
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism of non-phenolic substrate oxidation by the fungal laccase Type 1 copper site from Trametes versicolor: the case of benzo[a]pyrene and anthracene†
Carla Orlando ac,
Isabella Cecilia Rizzoa,
Federica Arrigonia,
Jessica Zampollia,
Marco Mangiagallia,
Patrizia Di Gennaroa,
Marina Lottia,
Luca De Gioiaa,
Tiziana Marinoc,
Claudio Grecob and
Luca Bertini*a
ac,
Isabella Cecilia Rizzoa,
Federica Arrigonia,
Jessica Zampollia,
Marco Mangiagallia,
Patrizia Di Gennaroa,
Marina Lottia,
Luca De Gioiaa,
Tiziana Marinoc,
Claudio Grecob and
Luca Bertini*a
aDepartment of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, 20126 Milan, Italy. E-mail: luca.bertini@unimib.it
bDepartment of Earth and Environmental Sciences, University of Milano-Bicocca, Piazza della Scienza 1, 20126 Milano, Italy
cDepartment of Chemistry and Chemical Technologies, Università della Calabria, Ponte Pietro Bucci, cubo 14c, 87036 Rende, CS, Italy
First published on 9th July 2024
Abstract
Laccases (EC 1.10.3.2) are multicopper oxidases with the capability to oxidize diverse phenolic and non-phenolic substrates. While the molecular mechanism of their activity towards phenolic substrates is well-established, their reactivity towards non-phenolic substrates, such as polycyclic aromatic hydrocarbons (PAHs), remains unclear. To elucidate the oxidation mechanism of PAHs, particularly the activation mechanism of the sp2 aromatic C–H bond, we conducted a density functional theory investigation on the oxidation of two PAHs (anthracene and benzo[a]pyrene) using an extensive model of the T1 copper catalytic site of the fungal laccase from Trametes versicolor.
Introduction
Laccases (EC 1.10.3.2. oxygen oxidoreductase or multicopper oxidase) belong to the group of phenol-oxidases and are widespread in bacteria,1 fungi,2–6 plants, insects,7 and marine species.8 These enzymes catalyze the one-electron oxidation of diverse substrates concomitantly with the reduction of molecular oxygen to water.9The active site of laccases contains four copper ions, which are distributed at three different types of copper centers (T1 Cu, T2 Cu, and T3 Cu, as shown in Fig. 1).
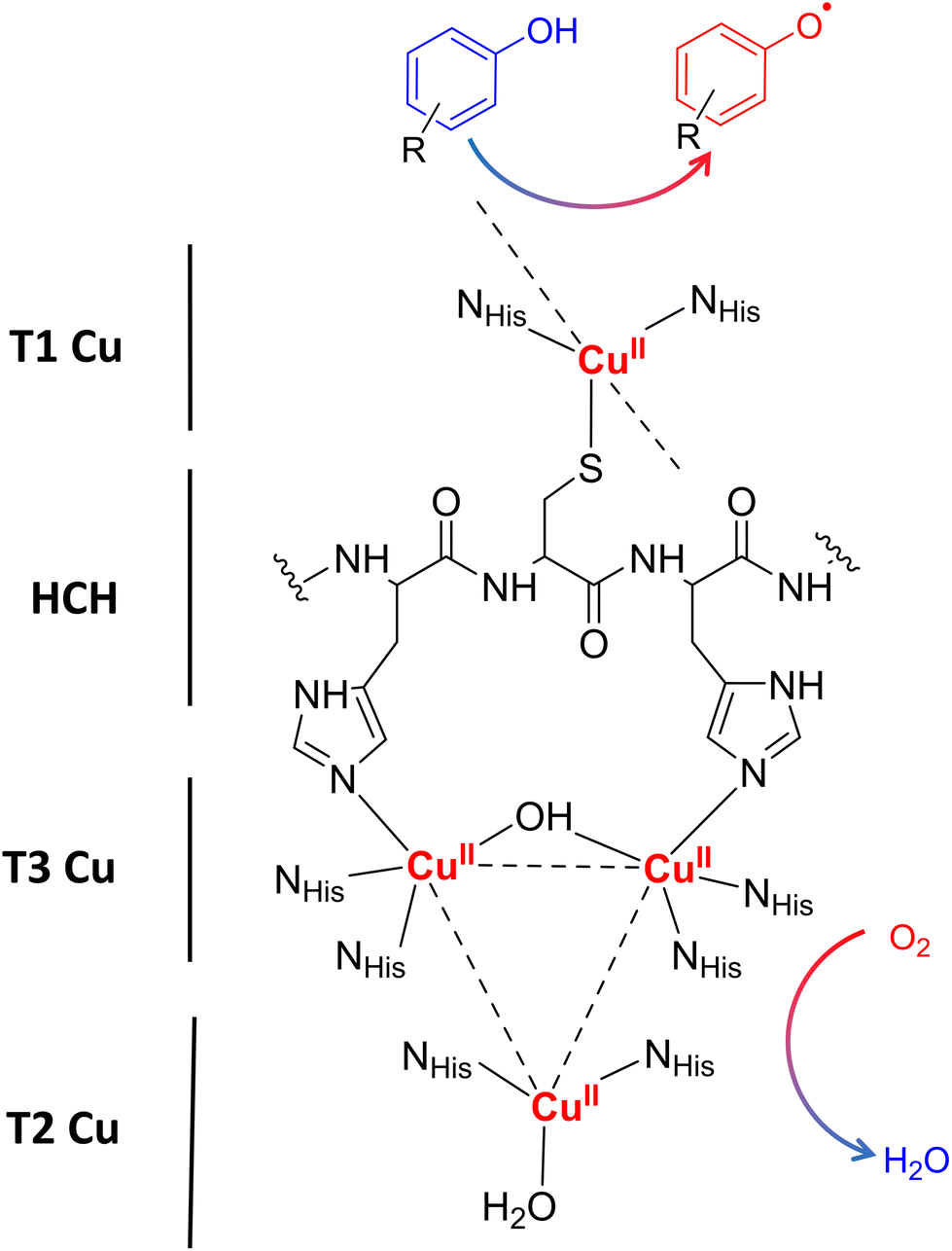 | ||
| Fig. 1 Schematic representation of the laccase catalytic site processing a phenolic compound. Type-1 Copper site (T1 Cu) located at the surface of the substrate binding pocket with one copper atom coordinating two histidine residues and one cysteine residue. In addition, T1 Cu has two weakly coordinating residues in the axial position that vary depending on the source of the enzyme (typically Phe or Leu for high-potential fungal laccases or Met for low-potential bacterial laccases). Type 2 (T2 Cu) and binuclear Type 3 (T3 Cu) copper ions form a trinuclear cluster (TNC) which is coordinated by eight histidine residues and connected with T1 Cu throughout the very conserved histidine–cysteine–histidine (HCH) motif. When the substrate is oxidized near the T1 Cu site, electrons are transferred one by one along an HCH pathway to the TNC, where molecular oxygen is bound and then reduced to water. | ||
Substrate oxidation occurs at T1 Cu, which determines the reduction potential of the enzyme. In particular, the physicochemical properties of the amino acid residue in the axial position of the T1 Cu have been shown to modulate the redox potential.10 Electrons are then transferred via the histidine–cysteine–histidine (HCH) pathway from the substrate to T3 Cu and T2 Cu (on average 12–13 Å afar from T1 site)11 where the reduction of oxygen to water takes place.12
The biological functions of laccases vary depending on their respective sources, ranging from lignin degradation in fungi to lignin biosynthesis in plants, as well as encompassing roles such as spore-coating and pigmentation in bacteria. Precisely, due to their different biological functions observed in nature, laccases are versatile enzymes known for their promiscuity and ability to oxidize a wide range of substrates.13 Laccases typical substrates are various phenolic and lignin-related compounds, but they are active also towards a variety of non-phenolic substrates such as amines, alcohol, dyes,14,15 carbohydrates, metal compounds,16 and some polymers.17,18 The range of substrates that laccases can process can be significantly expanded if redox mediators, i.e. small molecules that act as an electron shuttle, are employed, allowing laccases to oxidize substrates recalcitrant to direct oxidation.19,20
Although the oxidation mechanism of phenolic-type substrates has been comprehensively investigated and is well-documented,19,21–26 the oxidation mechanism for non-phenolic substrates still remains notably less elucidated. Among non-phenolic compounds, in recent years, numerous experimental investigations have focused on the degradation of highly toxic polycyclic aromatic hydrocarbons (PAHs).27,28 PAHs represent persistent organic pollutants commonly present in terrestrial ecosystems, originating from the incomplete combustion of organic substances such as oil, coal, and natural gas. PAH enzymatic bioremediation processes are promising for the treatment of these hazardous molecular systems. Following geminal studies reporting the oxidation of a PAH by the fungal laccases of Trametes versicolor,29,30 Pleurotus ostreatus,31 and Coriolopsis gallica,32 the number of investigations on this subject has grown extensively, in order to understand how to improve the percentage of oxidation of PAHs as the chemical nature of the mediator varies33–39 or using the immobilized enzyme.33,34,40–42 In addition to fungal laccases,43–50 recent studies on PHA degradation have also focused on bacterial laccases46,48,51,52 due to their significant advantages such as higher thermostability and tolerance to a broader pH range, both crucial factors in bioremediation processes.
Regarding the enzymatic oxidation mechanism, the rate-determining step is the substrate mono-electronic oxidation.53,54 The rate of this step is governed by the standard reduction potential (E°) difference between the T1 Cu55 and the substrate. In the case of phenolic substrates, this step employs a simultaneous electron/proton transfer to different acceptors, thereby circumventing the formation of high-energy charged intermediates that would inevitably arise if the electron or proton were transferred independently. In the case of laccase from Trametes versicolor (TvL), proton transfer is facilitated by the acidic character of the phenolic proton and by an Asp residue in proximity to T1 Cu.48,51 In contrast, for non-phenolic derivatives, such as PAHs, the acidity of the proton associated with an aromatic C–H bond is significantly lower than that of the phenolic proton and consequently proton transfer is inherently disfavoured. Nevertheless, fungal laccases exhibit the capability to oxidize PAHs even without a mediator, albeit with comparatively lower oxidation efficiencies. This result suggests that proton transfer is also accomplished in the case of PAHs. More in detail, the laccase PAH oxidation pathway requires six different ETs to oxidize the PAH to the corresponding quinone (see Fig. 2).
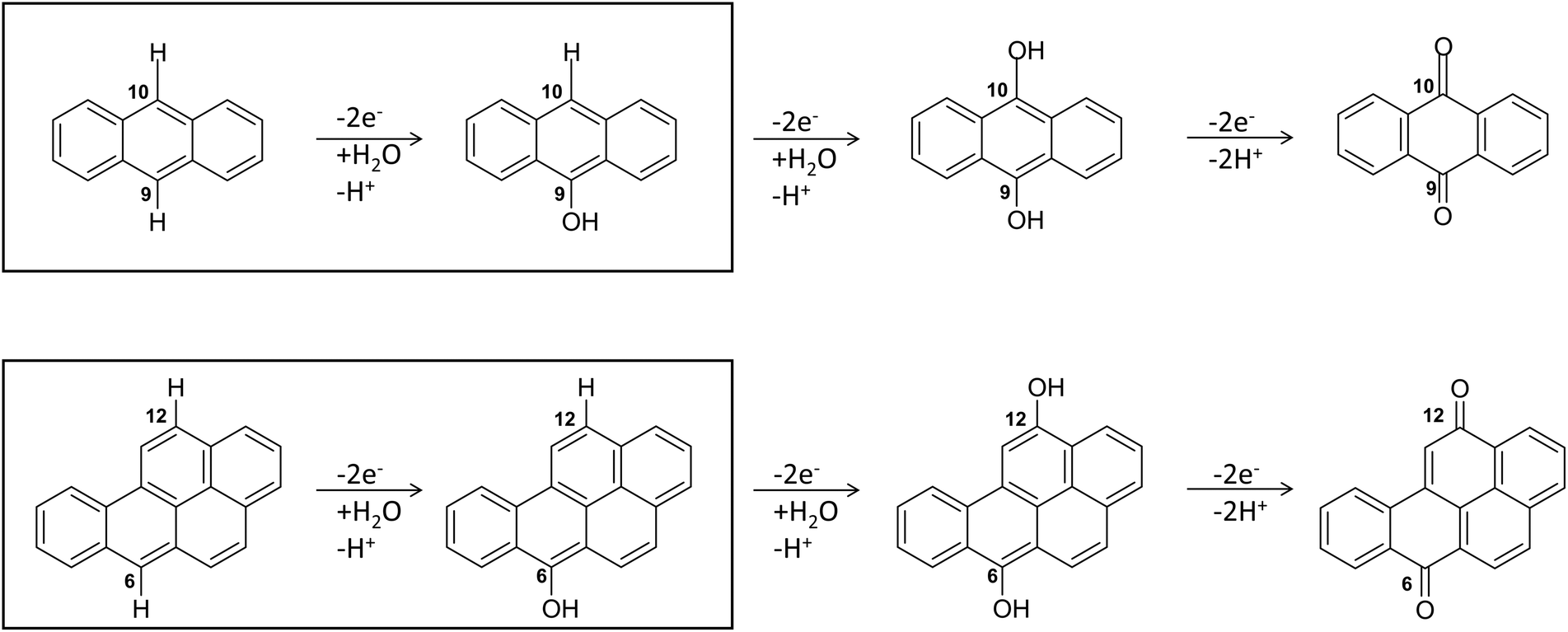 | ||
| Fig. 2 The six-electrons oxidation of ANT e BaP to quinones species catalyzed by laccases. The IUPAC numbering shown is that relating to the BaP.56 The carbon atom 6 and 10b in BaP correspond to the carbon 9 and 10 in ANT. The reaction evidenced in the box is the focal point of the present investigation. The whole conversion requires at least two water molecules. | ||
In this pathway, the first stage, i.e. direct oxidation of the C–H bond, is presumed to be rate limiting. Indeed, subsequent steps involve intermediates with at least one C–OH group, which are characterized by lower reduction potentials and thus exhibit greater reactivity toward oxidation.
Based on these considerations, the present work aims to elucidate the atomic-level mechanism of PAH oxidation by T1 Cu of TvL excluding the effect of the mediator on the catalytic activity. This laccase has high E° (785 mV) and it is well characterized from a functional and structural point of view. The high E° is due to the hydrophobic amino acid in the axial position of the T1 Cu.55 More in detail, we have focused on the crucial stage of their catalytic conversion, i.e. the direct oxidation of aromatic C–H bonds into the corresponding C–OH functionality. Our results shed light on the most likely mechanism associated with this transformation, considering different pathways previously discussed and proposed.52 This approach is of further interest to unveil potential mechanisms that can be transferred to the activation of sp3 aliphatic C–H bonds, in light of recent investigations on polyethylene degradation by other laccases.18
Calculations have been carried out considering two selected substrates: anthracene and benzo[a]pyrene (ANT and BaP hereafter). These substrates shows comparable reduction potential in CH3CN (886 mV and 712 mV for ANT and BaP, respectively)33,57,58 and low water solubility (0.045 and 0.0016 mg L−1, for ANT and BaP, respectively).44 The choice of these two PAHs was guided by the observation that fungal laccases, including TvL, are able to oxidize ANT, but consistently fail to oxidize BaP in the absence of mediators29,30,33,34 (Table S1†), while the mediator facilitates both oxidation processes. This is somewhat unexpected since BaP has a lower ionization potential (7.12 eV) than ANT (7.55 eV), as well as a less positive redox potential.32 Oxidation efficiency is highly dependent on reaction conditions, and in the case of TvL, the addition of Tween80, a surfactant that improves substrate solubility, increases oxidation (Table S1†). The prospect of delving deeper into this phenomenon is intriguing, especially considering the notable chemical similarities between the two substrates.
The investigation unfolds in two stages: in the first stage the binding of theaforementioned substrates to the enzyme is examined through Molecular Docking simulations; in the second stage the oxidation mechanism is then explored via Density Functional Theory (DFT) with a cluster approach,59,60 by using models of increasing complexity derived from representative ligand poses obtained during molecular docking.
Materials and methods
Molecular Docking was performed using Glide, as implemented in Maestro Schrödinger Suite software (Schrödinger Release 2020-3: Maestro, Schrödinger, LLC, New York, NY, 2021). We tested the reliability of the docking protocol by performing a molecular recognition of the known inhibitors, such as 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) and 2,6-dimethoxyphenol (2,6 DMP), into the binding site of TvL (PDB ID: 1KYA61) More in detail, a flexible ligand extra precision (XP) docking was carried out, using the OPLS3 force field (see Fig. S1 in ESI† for details). In order to have a more accurate estimation of ligand-binding affinities, Prime Molecular Mechanics Generalized Born and Surface Area (MM/GBSA) calculations were carried out.62 The stability of substrate positioning within the pocket, was assessed by Molecular Dynamics (MD) simulations. See ESI† for details about MD procedure and analysis.All DFT calculations have been performed with the TURBOMOLE 7.4 suite.63 Geometry optimizations were performed using the pure GGA BP8664,65 functional. The RI technique66 was adopted to speed up calculations. Single point energies on BP86-optimized geometries have been also re-calculated using the hybrid PBE067 functional. Basis sets of triple-ζ plus polarization split valence quality68 (def-TZVP) were adopted for all atoms. Solvent effect is accounted for by using the Conductor-like Screening Model (COSMO) approach.69 Water solvation has been considered by setting the dielectric constant equal to 80. In general, this computational setting provides ground state geometry parameters in good agreement with experimental X-ray values.70 Charge distribution was evaluated using Natural Bond Orbital (NBO) analysis using the PBE0 functional. Geometry optimizations were carried out with convergence criteria fixed to 10−6 Hartree for the energy and 0.001 Hartree·per bohr for the gradient norm vector. D3 Grimme empirical dispersion correction71 was adopted. This theoretical scheme has already been tested and successfully adopted in previous DFT investigations carried out in our laboratory on various bioinorganic copper systems.72–74
Results and discussion
Characterization of enzyme–substrate interactions
To reliably unravel the mechanism(s) of PAH oxidation, a preliminary study on PAHs recognition by the enzyme is needed. Thus, molecular docking of BaP and ANT to the TvL (1KYA61) was carried out allowing to characterize the nature of the enzyme–substrate interaction and to drive subsequent higher level calculations. Indeed, for each substrate, the best-energy pose was chosen as the starting point for subsequent DFT calculations. The analysis of enzyme–substrate interactions also allowed the selection of those residues to be included in our DFT model(s) (vide infra). Furthermore, the obtained docking poses/scores for ANT and BaP provided indications about similarities and/or differences in both their accommodation mode within the binding pocket and their affinity towards TvL. More in detail, the analyses of the five top-ranked poses for both ANT and BaP clearly indicate that they both bind at the same site, in close proximity to T1 Cu (see Fig. S1† for details). Almost all the poses for both ligands are superimposed to the first ranked one (Fig. 3 top, yellow ligand structures), except for a few less stable dispositions, in which the three-ring common scaffold of ANT and BaP is slightly rotated in the pocket.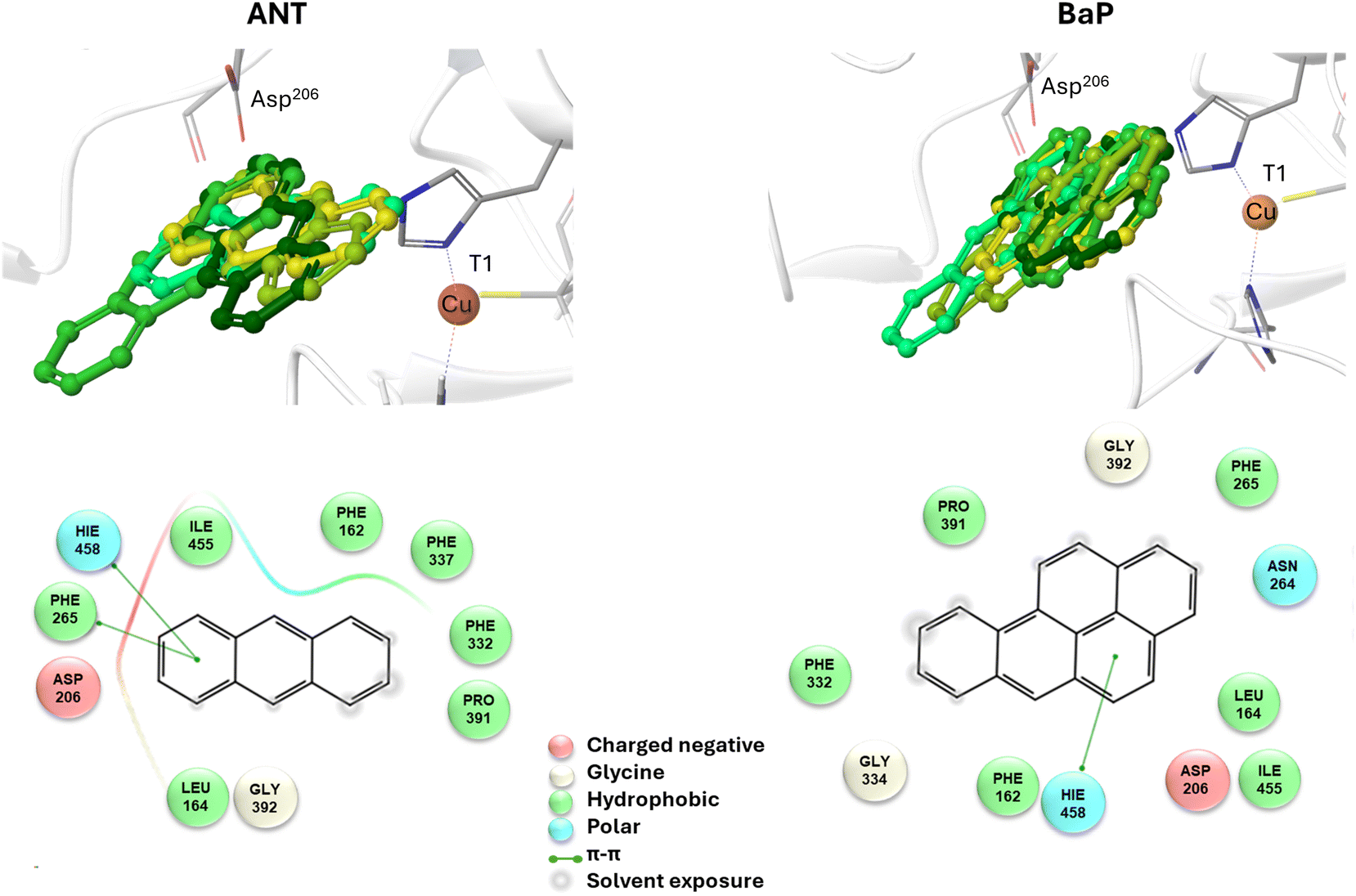 | ||
| Fig. 3 Top: a focus on the superimposition of the top ranked five docking poses of ANT (left) and BaP (right) to TvL (PDB: 1KYA). Bottom: interaction diagrams of the best pose obtained for each substrate to TvL. | ||
Considering the best pose resulting from the docking of each substrate (Fig. 3), we observed quite very similar docking scores (kcal mol−1), equal to −3.61 for ANT and −4.07 for BAP (Table S1†).
The binding is driven by hydrophobic interactions mainly involving the same residues in the two cases (Fig. 2). Indeed, both ligands form intermolecular interactions with residues His458, Asp206, Ile455, Leu164, Phe265, Gly392, Pro391, Phe332, and Phe162; for ANT, an additional interaction with Phe337 was detected, while BaP was also found to interact with Gly334 and Asn264. We recall that His458 is also coordinated with T1 Cu while Asp206 is responsible for proton transfer.
By using the MM/GBSA method, the binding energies of the two substrates slightly diverged, with BaP binding favored by 4.8 kcal mol−1 over ANT.
Exploration of the most likely mechanism with a minimal T1 Cu model
The structures of the experimentally observed quinone species, resulting from ANT and BaP oxidation by laccases, support the mechanistic proposal reported in Fig. 2, which involves six consecutive mono-electronic oxidation steps.52 The C–H bonds involved in oxidation are those at the centrally fused benzene rings, namely C10 and C9 for ANT, and C6 and C12 for BaP (according to IUPAC numbering, Fig. 3). This mechanism is similar to that reported for the degradation of BaP by P450 peroxidase.75The oxidation mechanism consists of three different stages. The first stage involves the two-electron oxidation of the C–H bond, resulting in the formation of phenolic analog intermediates (6-OH BaP and 10-OH ANT). During this stage, the oxidation state of the carbon atom goes from −1 to +1. Subsequently, in the second stage, a successive oxidation occurs, leading to the generation of a di-hydroxy intermediate, which then undergoes further oxidation to quinone species.
Our DFT investigation is focused on the first stage, i.e. the two-electron oxidation of the C–H bond to C–OH, since it is the most critical (and thus relevant) of the whole catalytic cycle (Fig. 3). This stage has been fully characterized following a two-step procedure, in which two T1 Cu models of increasing complexity have been used. In an explorative phase, a minimal model (Fig. 4) has been scrutinized, to provide a thermodynamic picture for the first oxidation, taking BaP as a case study. Indeed, different mechanistic alternatives can underlie this reaction, as also proposed in a previous investigation by Guan et al.52
 | ||
| Fig. 4 Right: structure of the minimal model used for our preliminary calculations, also providing details on the T1 Cu first coordination sphere. Left: structure of the extended cluster model used in this investigation. This model includes 14 residue side chains plus Cu(II), the ligand, and five water molecules (W1–5). The approach of the W4 oxygen atom to the BaP/ANT carbon atom involved in the reaction, defines the reaction coordinate for the first mono-electronic oxidation. | ||
The minimal model only includes the first coordination sphere of T1 Cu, along with the side chain of Asp206. Three different scenarios for the early oxidation were considered (Fig. 5):
(A) Oxidation of the ligand at C6 with the formation of a radical cation species;
(B) Hydrogen atom transfer (HAT) process from BaP with the protonation of the Asp206 side chain;
(C) Oxidation of BaP with the concomitant nucleophilic attack of a water molecule and the protonation of the Asp206 side chain.
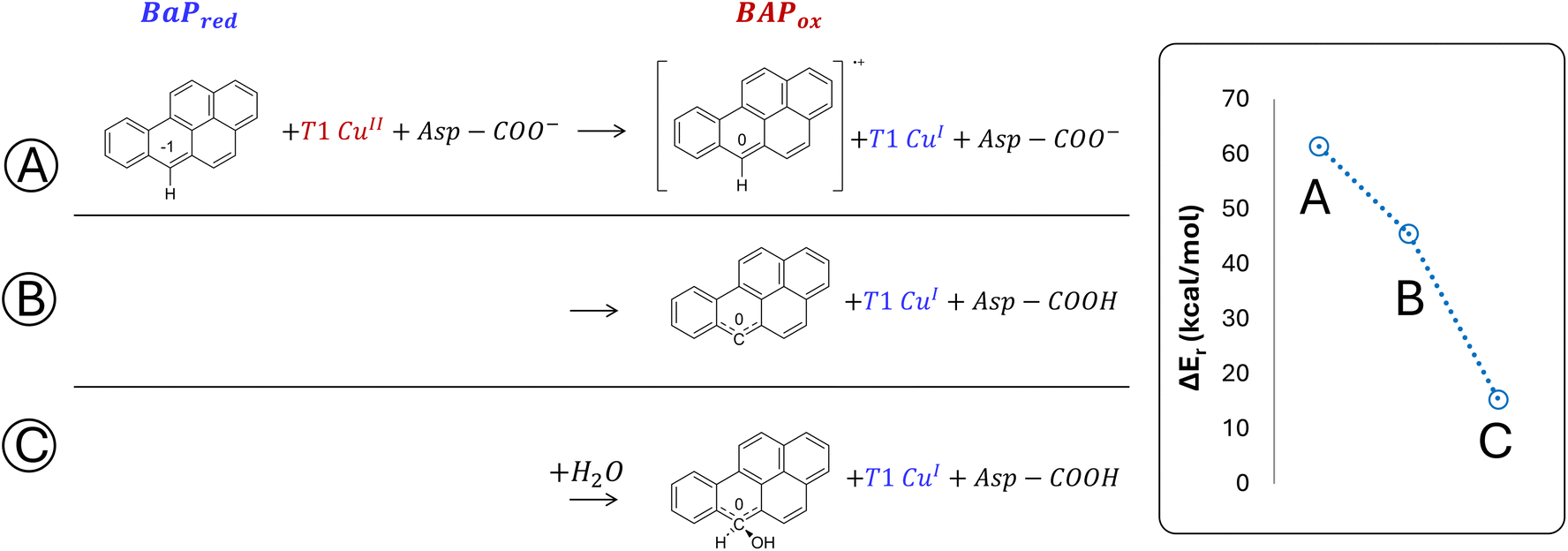 | ||
| Fig. 5 DFT energies (in kcal mol−1) for the first oxidation of BaP, computed using the minimal model, are reported in blue. Results are based on three mechanistic hypotheses: (A) monoelectronic oxidation with the formation of a carbocation, (B) HAT process with the formation of a neutral radical species and protonation of Asp206, (C) nucleophilic attack on the carbocation by a water molecule, and protonation of Asp206. All shown energy values are obtained from single point calculations at the PBE0-D3/TZVP/COSMO level on BP86-D3/TZVP/COSMO-optimized geometries. | ||
Considering both the first and second hypotheses, we calculated too high reaction energies (>45 kcal mol−1) for these reactive channels to be feasible, so these mechanistic possibilities have been ruled out and excluded from subsequent calculations. According to the third hypothesis, an intriguing intermediate was obtained in which the reactive carbon, undergoing the nucleophilic attack by water, transiently adopts a sp3 geometry. The estimated reaction energy of 15.6 kcal mol−1 suggests that this is the most likely reactive channel for the first oxidation, being thermodynamically feasible.
Detailed mechanism characterization using an extended model
In a second step, the investigation proceeded by switching to a more comprehensive T1 Cu cluster model (denoted as extended model herein, see Fig. 4), which was used to (i) recalculate and refine the most likely mechanism previously elucidated using the minimal model, (ii) investigate the second oxidation, (iii) complete the calculated energy profiles by also providing kinetic information, (iv) compare the results obtained for BaP with those for ANT to highlight eventual differences in reactivity between the two, and (v) provide a detailed characterization of the structures of all the species involved in the first two-electron oxidation. The extended model was built considering the interaction pattern of the bulkier substrate, i.e. BaP, with the enzyme, as obtained from molecular docking. It thus incorporates the eight residues mainly involved in the ligand–TvL interaction and additional six residues to ensure the complete coordination of T1 Cu, resulting in a total of 14 residues.For structural simplification, each residue was restricted to its side chain, originating from the Cα position, unless they exhibited contiguous connectivity (Asn264–Phe265 and Gly392–Pro391). The total charge of the model is zero. Since water is directly involved in the most likely mechanism, as suggested by preliminary calculations on the minimal model, our extended model also incorporates five water molecules (W1–5) strategically positioned between Asp206 side chain and the substrate forming a “Grotthuss-like” channel to assist proton transfer. Remarkably, the presence and persistence of water molecules in the T1 Cu proximity, in between the metal center and the substrate, was confirmed by MD simulations (see Fig. S2†). W4 is placed in proximity to the reactive C–H bond of both substrates. A visual examination of the model reveals that the C–H bond is in the vicinity of both the Cu T1 (which will undergo reduction by the substrate) and the side chain of Asp206 (which will accept the proton from the substrate).
By scanning the potential energy surface (PES) along the reaction coordinate for both ligands, we identified the transition state geometry and evaluated the activation barriers associated with both first and second one-electron oxidation (see Fig. S3 and S4† or details). The characterized mechanism is sketched in Fig. 6. The DFT optimized starting species (R–Cu(II)) are in S = ½ state and are substantially similar for the two substrates (see Table S2 and Fig. S3 in ESI†), as might be expected based on the small differences between BaP and ANT. Indeed, the two substrates accommodate similarly within the binding pocket, with the ANT molecular plane slightly bent compared to that of BaP by approximately 14 degrees. The T1 Cu site shows the typical trigonally distorted C3v structure, characterized by an unusually short Cu–SCys bond.76,77 The carbon atom belonging to the carboxylic acid group of the side chain of Asp206 is distant from T1 Cu 7.7 Å (BaP) and 7.1 Å (ANT).
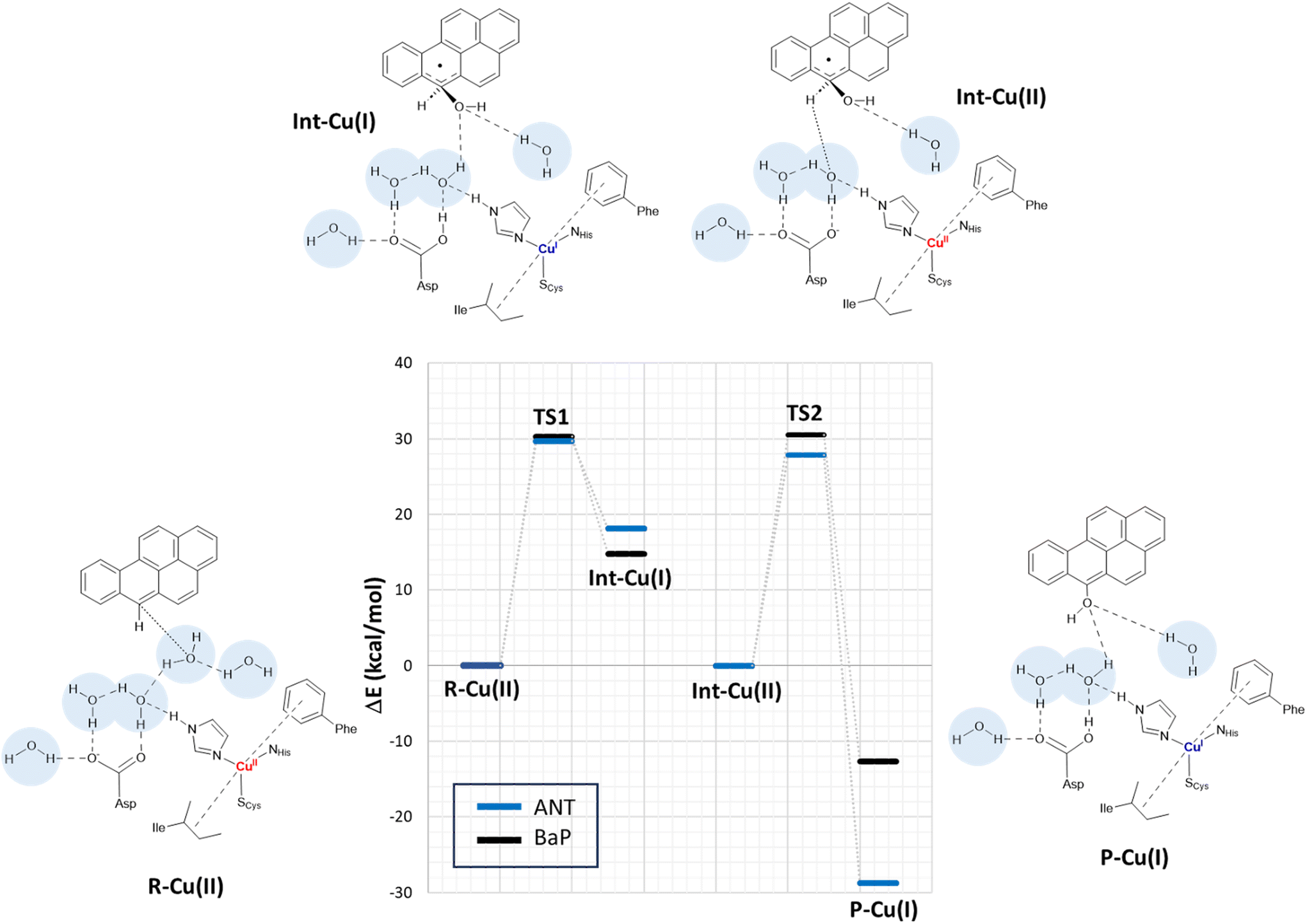 | ||
| Fig. 6 Proposed mechanism for the two-electron oxidation of ANT and BAP catalyzed by TvL based on DFT calculations on the extended model. Energy differences (in kcal mol−1) at the PBE0-D3/TZVP/COSMO level are derived from single-point calculations on BP86-D3/TZVP/COSMO optimized structures. The structures of the various forms (reactant R–Cu(II), Cu(I)/Cu(II) intermediate Int–Cu(I) and Int–Cu(II), and product P–Cu(I)) are depicted on the side, considering BaP as example TS1 and TS2 refer to the transition states along the pathway and are associated with the first and second ET from the ligand to T1 Cu. Initially, the ligand is located in the binding pocket near T1 Cu and Asp206. The latter assists a proton transfer from the substrate, facilitating the ET. The proton transfer is mediated by at least one solvent molecule. The 1e oxidized form of the ligand has a tetrahedral carbon carrying an alcohol function. In the second step, T1 Cu is re-oxidized and Asp206 deprotonated obtaining a S = 0 broken-symmetry state. Then, in the second electron/proton transfer, T1 Cu is reduced and the Asp206 side chain protonated again with the formation of a mono-hydroxy product. | ||
Cu and SCys spin densities in the Cu(II) equilibrium structures are on average 0.50 and 0.43, respectively, in line with previous DFT investigations. The Cu–C distances with the carbon atom formally under oxidation are 8.4 Å (ANT) and 8.5 Å (BaP) (Fig. 4).
The first oxidation step is a concomitant electron/proton transfer during which we observe the:
(1) electron transfer from the substrate to T1 Cu;
(2) nucleophilic attack of W4 to the substrate mono-oxidized carbon atom. W4 is in a suitable position for nucleophilic attack with a C–O internuclear distance of 3.851 Å and 3.218 Å for ANT and BaP, respectively;
(3) proton transfer from W4 to W3 and then from the latter to the side chain of Asp206;
(4) approaching W5 in close proximity to the substrate, positioning suitably to serve as a bridge in the subsequent oxidation process.
This initial step changes the hybridization of the central carbon atom of the substrate from sp2 to sp3, with the formation of a radical intermediate featuring a tetrahedral carbon carrying an alcohol function (Int-Cu(I)). This induces a slight bending of the system, as depicted in Fig. 7. This structural feature is in line with the Jahn–Teller distortion observed for the benzene radical cation by ab Initio molecular dynamics.78 T1 Cu is reduced with no spin population on Cu and SCys and with Cu–NHis and Cu–SCys distances similar to those of T1 Cu(II).79 The radical is entirely delocalized among the carbon atoms on the substrate, as illustrated in Fig. 7. In both substrates, particularly with ANT, we observe the localization of a substantial portion of the spin population on the C10 atom positioned opposite to the C6–OH, consistent with the electronic structure of the anthracene radical cation.77,78 This carbon atom will ultimately be responsible for the formation of the final quinone species in the 6-electron oxidation process (Fig. 2).
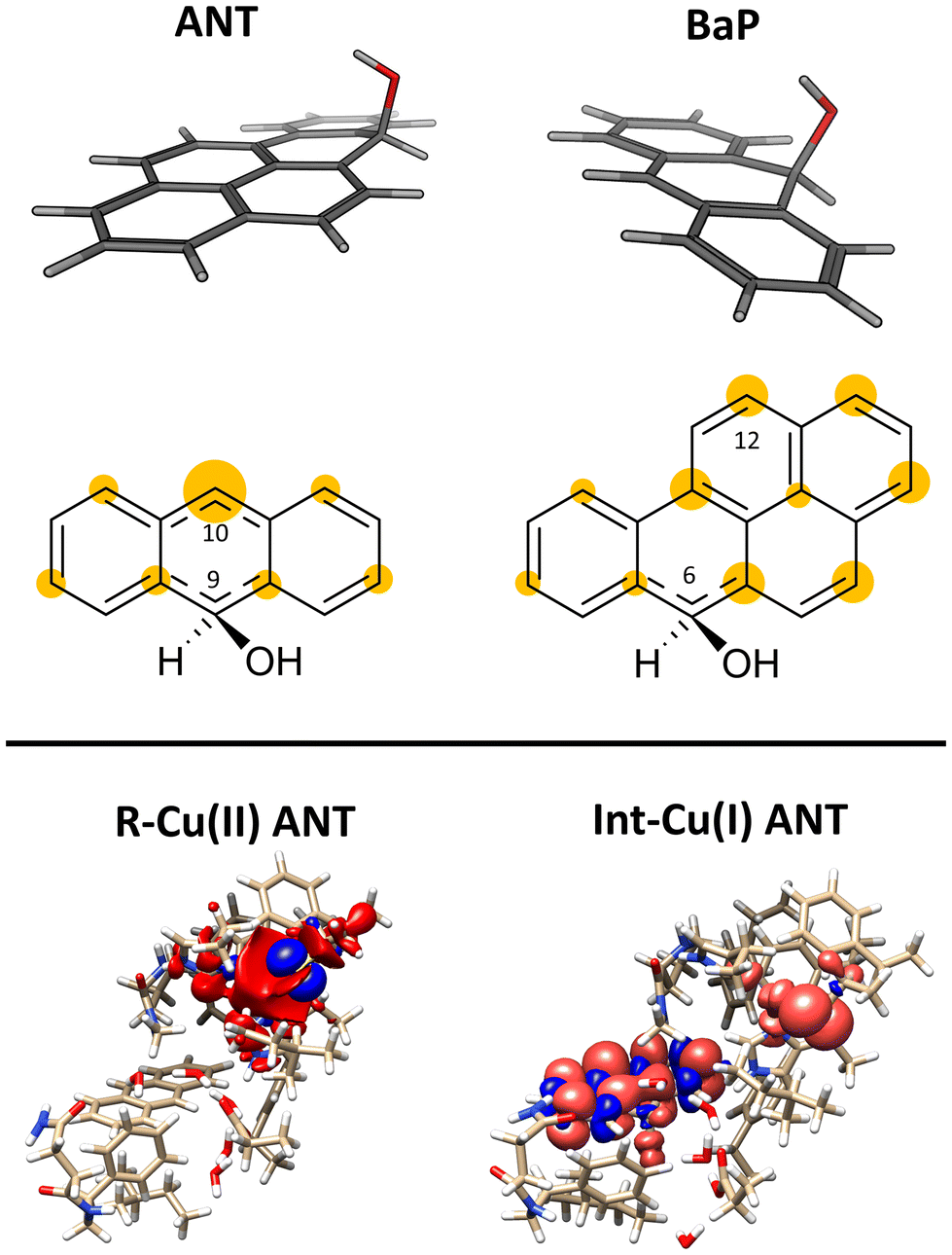 | ||
| Fig. 7 On the top: molecular shape and below spin populations of mono-oxidized ligand in the Int–Cu(I) intermediate for ANT and BaP. The highlighted areas in yellow on the carbon atoms are qualitatively proportional to the PBE0 spin population on that nucleus. On the bottom the total spin density for R–Cu(II) and Int–Cu(I) for ANT computed from the PBE0 level wave function at minimum geometry. | ||
The structures of the TS1 transition states (see Fig. S5† for details) for both substrates exhibit remarkable similarity, with their electronic structure being defined by a copper spin population, enabling us to attribute the redox state to Cu1.5.
The reaction profiles for ANT and BaP during the first ET exhibit remarkable similarity. The values show minimal deviation between BP86 to PBE0 results (Tables S3 and S4†). On average, the estimated activation energy remains constant at around 30 kcal mol−1, which is indicative of a kinetically impeded process. This result suggests that, even invoking the most likely mechanism among those investigated, PAH first oxidation is not effective.
At this stage, the model exhibits one fewer water molecule due to the dissociation of W4 (one proton migrates to the Asp206 side chain, while the OH– ion is now bound to the substrate). The second substrate oxidation requires the restoring of a Cu(II) center, with an electron transfer towards the TNC from the reduced T1 Cu(I), and the deprotonation of the Asp206 side chain. This step was modeled simply by removing one electron from the system and the proton from the Asp206 side chain, and finally re-optimizing the geometry. The resulting structure (Int–Cu(II)) is a S = 0 broken symmetry solution, in which one unpaired electron is localized on T1 Cu(II) and the other on the mono-oxidized substrate. The second 1e oxidation of the substrate, shown in Fig. 6, involves a hydrogen atom abstraction from the C–H bond of the tetrahedral carbon atom of the radical species during which we observe the:
(1) reduction of T1 Cu;
(2) proton transfer from the substrate to the Asp206 side chain via the W5 water molecule;
(3) formation of the hydroxy derivative of the substrate.
Again, we evaluated the activation energy of this step for both substrates by scanning the PES of each system along the reaction coordinate that identifies the overall process (Fig. S6 and S7†). During the second electron transfer process, the activation energy barrier for ANT, albeit slightly, is lowered by 2.1 kcal mol−1 compared to that computed for BaP. Furthermore, the reaction energy for ANT is significantly more negative, surpassing that of BaP by as much as 19.3 kcal mol−1.
The overall energy profiles depicted in Fig. 6 are compatible with a kinetically hindered process, since they entail activation barriers as high as 30 kcal mol−1 for both BaP and ANT. In general, extended cluster models such as those adopted here are inherently more precise in the description of the real system, encompassing first- and second-coordination sphere effects.60 Nevertheless, large models with a consistent number of constraints may result, in some cases, in artifactual rigidity during geometry optimizations, which could result in slightly overestimated energy barriers. In any case, even accounting for hypothetical overestimation of barriers, our calculations clearly suggest that direct PAH oxidation (i.e. in the absence of mediators) should be minimal. This agrees with the observation made by Johannes and Majcherczyk34 that the initial oxidation step (resulting in the formation of a radical species) is endergonic, as the standard reduction potential of the PAH˙+/PAH pair is more positive than that of T1 Cu, rendering it kinetically unfavorable. Such a process may remain chemically feasible if a subsequent irreversible step removes one of the reaction products, in this case, the reactive intermediate PAH˙+ radical species.
Our results nicely match the low yields reported in the literature for BaP oxidation by TvL (and by other fungal laccases as well, see Table S5†), that indicate that the process is reasonably characterized by high activation barriers approaching the threshold beyond which a reaction becomes kinetically unfeasible. Conversely, on the basis of our energy profiles, we could not rationalize the higher yields observed for ANT oxidation. Consequently, additional remarks are needed, specifically delving into the ANT and BaP oxidation experiments already present in the literature.81 For instance, as it follows from Table S4,† the presence of a surfactant (such as Tween 80) can enhance ANT oxidation yields by TvL and other laccases. This is intriguing in light of the recent suggestion by Yang et al.80 that Tween 80 may also act as a mediator, facilitating substrate oxidation. It is thus possible to suppose that in some cases, depending on different combinations of substrate nature and experimental conditions, oxidation is favored not so much by an effective positioning of the substrate in the ligand-binding pocket, but by the presence of the surfactant, which may act as a mediator.
Conclusions
This investigation provides a deeper understanding of the oxidation process of ANT and BaP by the T1 Cu catalytic site of TvL, elucidating the intricate molecular mechanisms involved. To date, most of the computational investigations on laccases have focused on T2/T3 reactivity7,52,79 rather than on that of T1. Furthermore, mechanisms for substrate oxidation have been in general unraveled without modeling its interaction with the enzyme pocket. In this scenario, our work represents a geminal study also providing, for the first time, hints on the nature of aromatic hydrocarbon oxidation by T1 Cu. The present investigation is articulated on different levels of computational techniques, starting from molecular docking and ending up with DFT calculations on an extended T1 Cu model. In a first stage, molecular docking was used to analyze the enzyme–PAHs interaction within the T1 Cu pocket. It revealed that both ANT and BaP can bind to the enzyme's binding pocket with comparable affinity, albeit slightly higher for BaP compared to ANT. Subsequent DFT calculations performed using a minimal cluster model of T1 Cu, allowed us to explore, for the first time, different mechanistic hypotheses previously proposed for the early 1e-oxidation (the most critical step of catalysis) and to pinpoint the most likely one. Our results clearly indicate that the only thermodynamically feasible mechanistic route requires the involvement of a water molecule that acts as a nucleophile towards the oxidized carbon of the ligand. The involvement of a water molecule is also supported by MD simulations, revealing a persistent presence of water in the T1 Cu pocket, between copper and ligand. Then, the kinetic accessibility of such a reactive pathway has been assessed by switching to an extended DFT model, that allowed us to provide full energy profiles for the whole 2e-oxidation of BaP and ANT. In the absence of mediators, oxidation of both substrates requires high activation barriers and no energetic preference has emerged towards either of the two substrates. In light of these results, there arises the necessity for future investigations regarding the specific role of mediators of different natures in the oxidative process, with a focused outlook on molecular details. Indeed, this aspect is still lacking in the literature, and the present computational study lays the groundwork for more in-depth developments on the oxidation of mediators by T1 Cu, and the subsequent electron transfer to various PAHs. Finally, the present work provides new insights on the C–H oxidation mechanism by laccases, which can be generalized inspiring future theoretical/experimental investigations focused on hydrocarbons of different nature, such as other aromatic pollutants or plastics.82Author contributions
C. O. and I. C. R. contributed equally. C. O., I. C. R., L. B. performed the majority of the computations; F. A., M. M., J. Z. performed the formal analysis and visualization; The manuscript was drafted by L. B. and F. A.; Conceptualization, resources and supervision by C. G., T. M., L. D. G., M. L., P. D. G., L. B.Data availability
The data that support the findings of this study are available on request from the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
For DFT calculations, the authors acknowledge CINECA for the availability of high-performance computing resources as part of the agreement with the University of Milano-Bicocca. This work was supported by Italian MUR, Project SEA-WAVE 2020BKK3W9, [CUP_E69J22001140005].References
- P. S. Chauhan, B. Goradia and A. Saxena, 3 Biotech, 2017, 7, 323 CrossRef PubMed.
- P. Baldrian, FEMS Microbiol. Rev., 2006, 30, 215–242 CrossRef CAS PubMed.
- J. Zeng, X. Lin, J. Zhang, X. Li and M. H. Wong, Appl. Microbiol. Biotechnol., 2011, 89, 1841–1849 CrossRef CAS PubMed.
- J. Zeng, Q. Zhu, Y. Wu and X. Lin, Chemosphere, 2016, 148, 1–7 CrossRef CAS PubMed.
- M. Loi, O. Glazunova, T. Fedorova, A. F. Logrieco and G. Mulè, J. Fungi, 2021, 7, 1048 CrossRef CAS PubMed.
- P. Aza and S. Camarero, Biomolecules, 2023, 13, 1716 CrossRef CAS PubMed.
- D. Singh and N. Gupta, Biologia, 2020, 75, 1183–1193 CrossRef CAS.
- A. B. da Rocha, R. de Aquino Saraiva, V. M. de Siqueira, G. T. Yogui, R. de Souza Bezerra, C. R. D. de Assis, M. S. B. Sousa and D. de Souza Buarque, Mar. Pollut. Bull., 2023, 194, 115445 CrossRef CAS PubMed.
- S. M. Jones and E. I. Solomon, Cell. Mol. Life Sci., 2015, 72, 869–883 CrossRef CAS PubMed.
- Y. Zhu, Y. Zhang, J. Zhan, Y. Lin and X. Yang, FEBS Open Bio, 2019, 9, 986–995 CrossRef CAS PubMed.
- A. C. Mot and R. Silaghi-Dumitrescu, Biochemistry, 2012, 77, 1395–1407 CAS.
- E. I. Solomon, A. J. Augustine and J. Yoon, Dalton Trans., 2008, 3921–3932 RSC.
- L. Arregui, M. Ayala, X. Gómez-Gil, G. Gutiérrez-Soto, C. E. Hernández-Luna, M. Herrera de Los Santos, L. Levin, A. Rojo-Domínguez, D. Romero-Martínez, M. C. N. Saparrat, M. A. Trujillo-Roldán and N. A. Valdez-Cruz, Microb. Cell Fact., 2019, 18, 200 CrossRef CAS PubMed.
- J. Yang, X. Yang, Y. Lin, T. B. Ng, J. Lin and X. Ye, PLoS One, 2015, 10, e0127714 CrossRef PubMed.
- J. Jeyabalan, A. Veluchamy, V. Priyan, A. Kumar, R. Chandrasekar and S. Narayanasamy, J. Taiwan Inst. Chem. Eng., 2023, 151, 105081 CrossRef CAS.
- M. Frasconi, G. Favero, H. Boer, A. Koivula and F. Mazzei, Biochim. Biophys. Acta, 2010, 1804, 899–908 CrossRef CAS PubMed.
- M. Santo, R. Weitsman and A. Sivan, Int. Biodeterior. Biodegrad., 2013, 84, 204–210 CrossRef CAS.
- J. Zampolli, M. Mangiagalli, D. Vezzini, M. Lasagni, D. Ami, A. Natalello, F. Arrigoni, L. Bertini, M. Lotti and P. Di Gennaro, Environ. Technol. Innovation, 2023, 32, 103273 CrossRef CAS.
- P. Astolfi, P. Brandi, C. Galli, P. Gentili, M. F. Gerini, L. Greci and O. Lanzalunga, New J. Chem., 2005, 29, 1308 RSC.
- J.-A. Majeau, S. K. Brar and R. D. Tyagi, Bioresour. Technol., 2010, 101, 2331–2350 CrossRef CAS PubMed.
- M. A. Tadesse, A. D'Annibale, C. Galli, P. Gentili and F. Sergi, Org. Biomol. Chem., 2008, 6, 868–878 RSC.
- C. Galli, C. Madzak, R. Vadalà, C. Jolivalt and P. Gentili, ChemBioChem, 2013, 14, 2500–2505 CrossRef CAS PubMed.
- E. Monza, M. F. Lucas, S. Camarero, L. C. Alejaldre, A. T. Martínez and V. Guallar, J. Phys. Chem. Lett., 2015, 6, 1447–1453 CrossRef CAS PubMed.
- I. Pardo, G. Santiago, P. Gentili, F. Lucas, E. Monza, F. J. Medrano, C. Galli, A. T. Martínez, V. Guallar and S. Camarero, Catal. Sci. Technol., 2016, 6, 3900–3910 RSC.
- M. F. Lucas, E. Monza, L. J. Jørgensen, H. A. Ernst, K. Piontek, M. J. Bjerrum, Á. T. Martinez, S. Camarero and V. Guallar, J. Chem. Theory Comput., 2017, 13, 1462–1467 CrossRef CAS PubMed.
- R. Mehra and K. P. Kepp, Phys. Chem. Chem. Phys., 2019, 21, 15805–15814 RSC.
- N. Premnath, K. Mohanrasu, R. Guru Raj Rao, G. H. Dinesh, G. S. Prakash, V. Ananthi, K. Ponnuchamy, G. Muthusamy and A. Arun, Chemosphere, 2021, 280, 130608 CrossRef CAS PubMed.
- T. Kadri, T. Rouissi, S. Kaur Brar, M. Cledon, S. Sarma and M. Verma, J. Environ. Sci., 2017, 51, 52–74 CrossRef CAS PubMed.
- P. J. Collins, M. Kotterman, J. A. Field and A. Dobson, Appl. Environ. Microbiol., 1996, 62, 4563–4567 CrossRef CAS PubMed.
- C. Johannes, A. Majcherczyk and A. Hüttermann, Appl. Microbiol. Biotechnol., 1996, 46, 313–317 CrossRef CAS PubMed.
- L. Bezalel, Y. Hadar and C. E. Cerniglia, Appl. Environ. Microbiol., 1996, 62, 292–295 CrossRef CAS PubMed.
- M. A. Pickard, R. Roman, R. Tinoco and R. Vazquez-Duhalt, Appl. Environ. Microbiol., 1999, 65, 3805–3809 CrossRef CAS PubMed.
- A. Majcherczyk, C. Johannes and A. Hüttermann, Enzyme Microb. Technol., 1998, 22, 335–341 CrossRef CAS.
- C. Johannes and A. Majcherczyk, Appl. Environ. Microbiol., 2000, 66, 524–528 CrossRef CAS PubMed.
- N. N. Pozdnyakova, J. Rodakiewicz-Nowak, O. V. Turkovskaya and J. Haber, Enzyme Microb. Technol., 2006, 39, 1242–1249 CrossRef CAS.
- A. I. Cañas, M. Alcalde, F. Plou, M. J. Martínez, A. T. Martínez and S. Camarero, Environ. Sci. Technol., 2007, 41, 2964–2971 CrossRef PubMed.
- S. Camarero, A. I. Cañas, P. Nousiainen, E. Record, A. Lomascolo, M. J. Martínez and A. T. Martínez, Environ. Sci. Technol., 2008, 42, 6703–6709 CrossRef CAS PubMed.
- M. T. Cambria, Z. Minniti, V. Librando and A. Cambria, Appl. Biochem. Biotechnol., 2008, 149, 1–8 CrossRef CAS PubMed.
- J. Zeng, Q. Zhu, Y. Wu, J. Shan, R. Ji and X. Lin, Environ. Pollut., 2018, 242, 462–469 CrossRef CAS PubMed.
- D. E. Dodor, H.-M. Hwang and S. I. N. Ekunwe, Enzyme Microb. Technol., 2004, 35, 210–217 CrossRef CAS.
- X. Hu, P. Wang and H.-M. Hwang, Bioresour. Technol., 2009, 100, 4963–4968 CrossRef CAS PubMed.
- L. F. Bautista, G. Morales and R. Sanz, Chemosphere, 2015, 136, 273–280 CrossRef CAS PubMed.
- S.-J. Cho, S. J. Park, J.-S. Lim, Y. H. Rhee and K.-S. Shin, Biotechnol. Lett., 2002, 24, 1337–1340 CrossRef CAS.
- A. M. Farnet, G. Gil, F. Ruaudel, A. C. Chevremont and E. Ferre, Geoderma, 2009, 149, 267–271 CrossRef CAS.
- Z. Vipotnik, M. Michelin and T. Tavares, Environ. Technol. Innovation, 2022, 28, 102737 CrossRef CAS.
- L. Wang, Y. Tan, S. Sun, L. Zhou, G. Wu, Y. Shao, M. Wang and Z. Xin, Biology, 2022, 11, 1129 CrossRef CAS PubMed.
- Y. Li, H. Zhao, L. Wang, Y. Bai, T. Tang, H. Liang and D. Gao, Environ. Technol., 2023, 1–12 Search PubMed.
- A. D. Behera, N. Chittoria, S. Kumari, S. Chatterjee and S. Das, Geomicrobiol. J., 2023, 1–13 Search PubMed.
- S. Humel, B. Führer, M. Svetitsch, P. Mayer and A. P. Loibner, J. Hazard. Mater., 2023, 443, 130286 CrossRef CAS PubMed.
- I. Sorrentino, M. Carrière, H. Jamet, I. Stanzione, A. Piscitelli, P. Giardina and A. Le Goff, Analyst, 2022, 147, 897–904 RSC.
- C. Bueno-Nieto, R. Cortés-Antiquera, G. Espina, J. Atalah, J. Villanueva, C. Aliaga, G. E. Zuñiga and J. M. Blamey, Catalysts, 2023, 13, 763 CrossRef CAS.
- Z.-B. Guan, Q. Luo, H.-R. Wang, Y. Chen and X.-R. Liao, Cell. Mol. Life Sci., 2018, 75, 3569–3592 CrossRef CAS PubMed.
- M. B. Agustin, D. M. de Carvalho, M. H. Lahtinen, K. Hilden, T. Lundell and K. S. Mikkonen, ChemSusChem, 2021, 14, 4615–4635 CrossRef CAS PubMed.
- R. Farran, Y. Mekmouche, N. T. Vo, C. Herrero, A. Quaranta, M. Sircoglou, F. Banse, P. Rousselot-Pailley, A. J. Simaan, A. Aukauloo, T. Tron and W. Leibl, iScience, 2021, 24, 102378 CrossRef CAS PubMed.
- D. M. Mate and M. Alcalde, Biotechnol. Adv., 2015, 33, 25–40 CrossRef CAS PubMed.
- L. C. Sander and S. A. Wise, Polycyclic Aromatic Hydrocarbon Structure Index, 1997 Search PubMed.
- H. Lund, C. Tegnér and B. Takman, Acta Chem. Scand., 1957, 11, 1323–1330 CrossRef CAS.
- A. P. Davis and A. J. Fry, J. Phys. Chem. A, 2010, 114, 12299–12304 CrossRef CAS PubMed.
- X. Sheng and F. Himo, Acc. Chem. Res., 2023, 56, 938–947 CrossRef CAS PubMed.
- F. Himo and S. P. de Visser, Commun. Chem., 2022, 5, 29 CrossRef PubMed.
- T. Bertrand, C. Jolivalt, P. Briozzo, E. Caminade, N. Joly, C. Madzak and C. Mougin, Biochemistry, 2002, 41, 7325–7333 CrossRef CAS PubMed.
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- C. Adamo, M. Cossi and V. Barone, J. Mol. Struct.: THEOCHEM, 1999, 493, 145–157 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- L. Bertini, M. Bruschi, M. Romaniello, G. Zampella, M. Tiberti, V. Barbieri, C. Greco, D. La Mendola, R. P. Bonomo, P. Fantucci and L. De Gioia, in Highlights in Theoretical Chemistry, 2012, pp. 255–269 Search PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- F. Arrigoni, F. Rizza, R. Tisi, L. De Gioia, G. Zampella and L. Bertini, Metallomics, 2020, 12, 1765–1780 CrossRef CAS PubMed.
- A. Rovetta, L. Carosella, F. Arrigoni, J. Vertemara, L. De Gioia, G. Zampella and L. Bertini, Inorganics, 2023, 11, 227 CrossRef CAS.
- F. Pirro, S. La Gatta, F. Arrigoni, A. Famulari, O. Maglio, P. Del Vecchio, M. Chiesa, L. De Gioia, L. Bertini, M. Chino, F. Nastri and A. Lombardi, Angew. Chem., 2023, 135, e202211552 CrossRef.
- S. Sen, P. Bhojnagarwala, L. Francey, D. Lu, T. M. Penning and J. Field, Chem. Res. Toxicol., 2012, 25, 2117–2126 Search PubMed.
- A. Singha, A. Sekretareva, L. Tao, H. Lim, Y. Ha, A. Braun, S. M. Jones, B. Hedman, K. O. Hodgson, R. D. Britt, D. J. Kosman and E. I. Solomon, J. Am. Chem. Soc., 2023, 145, 13284–13301 CrossRef CAS PubMed.
- D. W. Randall, D. R. Gamelin, L. B. LaCroix and E. I. Solomon, J. Biol. Inorg. Chem., 2000, 5, 16–29 CrossRef CAS PubMed.
- H. Tachikawa, J. Phys. Chem. A, 2018, 122, 4121–4129 CrossRef CAS PubMed.
- U. Ryde, M. H. Olsson, K. Pierloot and B. O. Roos, J. Mol. Biol., 1996, 261, 586–596 CrossRef CAS PubMed.
- Z. Yang, X. Mao, J. Cui, Y. Wang and Y. Zhang, Sci. Rep., 2021, 11, 13121 CrossRef CAS PubMed.
- N. N. Pozdnyakova, J. Rodakiewicz-Nowak and O. V. Turkovskaya, J. Mol. Catal. B: Enzym., 2004, 30, 19–24 CrossRef CAS.
- A. Rovaletti, L. De Gioia, P. Fantucci, C. Greco, J. Vertemara, G. Zampella, F. Arrigoni and L. Bertini, Int. J. Mol. Sci., 2023, 24, 6368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt01377h |
| This journal is © The Royal Society of Chemistry 2024 |