
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectricity-driven, oxidative C–H selenylative and tellurylative annulation of N-(2-alkynyl)anilines: sustainable synthesis of 3-selanyl/tellanylquinolines†
Ainala
Naresh‡
,
H. Sai
Keerthana‡
,
Nilanjana
Mukherjee
and
Tanmay
Chatterjee
 *
*
Department of Chemistry, Birla Institute of Technology and Science, Pilani (BITS Pilani), Hyderabad Campus, Jawahar Nagar, Kapra Mandal, Hyderabad-500078, Telangana, India. E-mail: tanmay@hyderabad.bits-pilani.ac.in
First published on 13th June 2024
Abstract
A metal- and oxidant-free, radical C–H selenylative and tellurylative annulation of N-(2-alkynyl)anilines with diorganyl dichalcogenides is developed under electrochemical conditions for the sustainable synthesis of valuable 3-selanyl/tellanylquinolines up to 92% yield at room temperature. The developed protocol required only electricity as the green reagent and offers high atom economy, broad substrate scope, and efficient scalability.
Quinoline is commonly found in many natural products, such as alkaloids,1 and bioactive compounds with potential anticancer, antiviral, antiprotozoal, antimalarial, antibacterial, and antifungal properties.2 Therefore, developing new synthetic techniques for efficient synthesis and modification of quinolines is a subject of great importance in synthetic organic and medicinal chemistry.3
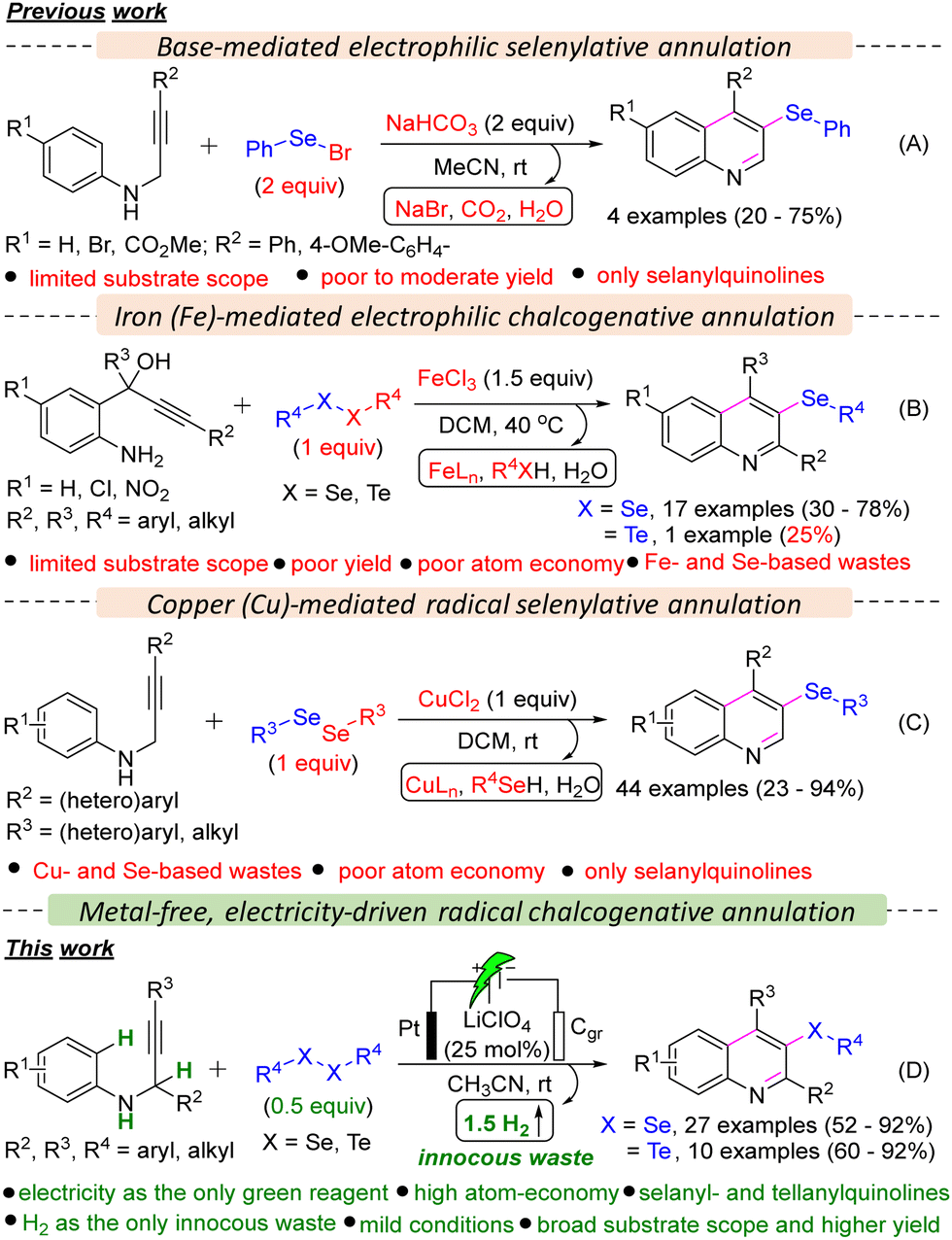
Organoselenides have recently gained significant attention from the scientific community due to the unique chemical and biological properties of selenium.4 These compounds have shown promise as synthetic intermediates and precursors to therapeutic compounds and as biologically active agents for treating various ailments, from cancer to cardiovascular disease.5 Additionally, the distinctive redox properties of selenium have sparked interest in the field of materials and nanochemistry.6 On the other hand, organotellurides are also found to have therapeutic applications in autoimmune diseases, enzymatic inhibition, and other biological activities.7 However, the synthesis of organotellurides is underdeveloped.7 Until now, limited methods have been documented for synthesizing selanylquinolines from acyclic starting materials. Larock and colleagues' seminal work showcased the use of relatively hazardous and unstable PhSeBr in over stoichiometric (2 equiv.) amounts to enable the electrophilic cyclization of N-(2-alkynyl)anilines, resulting in the formation of 3-selanylquinolines (Scheme 1A).8 Subsequently, the Zeni group introduced an alternative method utilizing 1.5 equiv. iron(III) chloride and diorganyl diselenides to produce 3-selanylquinolines through electrophilic selenylative annulation of 2-aminoaryl-2-ynols (Scheme 1B).9 Recently, Lukesh and co-workers reported a radical selenylative annulation of N-(2-alkynyl)anilines with diorganyl diselenides using a hazardous metal-salt, CuCl2 in stoichiometric (1 equiv.) amounts and hazardous chlorinated solvent, DCM (Scheme 1C).10 Despite notable advancement, the developed methods suffered from several serious limitations, such as (a) the requirement of hazardous PhSeBr or metallic reagents in over stoichiometric amounts, (b) limited substrate scope, (c) poor yield of products, (d) poor atom economy, (e) generation of Se- and/or Fe- and Cu-based stoichiometric waste. Moreover, these protocols were hardly employed for the tellurylative annulations and were only limited to synthesizing 3-selanylquinolines.
 | ||
| Scheme 1 Synthetic strategies to 3-chalcogenylquinolines. | ||
Only one example of the synthesis of 2,4-diphenyl-3-(phenyltellanyl)quinoline was reported by the Zeni group; however, the yield was poor (25%).9 Hence, developing a metal- and stoichiometric reagent-free, sustainable synthetic strategy for synthesizing both 3-selanyl- and 3-tellanylquinolines is highly desirable, and we became interested in developing the same as a part of our continued interest in developing sustainable chalcogenative C–H annulation and functionalization reactions.11 Recently, electro-organic synthesis has received tremendous attention from synthetic organic chemists.12 Consequently, several electrochemical seleno-functionalization reactions have been developed so far for the synthesis of organoselenides.13 Herein, we report a metal- and oxidant-free, highly atom-economic, scalable, and sustainable synthetic strategy for the oxidative C–H annulation of N-(2-alkynyl)anilines with diorganyl diselenides and diorganyl ditellurides under electrochemical conditions to access 3-selanyl- and 3-tellanylquinolines, which eventually has overcome the limitations of the previously developed protocols.
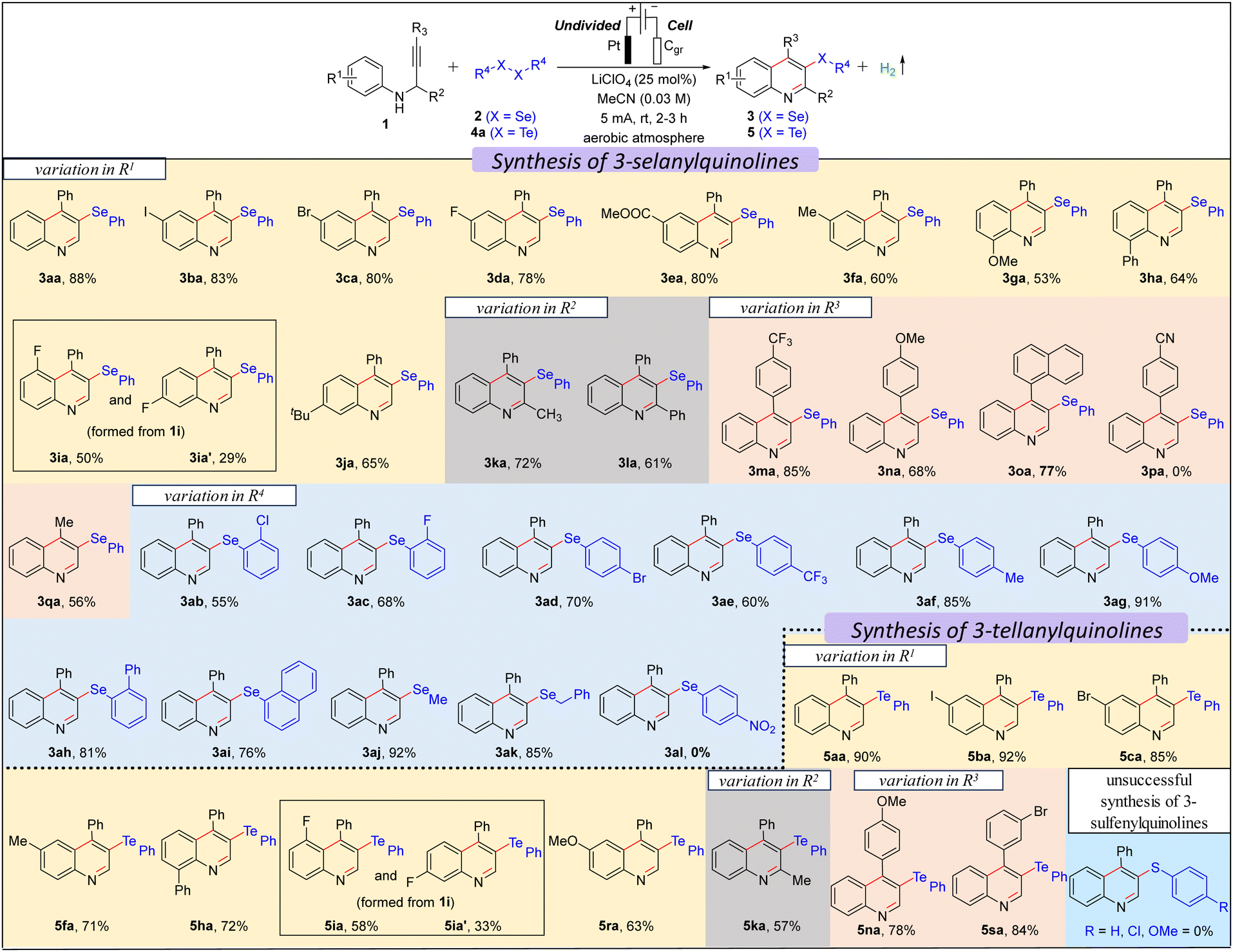
We commenced our investigation with N-(3-phenylprop-2-yn-1-yl)aniline 1a using 0.5 equiv. diphenyl diselenide 2a in an undivided cell under constant current electrolysis (Table S1 in ESI†). When the reaction mixture was electrolyzed using 5 mA constant current in the presence of a Pt-anode, graphite (Cgr) cathode, and LiClO4 (0.25 equiv.) in MeCN (0.03 M) for 3 h, gratifyingly, the desired product, 4-phenyl-3-(phenylselanyl)quinoline 3aa was formed in 92% yield (entry 1, Table S1, ESI†) with 49.32% faradaic efficiency and no other byproduct was formed except H2 as confirmed by the crude NMR (ESI†). While changing the cathode material from Cgr to Pt did not significantly affect the reaction outcome, switching the anode material from Pt to Cgr or Ni-foam had an adverse impact on the yield (entries 2–5 vs. 1, Table S1, ESI†). Altering the electrolyte from LiClO4 to nBu4NBr, Et4NBr, nBu4NClO4, nBu4NPF6, and KI, as well as changing the solvent from MeCN to EtOH, iPrOH, DMF, and water, resulted in a decrease in the reaction yield (entries 6–14 vs. 1, Table S1, ESI†). Furthermore, the optimal loading of 0.5 equiv. of 2a and 0.25 equiv. of LiClO4 was determined to be the most effective (entries 15–18 vs. 1, Table S1, ESI†). The optimal reaction time and constant current were identified as 3 hours and 5 mA, respectively (entries 19–21 vs. 1, Table S1, ESI†). Control experiments emphasized the essential roles of electrolyte and electricity in the reaction (entries 22 and 23, Table S1, ESI†). Next, we explored the scope of the selenylative C–H annulation reaction of N-(2-alkynyl)anilines 1 with diorganyl diselenides 2 under the optimized conditions (Table 1). The model reaction between 1a and 2a furnished 3aa in an 88% isolated yield. Ortho- and para-halogen (I, Br, Cl, and F), electron-withdrawing (Ph and CO2Et), and electron donating (Me and OMe) group substituted N-(2-alkynyl)anilines underwent the reaction with 2a smoothly to furnish the desired products (3aa–3ha) in moderate to good yield (53–88%). When meta-fluoro-substituted N-(2-alkynyl)aniline (1i) reacted with 2a, a mixture of regioisomers, i.e., 3ia and 3ia′, was formed in 52% and 30% yield, respectively. However, meta-tert-butyl-substituted N-(2-alkynyl)aniline (1j) furnished only 3ja regioselectively, as controlled by the bulk of the tBu-group. Highly substituted quinolines, i.e., 3ka and 3la, were formed from N-(1-substituted-2-alkynyl)anilines under standard conditions. Next, we explored the substrate scope with the variation in the alkyne part, and both electron-withdrawing (CF3) and electron-donating (OMe) group substituted N-(2-alkynyl-3-aryl)anilines furnished the desired products (3ma–3oa) in good yield. However, 4-(3-(phenylamino)prop-1-yn-1-yl)benzonitrile did not furnish 3pa. Moreover, 4-methyl-3-(phenylselanyl)quinoline 3qa was formed in 60% yield from N-(but-2-yn-1-yl)aniline. The scope of the diorganyl diselenides was found to be good as both the diaryl diselenide, bearing either a halogen (Br, Cl, F), or a strong electron-withdrawing (CF3) or electron-donating (OMe) group, and also dialkyl diselenides smoothly reacted with 1a to furnish the desired products (3ab–3ai and 3aj–3ak) in moderate to good yields, except for 1,2-bis(4-nitrophenyl)diselane (3al). Subsequently, we investigated the scope of the tellurylative C–H annulation of 1a with diphenyl ditelluride 4a, and, to our delight, the desired product, 4-phenyl-3-(phenyltellanyl)quinoline 5aa was formed in an excellent yield (90%) under standard conditions (Table 1). The versatility of the electrochemical tellurylative C–H annulation was notable as various substituted N-(2-alkynyl)anilines reacted with 4a to afford the desired 3-tellanylquinolines (5ba–5sa) in satisfactory to excellent yields (60–92%), regardless of the type and position of the substituents. As predicted, 1i yielded a mixture of regioisomers 5ia and 5ia′ in 58% and 33%, respectively. Unfortunately, the C–H sulfenylative annulation of 1a with various diaryl disulfides (R = H, Cl, OMe) under standard conditions did not furnish the desired 3-sulfenylquinolines.
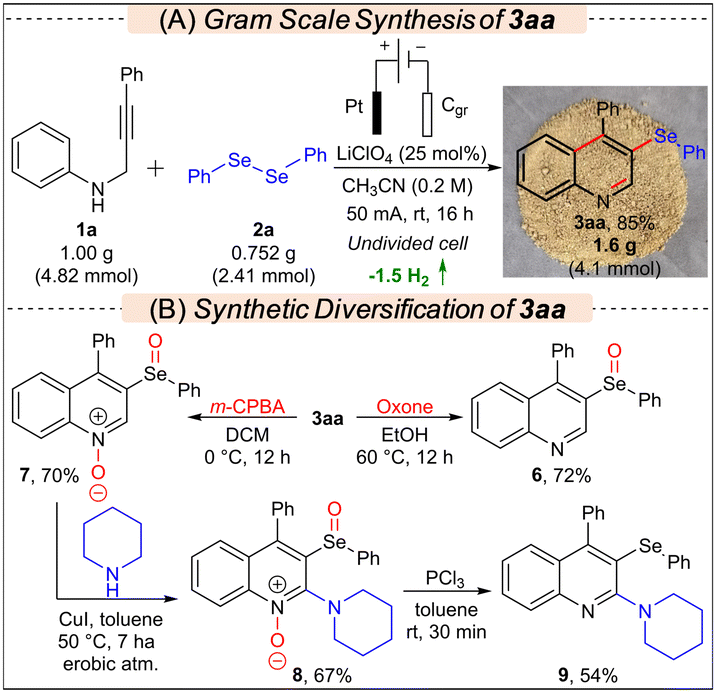
To showcase the practicality of the protocol, a gram-scale synthesis involving compounds 1a (1 g, 4.82 mmol) and 2a was conducted using the standard conditions, resulting in the formation of 1.6 g (4.1 mmol) 3aa in an 85% yield (Scheme 2A). To demonstrate the application of the developed protocol, 3aa was synthetically diversified to various new classes of molecules via controlled oxidation, C–H amination, and deoxygenation reactions, as presented in Scheme 2B.11d,14 When 3aa was treated with Oxone, only chemoselective oxidation of selenium occurred leading to the selective synthesis of 6 in 72% yield; however, in the presence of m-CPBA, 7 was formed selectively in 70% yield. Subsequently, 7 was further modified through a Cu-catalyzed C–H amination reaction with piperidine to access 8 in 67% yield, which after deoxygenation by PCl3 furnished 9 in 54% yield. Synthesis of 2-aminoquinolines is important as they are prevalent in various alkaloids and bioactive molecules.15
 | ||
| Scheme 2 Gram-scale synthesis of 3aa and synthetic diversification. | ||
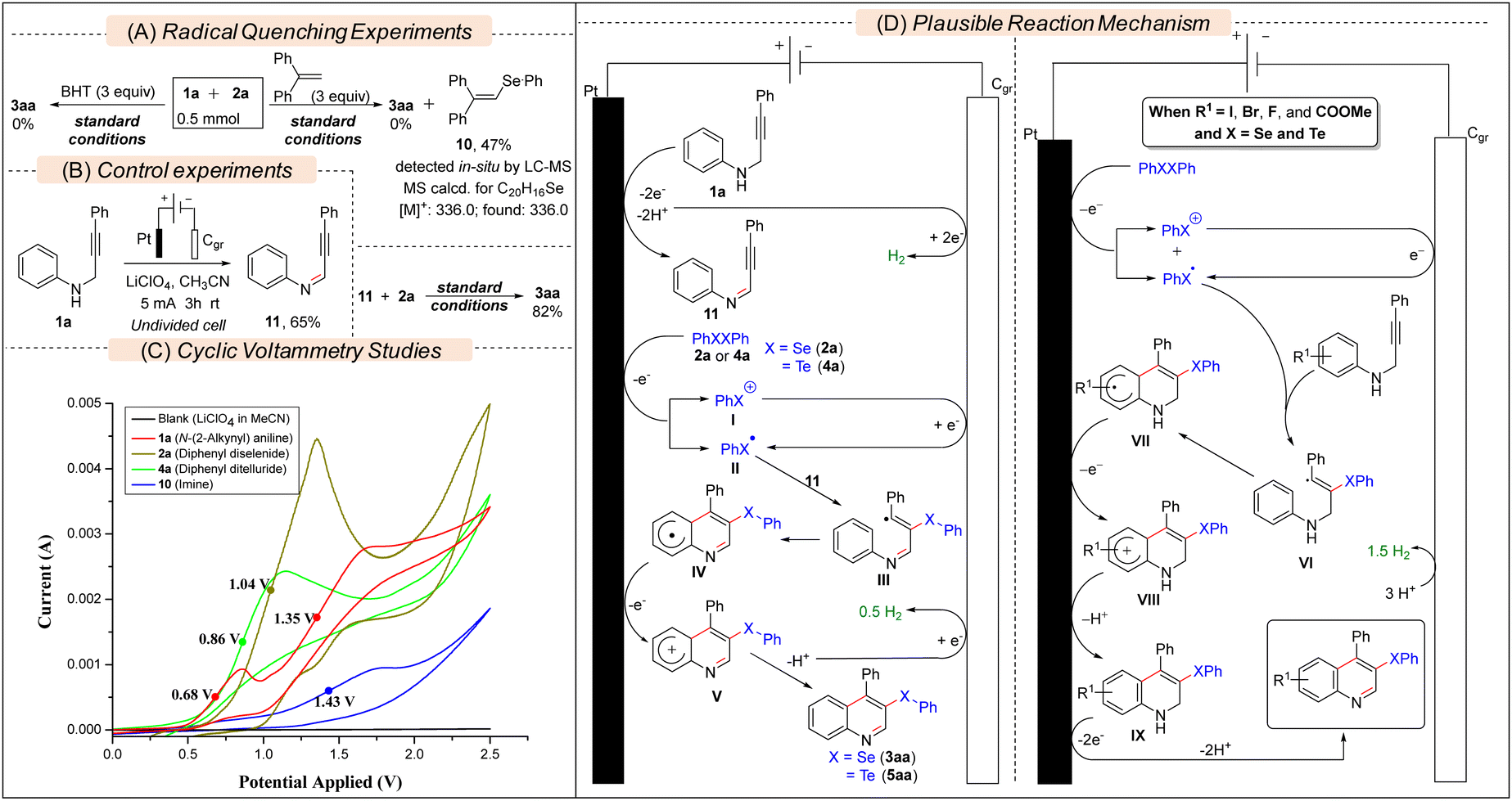
Several experiments have been conducted to get insights into the reaction mechanism. In the presence of a radical quencher, i.e., BHT or 1,1-diphenylethene (DPE), the model reaction furnished no 3aa, which suggested that the reaction proceeded via a free-radical pathway (Scheme 3A). Moreover, the detection and isolation of the radical adduct 10 revealed the involvement of a PhSe. radical as the key intermediate (Fig. S1, ESI†). In the absence of 2a, 1a was oxidized to (Z)-N,3-diphenylprop-2-yn-1-imine 11 (Scheme 3B). Then, 11 was subjected to reaction with 2a under electrochemical conditions, and 3aa was formed in 82% yield, revealing 11 as an intermediate of the reaction. The cyclic voltammetry (CV) experiments (Scheme 3C) revealed that the half-wave oxidation potentials (Ep/2) of diphenyl diselenide 2a (Ep/2 = 1.04 V) and diphenyl ditelluride 4a (Ep/2 = 0.86 V) are lower than that of the imine 11 (Ep/2 = 1.43 V). However, the half-wave first oxidation potential of 1a (Ep/2 = 0.68 V) is quite a bit lower than that of 2a and 4a. Based on these experimental results and previous reports,13b we proposed that 1a first oxidized to 11in situ and then 2a or 4a underwent preferential one-electron oxidation at the anode to furnish the key PhSe. radical II (Scheme 3D). Then II was added to 11 to form the vinyl radical III, which, after cyclization followed by anodic oxidation and deprotonation, furnished the desired product (3aa or 5aa). However, according to CV analysis of electron-withdrawing group (I, Br, F, and CO2Me) substituted N-(2-alkynyl)anilines (Fig S2, ESI†), it is more likely that 2a or 4a, having lower Ep/2 than that of these substrates, first underwent preferential oxidation at the anode to form the chalcogenyl radical (PhX.) which after radical addition and annulation formed IX. Finally, IX underwent electrochemical oxidation to form the desired product. The half-wave oxidation potential of diphenyl disulfide, (PhS)2 (Ep/2 = 1.53 V), is found to be much higher than that of 11 and 1a (Fig. S2, ESI†), which could be the cause of the failure of the reaction.
 | ||
| Scheme 3 Mechanistic studies and plausible reaction mechanisms. | ||
In conclusion, we developed a highly atom-economic, cost-effective, scalable, and sustainable synthetic strategy for the selenylative and tellurylative C–H annulation of readily available N-(2-alkynyl)anilines with diorganyl dichalcogenides to access a wide variety of valuable 3-selanyl/tellanylquinolines under electrochemical conditions. Electricity was used as the only green reagent, and hydrogen was formed as the only innocuous byproduct. Mechanistic studies revealed a radical pathway, and 3aa was synthetically diversified to other quinoline derivatives. This protocol also serves as a unique strategy to access 3-tellanylquinolines, which has been underdeveloped to date.
We thank SERB [File no. CRG/2023/003045] and CSIR [File no. 02(0390)/21/EMR-II], Govt. of India for financial support.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- J. P. Michael, Nat. Prod. Rep., 1997, 14, 605 RSC.
- (a) V. R. Solomon and H. Lee, Curr. Med. Chem., 2011, 18, 1488–1508 CrossRef CAS PubMed; (b) N. Shobeiri, M. Rashedi, F. Mosaffa, A. Zarghi, M. Ghandadi, A. Ghasemi and R. Ghodsi, Eur. J. Med. Chem., 2016, 114, 14–23 CrossRef CAS PubMed; (c) R. Kaur and K. Kumar, Eur. J. Med. Chem., 2021, 215, 113220 CrossRef CAS PubMed; (d) A. Dorababu, ChemistrySelect, 2021, 6, 2164–2177 CrossRef CAS; (e) C. W. Wright, J. A. Kyereme and A. G. Breen, J. Med. Chem., 2001, 44, 3187–3194 CrossRef CAS; (f) P. Grellier, L. Ramiaramanana and V. Millerioux, Phytother. Res., 1996, 10, 317–321 CrossRef CAS; (g) A. Dorababu, Arch. Pharm., 2021, 354, 2000232 CrossRef CAS.
- S. M. Prajapati, K. D. Patel, R. H. Vekariya, S. N. Panchal and H. D. Patel, RSC Adv., 2014, 4, 24463–24476 RSC.
- R. Naithani, Mini-Rev. Med. Chem., 2008, 8, 657–668 CrossRef CAS PubMed.
- D. Plano, D. N. Karelia, M. K. Pandey, J. E. Spallholz, S. Amin and A. K. Sharma, J. Med. Chem., 2016, 59, 1946–1959 CrossRef CAS PubMed.
- W. Huang, M. Wang, L. Hu, C. Wang, Z. Xie and H. Zhang, Adv. Funct. Mater., 2020, 30, 2003301 CrossRef CAS.
- (a) G. Halpret and B. Sredni, Autoimmun. Rev., 2014, 13, 1230–1235 CrossRef PubMed; (b) S. E. C. P. M. S. Maluf, F. P. Melo, M. L. Varotti, R. Gazarini, L. O. R. Cunha and A. K. Carmona, Parasitol. Int., 2016, 65, 20–22 CrossRef PubMed; (c) A. Albeck, H. Weitman, B. Sredni and M. Albeck, Inorg. Chem., 1998, 37, 1704–1712 CrossRef CAS.
- X. Zhang, M. A. Campo, X. T. Yao and R. C. Larock, Org. Lett., 2005, 7, 763–766 CrossRef CAS PubMed.
- X. Zhang, M. A. Campo, X. T. Yao and R. C. Larock, Tetrahedron, 2010, 66, 1177–1187 CrossRef CAS PubMed.
- C. Zhu, M. Nurko, C. S. Day and J. C. Lukesh, III, J. Org. Chem., 2022, 87, 8390–8395 CrossRef CAS PubMed.
- (a) N. Mukherjee and T. Chatterjee, J. Org. Chem., 2021, 86, 7881–7890 CrossRef CAS PubMed; (b) N. Mukherjee and T. Chatterjee, Green Chem., 2021, 23, 10006–10013 RSC; (c) N. Mukherjee, A. N. V. Satyanarayana, P. Singh, M. Dixit and T. Chatterjee, Green Chem., 2022, 24, 7029–7038 RSC; (d) A. N. V. Satyanarayana, N. Mukherjee and T. Chatterjee, Green Chem., 2023, 25, 779 RSC; (e) N. Mukherjee and T. Chatterjee, Green Chem., 2023, 25, 8798–8807 RSC.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (b) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS; (c) G. M. Martins, G. C. Zimmer, S. R. Mendes and N. Ahmed, Green Chem., 2020, 22, 4849 RSC; (d) J. Liu, J. P. Wan and Y. Liu, Org. Chem. Front., 2024, 11, 597 RSC; (e) J. Liu, M. Wang, L. Lia and L. Wang, Green Chem., 2021, 23, 4733–4740 RSC.
- (a) P. Qu, Y. Q. Jiang, Y. H. Wang and G. Q. Liu, Green Chem., 2023, 25, 7485 RSC; (b) N. Mukherjee and T. Chatterjee, Adv. Synth. Catal., 2023, 365, 2255–2263 CrossRef CAS; (c) A. B. Dapkekar and G. Satyanarayana, Chem. Commun., 2023, 59, 8719 RSC; (d) A. Chouhan, K. Ucheniya, L. Yadav, P. K. Jat, A. Gurjar and S. S. Badsara, Org. Biomol. Chem., 2023, 21, 7643 RSC; (e) R. Reddy and D. H. Kolgave, J. Org. Chem., 2021, 86, 17071–17081 CrossRef PubMed; (f) S. Mallick, T. Mandal, N. Kumari, L. Roy and S. D. Sarkar, Chem. – Eur. J., 2024, 30, e202304002 CrossRef CAS PubMed.
- (a) C. Zhu, M. Yi, D. Wei, X. Chen, T. Wu and X. Cui, Org. Lett., 2014, 16, 1840–1843 CrossRef CAS PubMed; (b) H. Hwang, J. Kim, J. Jeong and S. Chang, J. Am. Chem. Soc., 2014, 136, 10770–10776 CrossRef CAS PubMed.
- A. Vashistha, S. Kumar, S. Kirar, N. Sharma, B. Das, U. C. Banerjee, S. V. Pawar, R. Kumar and A. K. Yadav, Comput. Biol. Chem., 2023, 102, 107807 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Optimization table, experimental procedures, mass spectrum of 10, cyclic voltammograms, calculation of faradaic efficiency, and copies of NMR spectra. See DOI: https://doi.org/10.1039/d4cc01780c |
| ‡ These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |