
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
P(V)-bis(amidophenolate) ligand cooperation: stoichiometric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O-bond cleavage in aldehydes and ketones†
O-bond cleavage in aldehydes and ketones†
Simon B. H.
Karnbrock
,
Christopher
Golz
and
Manuel
Alcarazo
 *
*
Institute of Organic and Biomolecular Chemistry, University of Göttingen, Tammannstraße 2, 37077 Göttingen, Germany. E-mail: manuel.alcarazo@chemie.uni-goettingen.de
First published on 5th June 2024
Abstract
The cooperation between a geometrically constrained, highly electrophilic phosphorus(V) center, and an electronically rich tetradentate bis(amidophenolate) ligand enables the cleavage of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond from typical aldehydes and ketones delivering iminio phosphoramidate species. The amphiphilic nature of these products, which is demonstrated through their reaction with typical Lewis acids and bases, enables their use as a mild source of silylium cations from silanes, allowing the selective reductive coupling of aldehydes to ethers under catalytic conditions.
O bond from typical aldehydes and ketones delivering iminio phosphoramidate species. The amphiphilic nature of these products, which is demonstrated through their reaction with typical Lewis acids and bases, enables their use as a mild source of silylium cations from silanes, allowing the selective reductive coupling of aldehydes to ethers under catalytic conditions.
The design of catalysts that strategically make use of metal–ligand cooperativity as a tool to facilitate the bond-breaking/forming steps at the substrate during a catalytic cycle has become increasingly popular. Among other advantages, these catalysts often serve as environmentally benign alternatives to previously developed systems in which a possible active role of the ligand was not considered.1 However, and despite that undeniable impact, the exploitation of cooperativity in homogeneous catalysis has been mostly limited to transition metal-mediated processes; the transfer of such designing principles to p-block element-based catalysis is, in comparison, underdeveloped.2 Remarkable exceptions are Radosevich catalytic hydrogenations and hydroborations using geometrically distorted phosphoramidites and phosphorus triamides,3 the hydroboration of aldehydes and ketones mediated by N-heterocyclic germylenes,4 and the digallane-catalysed hydroamination of alkynes introduced by Fedushkin,5 among others.6
Cooperative behaviour between p-block elements and their surrounding ligands is more commonly found in stoichiometric transformations, which certainly have reduced synthetic utility, but serve to explore non-obvious modes of substrate activation and provide models for the development of new catalytic processes.2 In this regard, and due to their ubiquity, the activation of carbonyl moieties is very appealing. Numerous reports evidence that aldehydes or ketones readily bind to amphiphilic p-block complexes with concomitant cleavage of the C–O π-bond. For example, Erker described the reaction of a geminal alkylidene-bridged phosphine/borane Lewis pair with benzaldehyde to deliver compound A (Scheme 1b);7 Wu, Liu and Zhao demonstrated that benzaldehyde cooperatively binds to Driess’ N-heterocyclic germylene4 to yield the corresponding adduct B,8 and Greb and co-workers assessed the reactivity of a square planar aluminate towards aldehydes. Remarkably, the formation of adduct C is reversible and controllable by external stimuli.6c
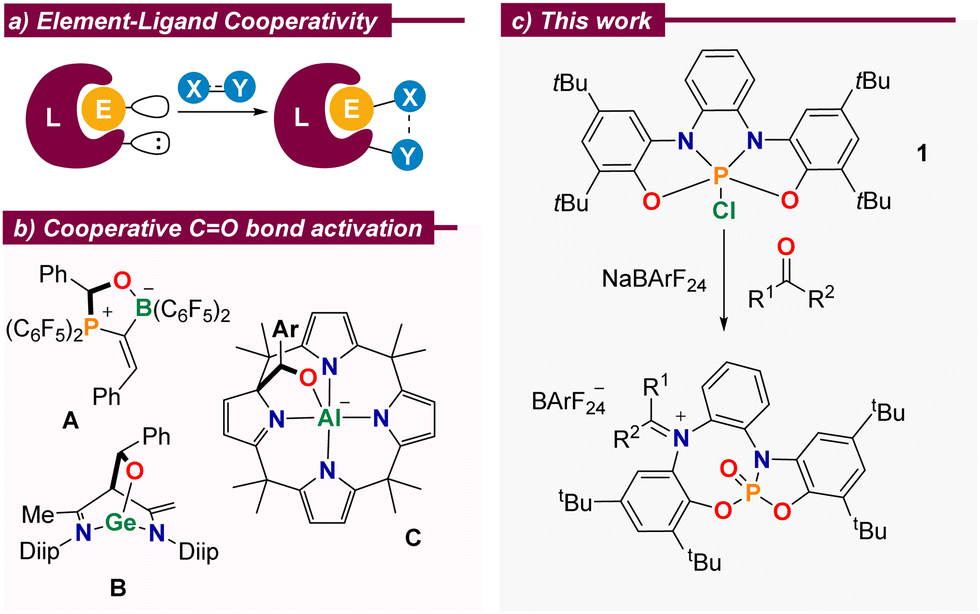 | ||
| Scheme 1 (a) Concept of element–ligand cooperativity; (b) selected literature examples; (c) CO bond cleavage in aldehydes and ketones. | ||
We recently showed that the embedment of a highly electrophilic P(V) centre into an electron-rich, redox non-innocent, tetradentate bis(amidophenolato) scaffold results in phosphorane 1. Once activated by chloride abstraction, this species is able to catalyse the disproportionation of 1,2-diphenylhydrazine through a mechanism that makes use of the electron reservoir character of the ligand.9 These structural features, namely strong Lewis acidity at the central P(V)-centre and a high-lying occupied molecular orbital in spatial proximity, qualifies the system for further modes of element–ligand cooperativity.2 Herein, we report our studies on the reaction of 1 towards aldehydes and ketones of different structure, the evaluation of the reactivity of the iminiophosphoramidate products obtained, and their subsequent use as promotors for the reductive etherification of aldehydes.
At the outset of our study, chlorophosphorane 19a was treated with acetone in the presence of Na[B(C6H3(CF3)2)4] (NaBArF24) as a chloride anion abstractor. The 1H NMR spectrum of the crude reaction mixture clearly indicated loss of the original CS-symmetry of 1 and the splitting of the acetone methyl groups into two singlets at 2.67 and 2.54 ppm, which suggested the involvement of the bis(amidophenolate) sidearms in the transformation. Moreover, the 31P NMR spectrum of the same reaction depicted a well-defined signal at −6.1 ppm, indicating the formation of a P(V)O moiety.10 Finally, compound 2 was isolated in a good yield by simple extraction from the reaction mixture with CH2Cl2 and subsequent washing with pentane; its connectivity as an iminiophosphoramidate was unambiguously determined by X-ray crystallography (Scheme 2).
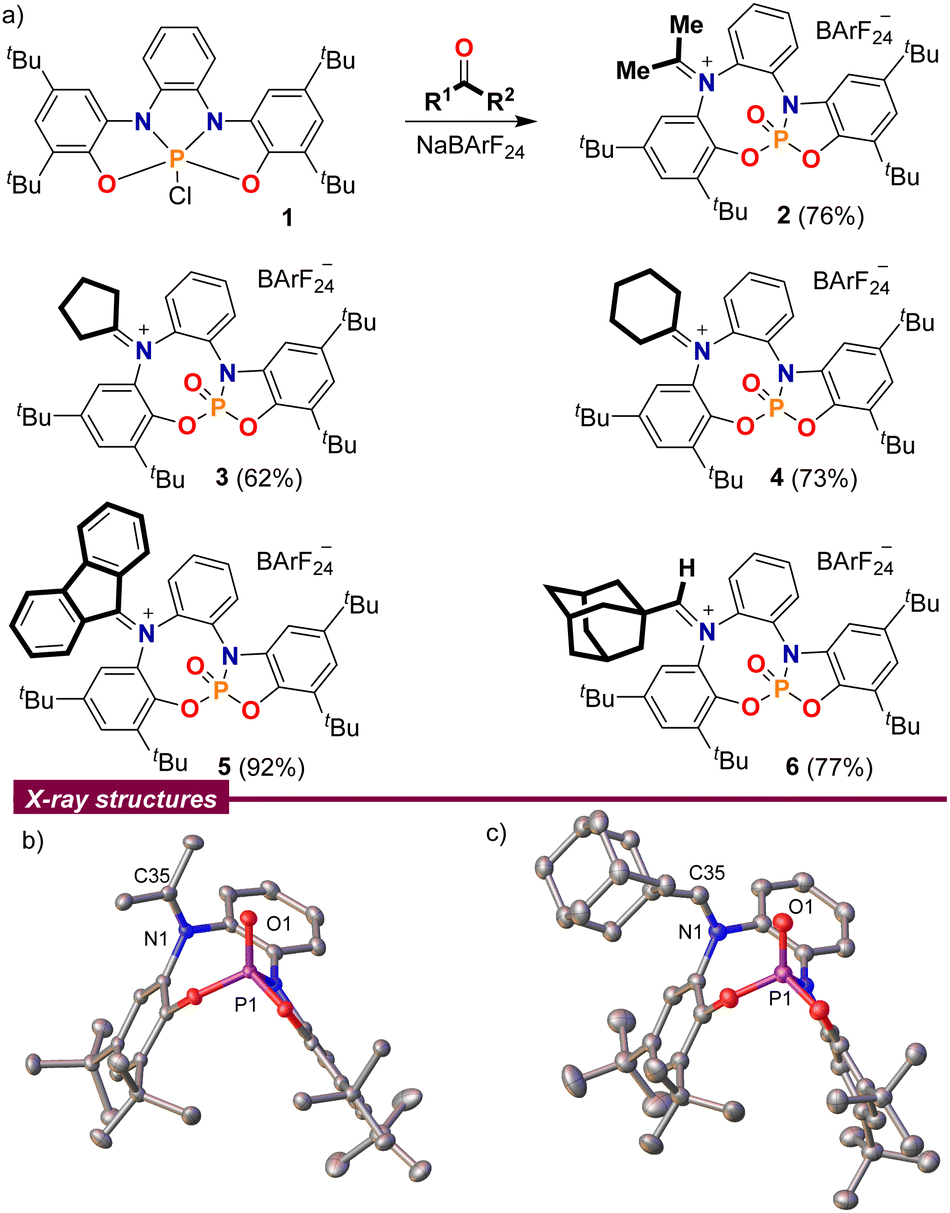 | ||
| Scheme 2 (a) Reaction of ketones and aldehydes with chlorophosphorane 1; (b) and (c) solid-state molecular structures of 2 (left) and 6 (right). Hydrogen atoms, BArF anions and solvent molecules omitted for clarity. Ellipsoids are set at the 50% probability level.11 | ||
This reaction is not limited to acetone. Other aliphatic and aromatic ketones such as cyclopentanone, cyclohexanone and fluorenone were also found to be suitable substrates 3–5; but unfortunately, non-symmetric ketones invariably delivered non-separable E/Z-product mixtures. 1-Adamantyl carbaldehyde also delivers a stable adduct 6, which was isolated as a pure (Z)-isomer after recrystallization from hot toluene, but the iminiophosphoramidates derived from less sterically demanding aldehydes were all prone to decompose slowly.
A closer look into the X-ray structures of the carbonyl adducts 2–6 is quite informative (Scheme 2b and c for compounds 2 and 6, and Fig. S2–S4 for 3–5, ESI†). The C–N bond lengths are in all adducts within the typical range for iminium moieties,12 and only vary slightly depending on the substituents at the carbon atom. Thus, the shortest C–N distance is observed for the aldiminium-derivative 6 (N1–C35; 1.297(2) Å) while the longest is found for the π-conjugated fluorenone derivative 5 (N1–C35; 1.3206(13) Å). A recurring feature observed throughout all the molecular structures is the close spatial proximity between the iminium carbon C35 and O1, with values ranging from 2.7028(14) Å in 4 to 3.0263(13) Å in 5, all shorter than the sum of their van der Waals radii.13 This is probably a consequence of the sum of two factors; namely, the structural constraints imposed by the rigid bis(amidophenolato) scaffold, and the polarity match between the electrophilic iminium carbon and the Lewis basic oxygen atom from the P(V)O moiety.
In order to gain a deeper understanding of the mechanism leading to the formation of 2–6, electronic structure calculations were carried out at the B3LYP-D3(BJ)/def2-TZVP//PBE-D3(BJ)/def-2SVP level of theory to evaluate the feasibility of the critical steps.14Fig. 1a shows the computed Gibbs free energies for intermediates and transition states using acetone as a model substrate. Once the chloride anion has been replaced by acetone Int I, an amino moiety of the ligand attacks the electrophilic carbonyl carbon of the coordinated acetone delivering oxazaphosphetane Int IIvia transition state TSI. This cooperative pathway requires a moderate Gibbs activation energy (ΔG‡) of 11.4 kcal mol−1. From Int I, cleavage of the C–O σ-bond proceeds with an even lower barrier (4.8 kcal mol−1) through TS2, which resembles the [2+2]-cycloreversion step of an aza-Wittig reaction.15 The formation of a strong P–O bond in Int III offers a qualitative explanation for the exergonic nature of this transformation.
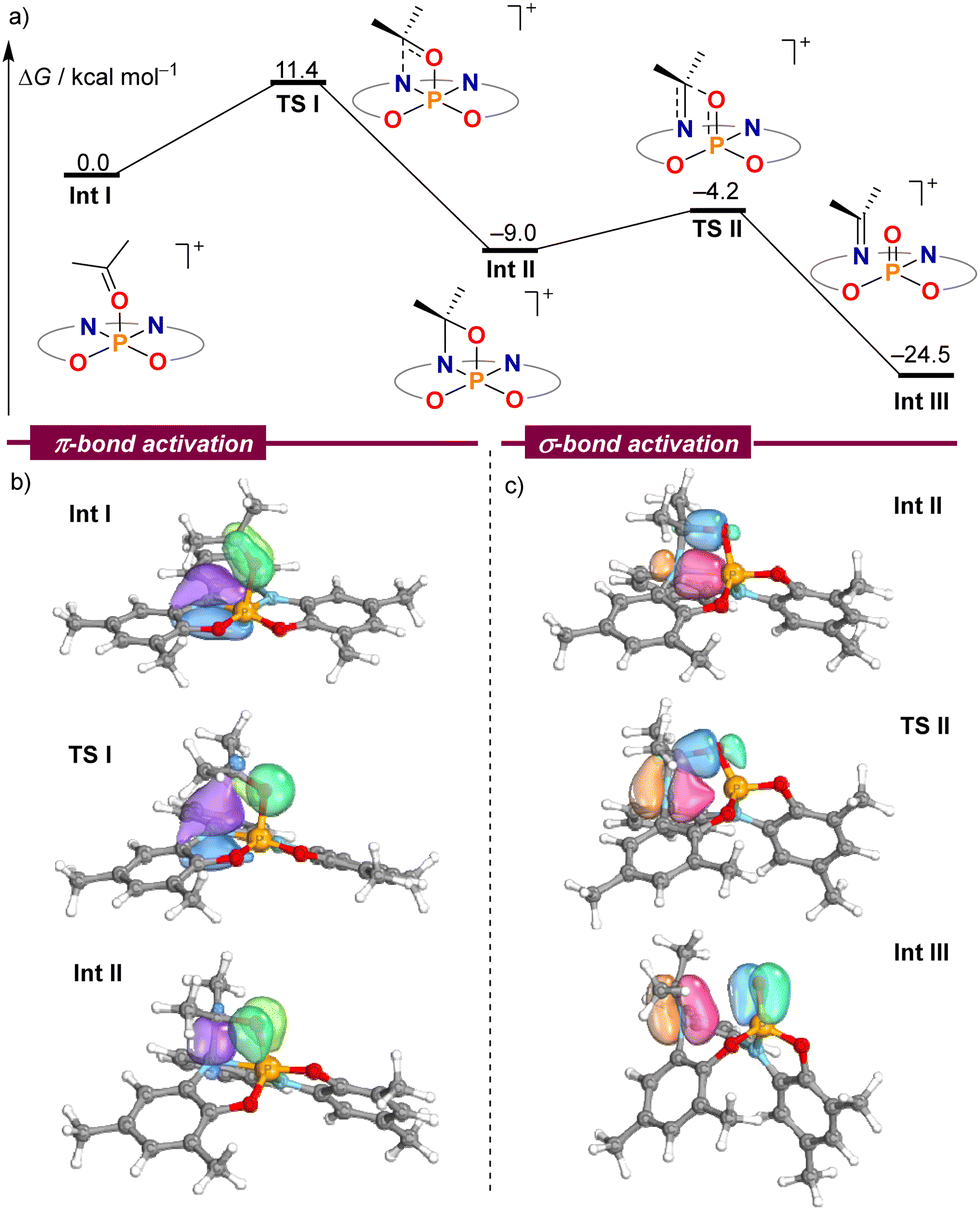 | ||
| Fig. 1 (a) DFT calculations at the B3LYP-D3(BJ)/def2-TZVP//PBE-D3(BJ)/def2-SVP level on the mechanism of the CO bond cleavage. (b) and (c) Selected IBO plots through the reaction coordinate. | ||
The analysis of electron flow for this reaction pathway using intrinsic bond orbitals (IBOs) beautifully pinpoints the phosphorus–ligand cooperative action.16 On the way to Int II the nitrogen lone pair attacks the carbonyl group to form the σ(N–C) bond of the oxazaphosphetane moiety, while simultaneously, the initial π(CO) bond is transformed into an oxygen-centred lone pair, thus breaking the π(CO) interaction (Fig. 1b). Subsequently, the dative σ(N–P) bond in Int II slowly breaks, giving rise to the π(N–C) bond in 2, while at the same time the σ(C–O) bond in Int II dissipates giving rise to the formation of the π(PO) interaction (Fig. 1c).16
Based on the spatial proximity of non-quenched Lewis acid and base sites in 2–6, we speculated that these species might exhibit amphiphilic reactivity. Hence, using 6 as a model substrate, we initially treated this compound with an archetypical Lewis base, 4-dimethylaminopyridine (DMAP). The expected attack at the iminium functionality took place and adduct 7 could be isolated by crystallization (Scheme 3a). Contrarily, the reaction of 6 with GaCl3 proved to be more complex. Instead of mere coordination of the Lewis acidic Ga-centre to O1, degradation of the BArF24 counterion was observed, and 8 crystallized as a [GaCl4]− salt. By employing three equivalents of GaCl3, we were able to obtain 8 in 46% yield as an orange crystalline solid. As expected, by formation of 7 the C35–N1 bond distance elongates from 1.297(2) Å in 6 to 1.487(2) Å, while in 8 it is the P1–O1 distance that gets elongated as a result of coordination to the Lewis acid, from 1.4545(14) Å in 6 to 1.4878(10) Å in 8. It is worth noting that the weak pyramidalization observed at the Ga centre (∑∠Ga = 345.8°) is indicative of a limited donor ability of 6.17
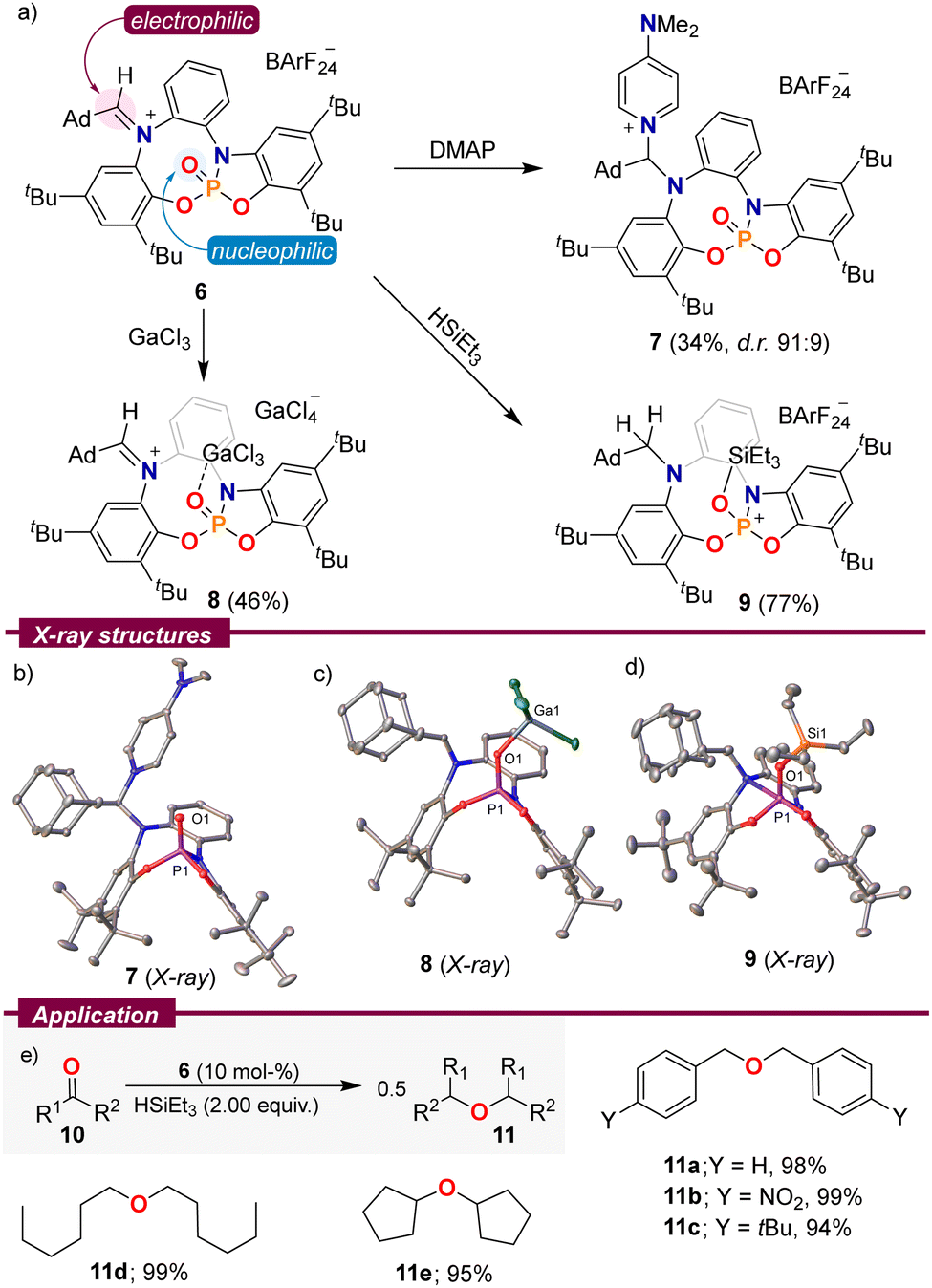 | ||
| Scheme 3 (a) Reactivity of 6; (b)–(d) solid-state molecular structures of 7–9. Hydrogen atoms, anions and solvent molecules in the solid-state structures were omitted. Ellipsoids are set at 50% probability;11 (e) reductive etherification of aldehydes and ketones. Yields were determined by quantitative NMR spectroscopy using hexamethylbenzene as an internal standard. | ||
Interestingly, both reactivity modes can be observed upon the addition of triethyl silane to a CDCl3 solution of 6. The aldiminium resonance at 8.38 ppm in the 1H NMR spectrum slowly disappears while two new signals at 3.66 and 3.25 ppm with a geminal 2JHH = 13.4 Hz arise, in agreement with the reduction of the iminium functionality. Simultaneously, a doublet in the 29Si-INEPT spectrum resulting from 2JPSi coupling verified binding of the silylium fragment to the PO moiety. On a preparative scale, 9 was isolated in a good yield of 77% and single crystals suitable for X-ray diffraction were grown by vapor diffusion of pentane into a toluene solution of 9. The solid-state structure confirms the proposed connectivity. Compound 9 depicts an exceptionally long O1–Si1 bond (1.7301(11) Å),15,18 which is indicative of the low basicity of the PO fragment in 6.
As a result of that weak O–Si interaction, it was rationalized that 9 might serve as a mild source of triethylsilylium cations, and therefore, it could be used as a pre-catalyst for transformations in which that cation plays an active role. Hence, we evaluated the ability of 6 to catalyse the reductive etherification of aldehydes and ketones to ethers using Et3SiH as a reductant.19 The reaction proved general, and benzaldehyde derivatives of different electronic properties 10a–c, aliphatic aldehydes 10d, and even cyclopentanone were selectively transformed into the corresponding symmetrical ethers in excellent yields (Scheme 3e).
In summary, we report the cleavage of the CO bond in aldehydes and ketones through the cooperation between a central P(V)-atom and its surrounding tetradentate ligand. This behaviour can be rationalized considering the high electrophilicity at the P-centre, which is a consequence of the structural constraints imposed by the multidentate bis(amidophenolate) scaffold, and the electron richness of that ligand. A preliminary application of the iminiophosphoramidate species such obtained as promotors for the reductive etherification of aldehydes and ketones is also described.
S. B. H. K. and M. A. conceived the project and designed the experiments. S. B. H. K. performed the experiments, analysed the results and carried out the computational studies. C. G. did the X-ray crystallographic studies. All authors discussed the results and S. B. H. K. and M. A. prepared the manuscript.
Financial support from the DFG through the projects INST 186/1237-1, INST 186/1318-1 and INST 186/1324-1 is gratefully acknowledged. We also thank the NMR and MS services at the Faculty of Chemistry (University of Göttingen) for technical assistance.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For recent reviews see: (a) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed; (b) P. A. Dub and J. C. Gordon, Nat. Rev. Chem., 2018, 2, 396–408 CrossRef CAS; (c) M. Wodrich and X. Hu, Nat. Rev. Chem., 2018, 2, 0099 CrossRef CAS; (d) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed; (e) T. Higashi, S. Kusumoto and K. Nozaki, Chem. Rev., 2019, 119, 10393–10402 CrossRef CAS PubMed; (f) M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC; (g) T. Shimbayashi and K.-i Fujita, Catalysts, 2020, 10, 635 CrossRef CAS; (h) W. M. Hollinsworth and E. A. Hill, J. Coord. Chem., 2022, 75, 1436–1466 CrossRef; (i) T.-F. Ramspoth, J. Kootstra and S. R. Harutyunyan, Chem. Soc. Rev., 2024, 53, 3216–3223 RSC; (j) S. Becker, ChemPlusChem, 2024, e202300619 CrossRef PubMed.
- For recent reviews see: (a) A. Hanft and C. Lichtenberg, Eur. J. Inorg. Chem., 2018, 3361–3373 CrossRef CAS; (b) Y. Bai, W. Chen, J. Li and C. Cui, Coord. Chem. Rev., 2019, 383, 132–154 CrossRef CAS; (c) I. V. Ershova and A. V. Piskunov, Russ. J. Coord. Chem., 2020, 46, 154–177 CrossRef CAS; (d) I. V. Ershova, A. V. Piskunov and V. K. Cherkasov, Russ. Chem. Rev., 2020, 89, 1157–1183 CrossRef CAS; (e) C. Lichtenberg, Chem. – Eur. J., 2020, 26, 9674–9687 CrossRef CAS PubMed; (f) J. Wang, H. S. Soo and F. Garcia, Commun. Chem., 2020, 3, 113 CrossRef CAS PubMed; (g) R. Zhang, Y. Wang,Y. Zhao, C. Redshaw, I. L. Fedushkin,B. Wu and X. J. Yang, Dalton Trans., 2021, 50, 13634–13650 RSC; (h) L. Greb, F. Ebner, Y. Ginzburg and L. M. Sigmund, Eur. J. Inorg. Chem., 2020, 3030–3047 CrossRef CAS; (i) L. Greb, Eur. J. Inorg. Chem., 2022, e202100871 CrossRef CAS PubMed; (j) S. H. B. Karnbrock and M. Alcarazo, Chem. – Eur. J., 2024, 30, e202302879 CrossRef CAS PubMed.
- (a) N. L. Dunn, M. Ha and A. T. Radosevich, J. Am. Chem. Soc., 2012, 134, 11330–11333 CrossRef CAS PubMed; (b) G. Zeng, S. Maeda, T. Taketsugu and S. Sakaki, Angew. Chem., Int. Ed., 2014, 53, 4633–4637 CrossRef CAS PubMed; (c) W. Zhao, S. M. McCarthy, T. Y. Lai, H. P. Yennawar and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 17634–17644 CrossRef CAS PubMed; (d) Y.-C. Lin, E. Hatzakis, S. M. McCarthy, K. D. Reichl, T.-Y. Lai, H. P. Yennawar and A. T. Radosevich, J. Am. Chem. Soc., 2017, 139, 6008–6016 CrossRef CAS PubMed; (e) J. M. Lipshultz, Y. Fu, P. Liu and A. T. Radosevich, Chem. Sci., 2021, 12, 1031–1037 RSC.
- S. Yao, C. van Wüllen and M. Driess, Chem. Commun., 2008, 5393–5395 RSC.
- (a) I. L. Fedushkin, A. S. Nikipelov and K. A. Lyssenko, J. Am. Chem. Soc., 2010, 132, 7874–7875 CrossRef CAS PubMed; (b) I. L. Fedushkin, A. S. Nikipelov, A. G. Morozov, A. A. Skatova, A. V. Cherkasov and G. Abakumov, Chem. – Eur. J., 2012, 18, 255–266 CrossRef CAS PubMed.
- For selected examples see: (a) W. Myers and L. A. Berben, J. Am. Chem. Soc., 2013, 135, 9988–9990 CrossRef PubMed; (b) L. Fedushkin, M. V. Moskalev, E. V. Baranov and G. A. Abakumov, J. Organomet. Chem., 2013, 747, 235–240 CrossRef; (c) F. Ebner, L. M. Sigmund and L. Greb, Angew. Chem., Int. Ed., 2020, 59, 17118–17124 CrossRef CAS PubMed; (d) F. Schön, L. M. Sigmund, F. Schneider, D. Hartmann, M. A. Wiebe, I. Manners and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202202176 CrossRef PubMed; (e) X. Chen, Y. Yang, H. Wang and Z. Mo, J. Am. Chem. Soc., 2023, 145(12), 7011–7023 CrossRef CAS PubMed.
- C. Rosorius, C. G. Daniliuc, R. Fröhlich, G. Kehr and G. Erker, J. Organomet. Chem., 2013, 744, 149–155 CrossRef CAS.
- Y. Wu, C. Shan, Y. Sun, P. Chen, J. Ying, J. Zhu, L. L. Liu and Y. Zhao, Chem. Commun., 2016, 52, 13799–13802 RSC.
- (a) S. B. H. Karnbrock, C. Golz, R. A. Mata and M. Alcarazo, Angew. Chem., Int. Ed., 2022, 61, e202207450 CrossRef CAS PubMed . The same system was simultaneously studied in: ; (b) S. Volodarsky, I. Malahov, D. Bawari, M. Diab, N. Malik, B. Tumanskii and R. Dobrovetsky, Chem. Sci., 2022, 13, 5957–5963 RSC.
- CRC Handbook of Phosphorus-31 Nuclear Magnetic Resonance Data, ed. J. C. Tebby, CRC Press, Boca Raton, 2018 Search PubMed.
- Deposition numbers 2353322 (2), 2353323 (3), 2353324 (4), 2353325 (5), 2353326 (6), 2353327 (7), 2353328 (8) and 2353329 (9) contain the supplementary crystallographic data for this paper.†.
- (a) A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, Appendix A: Typical Interatomic Distances in Organic Compounds and Organometallic Compounds and Coordination Complexes of the d- and f-block metals, Structure Correlations, ed. H.-B. Burgi and J. D. Dunitz, VCH Publishers, Weinheim, 1994, vol. 2 Search PubMed; (b) F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (c) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed; (d) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC; (e) J. P. Perdew, K. Burke and M. Enzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed; (f) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- L. Kürti and B. Czakó, Strategic Applications of Named Reactions in Organic Synthesis, Elsevier Academic Press, Cambridge, 2005 Search PubMed.
- (a) G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2015, 54, 5518–5522 CrossRef CAS PubMed; (b) G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- A. El-Hellani, J. Monot, S. Tang, R. Guillot, C. Bour and V. Gandon, Inorg. Chem., 2013, 52, 11493–11502 CrossRef CAS PubMed.
- Z. Rappoport and Y. Apeloig, The Chemistry of Organic Silicon Compounds, Wiley, 1998, vol. 2 Search PubMed.
- (a) B. A. Gellert, N. Kahlcke, M. Feurer and S. Roth, Chem. – Eur. J., 2011, 17, 12203–12209 CrossRef CAS PubMed; (b) L. Monsigny, P. Thuéry, J.-C. Berthet and T. Cantat, ACS Catal., 2019, 9, 9025–9033 CrossRef CAS; (c) F. S. Tschernuth, T. Thorwart, L. Greb, F. Hanusch and S. Inoue, Angew. Chem., Int. Ed., 2021, 60, 25799–25803 CrossRef CAS PubMed; (d) W. Lv, Y. Dai, R. Guo, Y. Su, D. A. Ruiz, L. L. Liu, C.-H. Tung and L. Kong, Angew. Chem., Int. Ed., 2023, 62, e202308467 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2353322–2353329. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02202e |
| This journal is © The Royal Society of Chemistry 2024 |