DOI:
10.1039/D3AN02164E
(Paper)
Analyst, 2024,
149, 2647-2654
Modulating the ion-transfer electrochemistry of perfluorooctanoate with serum albumin and β-cyclodextrin†
Received
14th December 2023
, Accepted 20th March 2024
First published on 28th March 2024
Introduction
Per- and polyfluoroalkyl substances (PFAS) are synthetic organofluorine compounds that have been widely used in industrial and commercial applications, such as food packaging, paper and textile coatings, clothing and furnishings, fire-fighting foams, and in cosmetics.1–3 Due to their strong carbon-fluorine bonds, PFAS are stable and are persistent and non-biodegradable when they enter the environment. PFAS are ubiquitous in distribution, having been detected at different concentrations in diverse environmental samples (e.g., drinking water, soils, biological species).4–8 Elevated PFAS concentrations have been detected in drinking waters in Australia and USA.9,10 Drinking water, food, air, and household dust are the common sources of exposure of humans to PFAS, with PFAS found in several samples of human hair, breast milk, blood, and urine.11–14 A study in Australia with over 2000 donors, mostly from urban and some industrial areas, revealed the presence of several PFAS in human blood serum at concentrations in the range 0.8–15.2 ng mL−1.11 Amongst all PFAS, perfluorooctane sulfonate (PFOS) was reported at the highest concentration. PFAS have also been detected in hair (2.40–233 pg g−1) and urine (0.18–2.97 ng L−1) samples collected from children (aged 4–6 years) in Hong Kong.12 Likewise, a study in China reported PFAS in human breast milk,14 showing that amongst all PFAS, perfluorooctanoate (PFOA) and PFOS were detected in the largest proportions. Many studies have shown that an accumulation of PFAS in the human body is associated with adverse health effects15–19 that ultimately result in economic and social impacts worldwide. For example, the USA has invested ca. US$37–59 billion annually for health-related costs of PFAS exposure.20
Serum albumin is the most abundant soluble protein found in humans and it has many physiological roles.21–25 It forms complexes with various other substances e.g., albumin-drug complexes,26 albumin-surfactant complexes.27 Studies have shown that serum albumin binds with some PFAS27–30 and that more than 90% of PFOA was bound with serum albumin (both human and rat).27 In some cases, over 99% of PFAS were bound with serum albumin.30,31 One study showed that the interaction of PFOS with serum albumin not only changed the protein's physiological functions but also its secondary structure.32 Wang et al.33 and Chen et al.34 also reported structural changes in serum albumin upon interaction with PFAS.
Cyclodextrins, cyclic carbohydrates composed of glucose monomers, are widely used as carriers of hydrophobic drugs due to their ability to encapsulate organic molecules within their macrocyclic cavity.35 β-cyclodextrin (β-CD), made up of seven glucose monomers, forms complexes with PFAS (e.g., PFOA and PFOS).36–40 One study revealed that in the presence of β-CD, the binding of PFOA with serum albumin was unfavourable.37 For example, it was found that serum albumin was completely dissociated from its complexes with PFOA if the concentration of β-CD was ca. 5-times higher than the PFOA concentration.37
In order to study the complexes resulting from interaction of PFAS with serum albumin, usually one or more of a variety of spectroscopic techniques (e.g., nuclear magnetic resonance (NMR) spectroscopy, fluorescence spectroscopy, circular dichroism spectroscopy), mass spectrometry, surface tension, and/or equilibrium dialysis have been used.31,41–44 These techniques have limitations, such as time-consumption, poor sensitivity, and necessity for an exceptionally pure reagents.45 Studies of PFAS by electrochemical methods has not been popular because their high stability makes direct redox electrochemistry impractical. However, some electroanalytical methods have been examined.46 One such approach is electrochemistry at the interface between two immiscible electrolyte solutions (ITIES), which enables the non-redox detection of ionised substances.47 Despite some disadvantages of the ITIES, such as instability and high capacitance, improvements have progressed by miniaturisation48,49 to microscale and by mechanical support with porous membranes or gelled organic phases.50–54 As a result, PFAS electrochemical detection and sensing has been investigated at the ITIES55–58 including the microITIES (μITIES).55,59 We have previously reported the sensitive detection of PFOA with this approach and identified the impact of matrix components on the electrochemical detection of PFOA.59 These matrix effects indicated that bovine serum albumin (BSA) and β-CD bind to PFOA.
Here, we aim to evaluate the capability of electrochemistry at a μITIES array as a means to characterise and study PFOA binding with BSA and β-CD, and to determine quantitative data on binding from the resulting electrochemical behaviour.
Experimental section
Materials
All chemicals were from Sigma-Aldrich Australia and used as received unless otherwise mentioned. Bis(triphenylphosphoranylidene)ammonium tetrakis(4-chlorophenyl)borate (BTPPATPBCl) was synthesised by metathesis of equimolar quantities of bis(triphenylphosphoranylidene)ammonium chloride (BTPPACl) and potassium tetrakis(4-chlorophenyl)borate (KTPBCl) (STREM Chemicals). A stock solution of pentadecafluorooctanoic acid (PFOA, purity: 98%) (STREM Chemical) was prepared in 10 mM lithium chloride (LiCl). Reference solution for the organic phase reference electrode was 1 mM BTPPACl in aqueous 10 mM LiCl. Organic phase and aqueous phase electrolyte solutions were 10 mM BTPPATPBCl in 1,2-dichloroethane (1,2-DCE) and aqueous 10 mM LiCl, respectively. Solutions (0.6 mM) of BSA and β-CD were prepared in aqueous 10 mM LiCl, unless otherwise mentioned. Purified water (resistivity 18.2 MΩcm) from a Milli-Q system (Millipore Pty. Ltd, Australia) was used to prepare aqueous solutions.
Methods
An Autolab PGSTAT302N electrochemical workstation (Metrohm, The Netherlands) supported with NOVA software was used for all electrochemical experiments. The electrochemical cell was placed in a Faraday cage, at ambient temperature. The μITIES arrays were prepared with glass membranes having 100 pores (10 × 10 square array, prepared by laser ablation in a ca. 130 μm thick borosilicate substrate).60 The membrane was attached to the mouth of ca. 6 cm long glass cylinder using silicone sealant (Selley, Australia and New Zealand) in such a way that the pores having wider diameter (i.e. 42 μm; laser entry side) were facing away from the glass cylinder, and the pores having a smaller diameter (i.e. 21 μm; laser exit side) were facing towards the glass tube.60 The inner side of the pores along with surface of the glass membrane that faced toward the glass tube was made hydrophobic by treatment with trichloro(1H,1H,2H,2H-perfluorooctyl)silane. These glass microporous membranes were prepared at the Australian National Fabrication Facility. The electrochemical cell set-up is summarised in Scheme 1. All experiments employed this two-electrode cell using a Ag/AgCl electrode on each side of the μITIES array. Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were employed. The CV scan rate was 10 mV s−1. The DPV waveform parameters were: modulation amplitude: 0.025 V, step potential: 0.005 V, interval time: 0.5 s, modulation time: 0.05 s, wait time: 20 s. A Langmuir binding isotherm model was used to estimate the PFOA-BSA or PFOA-β-CD association constants (K) using non-linear curve fitting with Microsoft Excel Solver.
 |
| | Scheme 1 Schematic representation of the electrochemical cell used in the experiments, where x = concentration of PFOA, y = concentration of BSA, and z = concentration of β-CD. O = organic phase, W = water phase. | |
Results and discussion
Effect of bovine serum albumin on the ion transfer electrochemistry of PFOA
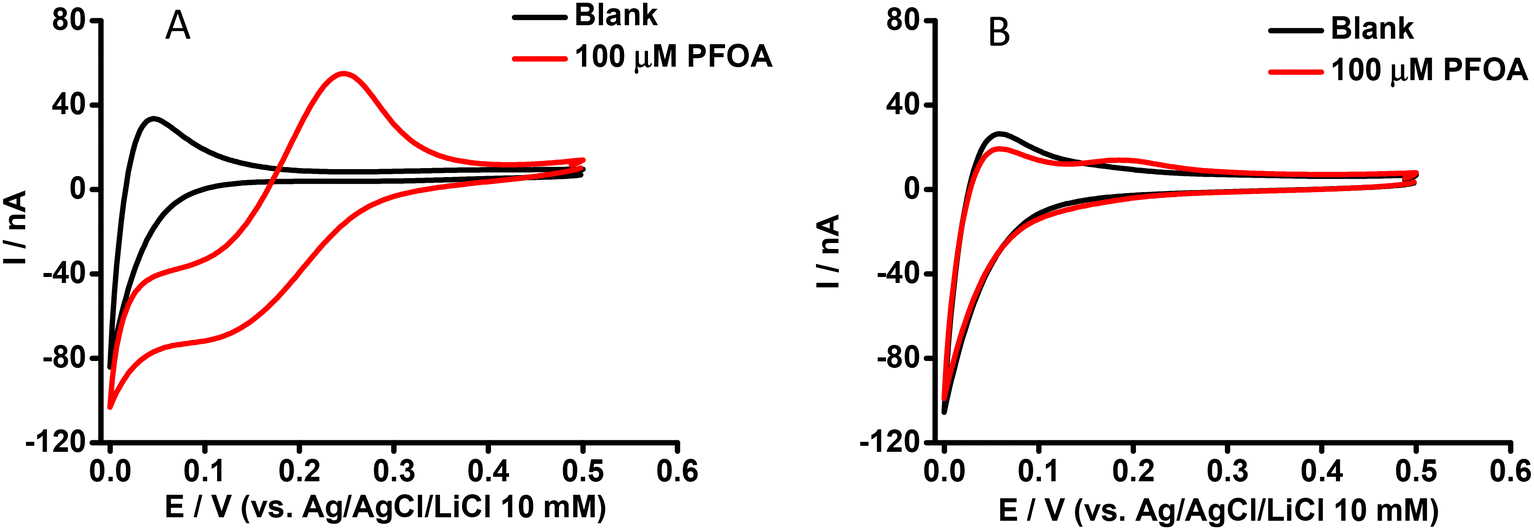
Our previous study reported that an aqueous phase at pH ≥ 4 was suitable for the ion transfer detection of PFOA at nanomolar concentrations using a μITIES array.59 Also, the matrix effect of different sample constituents (including BSA, and β-CD) was identified.59 Here, CV was employed to further examine the influence of BSA on the ion transfer electrochemistry of PFOA using the cell shown in Scheme 1. CVs of 100 μM PFOA in the presence and absence of BSA at physiological concentration (∼0.6 mM)61 are shown in Fig. 1A and B, respectively. These results show that in the absence of BSA, PFOA presents (Fig. 1A) a forward scan steady-state voltammetric response, due to the radial diffusion control of anionic PFOA transfer across the μITIES. However, the reverse scan presents a peak shape, due to linear diffusion control of anionic PFOA within the pores that hold the μITIES, in agreement with our previous report.59Fig. 1B shows the CV of 100 μM PFOA in the presence of the physiological concentration of BSA, indicating very poor detection of PFOA. These results show that BSA has a significant impact on the detection of PFOA; this is attributed to complexation of PFOA by BSA.29,30 Accordingly, we set out to further study and assess if electrochemistry at a μITIES array could be used for characterisation of PFOA-BSA complexation.
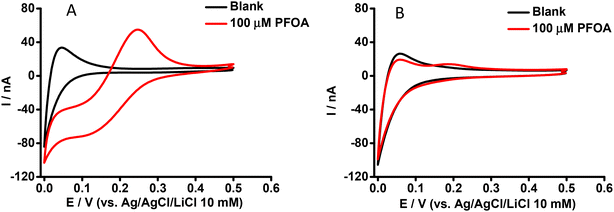 |
| | Fig. 1 CVs in the absence (black) and presence (red) of aqueous phase PFOA. Cell as shown in Scheme 1. Aqueous phase contains (A) 0 mM or (B) 0.6 mM BSA. Scan rate: 10 mV s−1. | |
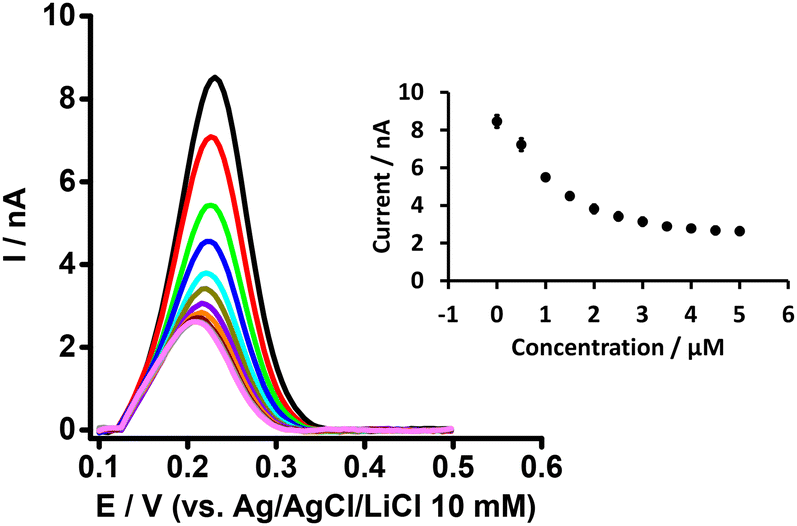
In order to understand the basis of PFOA-BSA complexation, we titrated a fixed concentration of PFOA (i.e. 5 μM) with different concentrations of BSA with detection by DPV of PFOA ion transfer (Fig. 2). It can be seen that the PFOA response decreases with an increase in BSA concentration, and that the peak response of PFOA remains unchanged above a certain concentration of BSA (inset, Fig. 2). These results confirm that the decrease in response of PFOA with the increase in BSA concentration is due to complexation of PFOA by BSA, in agreement with the literature.31,34 As a control experiment, the response of a model analyte, tetrapropylammonium (TPrA+), at the same concentrations of BSA was examined. Here, the peak responses of TPrA+ did not change with BSA concentrations (Fig. S1†), showing no complexation between BSA and TPrA+, in alignment with previous work,62 but also that the changing responses to PFOA in the presence of BSA were not due to an experimental artefact. Additionally, CV study of a model anionic analyte, hexafluorophosphate (PF6−), in the absence and presence of BSA revealed a hindrance of this anion transfer due to complex formation with serum albumin63 as illustrated in the Fig. S2A and S2B,† respectively.
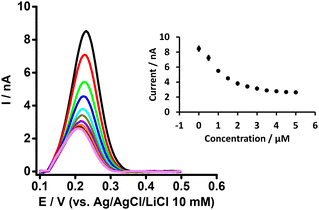 |
| | Fig. 2 DPVs at different concentrations of BSA at a fixed concentration of PFOA (5 μM) at the μITIES array. Cell composition as shown in Scheme 1. Black, red, green, blue, cyan, dark yellow, violet, orange, wine red, gray, and magneta, respectively, represent the DPVs at 0, 0.5, 1, 1.5, 2, 2.5,3, 3.5, 4, 4.5, and 5 μM of BSA. Inset: effect of BSA concentration on response of PFOA. Error bars indicate ±1 standard deviation from three independent measurements. DPVs are background-subtracted followed by baseline-corrected. | |
Effect of β-cyclodextrin on the ion transfer electrochemistry of PFOA
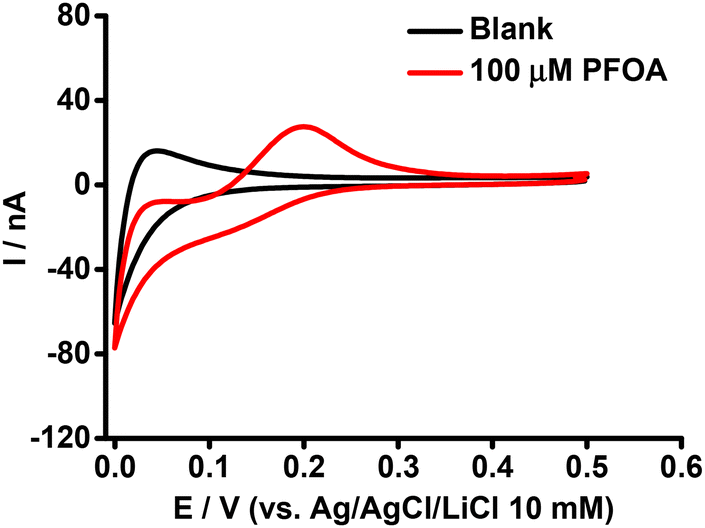
The purpose of this experiment was to understand the impact of β-CD on the ion transfer electrochemistry of PFOA at the μITIES array and to provide a direct comparison of the complexing tendencies of β-CD and BSA with PFOA. Here, the CV of 100 μM PFOA in the presence of 0.6 mM of β-CD (Fig. 3) showed a more pronounced signal than that obtained for the same concentration of PFOA in the presence of 0.6 mM BSA (Fig. 2). This means that BSA had a greater impact on the PFOA response than β-CD. From this, it can be suggested that the complexation of PFOA with BSA occurs to a greater extent than PFOA complexation with β-CD under the conditions employed here.
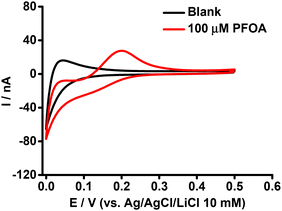 |
| | Fig. 3 CVs in the presence (red) and absence (black) of PFOA with 0.6 mM β-CD in the aqueous phase. Electrochemical cell as shown in Scheme 1. Scan rate: 10 mV s−1. | |
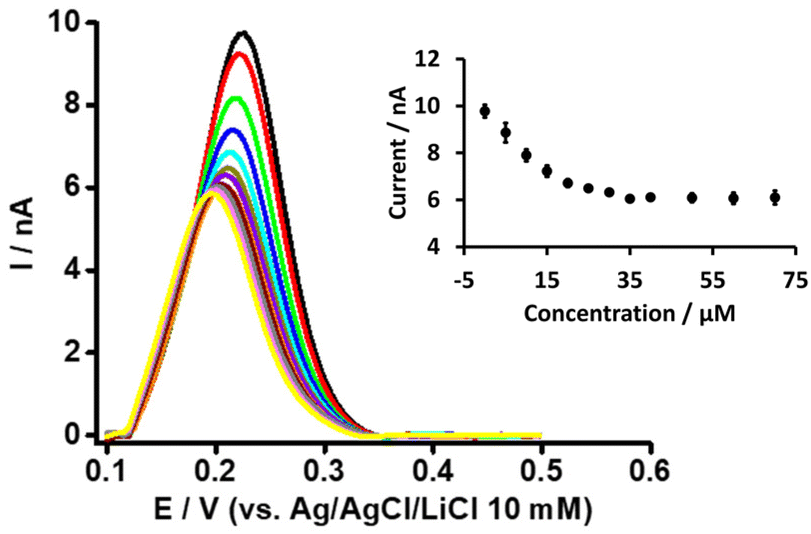
Likewise, in order to understand the basis of PFOA-β-CD complexation, we titrated PFOA (5 μM) with different concentration of β-CD (Fig. 4). As can be observed with the increase in β-CD concentration, the peak response of PFOA decreases up to a certain concentration and thereafter remains unchanged on further concentration increases. This decrease in PFOA signal is attributed to the formation of the complex between PFOA and β-CD, as reported in the literature.37,39 Furthermore, as a control experiment, the electrochemical response of TPrA+ at the μITIES array was studied in the presence of different concentrations of β-CD (Fig. S3†). This revealed that there was no change in the response to TPrA+ with different concentrations of β-CD and indicates that there was no complexation between them. Furthermore, CV of PF6− in the presence of β-CD revealed its negligible effect on the transfer of PF6− (Fig. S4†). Also, it indicates that the signal changes for PFOA in the presence of different β-CD concentrations were not due to an experimental artefact. It can also be said that under the same concentration conditions, the impact of BSA on the PFOA signal was more pronounced than that of β-CD.
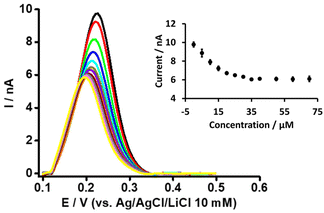 |
| | Fig. 4 DPVs at different concentrations of β-CD at a fixed concentration of PFOA (5 μM) at the μITIES array. Cell composition as shown in Scheme 1. Black, red, green, blue, cyan, dark yellow, violet, orange, wine red, gray, magenta, and yellow, respectively, represent the DPVs at 0, 5, 10, 15, 20, 25, 30, 35, 40, 50, 60, and 70 μM β-CD. Inset: relationship between current and concentration of β-CD. DPVs are background-subtracted followed by baseline-corrected. Error bars indicate ±1 standard deviation from three independent measurements. | |
Simultaneous effect of BSA and β-CD on the ion transfer electrochemistry of PFOA
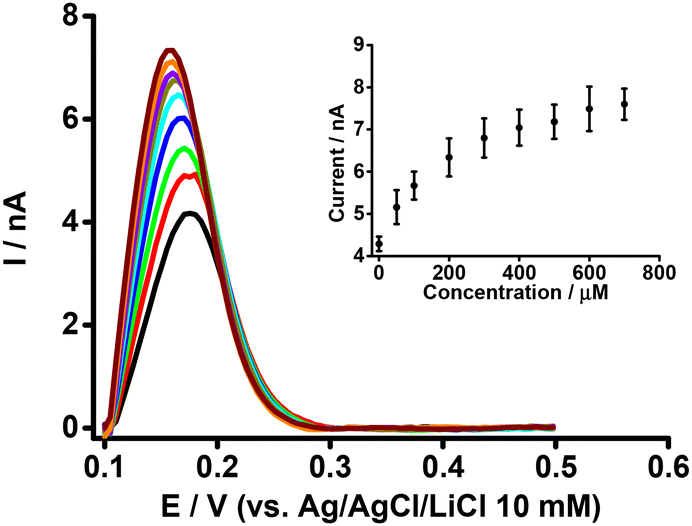
The simultaneous effect of both BSA and β-CD on the electrochemical response of PFOA at the μITIES array was examined, given the reports that indicate β-CD enables the de-complexation of PFOA from serum albumin.37Fig. 5 presents DPVs of 50 μM PFOA with 0.6 mM BSA and different concentrations of β-CD in the aqueous phase. The peak current initially increased on adding β-CD, and eventually reached a plateau (inset, Fig. 5). This initial increase in current on addition of β-CD is due to the release of PFOA anions from the complex with BSA. The release of PFOA anion is possible because β-CD interacts with BSA to form a β-CD-BSA complex,64–66 which results in BSA being unable to retain PFOA. However, at higher concentrations of β-CD, β-CD interacts with both BSA and PFOA and hence the peak response does not change with increasing β-CD concentration. These results suggest that β-CD can successfully de-complex PFOA from serum albumin, in agreement with other reports.37,66
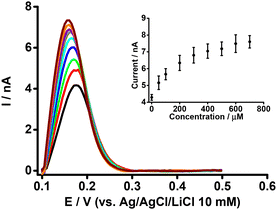 |
| | Fig. 5 DPVs at different concentrations of β-CD at fixed concentrations of BSA (0.6 mM) and PFOA (50 μM) at the μITIES array. Cell composition as given in Scheme 1. Black, red, green, blue, cyan, dark yellow, violet, orange, and wine red, respectively, represent the DPVs at 50, 100, 200, 300, 400, 500, 600, and 700 μM of β-CD. Inset: relationship between peak current and concentration of β-CD. DPVs were background-subtracted followed by baseline-corrected. Error bars indicate ±1 standard deviation from two independent measurements. | |
As a control experiment, DPV was performed using 0.6 mM BSA in the aqueous phase with different concentrations of β-CD, but without PFOA (Fig. S5†). This experiment did not reveal any ion transfer across the interface and confirmed that the DPV peak observed in the mixture of BSA, β-CD, and PFOA is only due to the uncomplexed PFOA, and not to any product of the BSA-β-CD interaction.
Calculation of association constants for PFOA complexation
The results presented above show that addition of BSA or β-CD to the aqueous phase containing PFOA influences the ion transfer current associated with PFOA at the μITIES array (Fig. 1B, 2, 3 and 4). These results show that a decrease in ion transfer current occurs on addition of BSA or β-CD to the aqueous phase containing PFOA. This decrease in current is due to the formation of complexes, and because only the free, uncomplexed PFOA anion transfers across the interface, the decrease in current is associated with the removal of PFOA from solution and its sequestration by BSA or β-CD. Similar observations for ion transfer at the ITIES have been reported by Horrocks and Mirkin,67 who studied the transfer of N-methylphenanthroline in the presence of DNA, and by Lopes and Kataky,68 who described the binding of propranolol with ∝1-acid-glycoprotein. The data in Fig. 2 and 4 show voltammetric titrations conducted by taking a fixed concentration of PFOA in the aqueous phase followed by addition of binding agent, BSA or β-CD, and measurement of the ion transfer current associated with the free, uncomplexed PFOA anion. This change in current may be used to calculate the binding constant for the reactions leading to BSA-PFOA or β-CD-PFOA complexes.
For this analysis, let us assume that each BSA or β-CD forms a complex with PFOA, as shown in eqn (1) and (2). The reactions are presented as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, although they could be more intricate; despite multiple binding sites in BSA and other serum albumins for small molecule ligands, a number of reports have indicated 1:1 or predominantly 1:1 complexes are present. This is reported not only for PFAS31,69,70 but also other small molecules such as flavonoids.71 Binding studies conducted in solution have shown that PFOA has one high-affinity binding site and three low affinity binding sites in serum albumin.69 Likewise, Bischel et al. have reported one to three primary binding sites in serum albumin for PFOA via equilibrium dialysis.31 Recently, Wu et al., have reported a 1:1 binding ratio of PFOA and serum albumin.70 Hence, we assume a 1:1 combination for our study.
:1, although they could be more intricate; despite multiple binding sites in BSA and other serum albumins for small molecule ligands, a number of reports have indicated 1:1 or predominantly 1:1 complexes are present. This is reported not only for PFAS31,69,70 but also other small molecules such as flavonoids.71 Binding studies conducted in solution have shown that PFOA has one high-affinity binding site and three low affinity binding sites in serum albumin.69 Likewise, Bischel et al. have reported one to three primary binding sites in serum albumin for PFOA via equilibrium dialysis.31 Recently, Wu et al., have reported a 1:1 binding ratio of PFOA and serum albumin.70 Hence, we assume a 1:1 combination for our study.
| | | PFOA + β-CD ⇌ PFOA-β-CD | (2) |
Then the appropriate mathematical expression relating the mole fraction of bound PFOA to the binding constant is given by the Langmuir equation,72,73eqn (3).
where,
Xb,
K and [P] are mole fraction of bound PFOA, the binding or association constant of the complex formed, and the concentration of BSA or β-CD, respectively. The mole fraction was calculated from the experimental data according to
eqn (4).
26,67,74| | | Xb = (IT-PFOA − IF-PFOA)/(IT-PFOA) | (4) |
where
IT-PFOA and
IF-PFOA are the peak currents in the absence of BSA or β-CD (
i.e. the current associated with the total concentration of free, ionised PFOA) and the peak currents in the presence of specific BSA or β-CD concentrations (
i.e. the current associated with free, uncomplexed ionised PFOA in the presence of complexing agents).
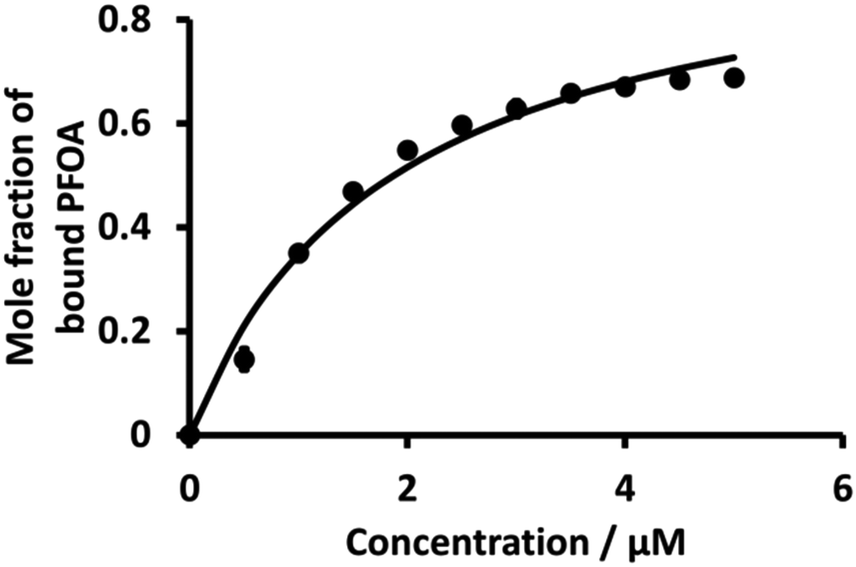
Fig. 6 (dot points) shows the relationship between the mole fraction of bound PFOA at different concentrations of BSA. This shows that with the increasing concentration of BSA, the mole fraction (from eqn (4)) of bound PFOA increases and reaches a plateau above a certain concentration. Nonlinear curve fitting to eqn (3) was undertaken (Fig. 6, solid line), and the association constant (K) of the complex was calculated as 5.3 (±0.22) × 105 M−1 (Table 1). This value of K is in close agreement with the literature values reported for PFOA-albumin complexes,31,41,44,75,76 which vary, e.g. 102–106 M−1.31,34,42,44,77
Table 1 Association constant of complexes formed using 5 μM PFOA by ion transfer electrochemistry at a μITIES array
| Complex types |
Combining ratio of PFOA with BSA or β-CD |
R
2
|
Association constant of complex (Ka) (M−1) |
|
The error values are the standard deviations determined from curve fitting to three independent sets of experimental data.
|
| PFOA-BSA |
1:1 (BSA:PFOA) |
0.98 |
5.3 (± 0.22) × 105 |
| PFOA-β-CD |
1:1 (β-CD:PFOA) |
0.97 |
2.0 (± 0.15) × 104 |
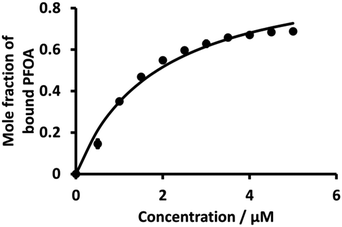 |
| | Fig. 6 Plot of mole fraction of bound PFOA obtained during titration of a fixed concentration of PFOA (5 μM) with different concentrations of BSA. The points are averages of three independent measurements with error bars of ±1 standard deviation. Where error bars are not visible, they are smaller than the symbol size. The solid line is a nonlinear fit of eqn (3) to the data. | |
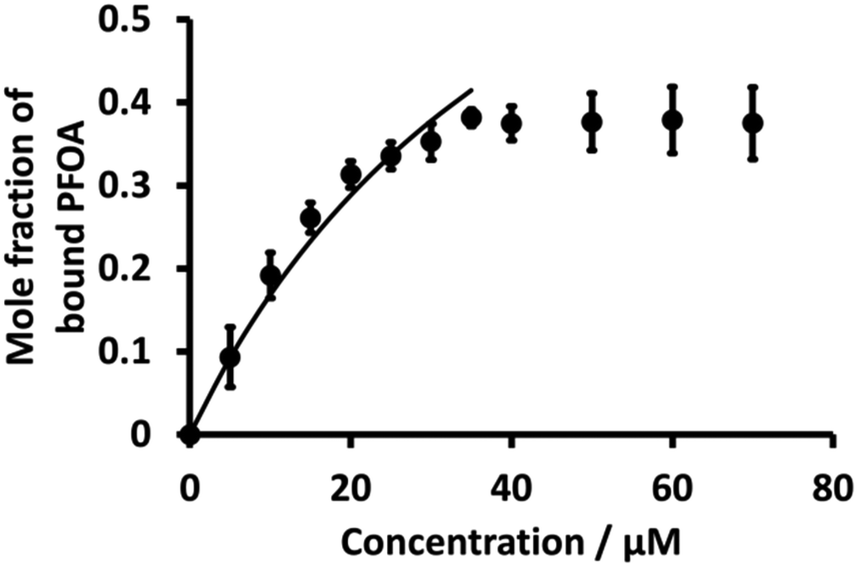
Literature shows that β-CD and PFOA interact with each other to form complexes.36,38,78 Here, K values of the 1:1 PFOA-β-CD complex using eqn (3) were evaluated as for BSA. Fig. 7 (dot points) shows the relationship between the mole fraction of bound PFOA and different concentrations of β-CD. The results reveal that, at the beginning, the mole fraction of bound PFOA increased with increasing concentration of β-CD, and eventually reached a plateau. However, poor curve fitting was obtained (Fig. S6†) when the full concentration range was considered. Instead, a better fit was achieved when the highest concentration points (upper four concentrations) were excluded, as shown in Fig. 7. This improved curve fitting provided a K value for β-CD-PFOA (1:1 complex) of 2.0 (±0.15) × 104 M−1 (Table 1). The reported K value for the 1:1 complex of β-CD-PFOA by Wilson and Verrall, as determined by NMR spectrometry, closely aligns with our calculated value.38 Similarly, various researchers have documented comparable values for the K of the 1:1 β-CD-PFOA.79,80 However, the reported K value (5.0 (±0.10) × 105) by Weiss-Errico and O'Shea was higher than our value.36
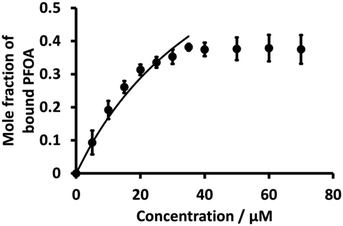 |
| | Fig. 7 Plot of mole fraction of bound PFOA obtained during titration of a fixed concentration of PFOA (5 μM) with different concentrations β-CD (dotted line). Nonlinear fitting using Langmuir binding isotherm (eqn (3)) (solid line) for 1:1 PFOA-β-CD complex. Error bars are ±1 standard deviation of three independent measurements. When error bars are not visible, they are smaller than the symbol size. | |
Conclusions
In conclusion, a μITIES array-based electrochemical method was assessed for the study of the interactions of PFOA with BSA or β-CD in aqueous phase. Results revealed that BSA has a more pronounced effect on PFOA transfer at the interface compared to β-CD. The combined effect of BSA and β-CD on the analytical signal of PFOA was also studied. Based on current changes with added concentration of either binding agent, the complex association constant was determined. For the BSA-PFOA and β-CD-PFOA complexes, the binding constants were of the order of 105 and 104 M−1, respectively, and in good agreement with literature values. In the mixture experiments, for suitable concentrations of β-CD, the PFOA-BSA complex was dissociated, leading to an increased current for PFOA transfer, indicating a direct interaction between BSA and β-CD. Overall, the results presented here show that ion transfer at a μITIES array is a useful strategy for study of the interaction between PFAS and proteins and/or carbohydrates.
Author contributions
HBL: conceptualisation, investigation, methodology, validation, visualisation, writing – original draft. DWMA: conceptualisation, funding acquisition, methodology, resources, supervision, writing – review and editing.
Conflicts of interest
D. W. M. Arrigan is an associate editor of this journal. There are no other conflicts of interest to declare.
Acknowledgements
HBL thanks Curtin University for the award of a PhD Scholarship. The authors thank the South Australian and OptoFab nodes of the NCRIS-enabled Australian National Fabrication Facility (ANFF-SA and ANFF-OptoFab, respectively) for fabrication of the glass microporous membranes used in this study.
References
- J. Glüge, M. Scheringer, I. T. Cousins, J. C. DeWitt, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, X. Trier and Z. Wang, Environ. Sci.: Processes Impacts, 2020, 22(12), 2345–2373, 10.1039/D0EM00291G.
- J. R. Lang, B. M. Allred, G. F. Peaslee, J. A. Field and M. A. Barlaz, Environ. Sci. Technol., 2016, 50(10), 5024–5032, DOI:10.1021/acs.est.5b06237.
- H. D. Whitehead, M. Venier, Y. Wu, E. Eastman, S. Urbanik, M. L. Diamond, A. Shalin, H. Schwartz-Narbonne, T. A. Bruton, A. Blum, Z. Wang, M. Green, M. Tighe, J. T. Wilkinson, S. McGuinness and G. F. Peaslee, Environ. Sci. Technol. Lett., 2021, 8(7), 538–544, DOI:10.1021/acs.estlett.1c00240.
- V. Boiteux, C. Bach, V. Sagres, J. Hemard, A. Colin, C. Rosin, J. F. Munoz and X. Dauchy, Int. J. Environ. Anal. Chem., 2016, 96, 705–728, DOI:10.1080/03067319.2016.1196683.
- W. Wang, G. Rhodes, J. Ge, X. Yu and H. Li, Chemosphere, 2020, 261, 127584, DOI:10.1016/j.chemosphere.2020.127584.
- C. Death, C. Bell, D. Champness, C. Milne, S. Reichman and T. Hagen, Sci. Total Environ., 2021, 774, 144795, DOI:10.1016/j.scitotenv.2020.144795.
- S. Kurwadkar, J. Dane, S. R. Kanel, M. N. Nadagouda, R. W. Cawdrey, B. Ambade, G. C. Struckhoff and R. Wilkin, Sci. Total Environ., 2022, 809, 151003, DOI:10.1016/j.scitotenv.2021.151003.
- J. S. C. Liou, B. Szostek, C. M. DeRito and E. L. Madsen, Chemosphere, 2010, 80(2), 176–183, DOI:10.1016/j.chemosphere.2010.03.009.
- J. Thompson, G. Eaglesham and J. Mueller, Chemosphere, 2011, 83, 1320–1325, DOI:10.1016/j.chemosphere.2011.04.017.
- X. C. Hu, D. Q. Andrews, A. B. Lindstrom, T. A. Bruton, L. A. Schaider, P. Grandjean, R. Lohmann, C. C. Carignan, A. Blum, S. A. Balan, C. P. Higgins and E. M. Sunderland, Environ. Sci. Technol. Lett., 2016, 3(10), 344–350, DOI:10.1021/acs.estlett.6b00260.
- L.-M. L. Toms, A. M. Calafat, K. Kato, J. Thompson, F. Harden, P. Hobson, A. Sjödin and J. F. Mueller, Environ. Sci. Technol., 2009, 43(11), 4194–4199, DOI:10.1021/es900272u.
- N. Li, G.-G. Ying, H. Hong and W.-J. Deng, Environ. Pollut., 2021, 270, 116219, DOI:10.1016/j.envpol.2020.116219.
- W. A. Gebbink and S. P. J. van Leeuwen, Environ. Int., 2020, 137, 105583, DOI:10.1016/j.envint.2020.105583.
- M. K. So, N. Yamashita, S. Taniyasu, Q. Jiang, J. P. Giesy, K. Chen and P. K. S. Lam, Environ. Sci. Technol., 2006, 40(9), 2924–2929, DOI:10.1021/es060031f.
- S. M. Bartell and V. M. Vieira, J. Air Waste Manage. Assoc., 2021, 71, 663–679, DOI:10.1080/10962247.2021.1909668.
- Z. Zeng, B. Song, R. Xiao, G. Zeng, J. Gong, M. Chen, P. Xu, P. Zhang, M. Shen and H. Yi, Environ. Int., 2019, 126, 598–610, DOI:10.1016/j.envint.2019.03.002.
- C. Lau, J. L. Butenhoff and J. M. Rogers, Toxicol. Appl. Pharmacol., 2004, 198, 231–241, DOI:10.1016/j.taap.2003.11.031.
-
M. Kirk, K. Smurthwaite, J. Bräunig, S. Trevenar, C. D'Este, R. Lucas, A. Lal, R. Korda, A. Clements, J. Mueller and B. Armstrong, The PFAS Health Study: Systematic Literature Review, The Australian National University, Canberra, 2018 Search PubMed.
- L. J. L. Espartero, M. Yamada, J. Ford, G. Owens, T. Prow and A. Juhasz, Environ. Res., 2022, 212, 113431, DOI:10.1016/j.envres.2022.113431.
- A. Cordner, G. Goldenman, L. S. Birnbaum, P. Brown, M. F. Miller, R. Mueller, S. Patton, D. H. Salvatore and L. Trasande, Environ. Sci. Technol., 2021, 55(14), 9630–9633, DOI:10.1021/acs.est.1c03565.
- M. Paar, C. Rossmann, C. Nusshold, T. Wagner, A. Schlagenhauf, B. Leschnik, K. Oettl, M. Koestenberger, G. Cvirn and S. Hallström, PLoS One, 2017, 12, e0182997, DOI:10.1371/journal.pone.0182997.
- F. W. Lam, M. A. Cruz, H.-C. E. Leung, K. S. Parikh, C. W. Smith and R. E. Rumbaut, Thromb. Res., 2013, 132, 69–76, DOI:10.1016/j.thromres.2013.04.018.
- M. Anraku, V. T. G. Chuang, T. Maruyama and M. Otagiri, Biochim. Biophys. Acta, 2013, 1830, 5465–5472 CrossRef CAS PubMed.
- M. Roche, P. Rondeau, N. R. Singh, E. Tarnus and E. Bourdon, FEBS Lett., 2008, 582, 1783–1787, DOI:10.1016/j.febslet.2008.04.057.
- W. J. Zhang and B. Frei, Cardiovasc. Res., 2002, 55, 820–829, DOI:10.1016/S0008-6363(02)00492-3.
- D. Ravelli, P. Isernia, A. Acquarulo, A. Profumo and D. Merli, Anal. Chem., 2019, 91(15), 10110–10115, DOI:10.1021/acs.analchem.9b02088.
- X. Han, T. A. Snow, R. A. Kemper and G. W. Jepson, Chem. Res. Toxicol., 2003, 16(6), 775–781, DOI:10.1021/tx034005w.
- S. Beesoon and J. W. Martin, Environ. Sci. Technol., 2015, 49(9), 5722–5731, DOI:10.1021/es505399w.
- Y.-D. Yang, R. Tian and N. Lu, Sci. Total Environ., 2023, 876, 162738, DOI:10.1016/j.scitotenv.2023.162738.
- H. N. Bischel, L. A. MacManus-Spencer, C. Zhang and R. G. Luthy, Environ. Toxicol. Chem., 2011, 30, 2423–2430, DOI:10.1002/etc.647.
- H. N. Bischel, L. A. MacManus-Spencer and R. G. Luthy, Environ. Sci. Technol., 2010, 44, 5263–5269, DOI:10.1021/es101334s.
- X. Zhang, L. Chen, X.-C. Fei, Y.-S. Ma and H.-W. Gao, BMC Mol. Biol., 2009, 10, 16, DOI:10.1186/1471-2199-10-16.
- Y. Wang, H. Zhang, Y. Kang and J. Cao, J. Photochem. Photobiol., B, 2016, 159, 66–73, DOI:10.1016/j.jphotobiol.2016.03.024.
- H. Chen, P. He, H. Rao, F. Wang, H. Liu and J. Yao, Chemosphere, 2015, 129, 217–224, DOI:10.1016/j.chemosphere.2014.11.040.
- A. Rasheed, C. K. Ashok Kumar and V. V. N. S. Srivanthi, Sci. Pharm., 2008, 76, 567–598, DOI:10.3797/scipharm.0808-05.
- M. J. Weiss-Errico and K. E. O’Shea, J. Hazard. Mater., 2017, 329, 57–65, DOI:10.1016/j.jhazmat.2017.01.017.
- M. J. Weiss-Errico, J. Miksovska and K. E. O’Shea, Chem. Res. Toxicol., 2018, 31(4), 277–284, DOI:10.1021/acs.chemrestox.8b00002.
- L. D. Wilson and R. E. Verrall, Langmuir, 1998, 14, 4710–4717, DOI:10.1021/la9802365.
- A. H. Karoyo, A. S. Borisov, L. D. Wilson and P. Hazendonk, J. Phys. Chem. B, 2011, 115(31), 9511–9527, DOI:10.1021/jp110806k.
- M. J. Weiss-Errico, I. Ghiviriga and K. E. O’Shea, J. Phys. Chem. B, 2017, 121, 8359–8366, DOI:10.1021/acs.jpcb.7b05901.
- Y.-M. Chen and L.-H. Guo, Arch. Toxicol., 2009, 83(3), 255–261, DOI:10.1007/s00204-008-0359-x.
- L. A. MacManus-Spencer, M. L. Tse, P. C. Hebert, H. N. Bischel and R. G. Luthy, Anal. Chem., 2010, 82(3), 974–981, DOI:10.1021/ac902238u.
- P. Messina, G. Prieto, V. Dodero, J. M. Ruso, P. Schulz and F. Sarmiento, Biopolymers, 2005, 79, 300–309, DOI:10.1002/bip.20353.
- Q. Chi, Z. Li, J. Huang, J. Ma and X. Wang, Chemosphere, 2018, 198, 442–449, DOI:10.1016/j.chemosphere.2018.01.152.
- X. Liu, M. Fang, F. Xu and D. Chen, TrAC, Trends Anal. Chem., 2019, 116, 177–185, DOI:10.1016/j.trac.2019.05.017.
- H. B. Lamichhane and D. W. M. Arrigan, Curr. Opin. Electrochem., 2023, 40, 101309, DOI:10.1016/j.coelec.2023.101309.
- F. Reymond, D. Fermı, H. J. Lee and H. H. Girault, Electrochim. Acta, 2000, 45, 2647–2662 CrossRef CAS.
- S. Liu, Q. Li and Y. Shao, Chem. Soc. Rev., 2011, 40, 2236–2253, 10.1039/c0cs00168f.
- M. D. Scanlon and D. W. M. Arrigan, Electroanalysis, 2011, 23(4), 1023–1028, DOI:10.1002/elan.201000667.
- T. Kakutani, T. Ohkouchi, T. Osakai, T. Kakiuchi and M. Senda, Anal. Sci., 1985, 1(3), 219–225, DOI:10.2116/analsci.1.219.
- T. Osakai, T. Kakutani and M. Senda, Bunseki Kagaku, 1984, 33, E371–E377 CrossRef CAS.
- M. M. Hossain, S. H. Lee, H. H. Girault, V. Devaud and H. J. Lee, Electrochim. Acta, 2012, 82, 12–18, DOI:10.1016/j.electacta.2012.03.127.
- H. B. Lamichhane, T. G. Henares, M. J. Hackett and D. W. M. Arrigan, Anal. Chem., 2021, 93(26), 9094–9102, DOI:10.1021/acs.analchem.1c00657.
- M. B. Garada, B. Kabagambe, Y. Kim and S. Amemiya, Anal. Chem., 2014, 86, 11230–11237, DOI:10.1021/ac5027836.
- B. N. Viada, L. M. Yudi and D. W. M. Arrigan, Analyst, 2020, 145(17), 5776–5786, 10.1039/D0AN00884B.
- P. Jing, P. J. Rodgers and S. Amemiya, J. Am. Chem. Soc., 2009, 131(6), 2290–2296, DOI:10.1021/ja807961s.
- G. J. Islam and D. W. M. Arrigan, ACS Sens., 2022, 7(10), 2960–2967, DOI:10.1021/acssensors.2c01100.
- R. Iwasaki, J. Uchida, Y. Yamana, Y. Nakamura, K. Maeda, S. Sotoma and Y. Yoshida, Electrochemistry, 2023, 91, 087004, DOI:10.5796/electrochemistry.23-00066.
- H. B. Lamichhane and D. W. M. Arrigan, Sens. Diagn., 2023, 2, 938–947, 10.1039/D3SD00080J.
- E. Alvarez de Eulate, J. Strutwolf, Y. Liu, K. O’Donnell and D. W. M. Arrigan, Anal. Chem., 2016, 88, 2596–2604, DOI:10.1021/acs.analchem.5b03091.
- L. R. S. Barbosa, M. G. Ortore, F. Spinozzi, P. Mariani, S. Bernstorff and R. Itri, Biophys. J., 2010, 98(1), 147–157, DOI:10.1016/j.bpj.2009.09.056.
- C. J. Collins, C. Lyons, J. Strutwolf and D. W. M. Arrigan, Talanta, 2010, 80, 1993–1998, DOI:10.1016/j.talanta.2009.10.060.
- J. E. Norne, S. G. Hjalmarsson, B. Lindman and M. Zeppezauer, Biochemistry, 1975, 14, 3401–3408, DOI:10.1021/bi00686a017.
- G. Ionita and V. Sahini, J. Inclusion Phenom. Macrocyclic Chem., 2004, 50, 183–186, DOI:10.1007/s10847-004-8842-3.
- R. Li, Z. Wu, Y. Wang, L. Ding and Y. Wang, J. Biotechnol., 2016, 220, 33–34, DOI:10.1016/j.jbiotec.2016.01.007.
- W. Zhang, Z. Liu, Y. Zhou, C. Lai, B. Sun, M. He, Z. Zhai, J. Wang, Q. Wang, X. Wang, F. Wang and Y. Pan, Chemosphere, 2022, 291, 132945, DOI:10.1016/j.chemosphere.2021.132945.
- B. R. Horrocks and M. V. Mirkin, Anal. Chem., 1998, 70(22), 4653–4660, DOI:10.1021/ac980718l.
- P. Lopes and R. Kataky, Anal. Chem., 2012, 84(5), 2299–2304, DOI:10.1021/ac2029425.
- L. Maso, M. Trande, S. Liberi, G. Moro, E. Daems, S. Linciano, F. Sobott, S. Covaceuszach, A. Cassetta, S. Fasolato, L. M. Moretto, K. De Wael, L. Cendron and A. Angelini, Protein Sci., 2021, 30, 830–841, DOI:10.1002/pro.4036.
- Y. Wu, J. Bao, Y. Liu, X. Wang, X. Lu and K. Wang, Toxics, 2024, 12, 46, DOI:10.3390/toxics12010046.
- N. Alexander, L. McDonald, C. Wesdemiotis and Y. Pang, Analyst, 2024, 149, 1929–1938, 10.1039/D3AN02070C.
- H. Swenson and N. P. Stadie, Langmuir, 2019, 35, 5409–5426, DOI:10.1021/acs.langmuir.9b00154.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323, 10.1039/C0CS00062K.
- J. A. Ribeiro, C. M. Pereira and F. Silva, Electrochim. Acta, 2015, 180, 687–694, DOI:10.1016/j.electacta.2015.08.074.
- J. L. Alesio, A. Slitt and G. D. Bothun, Chemosphere, 2022, 287(P1), 131979, DOI:10.1016/j.chemosphere.2021.131979.
- F. Allendorf, U. Berger, K.-U. Goss and N. Ulrich, Environ. Sci.: Processes Impacts, 2019, 21(11), 1852–1863, 10.1039/C9EM00290A.
- L. L. Wu, H. W. Gao, N. Y. Gao, F. F. Chen and L. Chen, BMC Struct. Biol., 2009, 9, 31, DOI:10.1186/1472-6807-9-31.
- H. Xing, S.-S. Lin, P. Yan, J.-X. Xiao and Y.-M. Chen, J. Phys. Chem. B, 2007, 111(28), 8089–8095, DOI:10.1021/jp070198a.
- E. Saint-Aman and D. Serve, J. Colloid Interface Sci., 1990, 138, 365–375, DOI:10.1016/0021-9797(90)90219-E.
- W. Guo, B. M. Fung and S. D. Christian, Langmuir, 1992, 8(2), 446–451, DOI:10.1021/la00038a022.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*