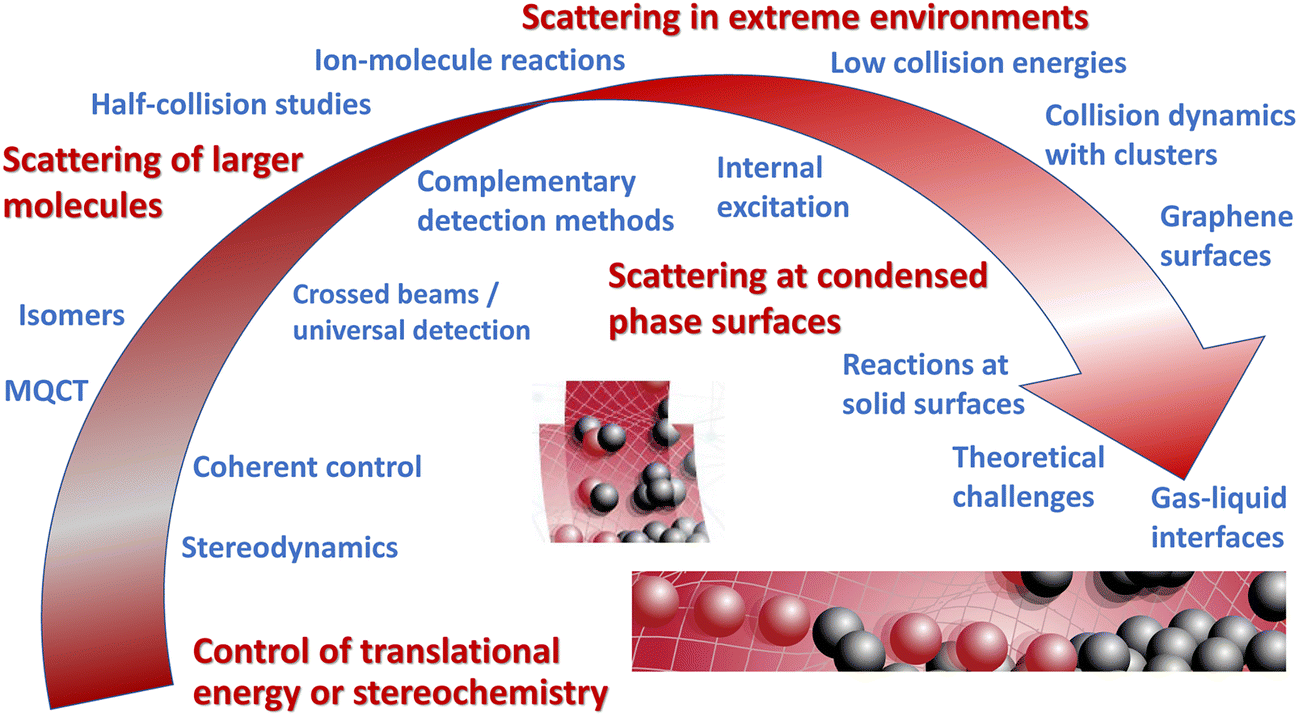 | ||
| Fig. 1 Some of the themes explored in the Faraday Discussion on New Directions in Molecular Scattering. | ||
The Faraday Discussion got off to an inspirational start with the Spiers Memorial Lecture presented by Alec Wodtke (https://doi.org/10.1039/D4FD00015C). He called for a new collaborative approach between experimentalists and theoreticians, empowered by the latest lab-based techniques and computational tools that would have been unheard of by our forbears. He illustrated the power of experiment/theory collaboration using benchmark examples from the literature, as well as current surface chemistry work from his own laboratory, an example of which featured later in the discussion (https://doi.org/10.1039/D3FD00174A). The spirit of Alec Wodtke's Spiers Memorial lecture filtered through much of the subsequent discussion, with the interplay between experiment and theory key to many of the new insights presented.
Control of translational energy or stereochemistry
This theme saw papers and discussion concerning the development of new experimental and theoretical methods required to investigate a wider range of collision systems. Innovations were presented in the use of existing control methods, principally involving applying external electric or magnetic fields, and the development of new methods to study phenomena such as the effect of molecular conformation on chemical reactivity. New methods exploiting quantum entanglement and coherent control were also highlighted.The control of collision geometry has been a long-standing challenge in the scattering dynamics community. Exciting work was presented by Chadwick et al. (https://doi.org/10.1039/D4FD00007B) on the use of hexapolar, dipolar and uniform magnetic fields to create controllable coherent superpositions of (mI, mj) reactant states of H2(j = 1). On collision with stepped or smooth Cu(511) and Cu(111) surfaces, the specularly scattered molecules were then analysed to reveal their preferred rotational alignment. The study revealed that cartwheel rotation was more likely to lead to specular scattering on Cu(511), with scattered molecules preferentially rotating like cartwheels irrespective of their initial alignment. Also highlighted were the differences in scattering behaviour between stepped Cu(511) and smooth Cu(111) surfaces, with the former being significantly more sensitive to the effects of increased temperature (https://doi.org/10.1039/D4FD00007B).
The surface scattering experiments by the Swansea team provided an example of a four vector correlation, in which the directions of the initial and final velocities (k, k′) of the H2 are defined, and the polarization of the initial and final rotational angular momenta (j, j′) are controlled or probed. For gas phase scattering, a major objective has been stereodynamical studies in which the mutual orientation and/or alignment of two molecular collision partners is controlled, by use of polarized light or combinations of electric and magnetic fields. The computational study by Jambrina et al. (https://doi.org/10.1039/D3FD00173C) showcased how such polarization offers the opportunity to control collision outcomes in the region of scattering resonances at low collision energies, revealing that what the ‘collision wants’ in terms of mutual molecular polarization can vary significantly in the region of a scattering resonance (https://doi.org/10.1039/D3FD00173C).
Methods for realizing optical control of molecular alignment were illustrated by the work of Costen et al. (https://doi.org/10.1039/D3FD00162H) who used laser excitation and resonantly enhanced multiphoton ionization (REMPI) coupled with velocity map imaging (VMI) methods to study collisions of NO(A) with CO2. Such experiments are in principle sensitive to the (k, j, k′, j′) four-vector correlation. This system is of interest as it undergoes electronic quenching to the ground electronic state, in competition with rotational energy transfer within the excited state of the system, and the experimental study was complemented, importantly, by detailed ab initio calculations. The absence of sideways scattering in the differential cross-sections (DCSs) was ascribed to electronic quenching, which is assumed to favour relatively low impact parameter collisions. An absence of polarization of j′ was observed, and attributed to the role of highly anisotropic attractive forces which dominate those collisions which remain in the excited electronic state and do not undergo electronic quenching.
The role of molecular conformation in chemical reactivity has been less fully explored in the literature. Willitsch et al. (https://doi.org/10.1039/D3FD00172E) presented new experimental data on the chemi-ionization of 1,2-dibromoethene with metastable Ne atoms which addressed this problem using a crossed molecular beam system coupled with time-of-flight (ToF) mass spectrometry (MS). The experiments demonstrated that both dissociative ionization and Penning ionization reaction pathways were sensitive to the initial conformer, i.e. cis or trans 1,2-dibromoethene.
As we saw throughout the Discussion, theory plays an increasingly important role in helping to interpret experimental results on complex molecular systems. Developing approximate theoretical methods that can cope with such complexity is clearly important and necessary, and the validation of approximate computational approaches is crucial. An interesting example of such work was provided by the paper and subsequent discussion on the mixed quantum/classical theory (MQCT) of Babikov et al. (https://doi.org/10.1039/D3FD00166K). In this approximation to the dynamics, the translational degrees of freedom are treated classically, whilst the internal modes are treated quantum mechanically. This leads to significant savings in computational effort over full quantum dynamical treatments. Results for integral cross sections (ICSs) and rate constants for p-H2O + p-H2 show promise in comparison with exact quantum scattering calculations, and it will be interesting to see how MQCT performs in the case of dynamical quantities, such as differential cross sections and polarization effects.
Moving into the quantum regime, there were stimulating papers and discussion concerning the manipulation of molecules as matter waves, and the development of diffraction mirrors for matter-wave optics. The work by Zhao et al. (https://doi.org/10.1039/D3FD00155E) demonstrated how relatively inexpensive holographic gratings can be used to reflect nearly 50% of He atoms under thermal energy conditions. Remarkably, reflection of weakly-bound He2 and He3 complexes was also observed. The work paves the way to other accessible matter-wave optical components, such as lenses for atoms and molecules.
A particularly thought-provoking paper was provided by Ni et al. (https://doi.org/10.1039/D3FD00175J), who considered the coherent control of the reaction
| 2KRb → K2 + Rb2. |
Scattering of larger, more complex molecules
Studying the scattering of larger, more complex systems brings with it new experimental and theoretical challenges. Full characterization of such systems requires the measurement of product branching ratios, as well as channel specific properties such as energy disposal and angular distributions. The need for the use of complementary experimental techniques was highlighted by a number of researchers, particularly in the paper by Suits et al. (https://doi.org/10.1039/D4FD00009A), in their study of the S(3P) reaction with 1,3-butadiene. They employed crossed molecular beam methods coupled with 157 nm laser ionization and VMI methods to elucidate the scattering dynamics, and complementary chirped-pulse Fourier-transform millimeter-wave spectroscopy measurements to provide spectroscopic identification of the thioketene product (https://doi.org/10.1039/D4FD00009A).Given the theme of the Faraday Discussion, it is of no surprise that most of the experimental papers presented involved some form of molecular scattering. Few considered the ‘half collision’ approach, in which the dissociation dynamics is followed subsequent to excitation of a van der Waals complex. In some cases, these experiments provide some control of specific reactant geometries and impact parameters, and can access specific energy regimes perhaps not easily accessible in full-collision studies. Kidwell et al. (https://doi.org/10.1039/D3FD00176H) provided an example of such a complementary approach to study the isomer-specific IR spectroscopy and vibrational predissociation dynamics of NO(X)–C2H6 collision complexes. Vibrational predissociation takes place on the same potential energy surface (PES) as that for inelastic scattering of NO(X) with C2H6, so in principle it is possible to model the dynamics of both processes with a unified theoretical treatment.
Two papers exemplified the strengths of the conventional crossed-molecular beam technique coupled with universal electron impact ionization and ToF MS detection (https://doi.org/10.1039/D3FD00159H and https://doi.org/10.1039/D3FD00181D). In addition to providing channel specific dynamical information (i.e. kinetic energy releases and angular distributions), this technique provides vital branching ratio information, something which is increasingly important for complex systems in which the existence of multiple reaction channels becomes prevalent. The study by Kaiser et al. (https://doi.org/10.1039/D3FD00159H) investigated the formation of polycyclic aromatic hydrocarbons (PAHs), of interest in astrochemistry, through the reaction of C6H6 with the phenylethynyl radical C6H5CC. Balucani et al. (https://doi.org/10.1039/D3FD00181D) on the other hand probed the reaction of O(3P) with toluene, and demonstrated through additional theoretical simulation that both singlet and triplet pathways need to be considered in order to reproduce the observed product yields.
Ion–molecule reactions featured significantly in a number of sessions of the meeting. Noteworthy examples included the crossed molecular beam–VMI experiments of Wester et al. (https://doi.org/10.1039/D3FD00164D) and of Meyer et al. (https://doi.org/10.1039/D3FD00171G), which highlighted the power of these techniques to interrogate the dynamics of more complex systems, particularly when using three-dimensional ion imaging methods. The two groups studied different aspects of reaction complexity, with that of Wester et al. (https://doi.org/10.1039/D3FD00164D) investigating the role of increased complexity of the anion in SN2 and proton transfer reactions of CH3O− with CH3I. Meyer et al., on the other hand, used a similar experimental approach to investigate the reaction
| Ta+ + CH4 → TaCH2+ + H2, |
As already noted, theory has a vital role to play in the interpretation of such experiments. The work of Czakó et al. (https://doi.org/10.1039/D3FD00161J) nicely illustrated this point, through their development of systematic methods for calculating PESs to be used for dynamical calculations. They investigated the SN2 and E2 reactions of OH− + CH3CH2Cl in 24 dimensions, calculating the PES and performing quasi-classical trajectory (QCT) calculations to obtain dynamical observables for the two channels, such as DCSs and energy disposals.
Scattering in extreme environments
This theme involved a number of different topics, some more familiar, involving cold molecular species capable of displaying uniquely quantum phenomena, and other less familiar topics, such as collisions involving highly internally excited species. The challenges in developing new theoretical methods to enable scattering calculations at high energies were also considered, with the need for appropriate approximate computational approaches discussed at several points throughout the meeting (https://doi.org/10.1039/D3FD00166K and https://doi.org/10.1039/D3FD00161J).Ion–molecule reactions at temperatures of a few Kelvin have long been a focus of attention in the kinetics and dynamics communities, because of their relevance to interstellar chemistry, as well as their fundamental importance in terms of the development of capture theory approaches to reaction rates. Lewandowski et al.’s novel approach, in work presented by Olivia Krohn, was to study such reactions by co-trapping Xe+ or O2+ ions in a Coulomb crystal of Ca+, and then reacting these ions with isomers of C3H4 (https://doi.org/10.1039/D4FD00005F). Product detection was achieved by extracting the ions in the crystal into a mass spectrometer. Lewandowski et al. showed us how the reactions of Xe+ and O2+ with isomers of C3H4 (allene and propyne) displayed markedly different kinetics, with those of Xe+ dominated by direct charge transfer reactions, whilst reactions of O2+ with C3H4 were shown to proceed primarily via complex forming pathways.
Molecular scattering under low-energy conditions was also explored, as exemplified by the beautiful crossed molecular beam experiments of Bergeat et al. (https://doi.org/10.1039/D3FD00168G). They investigated the inelastic scattering of HDO with Ne and n-H2. HDO and H2 are known to have relatively high abundances in interstellar media, so quantifying and elucidating their scattering dynamics is of keen interest. ICSs were measured for rotational excitation from HDO (JKaKc = 000) to the 110 or 111 rotational states over a range of collision energies. The results generally showed excellent agreement with theory, including even with the location of resonance structures observed in the ICSs. An exception was the absence of an experimentally observed transition from HDO(000) to the 101 rotational state in the case of scattering with n-H2, an anomaly compared with theory which will require further investigation.
Scattering of energized molecules was investigated by a number of researchers (https://doi.org/10.1039/D3FD00180F and https://doi.org/10.1039/D3FD00179B). Quantum dynamical scattering calculations by Loreau et al. (https://doi.org/10.1039/D3FD00180F) were used to investigate the role of vibrational relaxation of NH3 excited in the ν2 = 1 vibrational level of the umbrella mode by collision with He atoms under conditions relevant to astrophysical environments. Whilst vibrational relaxation was found to be slow compared to rotational relaxation at low collision energies, the vibrational relaxation rate constants showed significant increase with temperature, and thus could become competitive with rotational relaxation in warm astrophysical environments. Mullin et al.https://doi.org/10.1039/D3FD00179B) focussed their attention on the collision dynamics of very highly rotationally excited molecules, which is a relatively unexplored area of molecular collision dynamics. Their elegant experiments involve producing a pair of oppositely chirped 100 ps IR pulses, which were capable of inducing sequential Raman transitions in CO2, leading to highly rotating molecules with j ∼ 244–282. They termed the process an optical centrifuge. The highly rotating molecules were probed using transient IR spectroscopy, which revealed production of strongly polarized rotationally excited CO2, which was all but lost in the collisionally partially relaxed molecules observed in j ∼ 76–100. The experiments were used to measure collisional relaxation and depolarization rates, but the possibility of studying reactive collisions of very highly rotationally excited species remains a tantalizing prospect.
The study of the collision dynamics of atoms and molecules with clusters was perhaps under-represented at the Faraday Discussion, with the notable exception of the work of Fárník et al. (https://doi.org/10.1039/D3FD00160A). Using their bespoke cluster beam apparatus, they studied the take-up of alcohol molecules on hydrated nitric acid clusters, work which has potential to enhance fundamental understanding of molecule–cluster collisions, but also assist in the modelling of atmospheric new-particle formation and aerosol growth. They observed that the uptake cross sections of alcohols decreases with increasing alkyl chain length of the alcohol and with the chain-branching, consistent with the accessibility of the hydrophobic OH group, which decreases with increasing chain length of the alcohol and with steric hindrance. The data were very convincingly modelled using classical molecular dynamics simulations.
Scattering at condensed-phase surfaces
Gas–surface collisions received considerable attention at the Faraday Discussion, reflecting the diverse environments in which such processes are important, including atmospheric chemistry, heterogeneous catalysis, and biological respiration. Studies of both gas–solid and gas–liquid scattering were represented.In the former category, gas–solid interactions, Auerbach et al. (https://doi.org/10.1039/D3FD00174A), in their paper presented by Jan Fingerhut, showed us how to perform very accurate measurements of isotopologue specific thermal reaction rates for the decomposition of HCOOH on Pd. The experiments involved the use of the molecular beam–VMI techniques developed by the Göttingen team over recent years (https://doi.org/10.1039/D4FD00015C). With the help of detailed theory, these experiments enabled the authors to elucidate the complex network of elementary reaction steps that made up the catalytic process under consideration.
The experiments of Auerbach et al. (https://doi.org/10.1039/D3FD00174A) were performed using high vacuum conditions, under which it is possible to define very precisely the surface conditions. However, this can be seen as quite far removed from the ‘real world’ conditions under which catalysts are actually employed. To help bridge that gap, Harding et al. (https://doi.org/10.1039/D3FD00158J) have developed a modified molecular beam–VMI apparatus that enables the study of gas–surface reactions at higher pressures. They demonstrated this approach in a study of the scattering and oxidation of CO on Pd(110) at molecular oxygen pressures up to 1 × 10−5 mbar. The authors found that the production rate of CO2 was significantly dependent on the O2 pressure, being suppressed at higher pressures, shedding light on the catalytic processes involved at near-ambient pressures.
A different approach to studying gas–solid surface interactions was presented by Sibener et al. (https://doi.org/10.1039/D3FD00178D) in their exquisite scanning tunneling microscopy (STM) study of on-surface dynamics of monolayer, bilayer and many-layered graphene. In this work the authors used scattering of molecular beams of O2 and O(3P) onto graphene surfaces, coupled, crucially, with in situ STM. The team were able to resolve the site-specificity of atomic oxygen placement on the moiré lattice for both monolayer and bilayer graphene on Ru(0001) with atomic resolution, and investigated how that depends on incident beam angle and energy.
The philosophy underpinning the experiments of Walker and Polanyi et al. (https://doi.org/10.1039/D3FD00167A) was different, in that they attempted to use the surface as a template on which to initiate and guide molecular collisions of surface adsorbates. This approach is reminiscent of the study of reactions initiated within van der Waals complexes, which allow some control over impact parameter and collision geometry. In the Faraday Discussion paper, Walker and Polanyi et al. initiated scattering and monitor surface–collision outcomes under low-temperature, ultra high vacuum conditions using STM. The STM tip was used to generate ‘projectile’ F-atoms by electron-induced dissociation of CF3 adsorbed onto a Cu(110) surface. The directed F-atom projectiles could be directed at different ends of co-adsorbed allyl radical target molecules, to induce allyl adsorbate rotation either clockwise or counter-clockwise around the surface normal, depending on the angle of the F-atom attack.
The achievements of theoretical work in tackling problems of increasing complexity have been highlighted throughout the discussion and in this report. Within the theme of scattering at condensed-phase surfaces, the importance of theory was illustrated by papers from Kroes et al. (https://doi.org/10.1039/D3FD00165B) and Jiang et al. (https://doi.org/10.1039/D3FD00163F). The focus of the former work (https://doi.org/10.1039/D3FD00165B) was to develop robust density functional theory (DFT) PES tools to study chemical processes at surfaces. The authors calculated molecular beam sticking probabilities using a variety of different PESs and compared the results with experiment. In this way they were able to ascribe errors in their DFT PES to problems with the functional rather than errors in the density. The theoretical work of Jiang et al. (https://doi.org/10.1039/D3FD00163F) concerned the dynamics of an Eley–Rideal reaction between gaseous and adsorbed H/D-atoms on Cu(111). Their work nicely demonstrated that an accurate modelling of the quantum dynamics could only be achieved if the internal rovibrational motion of the product HD molecule were accurately described, requiring a six-dimensional dynamical treatment.
Finally, turning attention to scattering of molecules with liquid surfaces, the papers by Neumark et al. (https://doi.org/10.1039/D3FD00169E) and Nathanson et al. (https://doi.org/10.1039/D3FD00177F) provided a stimulating basis for discussion of the dynamics at the gas–liquid interface. Neumark et al. (https://doi.org/10.1039/D3FD00169E) presented work on the molecular beam scattering of deuterated ammonia, ND3, off flat liquid jets of dodecane, in which scattered molecules were detected using ToF MS. This enabled the team to measure the velocity distributions of scattered molecules as a function of incident and scattering angle. The study elucidated the impulsive scattering (IS) and thermal desorption (TD) contributions to the scattering of ND3 on the dodecane flat liquid jet. Along with previous work, this important study is likely to pave the way to further investigations of both non-reactive and reactive scattering from more volatile systems, such as water.
By contrast, the group of Nathanson et al. (https://doi.org/10.1039/D3FD00177F) took a somewhat different approach to probing the interfacial structure of surfactants by measuring the velocity distributions of evaporating He atoms dissolved in aqueous surfactants contained in a microjet. As with the Neumark et al. study, the velocity distributions were measured using ToF MS, with a chopper used to establish the timing for the ToF measurement. The proposition of the authors was that the measured super-Maxwellian velocity distributions of the evaporating He atoms provided a signature of their final interactions with the liquid surface. This is a novel approach to elucidating surface structure and dynamics, again qualitatively akin to the ‘half-collision’ approach discussed above concerning collisional processes initiated within van der Waals complexes. Further work is required to establish whether this method will yield the level of detailed information that might be anticipated from the more conventional molecular beam scattering approach.
Final remarks and future directions
The Faraday Discussion on New Directions in Molecular Scattering has provided confirmation that the scattering approach to investigating molecular collisions remains relevant and informative. Since the inception of molecular beam scattering methods, there have been numerous incremental improvements to the technique and to molecule detection methods. There have also been a more select number of transformational advances, some of which we have witnessed at this Discussion. We have observed particularly exciting developments in the control of reactants, including the use of laser based methods to prepare molecules in very highly rotationally and electronically excited states. Progress in studying the scattering at solid and liquid surfaces has been particularly striking. Significant advances in theory have also been showcased, and, in addition, it is noteworthy that most of the submitted work to the Discussion has included some consideration of theory.There have been some areas of molecular scattering which have been relatively unexplored. One might anticipate more work in the future involving scattering and reactions of clusters (see, for example, https://doi.org/10.1039/D3FD00160A) and aerosols. Such experimental work will impose great demands on the development of new theoretical tools. Another general area which has not been discussed a great deal at the meeting is the study of multi-body effects of solvent molecules on conventional ‘bimolecular’ reactions.
Notwithstanding the areas not featuring significantly at this meeting, it is clear from this Faraday Discussion that, in spite of the maturity of the field of molecular scattering, it still represents a highly active area of investigation. To those who wish to understand molecular collisions, it remains the case that the molecular scattering approach should be their first ‘port-of-call’. Moreover, there is an ever-expanding toolbox of theoretical methods to help interpret, or be challenged by, the arising experimental findings.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
I gratefully acknowledge support provided by the UK Engineering and Physical Sciences Research Council for funding through Programme Grants EP/V026690/1 and EP/T021675/1. I would also like to thank members of my research group in Oxford who helped with the preparation of this manuscript, particularly those involved in our crossed molecular scattering experiments, Josh Featherstone, Max McCrea and Matthew Strutton. A CC-BY license is applied to the author accepted manuscript arising from this submission, in accordance with UKRI open access conditions.References
- R. D. Levine and R. B. Bernstein, Molecular Reaction Dynamics and Chemical Reactivity, Oxford University Press, New York, USA, 1987 Search PubMed.
- M. S. Child, Molecular Collision Theory, Dover Publications, Inc., Mineola, New York, USA, 1997 Search PubMed.
- M. Brouard, Reaction Dynamics, Oxford University Press, Oxford, UK, 1998 Search PubMed.
- R. D. Levine, Molecular Reaction Dynamics, Cambridge University Press, Cambridge, UK, 2005 Search PubMed.
- Tutorials in Molecular Reaction Dynamics, ed. M. Brouard and C. Vallance, The Royal Society of Chemistry, Cambridge, UK, 2010 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |