
Evolution in the asymmetric synthesis of biaryl ethers and related atropisomers
Namrata
Kotwal
and
Pankaj
Chauhan
 *
*
Department of Chemistry, Indian Institute of Technology Jammu, Jagti, NH-44, Nagrota Bypass, Jammu, 181221 J&K, India. E-mail: pankaj.chauhan@iitjammu.ac.in; Web: https://www.pankajchauhanresearch.com
First published on 14th May 2024
Abstract
Axially chiral biaryl ethers and related compounds hold valuable potential in natural products, medicinal chemistry, and catalysis; however, their asymmetric syntheses have always been overlooked compared to other biaryl/hetero-biaryl atropisomers. Unlike the later class molecules bearing a single chiral axis, the former category possesses a unique type of atropisomerism bearing two potential axes. Due to their great importance in diverse research domains, catalytic atropselective biaryl ether synthesis has recently witnessed an upsurge. This highlight article provides an elaborated view on the developments of catalytic synthetic methods that have been explored to achieve dual axial chirality in biaryl ethers and related scaffolds.
 Namrata Kotwal | Namrata Kotwal was born in 1997 in Jammu, India. She obtained her BSc in 2017 and MSc Chemistry in 2019 from the University of Jammu. Since August 2020, she has been working as a PhD student in the research group of Dr Pankaj Chauhan at IIT Jammu. Her research interests include organocatalytic asymmetric synthesis of medium-sized carbo- and heterocyclic bridged biaryls, and the generation of stereogenic centres at the remote position of the unsaturated acceptors. |
 Pankaj Chauhan | Pankaj Chauhan obtained his BSc from Himachal Pradesh University in 2004 and MSc Chemistry from Guru Nanak Dev University, Amritsar, in 2007. He completed his PhD under the supervision of Prof. S. S. Chimni at Guru Nanak Dev University in 2012. In April 2013, he joined the research group of Professor Dieter Enders as a postdoctoral researcher at RWTH Aachen University, Germany. He worked as a sub-group leader in the same research group from April 2014 to September 2017. In October 2017, he started his independent research career as an Assistant Professor in the Department of Chemistry, Indian Institute of Technology Jammu, where he is currently working as an Associate Professor. His research interests include asymmetric synthesis, organocatalysis, photocatalysis and direct photoactivation, electroorganic synthesis and synergistic catalysis. |
1. Introduction
Biaryls/hetero-biaryls represent an essential class of organic molecules, frequently resolved as atropisomers due to the restricted rotation around the pivotal bond, specifically around the carbon–carbon, carbon–nitrogen, and nitrogen–nitrogen bonds. The significance of atropisomeric biaryls and related hetero-biaryls has long been recognised due to their widespread presence in natural products,1 drug molecules,2 privileged organocatalysts3 and chiral ligands in metal catalysis.4 Considering the diverse applications and interesting properties, substantial progress has been witnessed in the atropselective synthesis of these biaryls/hetero-biaryls bearing a C2 axis of symmetry.5 Besides, another essential and fascinating class of atropisomeric biaryls has been encountered, where a heteroatom is incorporated between the biaryl rings, providing unique structural features due to two adjacent chiral axes (Fig. 1A). In the case of biaryl ethers and related scaffolds such as biaryl amines and sulphides, the unsymmetrical substitution on both the aryl rings imposes the chiral element on the respective skeleton. The configurational stability of these dual-axial compounds with steady C–X (X = O, NR, S) stereogenic axes is mainly ascribed to the presence of 2,6-disubstitution on both the aromatic rings, which creates a substantial rotational barrier sufficient for the conformers to be resolved as atropisomers.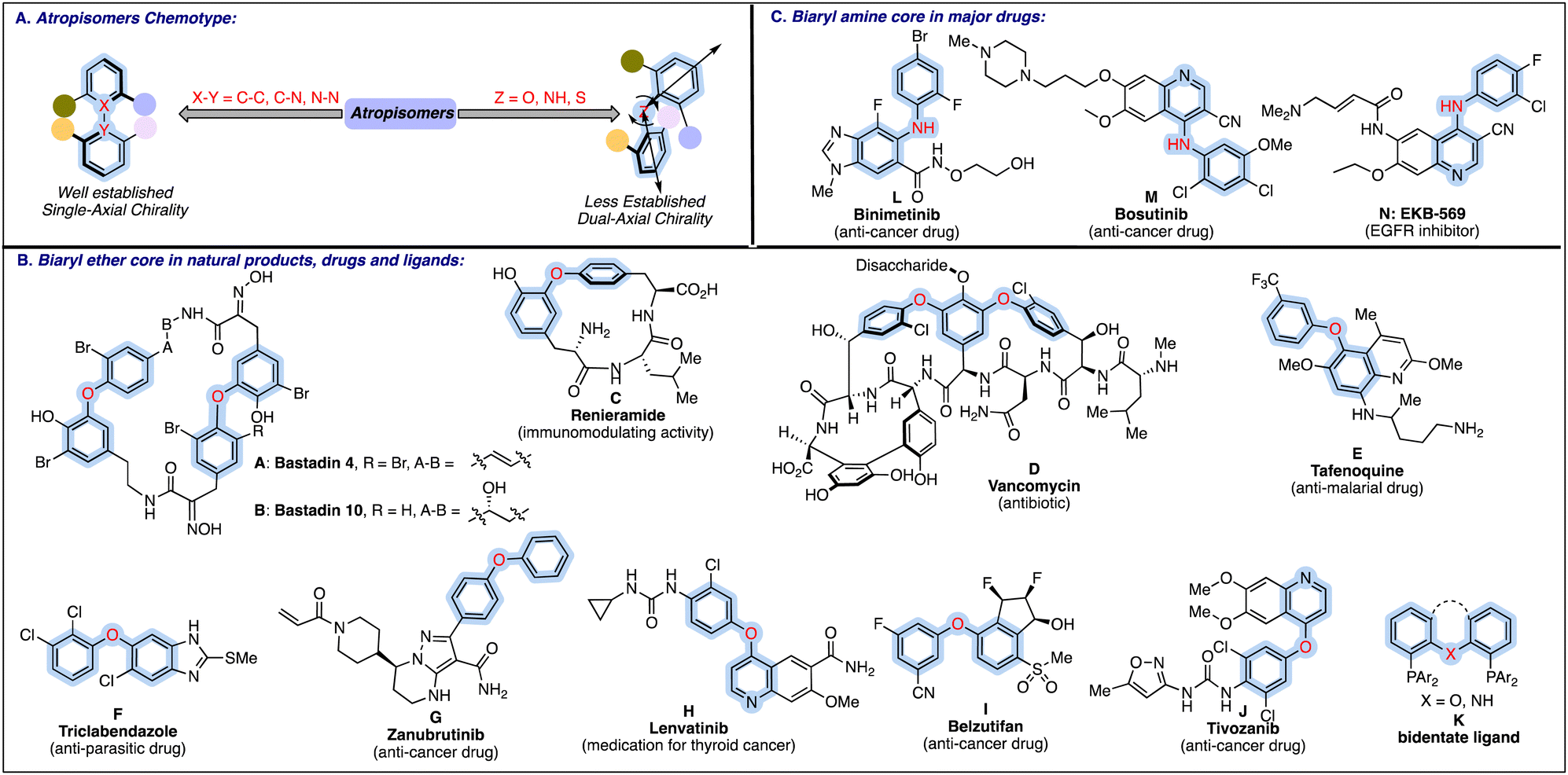 | ||
| Fig. 1 Types of atropisomerism in biaryls and functional molecules bearing biaryl ethers and related amines. | ||
Apart from distinctive structural properties, the above-mentioned potential dual axial chiral scaffolds are among the structural motifs in selected natural products, FDA-approved drug candidates, pharmaceuticals, and agrochemicals. Biaryl ether core is a privileged pharmacophore in major natural products A–C6 and antibiotics such as vancomycin D7 (Fig. 1B). Besides, biaryl ethers constitute the fundamental unit in certain other pharmaceuticals, viz., anti-malarial drug E,8 anti-parasitic drug F9 and anticancer medicine G–J.10 In an important class of bidentate diphosphine ligands, the biaryl ether (e.g.K) or biaryl amines serve as an important backbone.11 Biaryl amines are also key structural components in a broad range of anti-cancer drugs (L–M), and bioactive molecules like EKB-569 N (Fig. 1C).12
Based on the unique type of chirality leading to fascinating properties and wide applications, the development of synthetic routes to these biaxial atropisomers can be explored as an exciting research area. However, their atropselective synthesis is not well developed compared to biaryl compounds with a single axis. The innate difficulties encountered in generating flexible dual C–X axes could be rationalised based on the low rotational barrier (the half-life of racemization at least 1000 seconds at room temperature, corresponding to an energy barrier of 23.3 kcal mol−1 is generally required for the existence of atropisomerism)1 and more conformational space around dual axes that make the recognition and regulation of specific atropisomers relatively difficult. Additionally, compared to the conventional atropisomers with a single axis, rotational control around both potential axes is required to achieve atropselectivity. Incorporating a heteroatom between two axes not only reduces spatial proximity but also diminishes the electronic interactions among the different groups attached around the specific aryl rings.
Despite the difficulties in achieving atropselectivity in these biaxial compounds, recently, some advancements have been seen in developing catalytic methods for dual axial chiral compounds. In this context, mainly the catalytic potential of chiral organocatalysts has been explored for developing new methodologies leading to the synthesis of much-challenging dual axial chiral biaryl ethers, to a limited extent, related amines. This article highlights the seminal breakthroughs for the atropselective synthesis of biaryl ethers and other related atropisomers. For the convenience of readers, the early precedents describing the chirality in biaryl ethers, amines and sulfides have been thoroughly elucidated, and in the following sections, the recent breakthroughs for their catalytic atropselective synthesis have been described.
2. Early evidence for the atropisomerism in biaryl with C–X–C linkage
The first evidence for the restricted rotation around the C–O–C bond in biaryl ethers was illustrated by Gibbs and co-workers about 70 years back in 1954.13 Based on the wooden models, they observed a certain degree of steric hindrance in biaryl ethers such as 1 (Fig. 2). Four years later, Brewster and Dahlgard performed IR and UV spectral studies on more than three dozen differently substituted biaryl ethers (including compound 2) and found a significant weakening in the absorption bands in IR and UV regions for the larger substituent at the ortho positions.14 Other precedents also appeared later, where the variable temperature NMR investigations were carried out to determine the rotation barriers in biaryl ethers.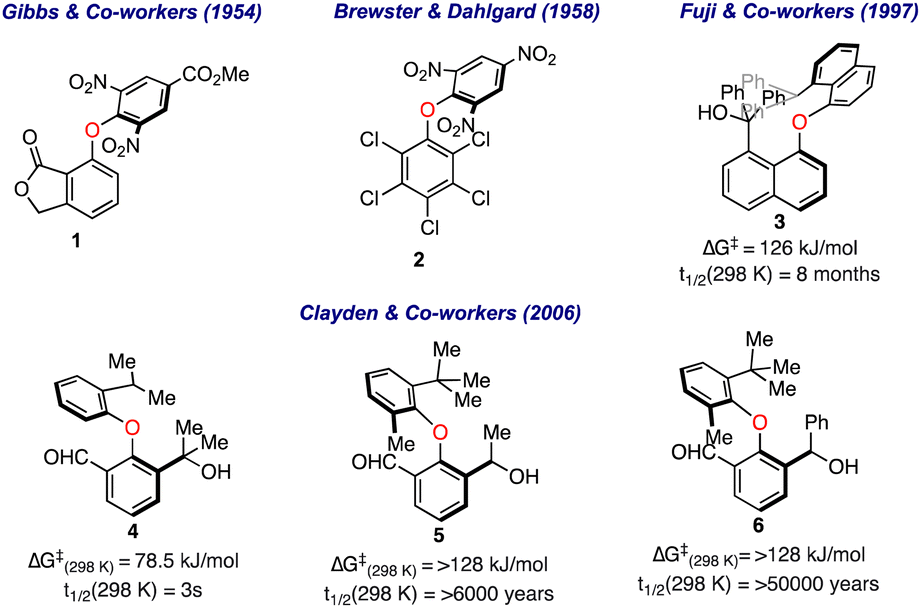 | ||
| Fig. 2 Early studies on the restricted rotation in biaryl ethers. | ||
In 1998, Fuji and co-workers reported the synthesis of optically active 8,8′-disubstituted 1,1′-binaphthyl ethers and calculated the rotation barrier based on the time-dependent alteration in the ee values.15 It was realized that the sterically crowded biaryl ether 3 has a half-life for racemization (t1/2) at 25 °C of 8 months and sufficient activation barrier (ΔGrot = 126 kJ mol−1) to be resolved as an atropisomer. Later, Clayden's group further investigated the phenomenon of atropisomerism among diphenyl ethers with unsymmetrical substitution.16 It was observed that the free energy barrier was enhanced with an increase in steric bulk around the aromatic rings, as shown for compounds 4 to 6, where a significant atropisomerism is only apparent when the tert-butyl group is present.
Clayden and co-workers also successfully established the phenomenon of atropsiomerism in biaryl sulfides and biaryl sulphones (Scheme 1).17 A separable diastereoisomeric sulfinyl sulfides 7 were detected in 1H NMR at 20 °C with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio; however, a rapid interconversion was observed due to a lower energy barrier at ambient temperature. The conformationally and enantio-pure sulfinyl sulfone 8 could be converted to bis sulfone 9, possessing only biaryl axes without central chirality by maintaining the high enantiomeric excess.
:1 ratio; however, a rapid interconversion was observed due to a lower energy barrier at ambient temperature. The conformationally and enantio-pure sulfinyl sulfone 8 could be converted to bis sulfone 9, possessing only biaryl axes without central chirality by maintaining the high enantiomeric excess.
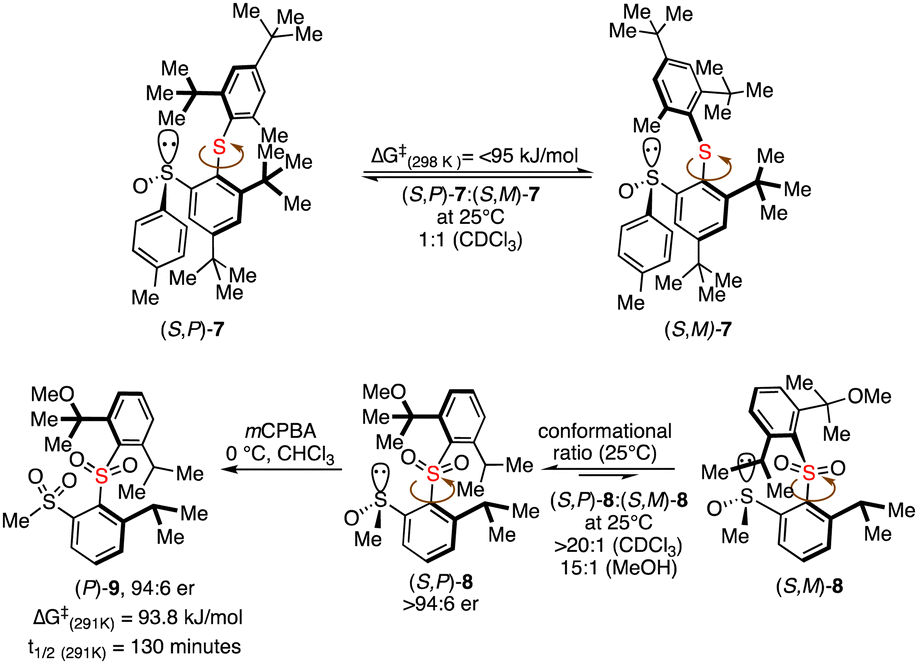 | ||
| Scheme 1 Atropsiomerism in biaryl sulfides and biaryl sulphones. | ||
A similar atropisomerism could be witnessed in biaryl amines.18 In this direction, Kawabata et al. investigated atropisomerism in a series of biaryl amines having the possibility of intramolecular H-bonding.19 It was revealed that in biaryl-substituted amine 10, a stable axial chirality arises due to intramolecular N–H–N hydrogen-bonding interactions, and the hydrogen bond gets stronger by substituting donor groups on the imine nitrogen and electron-withdrawing groups on the naphthyl ring (Scheme 2). Further, it was realized that substituting the naphthyl ring with sterically demanding groups effectively increased the rotational barrier around the C–N pivotal bond, thus enabling stable axial chirality among these molecules.
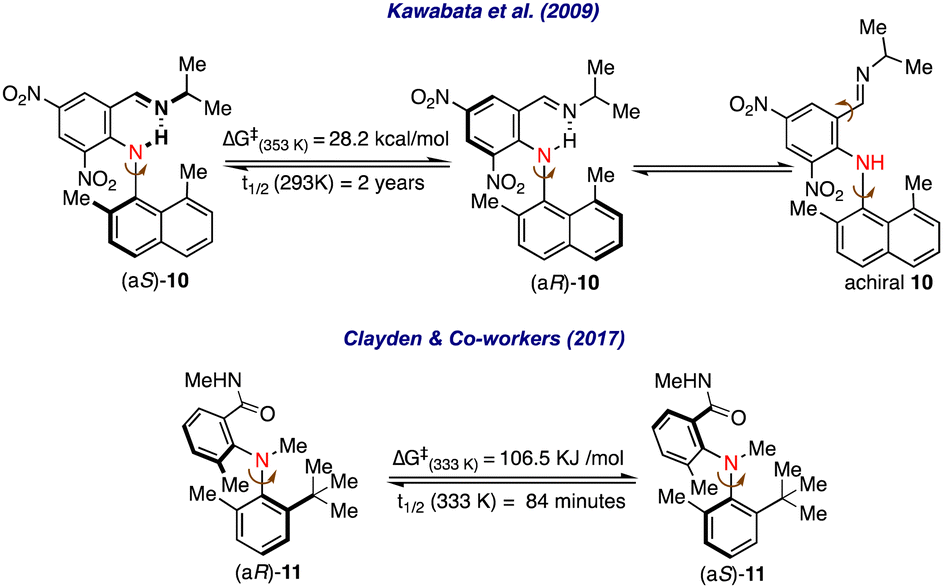 | ||
| Scheme 2 Atropsiomerism in biaryl amines. | ||
Clayden and co-workers were able to resolve the atropisomers in biaryl amines without intramolecular H-bonding on HPLC by using a chiral stationary phase.20 A significant rotation barrier and half-life of racemization were calculated by observing first-order decay in enantioselectivity for the biaryl amines 11.
3. Catalytic atropselective synthesis of biaryl ethers
3.1 Biocatalytic approach
Clayden's group provided the first catalytic pathway for accessing enantiopure biaryl ethers either via the biocatalytic oxidative or reductive desymmetrization of symmetrical diol 12 or dialdehyde 13, respectively (Scheme 3).21 With a variant of galactose oxidase (GOase), a benzylic diol 12 provided the biaryl ether (P)-14 with 88% ee in an hour. The ee value was further enriched to a maximum of 94% due to converting the minor enantiomer (M)-14 to the dialdehyde 13. This enantio-enrichment was attributed to combining both enantioselective desymmetrization and kinetic resolution (KR). Further, an initial attempt for the selective acylation of diol 12 with a lipase and vinyl acetate provided moderate enantioselectivity (60% ee) of 15 with approximately 50% conversion in 24 hours. Alternatively, a series of ketoreductases (KREDs) were screened for the reduction of dialdehyde 13 to reach the enantiomer of (P)-14 selectively. In the enzymatic reduction, most of the screened KREDs were found to be active; however, the one showing perfect enantio-induction resulted in poor conversion.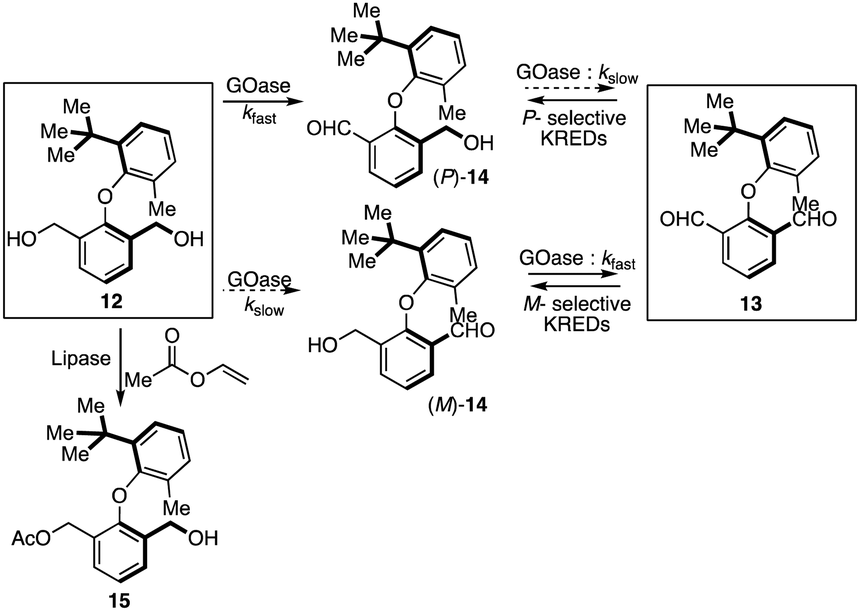 | ||
| Scheme 3 Biocatalytic oxidative or reductive desymmetrization of symmetrical biaryl ether diol or dialdehyde. | ||
3.2. Organocatalytic approaches
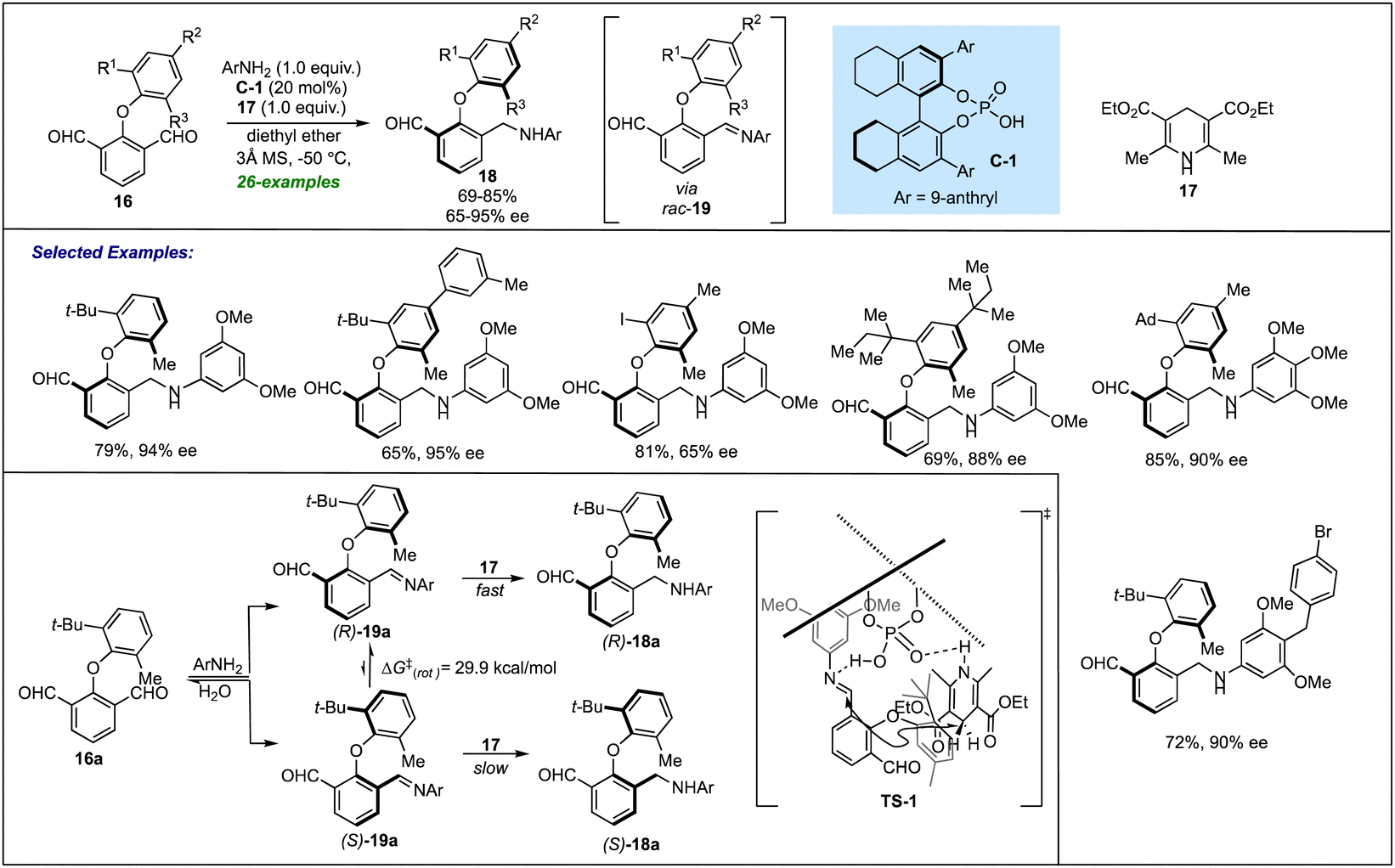 | ||
| Scheme 4 Phosphoric acid catalyzed DKR via ATH of the in situ generated imines for accessing atropisomeric biaryl ethers. | ||
A deep insight into the reaction mechanism revealed that in the absence of Hanztsch ester, the corresponding imine was generated in good yield but in <5% ee. This clearly indicated that even in the presence of a chiral catalyst, no enantio-induction was provided by the imine formation step. Further, computational studies combined with experimental observations led to the conclusion that in situ generated (R)-19 undergoes a fast ATH reaction catalyzed by C-1 to provide good enantioselectivity of the desired amine 18a. Conversely, another conformer (S)-19 with a slow reaction rate hydrolyzes to recover 16a dynamically. A stereochemical model (TS-1) was also proposed for the transformation, indicating the role of a chiral catalyst for the atropselective synthesis of biaryl ethers.
Yang's group explored the applications of chiral phosphoric acid catalysis to efficiently synthesise axially chiral biaryl ethers through a desymmetrization of symmetrical biaryl ether appended aryl amines 20 (Scheme 5).23 Using C-2 as a catalyst, the asymmetric electrophilic amination of the 2-aryloxy-substituted 1,3-benzenenedimaines 20 with azodicarboxylate 21 resulted in the broad range of enantiopure desymmetrized biaryl ethers 22. It was found that the substrates with various N-benzyl and N-alkyl substituents (with both cyclic and acyclic systems) at the R1 position resulted in excellent results in terms of yield and enantioselectivity. Interestingly, a free amine also undergoes amination reactions with high enantioselectivity (91% ee). Many alkyl and aryl groups, as well as a benzyl group on one of the aryl rings of biaryl ethers (at R2), were found to be tolerable, except the sterically crowded substrate (R2 = 2-MeOC6H4), which resulted in only 39% ee. Apart from a good acceptance of different substituents on different positions of the biaryl ethers, many commercially available azodicarboxylates were also examined, where the atropisomers of biaryl ethers were accessed in the highly enantioselective fashion except for di-tert butyl azodicarboxylates, which proved to be the unsuitable substrate for this transformation due to the steric reasons. The configurational stability of the biaryl ether products was further confirmed by thermal racemization experiments, where products were found to have sufficient racemization half-life and a high rotation barrier. Kinetic resolution of an unsymmetrical starting amine 23 occurs with high efficiency, suggesting the versatility of this amination process for delivering diverse atropisomeric biaryl ethers.
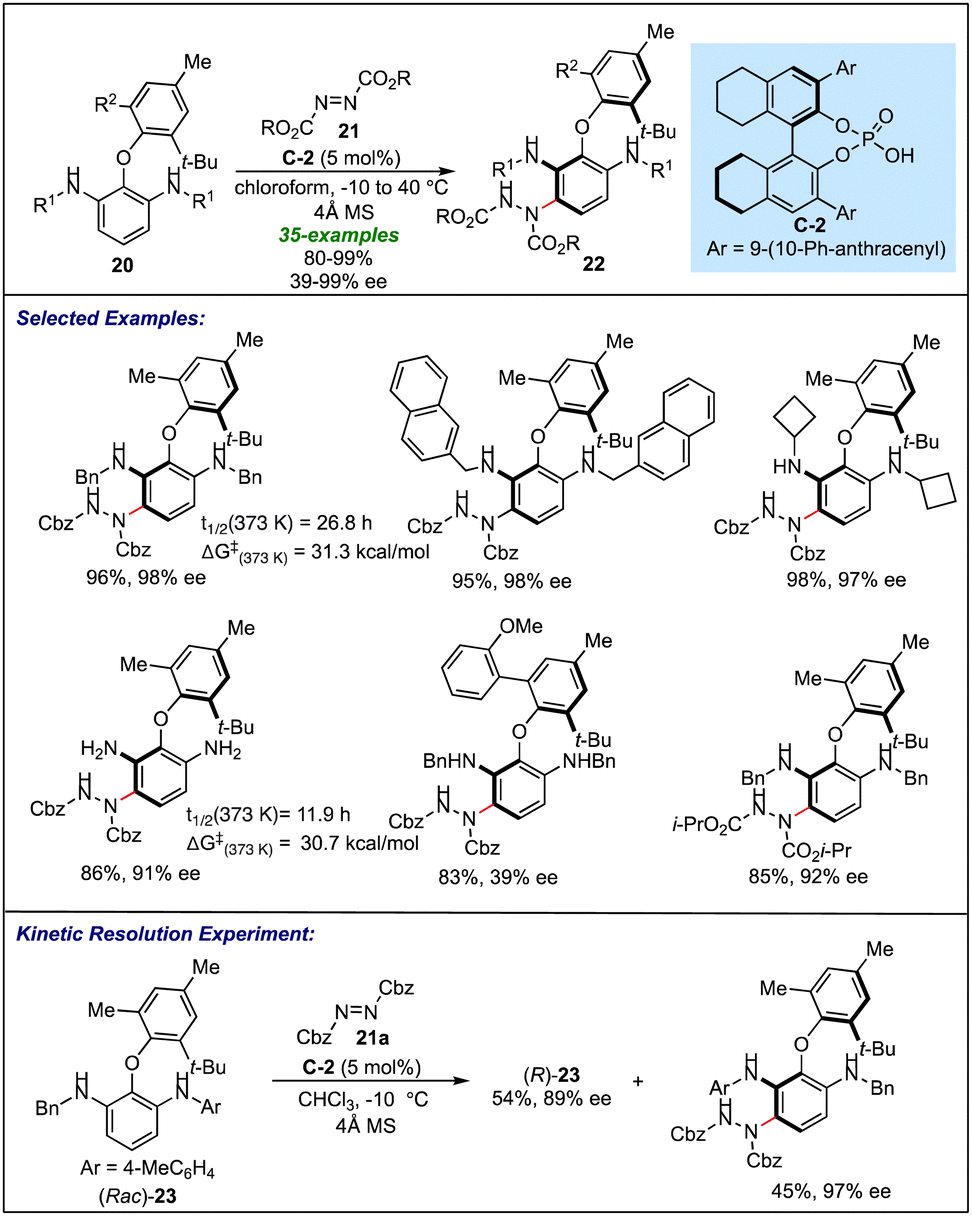 | ||
| Scheme 5 Phosphoric acid catalyzed desymmetrization via electrophilic amination of aryl amines for accessing atropisomeric biaryl ethers. | ||
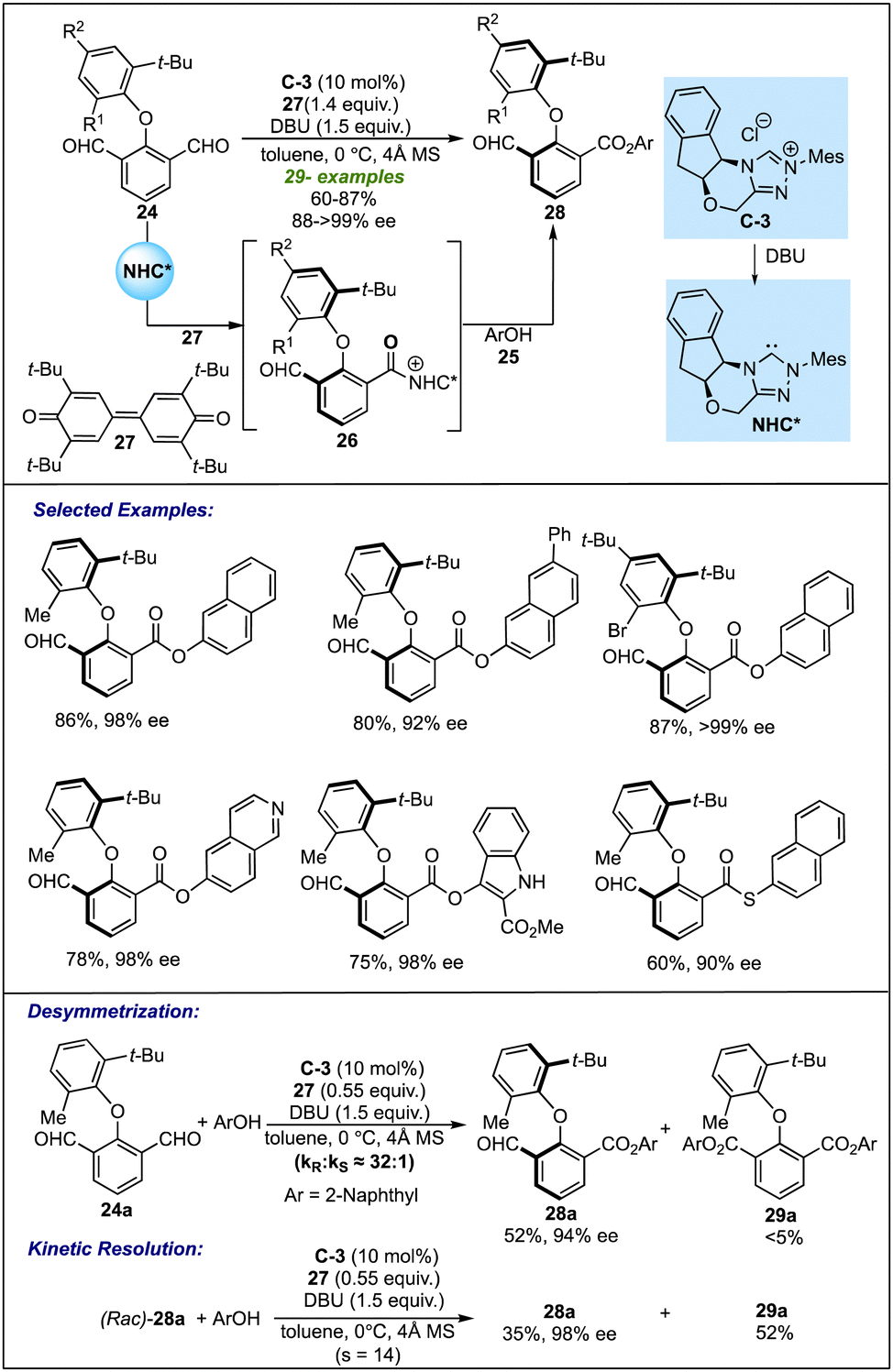 | ||
| Scheme 6 Biju's work on the NHC-catalyzed desymmetrization for atropselective synthesis of biaryl ethers. | ||
The configurational stability of the product was depicted with thermal stability experiments and DFT calculations. Both investigations revealed that compound 28a has a significant rotation barrier to be resolved as an atropisomer. Further insight into the mechanism revealed that lowering the equivalents of oxidant under optimized conditions led to the formation of the desired product in 52% yield and 94% ee, along with the formation of diester 29 as a by-product in traces. On the other hand, the reaction of racemic product 28a with 0.55 equivalent of oxidant resulted in kinetic resolution leading to enantiopure 28a in 35% yield and diester 29 in 52% yield. These observations suggested that desymmetrization plays a major role in controlling enantioselectivity; however, the possibility of further enantio-enrichment by a subsequent KR process could not be eliminated completely.25
Parallelly, Ye and co-workers disclosed similar NHC-catalyzed desymmetrization of 2-aryloxyisophthaladehydes 24 with alcohols 25 and amines 30 (Scheme 7).26 It was proposed that the NHC generated from C-4 or C-5 oxidatively provide azolium intermediate 26 from 24, which undergoes esterification or amidation to furnish asymmetrical biaryl ethers 28 and amides 31 with excellent enantioselectivities. By using CD3OD as a nucleophile, it was observed that deuterium incorporation did not occur in the remaining formyl group, which suggests the non-reversible formation of the Breslow intermediate. Based on similar controlled experiments as performed by Biju's group, the enantio-enrichment was proposed to be further assisted by a downstream kinetic resolution process in this desymmetrization process. The key features of this protocol include broad functional group tolerance and late-stage functionalizations under ambient reaction conditions. Apart from phenol derivatives as nucleophiles, the successful applications of aliphatic alcohols and amine nucleophiles, i.e. 2-aminobenzothiols 30, could be seen as further advancement in the NHC-catalyzed desymmetrization leading to biaxial biaryl ethers.
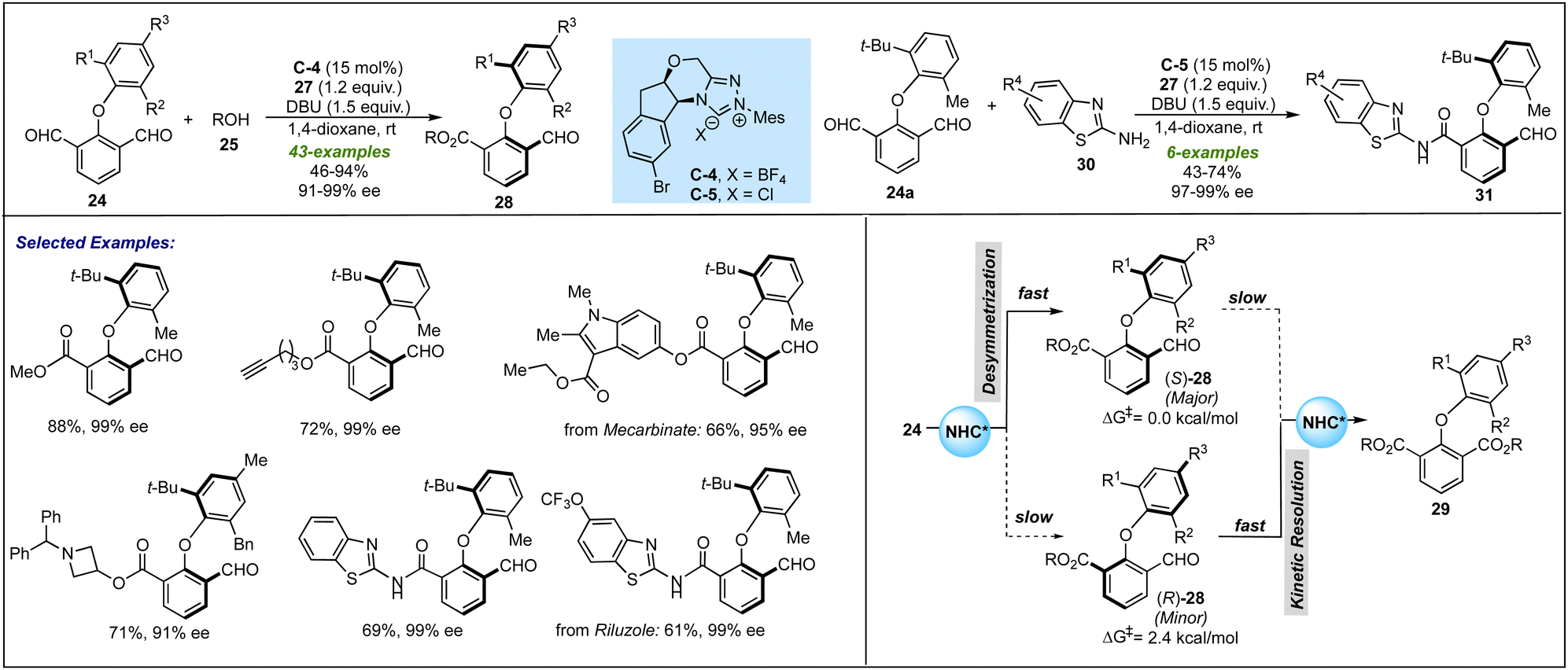 | ||
| Scheme 7 Ye's work on the NHC-catalyzed desymmetrization for atropselective synthesis of biaryl ethers. | ||
Soon after the works of Biju's and Ye's lab, the research groups of Zeng27 and Gao28 designed similar NHC-catalysed strategies to synthesize the biaxial biaryl ethers. Using C-3 as NHC-pre-catalyst and Cs2CO3 as a base assisted the above-described desymmetrization of isopthaladehyde derivatives 24 with a broad range of alcohols (Scheme 8).27 Again, an excellent level of enantioselectivity of esters 28 or alcohols 32 (formed by reduction of 28) was observed. With the triazolium salt C-6 as an NHC precursor, desymmetrization of isopthaladehyde derivatives 24 with a wide variety of aliphatic alcohols and some phenol derivatives yielded the desired biaryl ethers in good to excellent enantioselectivities (Scheme 9).28 The last two strategies are important not only in the context of synthetic methods leading to atropisomeric biaryl ethers but also in the utilization of corresponding biaxial ethers in chiral catalysis.
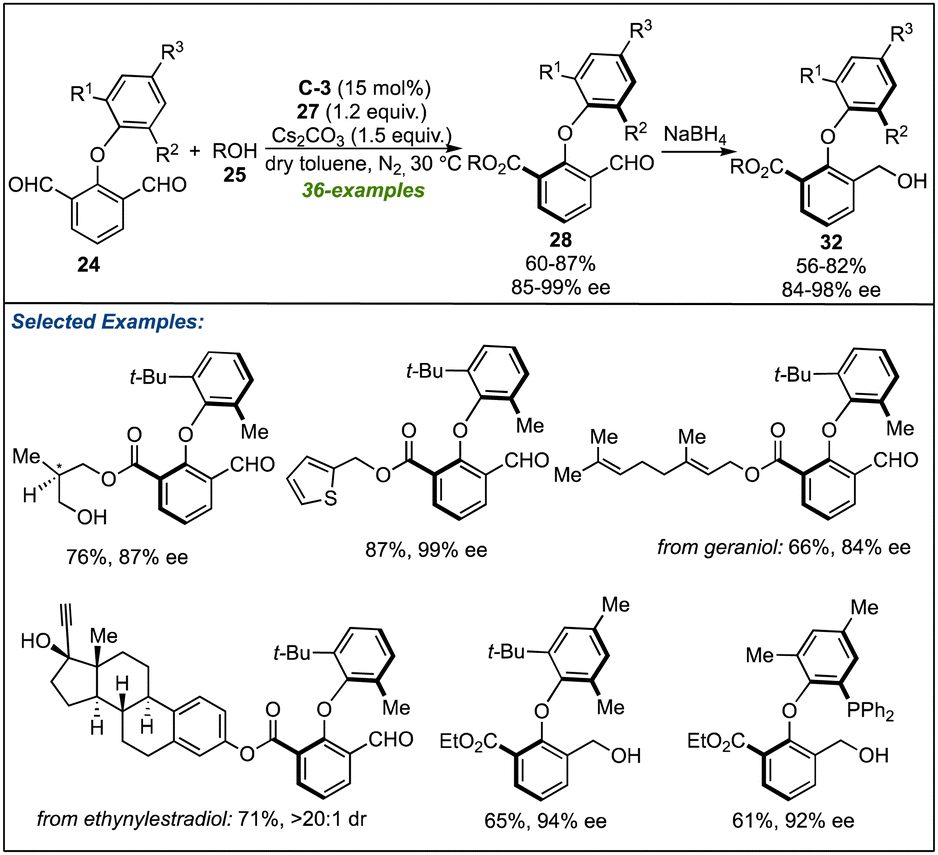 | ||
| Scheme 8 Zeng's work on the NHC-catalyzed desymmetrization for atropselective synthesis of axially chiral biaryl ethers. | ||
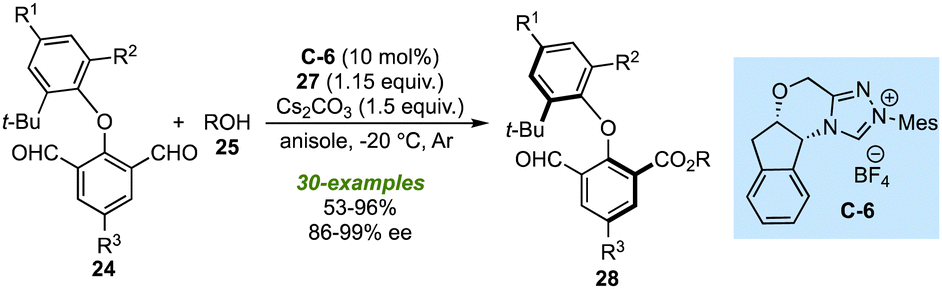 | ||
| Scheme 9 Gao's work on the NHC-catalyzed desymmetrization for atropselective synthesis of axially chiral biaryl ethers. | ||
3.3. Transition-metal catalyzed approach
Recently, Yao and co-workers applied transition-metal catalysis to provide direct access to the dual axial chirality among biaryl ether scaffolds. A new class of bisalkyne-based biaryl ethers 33 has been developed for Cu-catalyzed azide–alkyne Huisgen cycloaddition (CuAAC), leading to desymmetrization of the substrate (Scheme 10).29 An indane fused BOX ligand L-1, with the assistance of copper metal, catalyses the cycloaddition reaction between bis alkynes and azides to give a diverse array of axially chiral C–O atropisomers 34 in excellent yields and enantioselectivities. The developed methodology accommodates diverse substitution patterns irrespective of the nature and position of groups on the bisalkynes. The cycloaddition reaction proceeded smoothly with bio-active molecules such as zidovudine to furnish the desired desymmetrized product in 79% yield and 97:3 dr. It was observed that steric bulk at the C2 position is necessary to provide satisfactory results as replacing the bulky group (tert-Bu, tert-pentyl, etc.) with ethyl or isopropyl greatly affected the enantioselectivity of the corresponding products (18% ee with isopropyl and racemic with ethyl). The control experiment revealed that a prolonged reaction time of 24 hours resulted in the formation of di-triazole product 35 of 21% via a double CuAAC with a slight enantioselectivity enhancement (96% ee to 99% ee) of 34. This result suggests that the enantio-induction is primarily governed by the desymmetrization step, with little role of the KR.
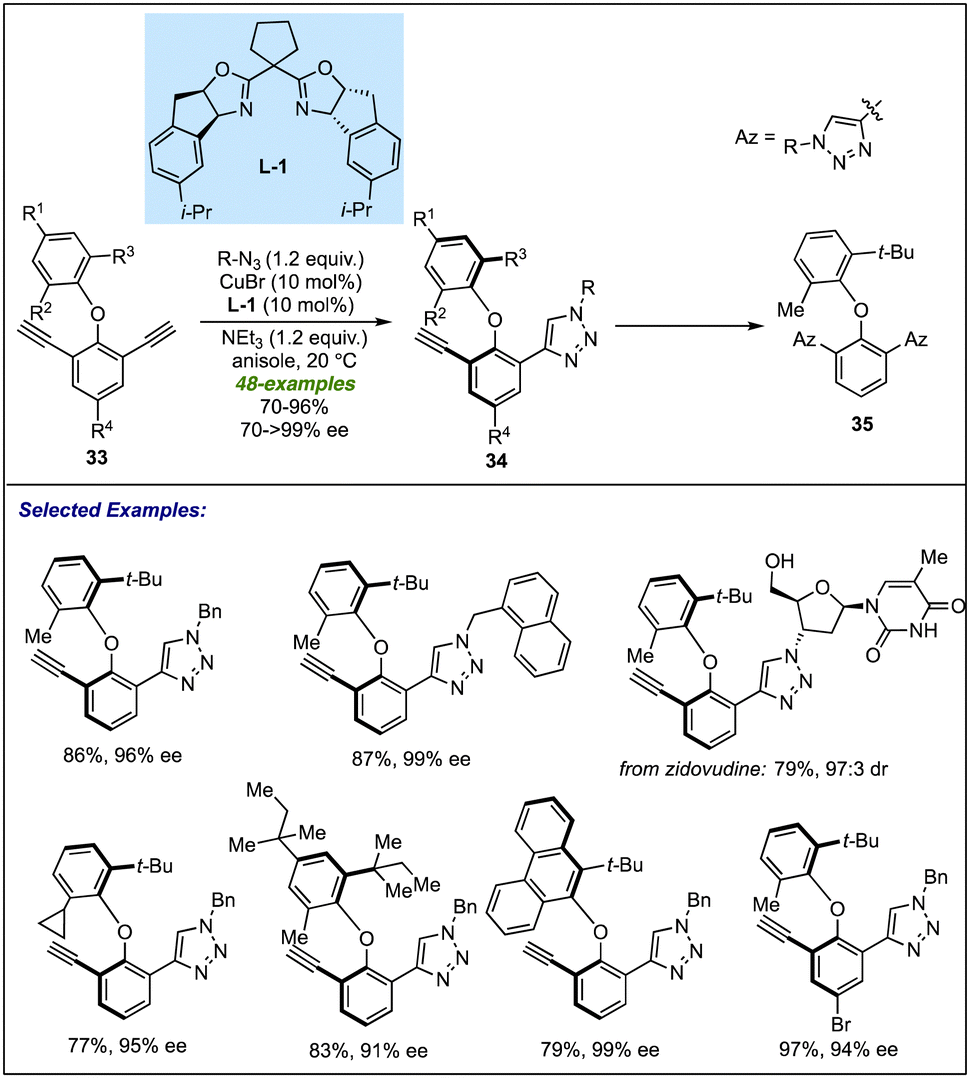 | ||
| Scheme 10 CuAAC for atropselective synthesis of axially chiral biaryl ethers. | ||
4. Catalytic atropselective synthesis of related ethers
Although a significant upsurge has been seen in the last year for procuring biaryl ethers, it was in 2018 when Gustafson's group laid a foundation for the catalytic atropselective synthesis of such biaxially chiral molecules. Their work was focused on the enantioselective synthesis of naphthoquinone-based aryl ethers (Scheme 11).30 By using cinchona-derived urea-bearing quaternary ammonium salt C-7, direct access to atropisomeric biaryl ethers 37 or 38 has been achieved through C(sp2)–H alkylation of substrate 36 with nitromethane or by a similar C(sp2)–H alkylation followed by second nitromethane addition. The C-7 assisted 1,4-nucleophilic addition of nitromethane to quinones 36, followed by the elimination of nitrous acid, resulting in atropselective synthesis of quinone-based biaryl ethers 37. Among the screened substrates, in some instances, the nitroethylated products 38 were isolated as a major product in decent yields and moderate enantioselectivities. In these cases, after eliminating nitrous acid from intermediate 39, the enone 40 (if having a sufficiently longer lifetime) undergoes another Michael-type addition to provide 41, which, after oxidation, results in the corresponding nitroethylated product 38. Notably, after one round of tituration, the enantioselectivity of both products has been enriched to an excellent level. The stereochemical model for the formation of the desired product revealed that there was hydrogen bonding between the urea moiety of the catalyst with the ether oxygen and one of the carbonyl carbons of the low-energy conformer of quinone substrate. As a result of this, the quinone-based biaxial biaryl ether locks into feasible (aS)-exo atropisomers.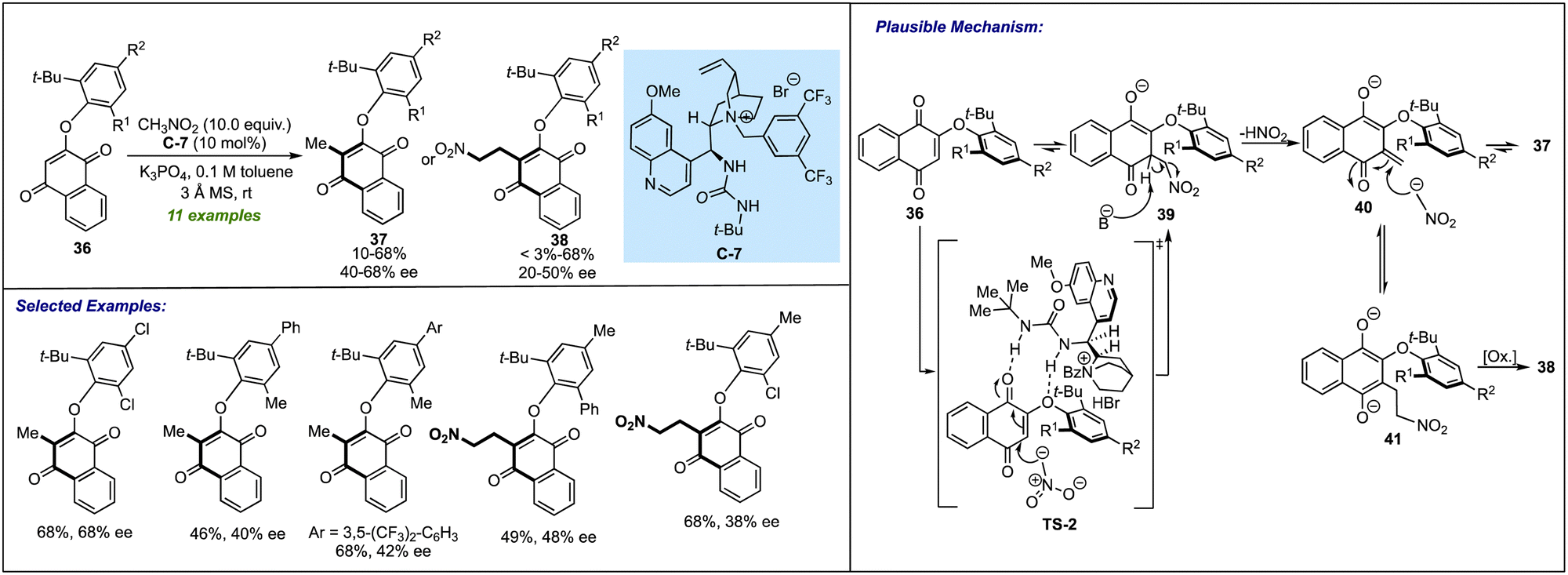 | ||
| Scheme 11 Atropselective synthesis of naphthoquinone-based aryl ethers. | ||
5. Catalytic atropselective synthesis of related amines
Despite being recognized as potential chemotypes in medicinal chemistry and many FDA-approved drugs,12 the atropselective synthesis of biaryl amines has been completely overlooked. Besides the early efforts of Kawabata19 and Clayden20 towards establishing the axial chirality among biaryl amines, Gustafson's group devised the atropselective synthesis of N-aryl quinoids-based biaryl amines 43 (Scheme 12).31 Their work disclosed a chiral phosphoric acid C-8 catalyzed halogenation of N-aryl quinoids 42, yielding the desired product 43 in excellent yields and enantioselectivities. Under ambient conditions, N-aryl quinoid-derived biaryl amines were found to be exhibiting strong N–H–O hydrogen bonding interactions, resulting in high stereochemical stability of the exo-product (ΔG≠= 30 kcal mol−1) over endo-product in both protic and aprotic solvents. During the reaction, it was observed that replacing aryl substitution at R1 and R2 by methyl groups suddenly resulted in an immense loss of ee value (20% ee). This is mainly attributed to the fact that π–π-stacking between the catalyst and aryl rings of the substrates at the R1 and R2 positions play a crucial role in leading to high enantiomeric excess. It was also found that decreasing the bulk by replacing the tert-butyl group with iso-propyl and trifluoromethyl groups drastically decreased the enantioselectivity to 28% ee and 12% ee, respectively, probably due to a low barrier of racemization. It was postulated that at onset, the C-8 catalyst is involved in hydrogen bonding with the stable exo-conformation of the quinoid substrate (TS-3). The resulting hydrogen bonding increases the energy of HOMO, i.e. the enamine-like amino naphthoquinone. The rotation around the C–N axis allows the positioning of the t-Bu group to be far away from the catalyst, thus organizing into (aS)-conformation. This conformation allows the π–π-stacking of the ortho-aryl group with the naphthyl group of the catalyst. Simultaneously, the acidic proton of C-8 activates the electrophilic halogenating reagent (NXS) via H-bonding, thus directing the halogenation towards the naphthoquinone site, and the axis will be further reinforced, leading to the selective formation of the product with (aS)-configuration, whereas the formation of (aR)-product is limited due to steric crowing and lack of π–π-stacking (TS-4).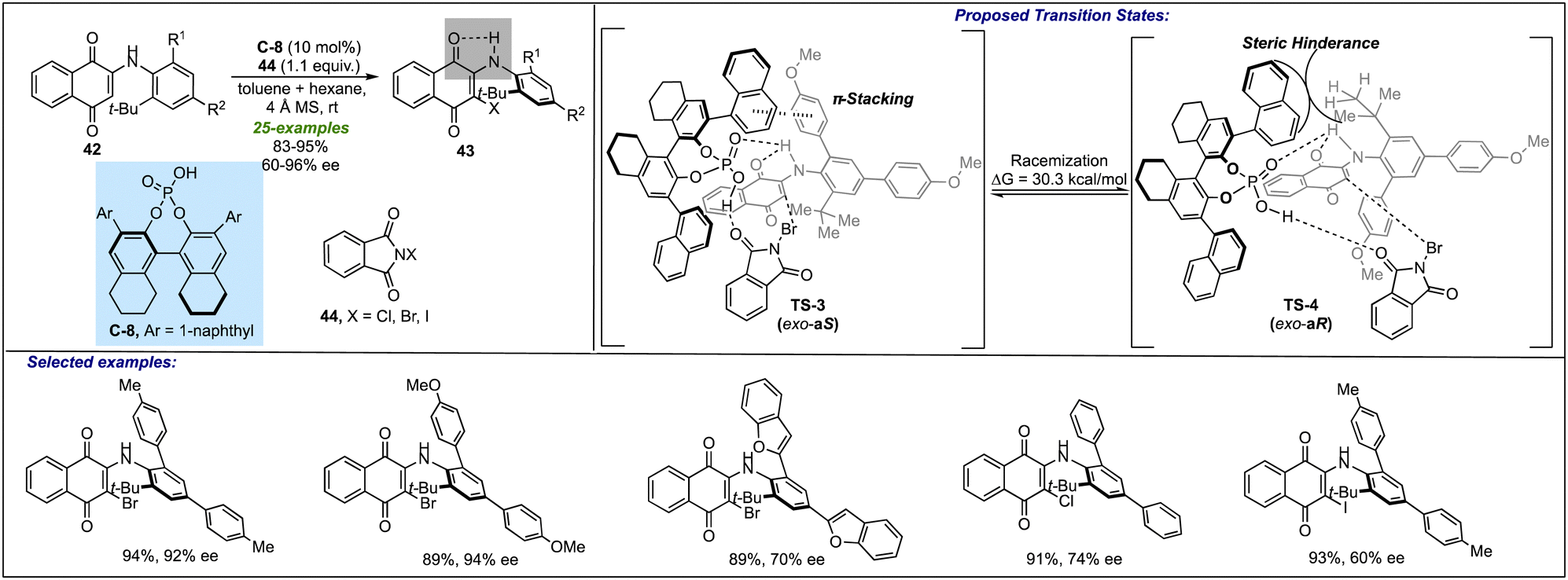 | ||
| Scheme 12 Atropselective synthesis of naphthoquinone-based aryl amines. | ||
Conclusions & outlook
Widespread applications and interesting properties of atropisomers have led to the discovery of new synthetic methods for selectively synthesizing these classes of compounds. This highlight article has summed up the importance, early precedents and asymmetric methods leading to a unique class of biaryl atropisomers viz biaryl ethers and related amines. Whereas the evidence of atropisomers in such molecules has long been recognized, their asymmetric synthesis has gained recognition only recently. In this context, desymmetrization reactions under the action of chiral catalysts have emerged as an efficient strategy. In most cases, the desymmetrization step imparts the desired enantio-differentiation; however, the role of KR has also been observed in improving the enantioselectivity. Apart from the first example on the catalytic pathway leading to enantioselective synthesis of biaryl ethers by using enzymes, recently, the potential of organocatalysts such as chiral Brønsted acid and NHC have been explored for accessing such biaxial biaryls. In addition, a transition metal-catalyzed desymmetrization/KR process has been developed to achieve dual axially chiral biaryl ethers. Regardless of the major challenges due to flexible C–X {X = O, N, S} axes and the prolonged distance between the groups imparting restricted rotations, all the seminal contributions discussed above provide excellent enantioselectivities of the biaryl ethers with a sufficiently longer half-life of racemization. Impressively, diverse precursors with wide functional group tolerance, including biologically relevant compounds, have been well accommodated under mild reaction conditions. In a couple of cases, the applications of newly developed enantiopure diaryl ethers have been successfully explored as chiral catalysts.Though the evidences of dual axial chirality due to the hindrance in rotation were apparent for related biaryl amines and sulfides, nothing much has been seen in terms of asymmetric methods for accessing these scaffolds. At this stage, we believe that these current developments will open a new area in asymmetric synthesis,32 which will not only be limited to the atropselective synthesis of biaryl ethers but may extend up to the enantioselective synthesis of dual axial biaryls bearing C–N–C, C–S–C, C–P–C linkage, etc. The enantioselective synthesis of these biaxial biaryls is also expected to extend beyond the desymmetrization process involving the site-specific modifications of a selected aryl ring or the substituent attached to the aryl ring. Beyond desymmetrization, at this stage, we can anticipate that the developing asymmetric methods relying on coupling reactions or direct C–H functionalization may also lead to the atropselective synthesis of biaryl ethers and related molecules.
Author contributions
N. Kotwal and P. Chauhan have contributed to the original draft preparation, editing, and proofreading of the manuscript of this highlight article.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are highly grateful to the researchers who have contributed to developing synthetic methods for accessing the enantiopure biaryl ethers and related scaffolds. This activity was supported by the Science & Engineering Research Board (SERB) India (File Number: CRG/2022/003212). N. K. thanks the Department of Science & Technology, India, for the INSPIRE fellowship.References
- J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562–1583 RSC.
- (a) S. R. LaPlante, P. J. Edwards, L. D. Fader, A. Jakalian and O. Hucke, ChemMedChem, 2011, 6, 505–513 CrossRef CAS PubMed; (b) S. R. LaPlante, L. D. Fader, K. R. Fandrick, D. R. Fandrick, O. Hucke, R. Kemper, S. P. F. Miller and P. J. Edwards, J. Med. Chem., 2011, 54, 7005–7022 CrossRef CAS PubMed.
- (a) J. M. Lassaletta, Atropisomerism and Axial Chirality, World Scientific Publishing, Singapore, 2019; (b) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- (a) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155–3212 CrossRef CAS PubMed; (b) Y. M. Li, F. Y. Kwong, W. Y. Yu and A. S. C. Chan, Coord. Chem. Rev., 2007, 251, 2119–2144 CrossRef CAS.
- For selected recent reviews on asymmetric synthesis of atropisomers: (a) J. W. Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC; (b) J. K. Cheng, S. H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS PubMed; (c) N. Kotwal, Tamanna and P. Chauhan, Chem. Commun., 2022, 58, 11031–11044 RSC; (d) P. R. Salamanca, R. Fernandez, V. Hornillos and J. M. Lassaletta, Chem. – Eur. J., 2022, 28, e202104442 CrossRef PubMed; (e) X. F. Bai, Y. M. Cui, J. Cao and L. W. Xu, Acc. Chem. Res., 2022, 55, 2545–2561 CrossRef CAS PubMed; (f) H. H. Zhang, T. Z. Li, S. J. Liu and F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202311053 CrossRef CAS PubMed.
- For selected examples see: (a) E. A. Couladouros and V. I. Moutsos, Tetrahedron Lett., 1999, 40, 7027–7030 CrossRef CAS; (b) L. Ciasullo, A. Casapullo, A. Cutignano, G. Bifulco, C. Debitus, J. Hooper, L. G. Paloma and R. Riccio, J. Nat. Prod., 2002, 65, 407–410 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, C. N. C. Boddy, S. Bräse and N. Winssinger, Angew. Chem., 1999, 111, 2230–2287 ( Angew. Chem., Int. Ed. , 1999 , 38 , 2096–2152 ) CrossRef; (b) K. C. Nicolaou and C. N. C. Boddy, J. Am. Chem. Soc., 2002, 124, 10451–10455 CrossRef CAS PubMed; (c) B. M. Crowley and D. L. Boger, J. Am. Chem. Soc., 2006, 128, 2885–2892 CrossRef CAS PubMed.
- R. D. Kavthe, J. R. A. Kincaid and B. H. Lipshutz, ACS Sustainable Chem. Eng., 2022, 10, 16896–16902 CrossRef CAS PubMed.
- M. Basilaia, M. H. Chen, J. Secka and J. L. Gustafson, Acc. Chem. Res., 2022, 55, 2904–2919 CrossRef CAS PubMed.
- (a) D. G. Brown and H. J. Wobst, J. Med. Chem., 2021, 64, 2313–2338 CrossRef PubMed; (b) R. K. Sadineni, R. K. Raplolu, V. V. N. K. V. P. Raju, S. Srinivasu, S. Malladi and N. Mulakayala, Chem. Pap., 2021, 75, 1475–1483 CrossRef CAS; (c) S. Guo, X. Pang, L. Peng, M. Zhan, L. Fan, Y. Gong, F. Kang and Y. Chen, Bioorg. Med. Chem. Lett., 2015, 25, 2425–2428 CrossRef CAS PubMed.
- (a) F. Fang, M. M. Xue, M. Ding, J. Zhang, S. Li and X. Chen, Chem. – Asian J., 2021, 16, 2489–2494 CrossRef CAS PubMed; (b) R. Wang, Y. Wu, J. Wang, H. Huang, Y. Wang, S. Xu and F. Zhao, J. Mol. Struct., 2022, 1257, 132642 CrossRef CAS.
- For selected examples, see: (a) N. M. Levinson and S. G. Boxer, PLoS One, 2012, 7, e29828 CrossRef CAS PubMed; (b) P. Koelblinger, J. Dornbierer and R. Dummer, Future Oncol., 2017, 13, 1755–1766 CrossRef CAS PubMed; (c) Q. Liu, Y. Sabnis, Z. Zhao, T. Zhang, S. J. Buhrlage, L. H. Jones and N. S. Gray, Chem. Biol., 2013, 20, 146–159 CrossRef CAS PubMed.
- J. A. McRae, R. Y. Moir, J. J. Ursprung and H. H. Gibbs, J. Org. Chem., 1954, 19, 1500–1508 CrossRef CAS.
- M. Dahlgard and R. Q. Brewster, J. Am. Chem. Soc., 1958, 80, 5861–5863 CrossRef CAS.
- K. Fuji, T. Oka, T. Kawabata and T. Kinoshita, Tetrahedron, 1998, 39, 1373–1376 CrossRef CAS.
- M. S. Betson, J. Clayden, C. P. Worrall and S. Peace, Angew. Chem., Int. Ed., 2006, 45, 5803–5807 ( Angew. Chem. , 2006 , 118 , 5935–5939 ) CrossRef CAS PubMed.
- J. Clayden, J. Senior and M. Helliwell, Angew. Chem., Int. Ed., 2009, 48, 6270–6273 ( Angew. Chem. , 2009 , 121 , 6388–6391 ) CrossRef CAS PubMed.
- (a) D. Casarini, L. Lunazzi and G. Placucci, J. Org. Chem., 1987, 52, 4721–4726 CrossRef CAS; (b) D. P. Curran, H. Qi, S. J. Geib and N. C. DeMello, J. Am. Chem. Soc., 1994, 116, 3131–3132 CrossRef CAS.
- (a) T. Kawabata, C. Jiang, K. Hayashi, K. Tsubaki, T. Yoshimura, S. Majumdar, T. Sasamori and N. Tokitoh, J. Am. Chem. Soc., 2009, 131, 54–55 CrossRef CAS PubMed; (b) K. Hayashi, N. Matubayasi, C. Jiang, T. Yoshimura, S. Majumdar, T. Sasamori, N. Tokitoh and T. Kawabata, J. Org. Chem., 2010, 75, 5031–5036 CrossRef CAS PubMed.
- R. Costil, H. J. A. Dale, N. Fey, G. Whitcombe, J. V. Matlock and J. Clayden, Angew. Chem., Int. Ed., 2017, 56, 12533–12537 CrossRef CAS PubMed.
- B. Yuan, A. Page, C. P. Worrall, F. Escalettes, S. C. Willies, J. J. W. McDouall, N. J. Turner and J. Clayden, Angew. Chem., Int. Ed., 2010, 49, 7010–7013 ( Angew. Chem. , 2010 , 122 , 7164–7167 ) CrossRef CAS PubMed.
- L. Dai, Y. Liu, Q. Xu, M. Wang, Q. Zhu, P. Yu, G. Zhong and X. Zeng, Angew. Chem., Int. Ed., 2023, 62, e202216534 ( Angew. Chem. , 2023 , 135 , e202216534 ) CrossRef CAS PubMed.
- H. Bao, Y. Chen and X. Yang, Angew. Chem., Int. Ed., 2023, 62, e202300481 CrossRef CAS PubMed.
- S. Shee, S. S. Ranganathappa, M. S. Gadhave, R. Gogoi and A. T. Biju, Angew. Chem., Int. Ed., 2023, 62, e202311709 CrossRef CAS PubMed.
- For reports on cooperative desymmetrization/kinetic resolution, see: (a) Y. Wu, M. Li, J. Sun, G. Zheng and Q. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202117340 CrossRef CAS PubMed; (b) M. Zhu, H. J. Jiang, I. Sharanov, E. Irran and M. Oestreich, Angew. Chem., Int. Ed., 2023, 62, e202304475 CrossRef CAS PubMed.
- B. A. Zhou, X. N. Li, C. L. Zhang, Z. X. Wang and S. Ye, Angew. Chem., Int. Ed., 2024, 63, e20231422 Search PubMed.
- Y. Liu, L. Yuan, L. Dai, Q. Zhu, G. Zhong and X. Zeng, ChemRxiv, 2023, preprint DOI:10.26434/chemrxiv-2023-78vb3.
- L. Li, W. Ti, T. Miao, J. Ma, A. Lin, Q. Chu and S. Gao, J. Org. Chem., 2024, 89, 4067–4073 CrossRef CAS PubMed.
- X. Han, L. Chan, Y. Yan, Y. Zhao, A. Lin, S. Gao and H. Yao, ACS Catal., 2024, 14, 3457–3481 CrossRef.
- A. N. Dinh, R. R. Noorbehesht, S. T. Toenjes, A. C. Jackson, M. A. Saputra, S. M. Maddox and J. L. Gustafson, Synlett, 2018, 2155–2160 CAS.
- S. D. Vaidya, S. T. Toenjes, N. Yamamoto, S. M. Maddox and J. L. Gustafson, J. Am. Chem. Soc., 2020, 142, 2198–2203 CrossRef CAS PubMed.
- During the preparation of this article, a few related works were published in the literature: (a) Y. Wu, X. Guan, H. Zhao, M. Li, T. Liang, J. Sun, G. Zheng and Q. Zhang, Chem. Sci., 2024, 15, 4564–4570 RSC; (b) L. Dai, X. Zhou, J. Guo, Q. Huang and Y. Lu, Chem. Sci., 2024, 15, 5993–6001 RSC; (c) Y. Wang, R. Mi, S. Yu and X. Li, ACS Catal., 2024, 14, 4638–4647 CrossRef CAS; (d) J. Xu, W. Lin, H. Zheng and X. Li, ACS Catal., 2024, 14, 6667–6673 CrossRef CAS; (e) Z. Ye, W. Xie, D. Wang, H. Liu and X. Yang, ACS Catal., 2024, 14, 4958–4967 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |