
Spectroelectrochemical determination of thiolate self-assembled monolayer adsorptive stability in aqueous and non-aqueous electrolytes†
Abdur-Rahman
Siddiqui‡
 a,
Jeanne
N'Diaye‡
ab,
Armando
Santiago-Carboney‡
a,
Kristin
Martin
a,
Rohit
Bhargava
abc and
Joaquín
Rodríguez-López
*ab
a,
Jeanne
N'Diaye‡
ab,
Armando
Santiago-Carboney‡
a,
Kristin
Martin
a,
Rohit
Bhargava
abc and
Joaquín
Rodríguez-López
*ab
aDepartment of Chemistry, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, USA. E-mail: joaquinr@illinois.edu
bThe Beckman Institute for Advanced Science and Technology, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, USA
cDepartment of Bioengineering and Cancer Center at Illinois, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, USA
First published on 2nd April 2024
Abstract
Self-assembled monolayers (SAM) are ubiquitous in studies of modified electrodes for sensing, electrocatalysis, and environmental and energy applications. However, determining their adsorptive stability is crucial to ensure robust experiments. In this work, the stable potential window (SPW) in which a SAM-covered electrode can function without inducing SAM desorption was determined for aromatic SAMs on gold electrodes in aqueous and non-aqueous solvents. The SPWs were determined by employing cyclic voltammetry, attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS), and surface plasmon resonance (SPR). The electrochemical and spectroscopic findings concluded that all the aromatic SAMs used displayed similar trends and SPWs. In aqueous systems, the SPW lies between the reductive desorption and oxidative desorption, with pH being the decisive factor affecting the range of the SPW, with the widest SPW observed at pH 1. In the non-aqueous electrolytes, the desorption of SAMs was observed to be slow and progressive. The polarity of the solvent was the main factor in determining the SPW. The lower the polarity of the solvent, the larger the SPW, with 1-butanol displaying the widest SPW. This work showcases the power of spectroelectrochemical analysis and provides ample future directions for the use of non-polar solvents to increase SAM stability in electrochemical applications.
Introduction
Self-assembled monolayers (SAM) are regarded as a convenient method for functionalizing metal electrode interfaces.1–5 Thiols spontaneously adsorb and organize themselves through intermolecular interactions into monolayers on metallic surfaces commonly used in electrochemistry such as Au, Cu, Pt, and others.1–5 Due to the ease of preparation and diversity of thiol molecules, thiolate SAMs allow the tailoring of electrode surfaces to many desired properties.1–5 Thus, SAM-covered electrodes apply to a plethora of electrochemical applications including electrochemical sensors,6–8 electrocatalysis,9 electrosynthesis,10,11 electronic devices,12,13 and batteries.14 The use of SAMs in such electrochemical applications requires a fundamental understanding of their adsorptive stability on the electrode, especially when under electrochemical stress. Yet, in the four decades since SAMs were discovered,15,16 the long-term adsorptive stability of SAMs remains one of their greatest vulnerabilities.17–19 The stability of SAMs is correlated to the formation of a high-quality well-packed layer on the electrode surface, with molecules exhibiting strong intermolecular interactions.17–19 Additionally, the vulnerability of SAMs to various environmental factors such as exposure to high temperatures, light, or air has been thoroughly established.17,20–23Despite these efforts to understand the factors contributing to SAM stability, there are only a handful of studies focusing on the electrochemical stability of SAMs.24–31 Ramos et al. established that the electrochemical stability varies by thiol types and pH for different metals, but the quality of the SAM remains the most relevant factor.25 More importantly, they demonstrated that for all electrodes there are discreet potential limits in which a SAM-covered electrode can operate and maintain its integrity.25 This stable potential window (SPW) is crucial to any electrochemical application utilizing SAMs as the SAM will desorb from the electrode at potentials beyond the SPW. However, these values have only been reported in a few electrolytic systems, primarily in alkaline aqueous electrolytes, as reductive desorption methods in these media have been conveniently used to quantify SAM surface coverage.32–34 The anodic limits have been explored sparsely.34–36 Thus, expanding the reporting of SPWs of SAMs for a variety of electrolyte environments is an essential analysis that is sorely underrepresented in literature. Such analysis would create convenient guidelines for any electrochemical application that uses SAMs.
The SPWs of various well-known SAMs were determined utilizing cyclic voltammetry, attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS), and surface plasmon resonance (SPR). Cyclic voltammetry allows a direct assessment of the SAM's electrochemical stability since the SAM passivates the electrode surface inducing solely capacitive anodic and cathodic currents.24,37–39 The stability of the SAM can be assessed by maintaining a capacitive current during continuous cycling.24,25 In contrast, if the CV profile tends to return to the bare gold baseline, it is taken as an indication that the SAM has desorbed from the electrode surface.24,25 Electrochemical SEIRAS (EC-SEIRAS) enables precise monitoring of the SAMs at an electrode interface due to signal enhancement induced by surface plasmon polaritons.40,41 Similarly, SPR is a plasmon-driven surface-sensitive measurement that is dependent upon the refractive index of material or solution near the metal surface.42 SPR offers the opportunity to measure in situ changes occurring at the gold interface and is easily coupled with electrochemistry.42 Therefore, electrochemical-SPR (EC-SPR) can be utilized to probe the stability of SAMs within their respective SPWs.43
In this work, we expand on the capabilities offered by EC-SEIRAS and EC-SPR to systematically probe SPWs in various electrolytes of interest to the broad electrochemistry community. We report the SPWs of 4 aromatic SAMs in aqueous and non-aqueous solvents on gold electrodes, showing that despite their structural differences, their SPWs remain similar. The aromatic SAMs included 4-mercaptopyridine (4-PySH), 4-mercaptoaniline (4-MA), 4-nitrothiolphenol (4-NTP), and 2-mercaptobenzothiazole (2-MBT). 4-PySH was chosen due to its extensive use and characterization in electrochemical literature.32,44–47 4-MA and 4-NTP were utilized due to their similar structure and unique IR signals stemming from their amine48–50 and nitro51–55 group, respectively. 2-MBT was utilized to act as a control to validate how universal the determined SPWs are for other aromatic SAMs with differing structures, adsorption methods, and surface coverage.56–58 The SPWs were investigated at acidic, neutral, and basic conditions for the aqueous systems. The non-aqueous systems studied were commonly used aprotic solvents and alkyl alcohols.
Materials and methods
Chemicals
All chemicals were purchased from commercial sources and used as received. 4-Mercaptopyridine (95%), 4-mercaptoaniline (97%), 2-mercaptobenzothiazole (97%), potassium hydroxide (KOH, >97%), sodium chloride (NaCl), perchloric acid (HClO4, 70%), acetonitrile (MeCN, 99%), propylene carbonate (PC, 99%), dimethyl formamide (DMF, 99%), tetrabutylammonium hexafluorophosphate (TBAPF6, 98%), and gold(III) chloride trihydrate (99.9%) were purchased from Sigma-Aldrich. 4-Nitrothiophenol (96%), methanol (MeOH, 99%), butanol (BuOH, 99%), lithium chloride (LiCl, 99.5%), and sulfuric acid (H2SO4, trace metal) were purchased from Thermo Fisher Scientific. Molecules from other suppliers include ethanol (EtOH, 200 proof, Decon Labs), and isopropanol (IPA, 99%, Honeywell). Aqueous solutions were made using deionized water (DI) from a Millipore Sigma Direct-Q 8 (resistivity of 18.2 MΩ).Electrode preparation and polishing
All electrochemical experiments were performed using a CHI potentiostat (CH Instruments Inc.) with a polycrystalline 2 mm gold working electrode (CH Instruments Inc.), a platinum wire for the counter electrode, and the Ag/AgCl (3 M KCl) reference electrode placed in the cell via a 0.1 M NaClO4|agar salt bridge. The gold electrode was polished using Buehler MicroCloth polishing pads with 1 μm, 0.3 μm, and 0.05 μm alumina suspensions with DI rinse in between. After the last polishing step, the electrode was sonicated for 5 minutes in acetone, IPA, and DI water. A further electrochemical cleaning step was conducted by cycling from 0 V to 1.3 V (vs. Ag/AgCl) for 30 cycles in 0.5 M H2SO4 at 500 mV s−1.Gold surface modification and surface coverage calculation
All SAMs were formed by submerging the gold electrode in 1 mM solutions of the thiols in EtOH for 10 minutes. Cyclic voltammetry was utilized to assess the surface coverage of the formed thiolate SAMs. Reductive desorption curves (RDC) were performed by cycling between 0 V and −1.3 V in degassed 1 M KOH (Fig. S1a†).1,32–34 Fig. S1b† shows the average surface coverage of each SAM calculated using eqn (1):| Γ = Q/nFA | (1) |
Determining the stable potential window (SPW)
Cyclic voltammograms (CV) were acquired from bare gold electrodes in every solvent tested to act as a baseline to compare against SAM-covered electrodes. Any mention of a baseline is in reference to the CV of a bare gold electrode. The aqueous solvents investigated were 1 M KOH, 0.1 M NaCl, and 0.1 M HClO4. For the aqueous systems tested: the SAM-covered electrode was cycled from open circuit potential (OCP) negatively until reductive desorption occurred. After another SAM adsorption, the SAM-covered electrode was cycled from OCP positively until oxidative desorption occurred. The non-aqueous solvents investigated were 0.1 M TBAPF6 in MeCN, PC, or DMF, and 0.1 M LiCl in MeOH, EtOH, IPA, or BuOH. For the non-aqueous solvents tested: the SAM-covered electrode was cycled from OCP to increasingly negative potentials in increments of 100 mV. The SAM was determined to have desorbed when the current increased to baseline levels when comparing the same potential window. The same procedure was followed for cycling from OCP to increasingly positive potential. Once the SPWs were established in both aqueous and non-aqueous systems, they were tested with the 4 thiol molecules (4-MA, 4-NTP, 4-PySH, 2-MBT) in each solvent in triplicate. To monitor the integrity of the SAM, the average charge cycled during the cycle was determined by integrating under the anodic sweeps of the triplicate CVs and averaging the attained charge. The average charge and its standard deviation were plotted versus the cycle number for easier visualization of the continual passivation of the electrode due to the SAM on the electrode surface.IR instrumentation and SEIRAS substrate preparation
EC-SEIRAS experiments were carried out using a PerkinElmer Spectrum 3 FTIR with a liquid nitrogen-cooled MCT detector. The FTIR was equipped with a VeeMax III (PIKE technologies) specular reflection accessory with a Jackfish electrochemical cell (PIKE technologies) for housing the specialized low-depth micro-grooved ATR crystal (PIKE technologies) internal reflection element (IRE).59 The angle of incidence used was 35° with the spectra collected between 4000–600 cm−1 at 4 cm−1 resolution using an 8.94 mm J-stop at a scan speed of 1 cm−1 s−1. The spectra were taken with an accumulation of 55 scans. The SEIRAS substrate was constructed by vacuum depositing 20 nm of gold onto the IRE using a Temescal electron-beam evaporator at 0.1 Å s−1. The gold-plated IRE was roughened following an electrochemical plating of gold islands from our earlier SEIRAS studies.60 In short using a 10 mM solution of gold(III) chloride trihydrate the potential was pulsed three times via chronoamperometry between open circuit potential and −0.35 V with a pulse width of 0.25 seconds for 300 steps.EC-SEIRAS verification of SPW
All EC-SEIRAS experiments used the gold-plated IRE as the working electrode, a carbon rod as the counter electrode, and an Ag/AgCl (3 M) as the reference electrode. The corresponding electrolytes were used as background for their respective experiments, followed by the modification of the gold-plated IRE with the SAMs dissolved in EtOH. After modification, the solution was removed and the EC-SEIRAS cell was washed thoroughly with EtOH. The IRE was then purged with Argon to eliminate any EtOH contaminates, and the background electrolyte was added back. Potential-dependent spectra were recorded starting at OCP, by holding increasingly negative potentials in increments of 100 mV for 1 minute until the signal from the SAM was lost. After subsequent adsorption of SAMs, the same procedure was followed by holding increasingly positive potentials from OCP.SPR instrumentation
All EC-SPR experiments were carried out using a BI-2500 SPR (Biosensing Instrument Inc.) and utilizing commercially available planar (111) gold single crystal sensor chips (Biosensing Instrument Inc.) in a custom-built static EC-SPR cell (Scheme S1†). The SPR setup is based on the Kretschmann configuration, using a 670 nm light source and a detection speed of 4 milliseconds.EC-SPR validation of SPW
All EC-SPR experiments used the gold sensor chip as the working electrode, a platinum wire as the counter electrode, and an Ag/AgCl (3 M) as the reference electrode. For all SPR experiments, both the bare gold baseline and SAM-modified sensor chips were cycled within the SPWs for a fair comparison. The baseline sensorgrams were obtained in the corresponding electrolytes for each SAM and electrolyte pairing. The gold sensor chip was then modified with SAMs for 15 minutes. The solution was removed, and the cell was rinsed to eliminate any excess electrolyte. Then, beginning at OCP, the systems were cycled within the determined SPWs to monitor refractive index signal shifts. Due to the gold sensor chip being planar (111) and of nanometer thickness, an added level of mindfulness is taken to ensure that Cl− ions did not etch away gold when working with all chloride-based electrolytes.61Results and discussion
Establishing stable potential windows in aqueous systems
The reductive and oxidative desorption of SAMs establishes the two potential limits in which a SAM can remain adsorbed to the gold. Thus, the electrochemical stability of SAMs in aqueous systems is strictly dependent on mitigating these two desorption reactions. Scheme 1a depicts the potential range between both desorption reactions as the SPW of the SAMs. Scheme 1b displays the 4 different thiol molecules used in the investigation of SPWs.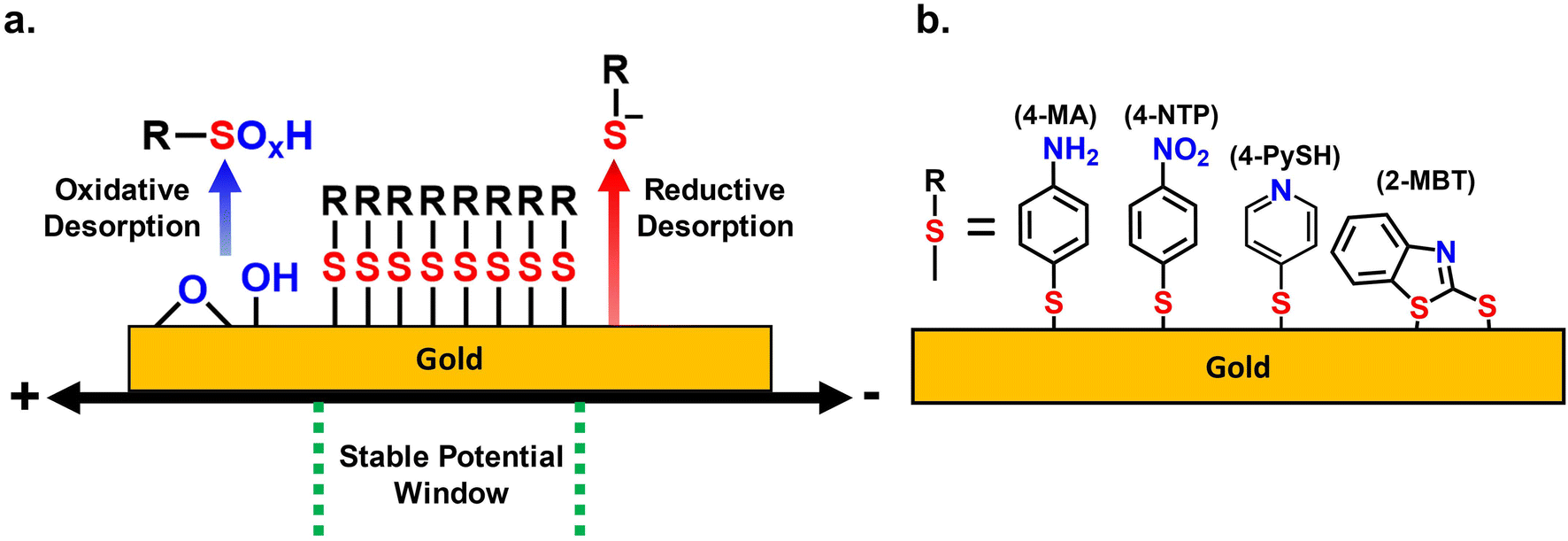 | ||
| Scheme 1 (a) Depiction of the stable potential window (SPW) of thiolate SAMs on gold in aqueous solvents. (b) The thiol molecules used to investigate the SPWs: 4-mercaptoaniline (4-MA), 4-nitrothiolphenol (4-NTP), 4-mercaptopyridine (4-PySH), and 2-mercaptobenzothiazole (2-MBT). | ||
Fig. 1a and b depict the CVs taken in degassed 1 M KOH with a bare gold electrode and a 4-PySH-covered electrode. When cycling negatively with a bare gold electrode, the CV is solely capacitive until the hydrogen evolution reaction (HER) occurs at potentials beyond −1 V (Fig. 1a).33,35 Conversely, on the cathodic sweep with a SAM-covered electrode, the stripping of the 4-PySH from the electrode surface occurs at approximately −0.5 V.32 The reductive desorption of the SAM is represented in eqn (2) and Scheme 1a, where the Au–S bond cleaves and the SAM desorbs as thiolate species.1,32–34 Additional peaks are observed between −0.8 V and −1.1 V, both corresponding to the desorption of sulfur species on terraces and steps of gold, respectively (eqn (3)).32
| Au − SR + e− → Au + RS− | (2) |
| H2O + Au − S + 2e− → HS− + OH− + Au | (3) |
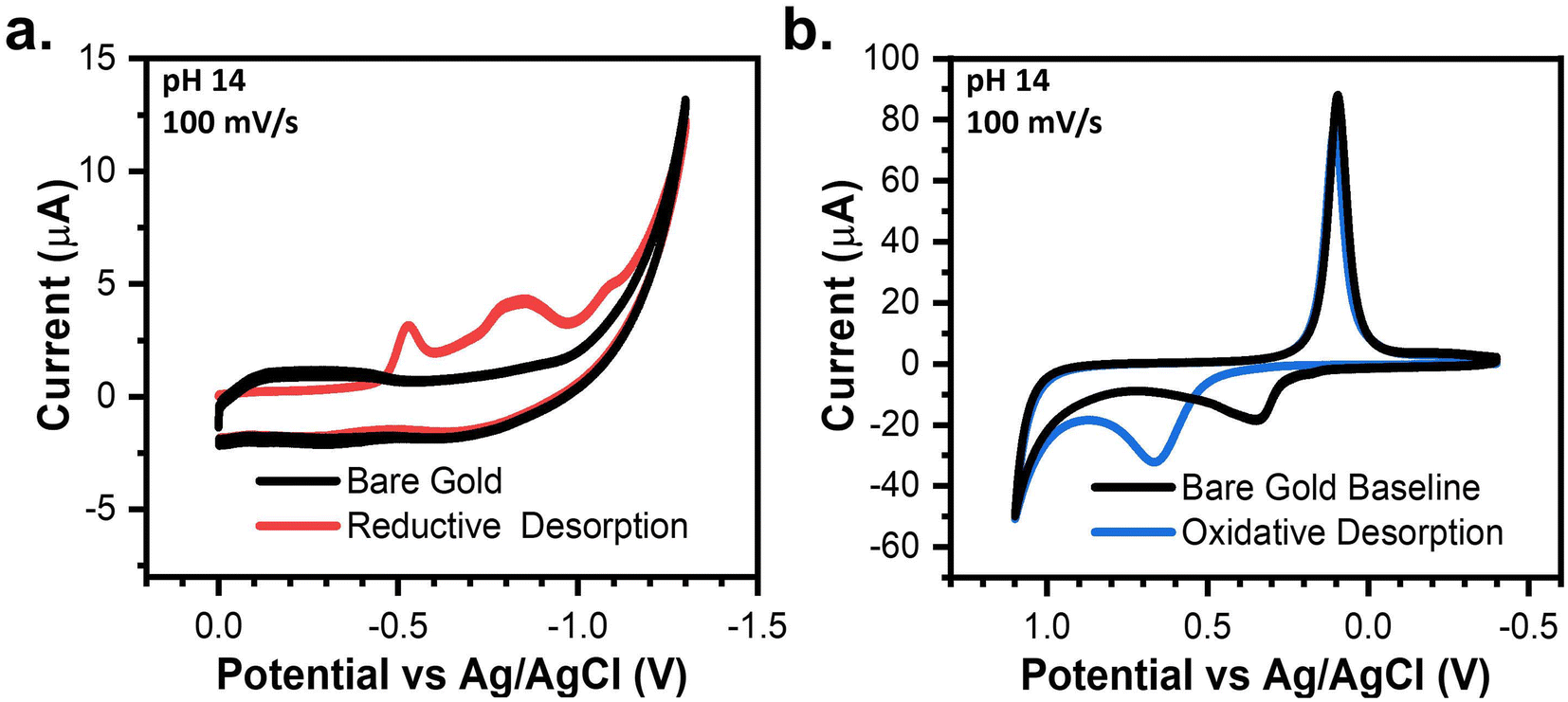 | ||
| Fig. 1 CVs in degassed 1 M KOH showing the (a) reductive desorption and (b) oxidative desorption of 4-PySH compared to baseline bare gold CVs. | ||
In contrast, when cycling positively with a bare gold electrode, the anodic sweep comprises the formation of gold oxides (eqn (4)) at ∼0.3 V and the oxygen evolution reaction (OER) at 1.1 V (Fig. 1b).35,36 On the returning cathodic sweep, the stripping of the gold oxides can be observed at ∼0.1 V.35,36 When cycling with a SAM-covered electrode, the oxidative desorption (presumably eqn (5)) and gold oxidation coincide. Fig. 1b shows the presence of a SAM delays the onset of gold oxidation by 300 mV and the peak current is doubled due to the simultaneous desorption of the SAM and oxidation of gold.34–36
| Au + 3H2O → Au2O3 + 6e− + 6H+ | (4) |
| Au − SR + 2H2O → RSO2H + 3e− + 3H− (5) | (5) |
Determining the effects of pH on the aqueous stable potential windows
Fig. 2 establishes the SPWs within aqueous systems at different pH values, including alkaline, neutral, and acidic environments. At pH 14, reductive desorption begins to onset at −0.5 V and oxidative desorption occurs at 0.4 V. However, we observed that cycling between these limits leads to an increase in the capacitive current and gold oxidation and stripping are observed, indicating degradation of the SAM (Fig. S2†). Cycling in a narrower window allowed us to identify a more conservative SPW. For example, cycling between 0.3 V and −0.4 V for 50 cycles showed no changes in the voltametric response, thus defining the viable SPW (Fig. 2a).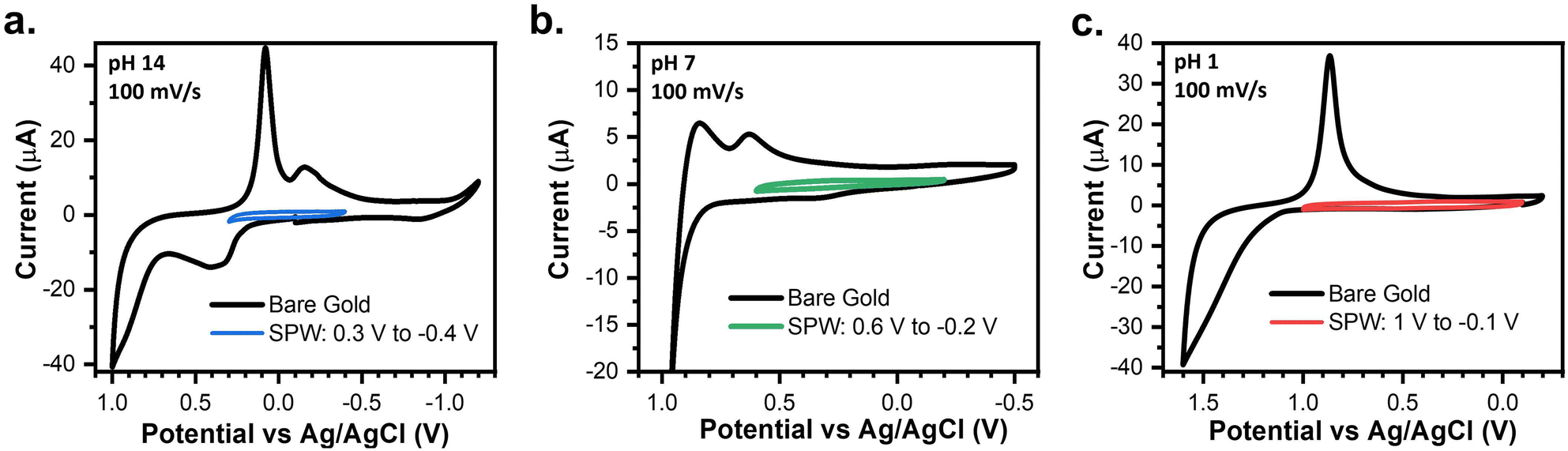 | ||
| Fig. 2 CVs of a bare gold electrode compared to a 4-PySH-covered electrode cycled within the SPW in (a) 1 M KOH (pH 14), (b) 0.1 M NaCl (pH 7), and (c) 0.1 M HClO4 (pH 1). | ||
Gold oxidation and oxidative desorption are both proton-coupled electron transfer (PCET) reactions (eqn (4) and (5)).34–36 Thus as pH decreases, the onset potentials of both reactions shift a positive 59 mV per unit of pH.25,35,36 The onset of gold oxide formation is ∼0.3 V, ∼0.6 V, and ∼1 V at pH 14, 7, and 1, respectively (Fig. 2a–c).25,36 Thus, in aqueous systems as the pH lowers, the anodic limit of the SPW expands into more positive potentials.25 Conversely, the reductive desorption is pH-independent except when the pH < pKa of the SAM.25,34 The pKa of the SAMs used in this study are reported in Table S1.† For every SAM the reductive desorption of the SAMs is pH-independent and pH-dependent at pH 14 and pH 1, respectively. Thus, as the pH lowers the reductive desorption will shift positively 59 mV pH−1. This effectively shortens the reductive limit of the SPW to −0.1 V, −0.2 V, and −0.4 V at pH 1, 7, and 14, respectively (Fig. 2a–c). Moreover, other PCET reactions such as the oxygen reduction reaction (ORR) and HER, disrupt SAM stability.25,34 Thus, the SPW is 800 mV at pH 7 (−0.2 V to 0.6 V) and 1100 mV at pH 1 (1 V to −0.1 V) (Fig. 2b and c).
Electrochemical assessment of the aqueous stable potential windows
To verify if the SPW is universal to all SAMs tested, 4-MA, 4-NTP, 4-PySH, and 2-MBT covered electrodes were cycled within the SPWs identified at pH 14, 7, and 1 for 50 cycles in triplicate (Fig. 3). The SAM-covered electrodes were compared to that of the gold baseline cycled within SPW for a clear demonstration of SAM stability.24,37 Remarkably, for every SAM at the studied pHs, the CVs retain their capacitive profile.24,37 Since the adsorption of SAMs passivates the electrode surface, with a reduced measured current.24,37 The integrity of the SAM can be assessed based on the current levels, however, when the SAM remains intact it is harder to visualize and compare the differences in current strictly based on the capacitive CVs (Fig. 3a–c). To establish an easier method to monitor the integrity of the SAM, the average charge cycled during the cycle was determined by integrating under the anodic sweeps of the triplicate CVs. The electrochemical stability of the SAMs can be better assessed by plotting the charge from the triplicate CVs versus the cycle number (Fig. 3d–f). These plots reiterate the consistent electrochemical stability of the SAM within the SPW for every SAM at every pH.24,25 Regardless of the SAM, the pH of the solution remains the main factor for the electrochemical stability of these aromatic SAMs.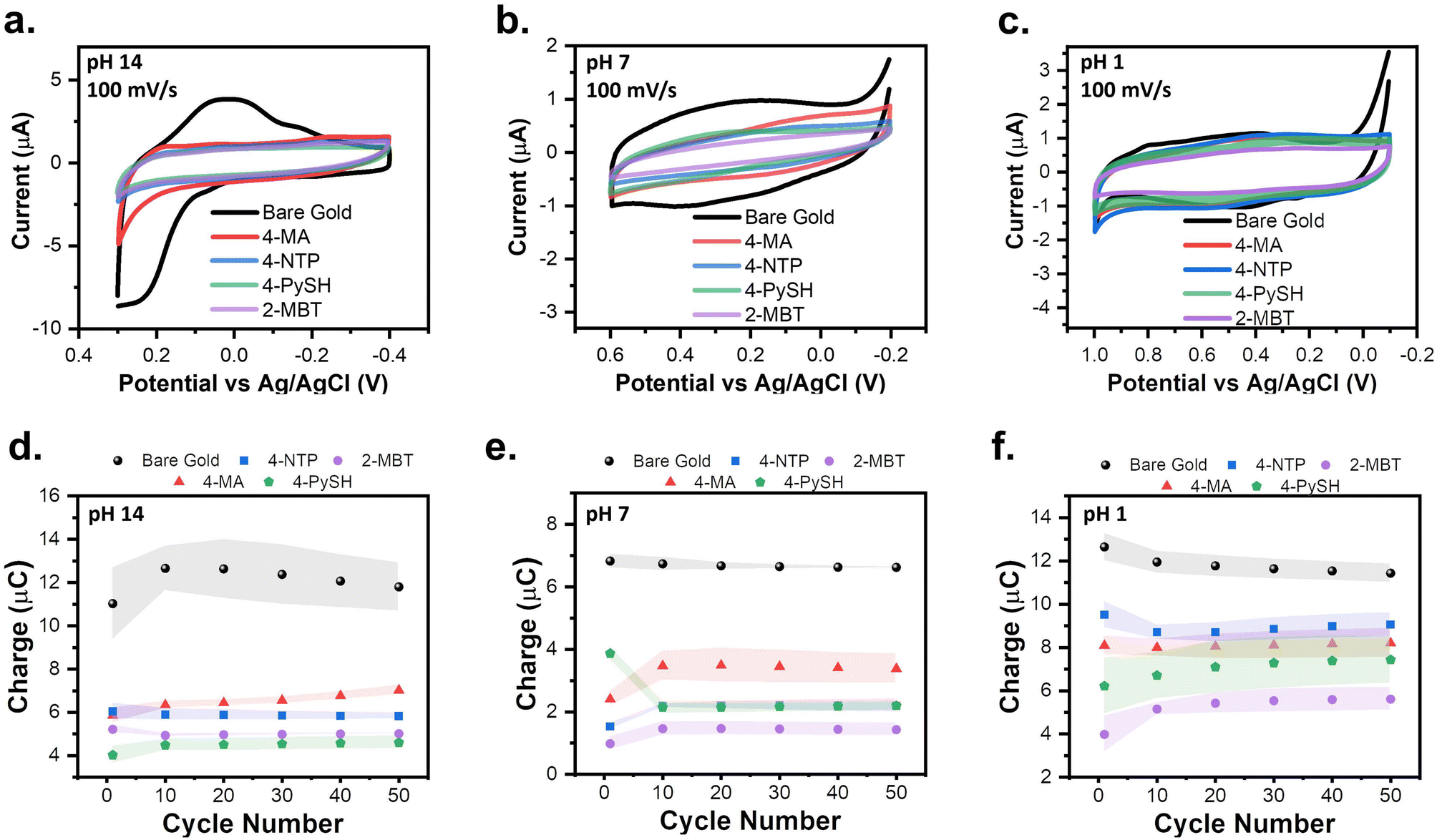 | ||
| Fig. 3 CVs of 4-MA, 4-NTP, 4-PySH, and 2-MBT modified electrodes compared to baseline CVs cycled within the SPWs established for (a) 1 M KOH (pH 14), (b) 0.1 M NaCl (pH 7), and (c) 0.1 M HClO4 (pH 1). The average integrated charge from the anodic sweeps of the baseline and modified electrodes CVs plotted versus the cycle number in (d) KOH, (e) NaCl, and (f) HClO4. The shaded regions depict the standard deviation of each measurement for three independent measurements. | ||
Spectroscopic confirmation of the aqueous stable potential windows
Spectroscopic assessment of the SAM stability was conducted using EC-SEIRAS and EC-SPR to monitor the electrochemical stability of SAMs at interfaces. Fig. 4 shows the potential-dependent spectra obtained for gold-plated IREs modified with 4-NTP at pH 14, 7, and 1. The spectra were acquired while applied potentials were held for 1 minute. The background of the spectra was the spectrum of gold-plated IRE in contact with the electrolyte of interest. Using such a background distinguishes the changes in the IR bands corresponding to the adsorption of 4-NTP and the effect of applied potentials. Negative peaks indicate desorption of the 4-NTP and/or changes in conformation of the molecules causing vibrational modes to fall outside of selection rules, while positive peaks correlate to the opposite.62 The two main peaks observed from 4-NTP correspond to the asymmetric and symmetrical NO2 stretches at 1520 cm−1 and 1330 cm−1, respectively.63–65 Other peaks observed are the aromatic C–C stretching at 1570 cm−1, a C–H twist at 1497 cm−1, and the H–O–H bend at 1650 cm−1.51–55 As seen in Fig. 4a and b, the 4-NTP peaks remain consistent at potentials within the SPW but abruptly disappear when cycled beyond the SPW. However, in acidic conditions at potentials >0.9 V, the 4-NTP signal completely disappears (Fig. 4c). This is perplexing as CV clearly shows retention of the SAM at these potentials. It is possible that partial degradation of the SAM could lead to structural rearrangement, with the SAM either bending out selection rules at positive potentials62 or partially desorbing from the electrode (Fig. S3†).66 Moreover, the fast nature of cycling mitigates the desorption of the SAM at potentials where desorption should occur (Fig. S4†). Hence, there is a discrepancy with the potential-dependent spectrum due to holding desorbing potentials for longer amounts of time compared to the CVs. To this point, the IR-based SPWs are more accurate than CV-based SPWs. These spectra confirm and validate the SPWs and the importance of pH on the electrochemical stability of the SAMs.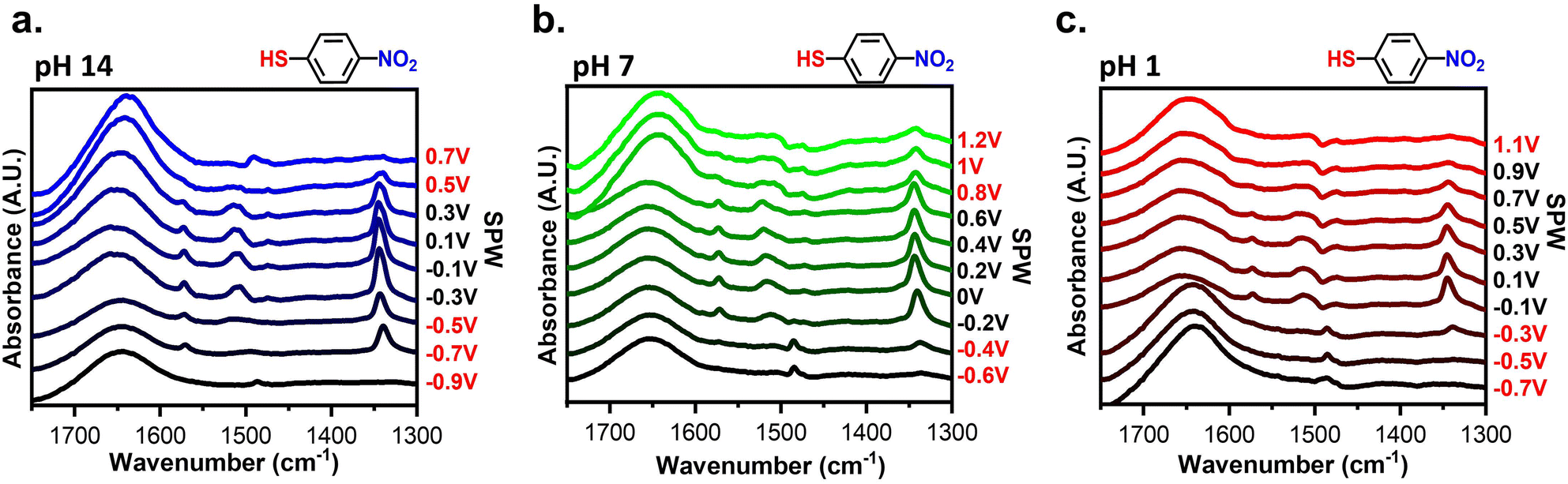 | ||
| Fig. 4 Spectra of a 4-NTP modified gold-plated IRE at different applied potentials within (a) 1 M KOH (pH: 14), (b) 0.1 M NaCl (pH: 7), and (c) 0.1 M HClO4 (pH: 1). The potentials marked in red denote those beyond the CV-based SPW. | ||
Fig. S5† compares the EC-SPR sensorgrams of a bare gold sensor chip and a 4-NTP-covered gold sensor chip at pH 14, 7, and 1 when cycled within their respective SPWs. The sensorgrams exhibit sinusoidal profiles that display the shifts in the refractive index of the gold chip over time as a function of the applied potential.42,43 A clear difference between the sensorgrams of the bare and SAM-modified electrodes was observed. The presence of SAMs decreases the magnitude of the shifts in the refractive index, however, the incidence angle increases, a phenomenon that is consistent with the adsorbed species effectively modifying the electrode surface.24,42,43 In NaCl electrolyte, the sensorgram of the SAM-covered shows a continual increase in the refractive index which can be attributed to the reported attraction of chlorides to the gold surface (Fig. S5b†).61 These sensorgrams confirm the validity of the SPWs and show the electrochemical stability of the SAMs in aqueous solvents.
Establishing stable potential windows in non-aqueous solvents
As established by the results above, the SPW is defined by the cathodic and anodic desorption events. Despite increases in the SPW as the pH decreases, the SPW of aqueous systems are still limited. The ideal system would be a solvent that induces no gold oxides, does not react with the SAM, and hinders the onset of reductive desorption. Aprotic non-aqueous solvents and alcohols seem like good candidates for expanding the SPW beyond those in aqueous systems. For those reasons, the electrochemical stability of thiolate SAMs was assessed in three aprotic solvents and four alcohols. MeCN, PC, and DMF with 0.1 M TBAPF6 supporting electrolyte were assessed due to their abundance in the electrochemistry literature.67 MeOH, EtOH, IPA, and BuOH with a 0.1 M LiCl supporting electrolyte were the four alcohols investigated in this study.In every case the desorption of the entire SAM did not occur instantly nor at a set potential limit. Instead, a SAM-covered electrode is capable of being cycled over large potential ranges with slow degradation after each cycle (Fig. 5). The tradeoff to this greater potential range is a decrease in the duration of the electrochemical stability. Because of its larger SPW range, we first discuss the case for 0.1 M LiCl in BuOH. As seen in Fig. 5a, a baseline was established by cycling between 0.3 V and −1 V. The resulting CV exhibits a capacitive profile with solvent breakdown occurring at negative potentials beyond −0.5 V. Comparing that baseline to the initial cycle of a 4-MA-covered electrode, it is obvious the SAM is passivating the electrode due to lowered anodic and cathodic currents. Yet, with each consecutive cycle, both currents continually increase back to the baseline. This indicates that the SAM is slowly desorbing from the electrode with each passing cycle.25 A similar phenomenon is observed when cycling positively from −0.3 V to 1 V (Fig. 5b). The baseline contains solvent breakdown at potentials greater than 0.7 V. Again, the initial cycle contains lowered currents but after the 25th cycle, the CV of the 4-MA-covered electrode is equivalent to the baseline. Fortunately, there exists an SPW in which the desorption of SAMs can be minimized (Fig. 5c). The SPWs within non-aqueous solvents differ from those in aqueous systems as there is no clear voltametric desorption in the solvents evaluated. The SPW in non-aqueous solvents is denoted as the potential range in which the electrode remains passivated for at least 50 cycles (Scheme 2).
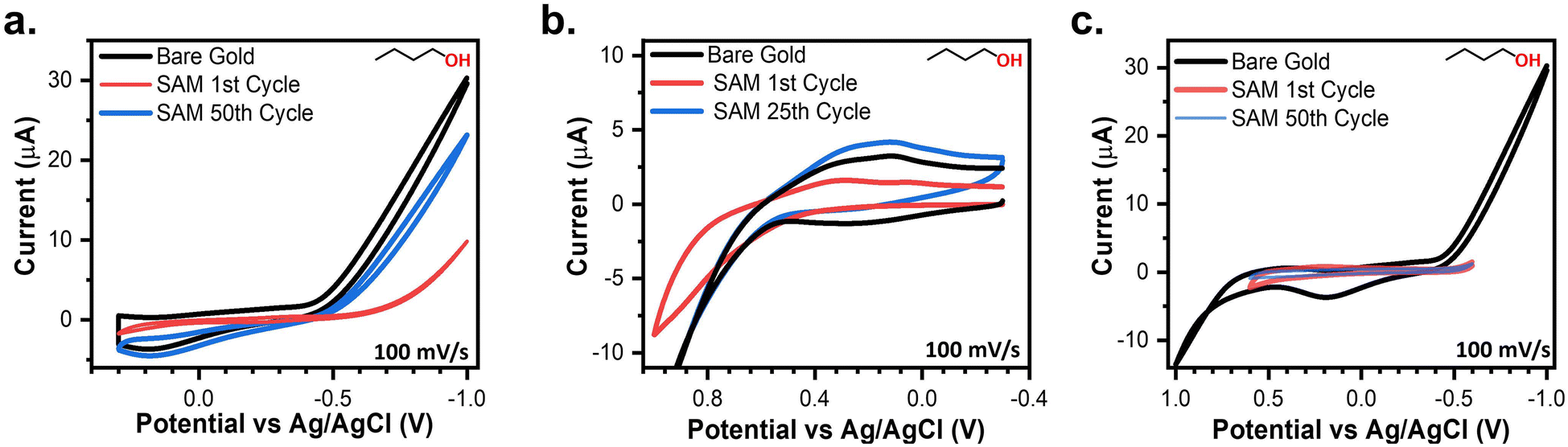 | ||
| Fig. 5 Comparing a baseline CV in 0.1 M LiCl BuOH against a 4-MA covered electrode when (a) cycled negatively beyond the SPW and (b) when cycled positively beyond SPW. (c) A scheme of the SPW of thiolate SAMs in non-aqueous solvents. | ||
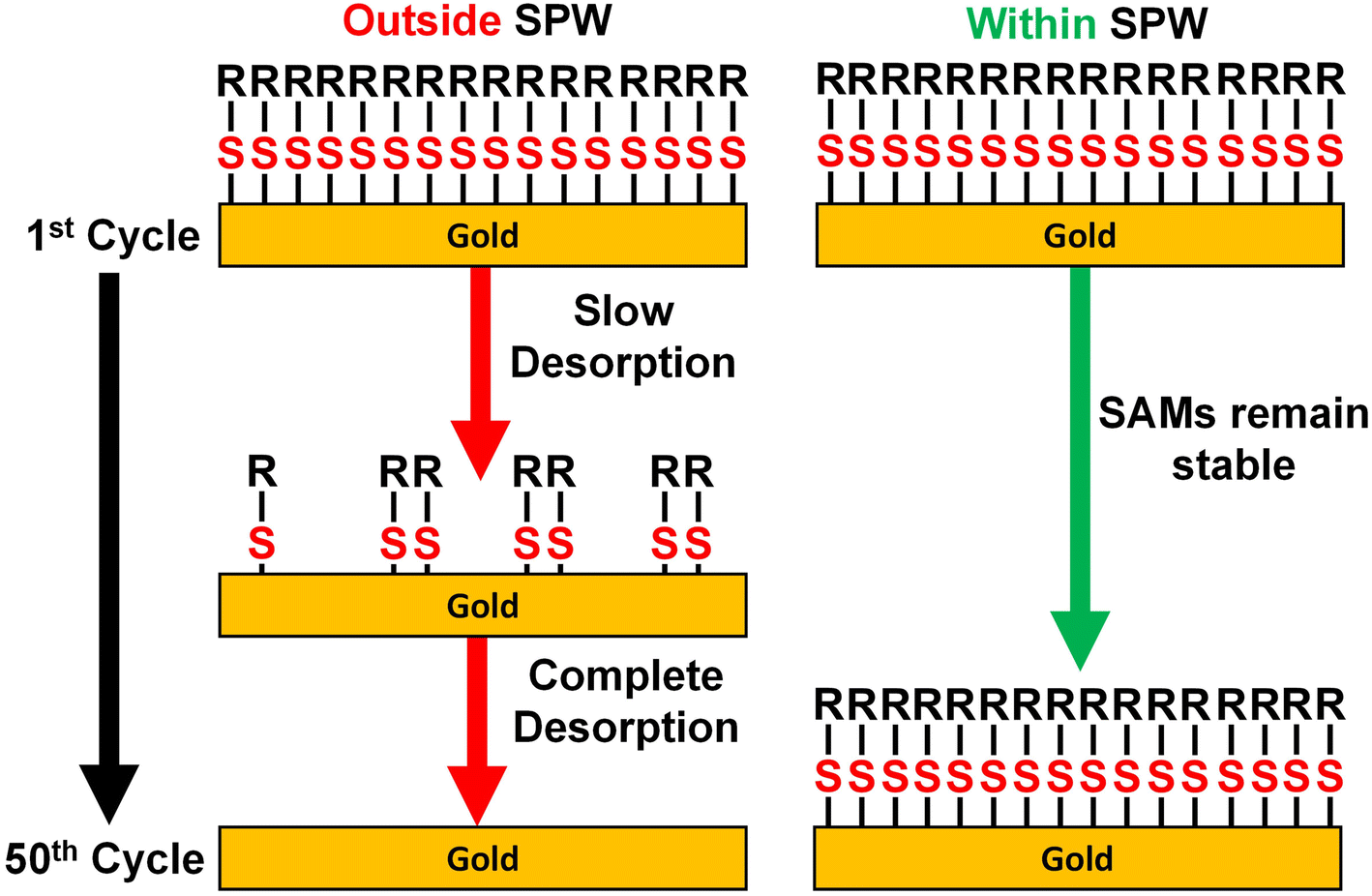 | ||
| Scheme 2 Depiction of slow desorption of thiolate SAMs on gold in non-aqueous solvents when cycling to potentials outside the SPW and the retention of the SAM when cycled within the SPW. | ||
Electrochemical assessment of the non-aqueous stable potential windows
Following the procedure described above, SPWs were identified in the three aprotic solvents (Fig. S6†) and evaluated based on a consistently passivated CV compared to the bare gold baseline for 50 cycles. Every aprotic system was assessed in triplicate for each SAM following the procedures used for the aqueous systems (Fig. S7†). The SPW identified were 600 mV in PC (−0.1 V to 0.5 V), 500 mV in DMF (−0.3 V to 0.2 V), and 400 mV in MeCN (−0.2 V to 0.2 V). For MeCN, PC, and DMF, the initial oxidative sweeps contained an oxidation wave consistently larger than any current in the baseline (Fig. S7†). For PC and DMF after the initial sweep, the currents lowered and remained below the baseline for the remaining 49 cycles (Fig. S7e and f†). For MeCN, only a 4-NTP-modified electrode exhibited oxidative current below the baseline for all 50 cycles (Fig. S7d†). It is apparent long-term electrochemical stability within MeCN is inconsistent. It is important to reiterate that modified electrodes in these solvents can still operate within large potential windows but only for short durations.The same procedure used for evaluating the aprotic solvents was used for identifying SPWs in the alcohols (Fig. S8†) and evaluating them. The SPWs are equivalent for both MeOH and EtOH at 400 mV (−0.2 V to 0.2 V) (Fig. 6a and b). The SPW increases to 900 mV in IPA (−0.6 V to 0.3 V) and further expands to 1200 mV in BuOH (−0.6 V to 0.6 V) (Fig. 6c and d). Fig. 6e–h show the SAM-covered electrode consistently remained passivated for all 50 cycles when cycled within the SPW for every alcohol. The only two outliers are the initial cycle of 4-MA and 4-PySH in BuOH, yet the current remained below the baseline (Fig. 6h).
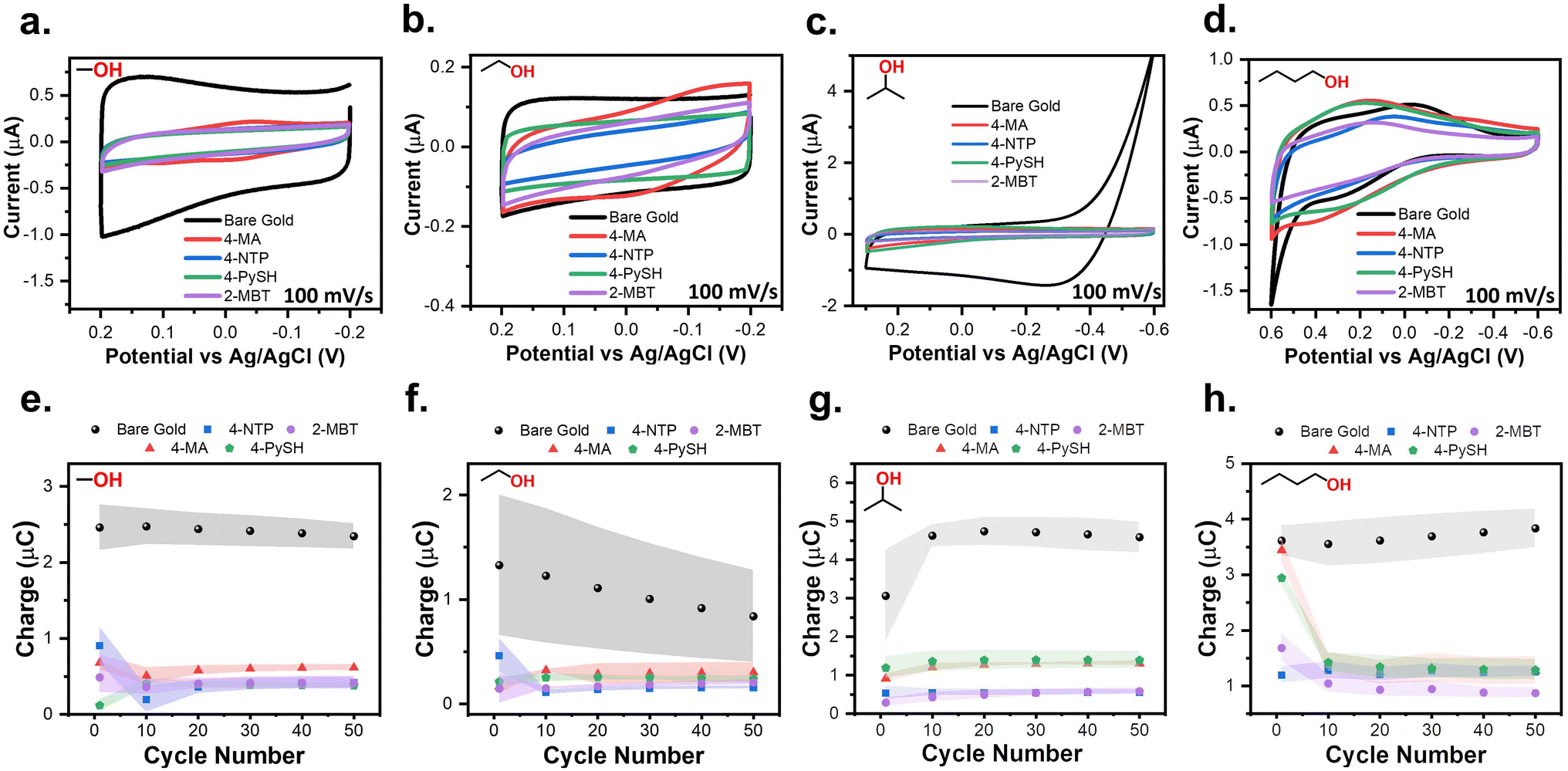 | ||
| Fig. 6 CVs of 4-MA, 4-NTP, 4-PySH, and 2-MBT modified electrodes compared to baseline CVs cycled within the SPWs established for 0.1 M LiCl (a) MeOH, (b) EtOH, (c) IPA, and (d) BuOH. The average integrated charge from the anodic sweeps of the baseline and modified electrodes CVs plotted versus the cycle number in (e) MeOH, (f) EtOH, (g) IPA, and (h) BuOH. The shaded regions depict the standard deviation of each measurement for three independent measurements. | ||
An interesting trend emerges that the SPW expands with increasing alkyl chain length of the alcohols. Just as pH was the main factor for SPWs in aqueous systems, we posit that the solubility of the SAM is the main factor in non-aqueous systems. With increasing alkyl chain length the alcohol becomes less polar.68 The polar SAMs used in this study are less soluble in non-polar solvents, therefore, due to the sluggish kinetics for dissolution of thiolates in these solvents, the SAMs remain attached to the electrode for larger potential windows. This explanation is confirmed by both the slow desorption of SAMs in non-aqueous systems (Fig. 5) and by the observation that the SPW increases with decreasing polarity index (Table S2†).68 These observations line up with established literature that thiolate-capped gold nanoparticles remain stable in toluene and hexane which have polarity indexes of 2.4 and 0.1, respectively.4,68 Moreover, the electrochemical stability of alkanethiols increases with increasing alkyl chain length.1,25 Further conformation is found in the observation that the aprotic solvents used here are the most polar non-aqueous solvents used, while the electrochemical stability of the SAMs is most inconsistent in these solvents (Fig. S7e and f†).
Spectroelectrochemical validation of non-aqueous stable potential windows
EC-SEIRAS was used to verify the non-aqueous SPWs and evaluate the slow desorption. Potential-dependent spectra were measured for gold-plated IREs modified with 4-MA. The background of the spectra was the spectrum of gold-plated IRE in contact with the electrolyte being assessed. The two main peaks observed from 4-MA correspond to the C–C stretch of the aryl ring at ∼1590 cm−1 and the N–H stretch of the amine at 1620 cm−1.48–50 In MeCN and PC the 4-MA peaks remain observable even at potentials beyond the CV-based SPW (Fig. S9a and b†). For DMF the 1590 cm−1 peak is only observed for positive potential close to the CV-based SPW (Fig. S9c†). These spectra further display the slow desorption in non-aqueous solvents and exemplify the inconsistencies of the SPWs in the polar aprotic electrolytes.This trend of the 1590 cm−1 peak shrinking at negative potentials and growing at positive potentials could be due to either desorption at negative potentials or the SAM bending out of selection rules due to potential-induced conformation changes (Scheme S2†).69 When applying negative potentials, a SAM can undergo a conformation change induced by the permeation of ions into the SAM while at positive potentials these changes are reported to be smaller or not observed.69 Thus, conformation changes will only occur at negative potentials and should be reversible when applying positive potentials.69 The reversibility of this potential-induced conformation change is best exemplified within BuOH (Fig. S10†). The signal derived from the 4-MA lowers with increasingly negative applied potential. Yet, upon applying positive potential immediately after, the 4-MA signal returns and grows with progressively positive applied potential.
Fig. 7 shows the potential-dependent spectra of a 4-MA modified gold-plated IRE in every alcohol. The spectra of the alcohols all display signals from the 1590 cm−1 C–C stretch and vary in capacity to resolve the 1620 cm−1 N–H stretch. In MeOH, EtOH, and IPA the 4-MA peaks alter significantly even within their voltametric SPW (Fig. 7a–c). Like the aprotic electrolytes, for potentials within the SPW, the 1590 cm−1 peak shrinks at negative potentials and grows at positive potentials. The same explanation of the SAM bending it out of selection rules can apply to these systems as well (Scheme S2†).69Fig. 7d confirms SAMs can exist on an electrode cycled in BuOH negatively longer than cycling positively as seen in Fig. 5. The 4-MA signal slowly diminishes when cycled beyond −0.2 V, but the signal abruptly disappears past 0.4 V.
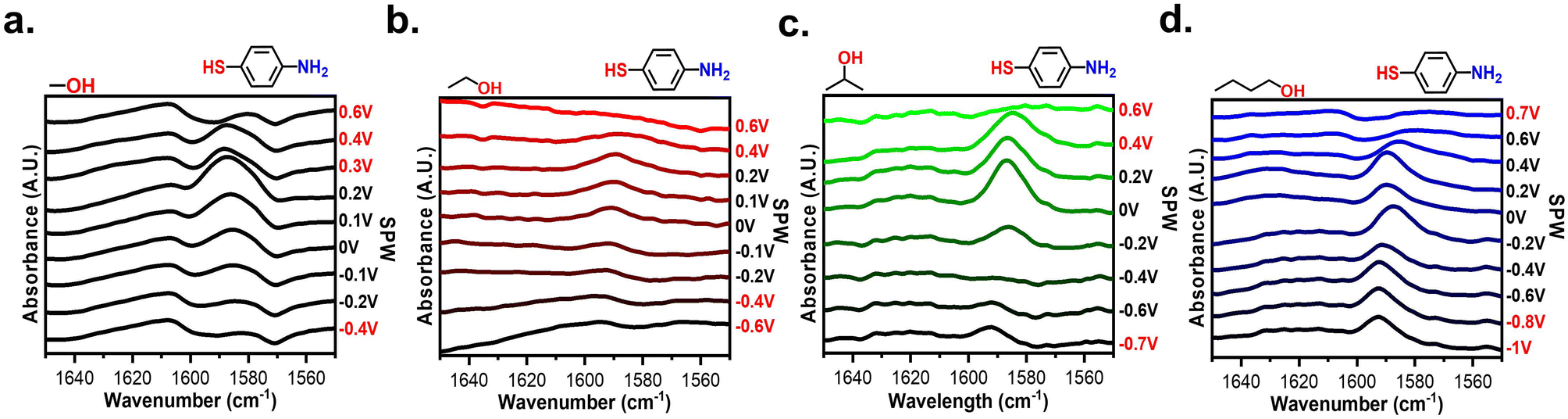 | ||
| Fig. 7 Spectra of a 4-MA modified SEIRAS substrate at different applied potentials within 0.1 M LiCl (a) MeOH, (b) EtOH (c) IPA, and (d) BuOH. The voltages marked in red denote applied potentials beyond the CV-based SPW. | ||
EC-SPR was used to further study the spectroscopic interactions of the SAMs in non-aqueous solvents (Fig. S11 and S12†). Both the baseline and 4-MA-covered gold sensor chips were cycled within the SPWs and monitored with the SPR.24,42,43 Baseline measurements for the non-aqueous solvents were not possible as the BI-2500 reached detection limits due to their high refractive indexes’. Yet, the 4-MA modified sensor chips were able to obtain SPR signals from the SAM interacting with these solvents while cycling within their respective SPWs. (Fig. S11 and S12a†). The key takeaway is this signal is observed solely when the SAMs are modifying the surface. Therefore, since this signal remains while cycling within the respective SPW in each solvent, the SAMs are stable. The sensorgrams of the alcohols demonstrate a sloping profile which is attributed to the attraction of the Cl− ions with the gold surface.61 MeCN demonstrated a similar sensorgram to the alcohols which could be attributed to the adsorption of MeCN onto gold.70,71 There were no clear signals from either DMF or PC (Fig. S12b and c†).
A summary of these findings is shown in Scheme 3. These SPWs have been confirmed to work for both types of gold electrodes used in this work (Fig. S13†). Interesting future directions could investigate the applicability of these findings with a wide variety of SAMs, explore the stability in more non-polar solvents, and study the effects of supporting electrolytes on electrochemical stability.
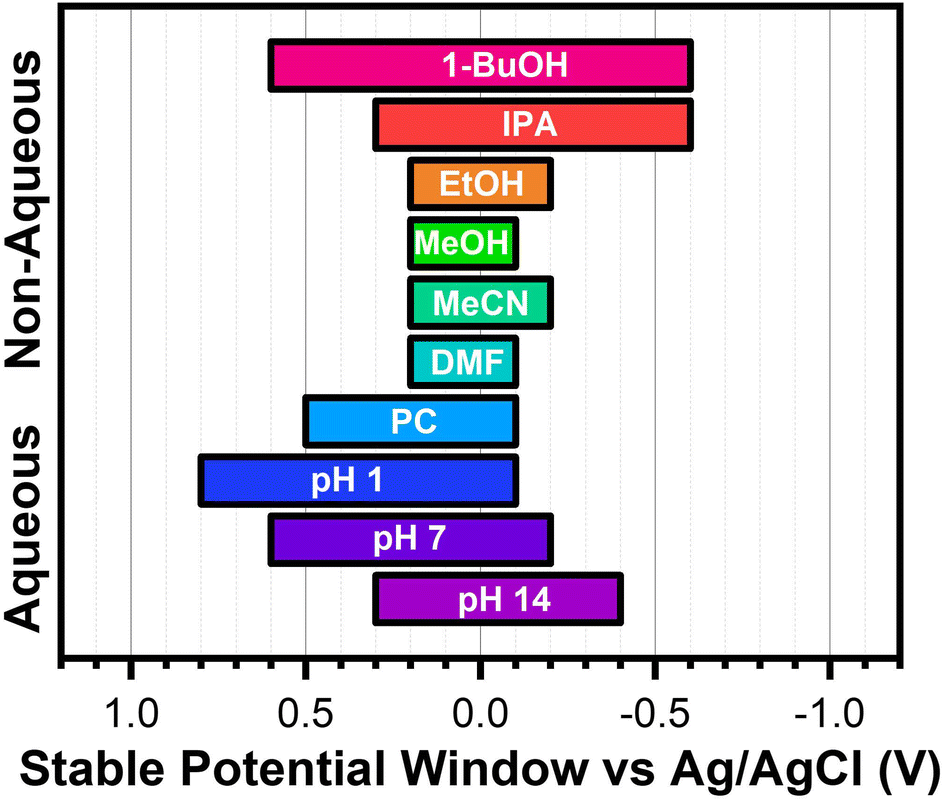 | ||
| Scheme 3 Summary of the identified SPWs for aromatic SAMs on gold in each aqueous and non-aqueous solvent investigated. | ||
Conclusions
In this study, we successfully used CV and ATR-SEIRAS to detect changes associated with SAM desorption events on modified gold electrodes. Using these techniques, we determined the stable potential windows of electrodes modified with aromatic SAMs in aqueous electrolytes as a function of pH and non-aqueous electrolytes for selected alcohols and aprotic polar solvents. Our results demonstrate that the SPWs determined universally apply to all four aromatic SAMs in every system tested, thus greatly simplifying our determined results (Scheme 3). In aqueous systems, the potential range between the two reductive desorption and oxidative desorption/gold oxidation defined the stable potential window. Clear voltametric features and well-defined changes in the SEIRAS signatures were measured for all cases. The pH was the largest factor for determining the range of the SPW as it directly delayed the onset of oxidative desorption to higher potentials. Since gold oxidation is pH-dependent, the SPW increases with decreasing pH. The largest SPW (1.1 V wide) was observed for pH 1. In the non-aqueous solvents, instead of being limited to potentials between desorption events, SAM slowly dissolves into solution when cycled at large potential ranges. The non-aqueous SPW was defined as the potential range the SAM-covered electrode could be cycled for 50 cycles without degradation of the SAM. It was determined that the polarity of the solvent was the main indicator for the SPW. The lower the polarity index of the solvent, the larger the SPW. The largest SPW (1.2 V wide) was observed for 0.1 M LiCl in 1-butanol. Our findings unambiguously demonstrate that the solvent utilized is the biggest decider in the electrochemical stability of aromatic SAMs. This work opens a new opportunity for further investigations into the uses of non-polar solvents in electrochemical applications involving SAMs, such as electrosynthesis, catalysis, battery research, and CO2 capture and conversion, the latter of which our laboratory is exploring using modified electrodes.Author contributions
A. S. and J. N. contributed to conceptualization, investigation, methodology, formal analysis, validation, visualization, writing the original draft, reviewing, and editing. A. S. C. and K. M. contributed to formal analysis, investigation, validation, visualization, writing the original draft, and reviewing and editing. R. B. contributed to funding acquisition, resources, reviewing and editing. J. R. L. contributed to conceptualization, funding acquisition, resources, supervision, project administration, reviewing and editing.Conflicts of interest
There are no conflicts to declare. Disclaimer: This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Govenrment nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Materials Chemistry program under Award Number DE-SC0022173. This work was carried out in part in the Materials Research Laboratory Central Research Facilities, University of Illinois. J. N. thanks the Arnold and Mabel Beckman Foundation for supporting this work through a Beckman-Brown Postdoctoral Fellowship at the Beckman Institute, University of Illinois at Urbana-Champaign. We acknowledge helpful discussions with Dr Adolfo I. B. Romo and Dr Md. Sazzad Hossain about this project.References
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Self-Assembled Monolayers of Thiolates on Metals as a Form of Nanotechnology, Chem. Rev., 2005, 105(4), 1103–1170, DOI:10.1021/cr0300789.
- A. Tverdokhlebova, I. Sterin, O. Smutok and E. Katz, Modification of Electrodes with Self-Assembled Monolayers—General Principles, J. Solid State Electrochem., 2023, 28, 711–755, DOI:10.1007/s10008-023-05700-w.
- F. Ghorbanizamani, E. Guler Celik, H. Moulahoum and S. Timur, Chapter 1 - Self-Assembled Monolayer–Based Nanoscaled Surfaces, in Biophysics At the Nanoscale, ed. A. Denizli,Academic Press, 2024, pp. 1–25. DOI:10.1016/B978-0-443-15359-4.00001-2.
- C. Vericat, M. E. Vela, G. Benitez, P. Carro and R. C. Salvarezza, Self-Assembled Monolayers of Thiols and Dithiols on Gold: New Challenges for a Well-Known System, Chem. Soc. Rev., 2010, 39(5), 1805–1834, 10.1039/B907301A.
- C. Vericat, M. E. Vela, G. Corthey, E. Pensa, E. Cortés, M. H. Fonticelli, F. Ibañez, G. E. Benitez, P. Carro and R. C. Salvarezza, Self-Assembled Monolayers of Thiolates on Metals: A Review Article on Sulfur-Metal Chemistry and Surface Structures, RSC Adv., 2014, 4(53), 27730–27754, 10.1039/C4RA04659E.
- A. Markovic, L. Buschbeck, I. Brand, C. Dosche, J. Christoffers and G. Wittstock, Electrochemical Activation of Self-Assembled Monolayers for the Binding of Effectors, Langmuir, 2020, 36(48), 14623–14632, DOI:10.1021/acs.langmuir.0c02426.
- D. Mandler and S. Kraus-Ophir, Self-Assembled Monolayers (SAMs) for Electrochemical Sensing, J. Solid State Electrochem., 2011, 15(7), 1535–1558, DOI:10.1007/s10008-011-1493-6.
- A. Shaver, S. D. Curtis and N. Arroyo-Currás, Alkanethiol Monolayer End Groups Affect the Long-Term Operational Stability and Signaling of Electrochemical, Aptamer-Based Sensors in Biological Fluids, ACS Appl. Mater. Interfaces, 2020, 12(9), 11214–11223, DOI:10.1021/acsami.9b22385.
- H. Shang, S. K. Wallentine, D. M. Hofmann, Q. Zhu, C. J. Murphy and L. R. Baker, Effect of Surface Ligands on Gold Nanocatalysts for CO2 Reduction, Chem. Sci., 2020, 11(45), 12298–12306, 10.1039/D0SC05089J.
- M. Li and Y. Li, Solid-Phase Electrosynthesis, Acc. Chem. Res., 2023, 56(24), 3694–3703, DOI:10.1021/acs.accounts.3c00620.
- J. Heo, H. Ahn, J. Won, J. G. Son, H. K. Shon, T. G. Lee, S. W. Han and M.-H. Baik, Electro-Inductive Effect: Electrodes as Functional Groups with Tunable Electronic Properties, Science, 2020, 370(6513), 214–219, DOI:10.1126/science.abb6375.
- M. Singh, N. Kaur and E. Comini, The Role of Self-Assembled Monolayers in Electronic Devices, J. Mater. Chem. C, 2020, 8(12), 3938–3955, 10.1039/D0TC00388C.
- S. Kim and H. Yoo, Self-Assembled Monolayers: Versatile Uses in Electronic Devices from Gate Dielectrics, Dopants, and Biosensing Linkers, Micromachines, 2021, 12(5), 565, DOI:10.3390/mi12050565.
- R. Yi, Y. Mao, Y. Shen and L. Chen, Self-Assembled Monolayers for Batteries, J. Am. Chem. Soc., 2021, 143(33), 12897–12912, DOI:10.1021/jacs.1c04416.
- R. G. Nuzzo and D. L. Allara, Adsorption of Bifunctional Organic Disulfides on Gold Surfaces, J. Am. Chem. Soc., 1983, 105(13), 4481–4483, DOI:10.1021/ja00351a063.
- L. Strong and G. M. Whitesides, Structures of Self-Assembled Monolayer Films of Organosulfur Compounds Adsorbed on Gold Single Crystals: Electron Diffraction Studies, Langmuir, 1988, 4(3), 546–558, DOI:10.1021/la00081a009.
- L. Srisombat, A. C. Jamison and T. R. Lee, Stability: A Key Issue for Self-Assembled Monolayers on Gold as Thin-Film Coatings and Nanoparticle Protectants. Colloids Surf. Physicochem, Eng. Aspects, 2011, 390(1–3), 1–19, DOI:10.1016/j.colsurfa.2011.09.020.
- N. Chen, S. Li, P. Zhao, R. Liu, Y. Xie, J.-L. Lin, C. A. Nijhuis, B. Xu, L. Zhang, H. Xu and Y. Li, Extreme Long-Lifetime Self-Assembled Monolayer for Air-Stable Molecular Junctions, Soft Sci., 2023, 9(42) DOI:10.1126/sciadv.adh3412.
- T. Yu, M. D. Marquez, H.-V. Tran and T. R. Lee, Crosslinked Organosulfur-Based Self-Assembled Monolayers: Formation and Applications, Soft Sci., 2022, 2(5) DOI:10.20517/ss.2022.04.
- J. B. Schlenoff, M. Li and H. Ly, Stability and Self-Exchange in Alkanethiol Monolayers, J. Am. Chem. Soc., 1995, 117(50), 12528–12536, DOI:10.1021/ja00155a016.
- A. Asyuda, S. Das and M. Zharnikov, Thermal Stability of Alkanethiolate and Aromatic Thiolate Self-Assembled Monolayers on Au(111): An X-Ray Photoelectron Spectroscopy Study, J. Phys. Chem. C, 2021, 125(39), 21754–21763, DOI:10.1021/acs.jpcc.1c06984.
- M. H. Schoenfisch and J. E. Pemberton, Air Stability of Alkanethiol Self-Assembled Monolayers on Silver and Gold Surfaces, J. Am. Chem. Soc., 1998, 120(18), 4502–4513, DOI:10.1021/ja974301t.
- E. Cortés, A. A. Rubert, G. Benitez, P. Carro, M. E. Vela and R. C. Salvarezza, Enhanced Stability of Thiolate Self-Assembled Monolayers (SAMs) on Nanostructured Gold Substrates, Langmuir, 2009, 25(10), 5661–5666, DOI:10.1021/la804251a.
- M. W. J. Beulen, M. I. Kastenberg, F. C. J. M. van Veggel and D. N. Reinhoudt, Electrochemical Stability of Self-Assembled Monolayers on Gold, Langmuir, 1998, 14(26), 7463–7467, DOI:10.1021/la981031z.
- N. C. Ramos, J. W. Medlin and A. Holewinski, Electrochemical Stability of Thiolate Self-Assembled Monolayers on Au, Pt, and Cu, ACS Appl. Mater. Interfaces, 2023, 15(11), 14470–14480, DOI:10.1021/acsami.3c01224.
- M. Hakamada, M. Takahashi, T. Furukawa, K. Tajima, K. Yoshimura, Y. Chino and M. Mabuchi, Electrochemical Stability of Self-Assembled Monolayers on Nanoporous Au, Phys. Chem. Chem. Phys., 2011, 13(26), 12277–12284, 10.1039/C0CP02553D.
- A. Kolodziej, F. Fernandez-Trillo and P. Rodriguez, Determining the Parameters Governing the Electrochemical Stability of Thiols and Disulfides Self-Assembled Monolayer on Gold Electrodes in Physiological Medium, J. Electroanal. Chem., 2018, 819, 51–57, DOI:10.1016/j.jelechem.2017.07.039.
- R. C. Salvarezza and P. Carro, The Electrochemical Stability of Thiols on Gold Surfaces, J. Electroanal. Chem., 2018, 819, 234–239, DOI:10.1016/j.jelechem.2017.10.046.
- I. Thom and M. Buck, Electrochemical Stability of Self-Assembled Monolayers of Biphenyl Based Thiols Studied by Cyclic Voltammetry and Second Harmonic Generation, Surf. Sci., 2005, 581(1), 33–46, DOI:10.1016/j.susc.2005.02.029.
- J. Li, Y. Shen, Y. Zhang and Y. Liu, Room-Temperature Ionic Liquids as Media to Enhance the Electrochemical Stability of Self-Assembled Monolayers of Alkanethiols on Gold Electrodes, Chem. Commun., 2005, 3, 360–362, 10.1039/B412412J.
- D. Badgurjar, M. Huynh, B. Masters and A. Wuttig, Non-Covalent Interactions Mimic the Covalent: An Electrode-Orthogonal Self-Assembled Layer, J. Am. Chem. Soc., 2023, 145(32), 17734–17745, DOI:10.1021/jacs.3c04387.
- E. A. Ramírez, E. Cortés, A. A. Rubert, P. Carro, G. Benítez, M. E. Vela and R. C. Salvarezza, Complex Surface Chemistry of 4-Mercaptopyridine Self-Assembled Monolayers on Au(111), Langmuir, 2012, 28(17), 6839–6847, DOI:10.1021/la204951u.
- M. M. Walczak, D. D. Popenoe, R. S. Deinhammer, B. D. Lamp, C. Chung and M. D. Porter, Reductive Desorption of Alkanethiolate Monolayers at Gold: A Measure of Surface Coverage, Langmuir, 1991, 7(11), 2687–2693, DOI:10.1021/la00059a048.
- C. A. Widrig, C. Chung and M. D. Porter, The Electrochemical Desorption of N-Alkanethiol Monolayers from Polycrystalline Au and Ag Electrodes. J. Electroanal. Chem, Interfacial Electrochem., 1991, 310(1), 335–359, DOI:10.1016/0022-0728(91)85271-P.
- L. D. Burke and P. F. Nugent, The Electrochemistry of Gold: I the Redox Behaviour of the Metal in Aqueous Media, Gold Bull., 1997, 30(2), 43–53, DOI:10.1007/BF03214756.
- S. Yang and D. G. H. Hetterscheid, Redefinition of the Active Species and the Mechanism of the Oxygen Evolution Reaction on Gold Oxide, ACS Catal., 2020, 10(21), 12582–12589, DOI:10.1021/acscatal.0c03548.
- C. A. Canaria, J. So, J. R. Maloney, C. J. Yu, J. O. Smith, M. L. Roukes, S. E. Fraser and R. Lansford, Formation and Removal of Alkylthiolate Self-Assembled Monolayers on Gold in Aqueous Solutions, Lab. Chip, 2006, 6(2), 289, 10.1039/b510661c.
- M. D. Porter, T. B. Bright, D. L. Allara and C. E. D. Chidsey, Spontaneously Organized Molecular Assemblies. 4. Structural Characterization of n-Alkyl Thiol Monolayers on Gold by Optical Ellipsometry, Infrared Spectroscopy, and Electrochemistry, J. Am. Chem. Soc., 1987, 109(12), 3559–3568, DOI:10.1021/ja00246a011.
- B. Liu, A. J. Bard, M. V. Mirkin and S. E. Creager, Electron Transfer at Self-Assembled Monolayers Measured by Scanning Electrochemical Microscopy, J. Am. Chem. Soc., 2004, 126(5), 1485–1492, DOI:10.1021/ja038611p.
- M. Osawa, Surface-Enhanced Infrared Absorption, in Near-Field Optics and Surface Plasmon Polaritons, ed. S. Kawata, Topics in Applied Physics; Springer, Berlin, Heidelberg, 2001, pp. 163–187. DOI:10.1007/3-540-44552-8_9.
- B. Unni and I. J. Burgess, Electrochemical and Surface Enhanced Infrared Absorption Spectroscopy Studies of TEMPO Self-Assembled Monolayers, Electrochim. Acta, 2021, 381, 138263, DOI:10.1016/j.electacta.2021.138263.
- Y. Feng, E. R. Dionne, V. Toader, G. Beaudoin and A. Badia, Odd–Even Effects in Electroactive Self-Assembled Monolayers Investigated by Electrochemical Surface Plasmon Resonance and Impedance Spectroscopy, J. Phys. Chem. C, 2017, 121(44), 24626–24640, DOI:10.1021/acs.jpcc.7b08053.
- X. Shan, U. Patel, S. Wang, R. Iglesias and N. Tao, Imaging Local Electrochemical Current via Surface Plasmon Resonance, Science, 2010, 327(5971), 1363–1366, DOI:10.1126/science.1186476.
- T. Sawaguchi, F. Mizutani, S. Yoshimoto and I. Taniguchi, Voltammetric and in Situ STM Studies on Self-Assembled Monolayers of 4-Mercaptopyridine, 2-Mercaptopyridine and Thiophenol on Au(111) Electrodes, Electrochim. Acta, 2000, 45(18), 2861–2867, DOI:10.1016/S0013-4686(00)00360-1.
- W. Zhou, T. Baunach, V. Ivanova and D. M. Kolb, Structure and Electrochemistry of 4,4′-Dithiodipyridine Self-Assembled Monolayers in Comparison with 4-Mercaptopyridine Self-Assembled Monolayers on Au(111), Langmuir, 2004, 20(11), 4590–4595, DOI:10.1021/la049903m.
- S. Herrera, F. Tasca, F. J. Williams, E. J. Calvo, P. Carro and R. C. Salvarezza, Surface Structure of 4-Mercaptopyridine on Au(111): A New Dense Phase, Langmuir, 2017, 33(38), 9565–9572, DOI:10.1021/acs.langmuir.7b01627.
- J. F. Silva, J. Pavez, C. P. Silva and J. H. Zagal, Electrocatalytic Actvity of Modified Gold Electrodes Based on Self-Assembled Monolayers of 4-Mercaptopyridine and 4-Aminothiophenol on Au(111) Surfaces Chemically Functionalized with Substituted and Unsubstituted Iron Phthalocyanines, Electrochim. Acta, 2013, 114, 7–13, DOI:10.1016/j.electacta.2013.10.017.
- N. Karthik, S. Asha and M. G. Sethuraman, Influence of pH-Sensitive 4-Aminothiophenol on the Copper Corrosion Inhibition of Hybrid Sol–Gel Monolayers, J. Sol-Gel Sci. Technol., 2016, 78(2), 248–257, DOI:10.1007/s10971-015-3944-5.
- W. A. Hayes and C. Shannon, Electrochemistry of Surface-Confined Mixed Monolayers of 4-Aminothiophenol and Thiophenol on Au, Langmuir, 1996, 12(15), 3688–3694, DOI:10.1021/la9507390.
- C. R. Raj, F. Kitamura and T. Ohsaka, Electrochemical and in Situ FTIR Spectroscopic Investigation on the Electrochemical Transformation of 4-Aminothiophenol on a Gold Electrode in Neutral Solution, Langmuir, 2001, 17(23), 7378–7386, DOI:10.1021/la010746q.
- L. Dong, X. Yang, C. Zhang, B. Cerjan, L. Zhou, M. L. Tseng, Y. Zhang, A. Alabastri, P. Nordlander and N. J. Halas, Nanogapped Au Antennas for Ultrasensitive Surface-Enhanced Infrared Absorption Spectroscopy, Nano Lett., 2017, 17(9), 5768–5774, DOI:10.1021/acs.nanolett.7b02736.
- L.-B. Zhao, J.-L. Chen, M. Zhang, D.-Y. Wu and Z.-Q. Tian, Theoretical Study on Electroreduction of P-Nitrothiophenol on Silver and Gold Electrode Surfaces, J. Phys. Chem. C, 2015, 119(9), 4949–4958, DOI:10.1021/jp512957c.
- Z. Zhang and T. Imae, Study of Surface-Enhanced Infrared Spectroscopy: 1. Dependence of the Enhancement on Thickness of Metal Island Films and Structure of Chemisorbed Molecules, J. Colloid Interface Sci., 2001, 233(1), 99–106, DOI:10.1006/jcis.2000.7220.
- M. Baillieul, E. Rinnert, J. Lemaitre, K. Michel, F. Colas, L. Bodiou, G. Demésy, S. Kakuta, A. Rumyantseva, G. Lerondel, K. Boukerma, G. Renversez, T. Toury, J. Charrier and V. Nazabal, Surface Functionalization with Polymer Membrane or SEIRA Interface to Improve the Sensitivity of Chalcogenide-Based Infrared Sensors Dedicated to the Detection of Organic Molecules, ACS Omega, 2022, 7(51), 47840–47850, DOI:10.1021/acsomega.2c05502.
- F. Verger, T. Pain, V. Nazabal, C. Boussard-Plédel, B. Bureau, F. Colas, E. Rinnert, K. Boukerma, C. Compère, M. Guilloux-Viry, S. Deputier, A. Perrin and J. P. Guin, Surface Enhanced Infrared Absorption (SEIRA) Spectroscopy Using Gold Nanoparticles on As2S3 Glass, Sens. Actuators, B, 2012, 175, 142–148, DOI:10.1016/j.snb.2012.01.038.
- R. K. Shervedani, A. Hatefi-Mehrjardi and M. K. Babadi, Comparative Electrochemical Study of Self-Assembled Monolayers of 2-Mercaptobenzoxazole, 2-Mercaptobenzothiazole, and 2-Mercaptobenzimidazole Formed on Polycrystalline Gold Electrode, Electrochim. Acta, 2007, 52(24), 7051–7060, DOI:10.1016/j.electacta.2007.05.030.
- S. Bharathi, V. Yegnaraman and G. P. Rao, Potential-Dependent “Opening” and “Closing” of Self-Assembled 2-Mercaptobenzthiazole on Gold Substrates, Langmuir, 1993, 9(7), 1614–1617, DOI:10.1021/la00031a002.
- N. Sandhyarani, G. Skanth, S. Berchmans, V. Yegnaraman and T. Pradeep, A Combined Surface-Enhanced Raman–X-Ray Photoelectron Spectroscopic Study of 2-Mercaptobenzothiazole Monolayers on Polycrystalline Au and Ag Films, J. Colloid Interface Sci., 1999, 209(1), 154–161, DOI:10.1006/jcis.1998.5882.
- T. A. Morhart, B. Unni, M. J. Lardner and I. J. Burgess, Electrochemical ATR-SEIRAS Using Low-Cost, Micromachined Si Wafers, Anal. Chem., 2017, 89(21), 11818–11824, DOI:10.1021/acs.analchem.7b03509.
- A. Siddiqui, J. N’Diaye, K. Martin, A. Baby, J. Dawlaty, V. Augustyn and J. Rodríguez-López, Monitoring SEIRAS on a Graphitic Electrode for Surface-Sensitive Electrochemistry: Real-Time Electrografting, Anal. Chem., 2024, 96(6), 2435–2444, DOI:10.1021/acs.analchem.3c04407.
- F. A. de Carvalho, A. Resende and V. A. Leão, Gold Leaching by Sodium Chloride and Calcium Hypochlorite Solutions, in Extraction 2018, The Minerals, Metals & Materials Series, Springer International Publishing, Cham, 2018, pp. 1787–1796. DOI:10.1007/978-3-319-95022-8_148.
- A. Cuesta, ATR-SEIRAS for Time-Resolved Studies of Electrode–Electrolyte Interfaces, Curr. Opin. Electrochem., 2022, 35, 101041, DOI:10.1016/j.coelec.2022.101041.
- H. A. Girard, T. Petit, S. Perruchas, T. Gacoin, C. Gesset, J. C. Arnault and P. Bergonzo, Surface Properties of Hydrogenated Nanodiamonds: A Chemical Investigation, Phys. Chem. Chem. Phys., 2011, 13(24), 11517–11523, 10.1039/C1CP20424F.
- A. K. Farquhar, H. M. Dykstra, M. R. Waterland, A. J. Downard and P. A. Brooksby, Spontaneous Modification of Free-Floating Few-Layer Graphene by Aryldiazonium Ions: Electrochemistry, Atomic Force Microscopy, and Infrared Spectroscopy from Grafted Films, J. Phys. Chem. C, 2016, 120(14), 7543–7552, DOI:10.1021/acs.jpcc.5b11279.
- F. A. E. Hadj, A. Amiar, M. Cherkaoui, J.-N. Chazalviel and F. Ozanam, Study of Organic Grafting of the Silicon Surface from 4-Nitrobenzene Diazonium Tetrafluoroborate, Electrochim. Acta, 2012, 70, 318–324, DOI:10.1016/j.electacta.2012.03.072.
- E. H. J. Wong, G. L. May and C. P. Wilde, Oxidative Desorption of Thiols as a Route to Controlled Formation of Binary Self Assembled Monolayer Surfaces, Electrochim. Acta, 2013, 109, 67–74, DOI:10.1016/j.electacta.2013.07.072.
- K. Gong, Q. Fang, S. Gu, S. F. Y. Li and Y. Yan, Nonaqueous Redox-Flow Batteries: Organic Solvents, Supporting Electrolytes, and Redox Pairs, Energy Environ. Sci., 2015, 8(12), 3515–3530, 10.1039/C5EE02341F.
- L. R. Snyder, Classification off the Solvent Properties of Common Liquids, J. Chromatogr. Sci., 1978, 16(6), 223–234, DOI:10.1093/chromsci/16.6.223.
- N. Darwish, P. K. Eggers, S. Ciampi, Y. Zhang, Y. Tong, S. Ye, M. N. Paddon-Row and J. J. Gooding, Reversible Potential-Induced Structural Changes of Alkanethiol Monolayers on Gold Surfaces, Electrochem. Commun., 2011, 13(5), 387–390, DOI:10.1016/j.elecom.2011.01.025.
- P. H. Reinsberg and H. Baltruschat, Potential- and Cation-Dependent Adsorption of Acetonitrile on Gold Investigated via Surface Enhanced Infrared Absorption Spectroscopy, Electrochim. Acta, 2020, 334, 135609, DOI:10.1016/j.electacta.2019.135609.
- T. Solomun, K. Christmann and H. Baumgaertel, Interaction of Acetonitrile and Benzonitrile with the Gold (100) Surface, J. Phys. Chem., 1989, 93(20), 7199–7208, DOI:10.1021/j100357a035.
Footnotes |
| † Electronic supplementary information (ESI) available: The SPR data, additional schemes, tables, and experimental results. Surface coverages of the SAMs, identification of the SPWs in non-aqueous systems, CVs, and SEIRAS spectra in non-aqueous systems. See DOI: https://doi.org/10.1039/d4an00241e |
| ‡ Theses authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |