
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLipoic acid-based vitrimer-like elastomer†
Xiaohong
Lan
,
Laura
Boetje
,
Théophile
Pelras
,
Chongnan
Ye
,
Fitrilia
Silvianti
 and
Katja
Loos
*
and
Katja
Loos
*
Macromolecular Chemistry & New Polymeric Materials, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747AG, Groningen, The Netherlands. E-mail: k.u.loos@rug.nl
First published on 20th October 2023
Abstract
Dynamic covalent networks (DCNs) are materials that feature reversible bond formation and breaking, allowing for self-healing and recyclability. To speed up the bond exchange, significant amounts of catalyst are used, which creates safety concerns. To tackle this issue, we report the synthesis of a lipoic acid-based vitrimer-like elastomer (LAVE) by combining (i) ring-opening polymerization (ROP) of lactones, (ii) lipoic acid modification of polylactones, and (iii) UV crosslinking. The melting temperature (Tm) of LAVE is below room temperature, which ensures the elastic properties of LAVE at service temperature. By carefully altering the network, it is possible to tune the Tm, as well as the mechanical strength and stretchability of the material. An increase in polylactone chain length in LAVE was found to increase strain at break from 25% to 180% and stress at break from 0.34 to 1.41 MPa. The material showed excellent network stability under cyclic strain loading, with no apparent hysteresis. The introduction of disulfide bonds allows the material to self-heal under UV exposure, extending its shelf life. Overall, this work presents an environmentally friendly approach for producing a sustainable elastomer that has potential for use in applications such as intelligent robots, smart wearable technology, and human–machine interfaces.
Introduction
Self-healing materials have gained popularity as an exciting and rapidly growing area of research due to their unique ability to repair themselves after being damaged.1 This property is achieved through the incorporation of healing agents or physical and/or chemical dynamic bonding within the material's structure.2,3 Dynamic covalent networks (DCNs), which combine the processing advantages of thermoplastics with permanent bonds of thermosets, have rapidly gained popularity and have been the subject of numerous studies. DCNs make use of dynamic covalent bonds, which can be broken and reformed at elevated temperatures or other stimuli, allowing for their dynamic behavior.4 In general, the bond exchange process of DCNs can proceed via two mechanisms. The first type of DCNs, also called vitrimers, simultaneously break and reform their bonds, resulting in a material that is highly stable.5 The other type follows a dissociative exchange mechanism, which means that the breaking and rebuilding of chemical bonds take place at different locations and times, resulting in a material that is highly adaptable; these materials are often referred to as nontraditional vitrimers. From a chemistry point of view, various dynamic bonds, including transesterification,6,7 transamination of vinylogous urethane,8 transalkylation of triazolium salts,9 olefin metathesis, dioxaborolane exchange, disulfide exchange, Diels–Alder reactions, and imine formation, have been featured in designing DCNs.10–14 In recent years, there has been intensive research to push the boundaries of polymer science and broaden the application of DCNs. For example, DCNs have been investigated for use in oil-spill remediation,15 fast healing strain sensors,16 sustainable 3D printing,17–19 and stimulus-responsive materials.20,21While DCNs have shown great promise in their ability to self-heal and be recycled, the use of a large amount of catalyst in their production is a major concern. The high level of catalyst required to boost the bond exchange reaction has negative environmental impacts, and the monomers used in their production are not sustainable, leading to questions about their long-term viability as a sustainable material choice.22 To address these issues, researchers have developed several catalyst-free networks to create DCNs, but these systems have their own limitations. For instance, a system developed by Zhang et al.23 utilized glycerol as an aid, which resulted in material degradation and reduced robustness, leading to a more thermoplastic material. Similarly, a transamination-type vitrimer developed by Du Prez et al.,8 used a vinylogous urethane moiety but the free amines in the network could pose concerns in long-term practical use due to oxidative damage and environmental issues. DCNs based on oxime–ester bonds also face similar limitations.7 These challenges underline the need for further research and development of sustainable and environmentally friendly methods of synthesizing DCNs with lower catalyst amounts and through more sustainable production processes.
Lipoic acid (LA), also known as thioctic acid, is a naturally occurring small molecule with a complex structure. It consists of a terminal carboxylic acid and a terminal dithiolane ring, which is a fully saturated, five-membered, sulfur heterocycle. This structure imparts distinctive chemical properties to LA that can undergo self-polymerization and depolymerization when exposed to heat, UV, or chemical stimuli.24–27 Due to this characteristic, LA has the potential to be used in the construction of DCNs in a sustainable manner. However, polymerized lipoic acid exhibits metastable properties because of an inverse ring-closing depolymerization process. To overcome the problem of this undesired metastability, grafting-through polymerization technology was used to incorporate lipoic acid into the network using a “phase-locked disulfide bond”.28,29 These bonds provide both elasticity and healing capability, with the soft segment ensuring elasticity and the hard backbone imparting healing capability.2 This approach allows for the mechanical properties and stability of the network to be tailored to meet specific requirements, making LA an attractive choice for the development of advanced materials with unique functionalities.
In this study, a straightforward method was designed to develop LA-based vitrimer-like elastomers (LAVE) by transforming a small molecule of biological origin into a high-performance elastomer with self-healing properties (Scheme 1). First, a diol primer was synthesized by ring-opening polymerization (ROP) of lactones, followed by esterification with LA, which ensures crosslinking and introduces DCNs. LA can undergo UV-induced ROP, allowing for crosslinking without the involvement of initiators or catalysts. Additionally, the presence of a polylactone network in the elastomer enhances its biodegradability in specific natural biological environments, thereby enhancing the overall sustainability of the materials.30 The resulting LAVE material exhibits high elasticity and low hysteresis and is able to self-heal under UV irradiation. The design flexibility of LAVE facilitates the tunability of mechanical properties, making it a potential candidate for sustainable elastomers used in soft robotics and electronics.
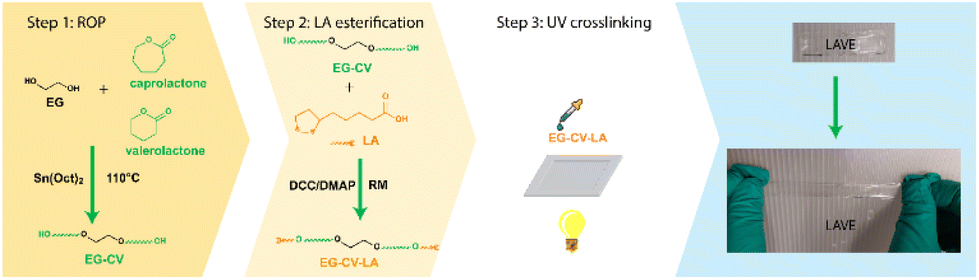 | ||
| Scheme 1 Schematic illustration of the LAVE preparation process. | ||
Results and discussion
LAVE was produced through a combination of (i) ROP of lactones, (ii) esterification of polylactones with LA, and (iii) UV crosslinking (see Scheme 1). As a first step, a stannous octoate (Sn(Oct)2)-catalyzed ROP of lactones using ethylene glycol (EG) as the initiator was performed. ε-CL and δ-VL were chosen because of their mutual interference with their crystallization profile and their ability to form copolymers with desirable properties, with a mole ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.31 We produced a series of diol primers (EG-CV-X, X = 10, 20, or 30) by varying the monomer to initiating site ratio (M/I, Table S1†) and achieved conversions as high as 98% after 240 min at 110 °C. To better understand the ROP, a kinetic study of EG-CV-10 was performed using 1H-NMR. Unlike most macroinitiators,30 EG does not impart any induction time during polymerization. With time, a decrease in the monomer fraction and an increase in the polymer fraction were observed. 1H-NMR analyses performed on EG-CV-X feature the characteristic signals of PCL and PVL at 4.03, 2.28, 1.61, and 1.35 ppm (see Fig. 1A and Fig. S1†). After ROP, the characteristic peak of EG at 3.63 ppm (f) shifted downfield to 4.27 ppm (j) due to the addition of the electron withdrawing polylactone chains. Furthermore, the peak at 3.60 ppm represents the terminal CH2, and was used to calculate the degree of polymerization of the polylactone chains (Fig. S1†).32 Since the polymerization was conducted in bulk, a minor amount of lactones was left in the system. Both catalyst Sn(Oct)2 (0.91 ppm) and monomer will not influence further modification since they are inactive at room temperature,33 and therefore, EG-CV-X was used without further purification.
:1.31 We produced a series of diol primers (EG-CV-X, X = 10, 20, or 30) by varying the monomer to initiating site ratio (M/I, Table S1†) and achieved conversions as high as 98% after 240 min at 110 °C. To better understand the ROP, a kinetic study of EG-CV-10 was performed using 1H-NMR. Unlike most macroinitiators,30 EG does not impart any induction time during polymerization. With time, a decrease in the monomer fraction and an increase in the polymer fraction were observed. 1H-NMR analyses performed on EG-CV-X feature the characteristic signals of PCL and PVL at 4.03, 2.28, 1.61, and 1.35 ppm (see Fig. 1A and Fig. S1†). After ROP, the characteristic peak of EG at 3.63 ppm (f) shifted downfield to 4.27 ppm (j) due to the addition of the electron withdrawing polylactone chains. Furthermore, the peak at 3.60 ppm represents the terminal CH2, and was used to calculate the degree of polymerization of the polylactone chains (Fig. S1†).32 Since the polymerization was conducted in bulk, a minor amount of lactones was left in the system. Both catalyst Sn(Oct)2 (0.91 ppm) and monomer will not influence further modification since they are inactive at room temperature,33 and therefore, EG-CV-X was used without further purification.
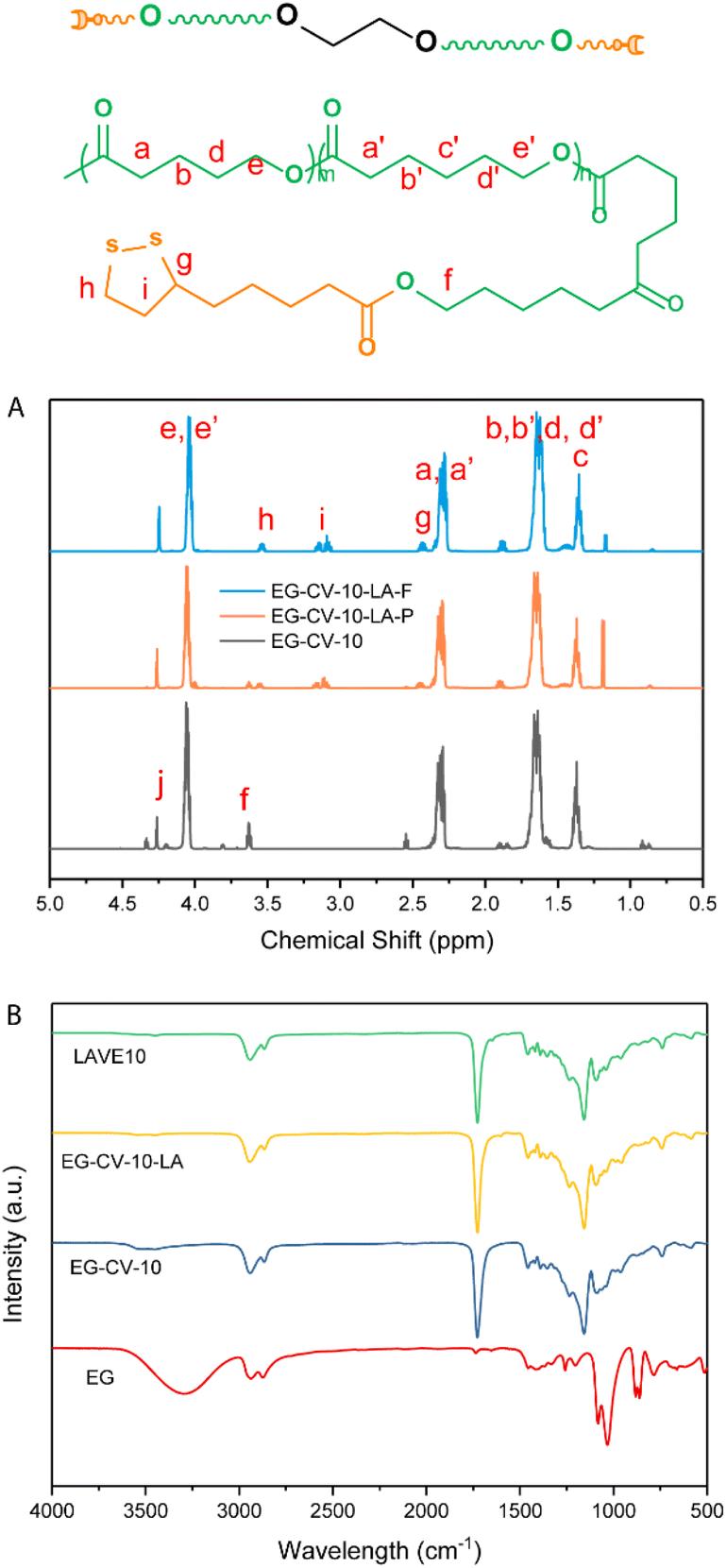 | ||
| Fig. 1 (A) 1H-NMR spectra of EG-CV-10, EG-CV-10-LA-F (full substituted), and EG-CV-10-LA-P (partly substituted); (B) FTIR of EG, EG-CV-X, EG-CV-X-LA, and LAVE. | ||
Size exclusion chromatography (SEC, Fig. 2A) confirmed the polymeric nature of EG-CV-X, displaying a single polymer peak for each sample. With increasing M/I ratios, a significant shift of the polymer peak toward lower retention times was observed, indicating an increase in the molecular weight of EG-CV-X, from 3100 Da to 6000 Da. However, the molecular weight distributions (Đ, Table S2†) of EG-CV-X were quite broad and increased from 2.22 to 2.83 with increasing M/I ratios. This is very common for ROP, likely due to side reactions such as transesterification, racemization, and the formation of macrocycles.34,35 The molecular weights of EG-CV-X agreed well with the calculated values from 1H-NMR spectroscopy and theoretical values based on the chemical composition of the system and increased linearly with the M/I ratios. Differential scanning calorimetry (DSC, Fig. 3A and Table S3†) analyses of EG-CV-X showed that all samples possess a melting temperature (Tm), which increased from 13 °C for EG-CV-10 to 18 °C for EG-CV-30, correlating to the increased length of the PCL-co-PVL. It is worth noting that the Tm of the pure PCL-co-PVL (1:1) is approximately 20 °C, higher than our systems, which may originate from EG hampering/limiting crystallization.31 With an increased M/I ratio, EG-CV-X is closer to pure PCL-co-PVL without much steric restriction; hence an increased Tm was observed. The cooling cycles show two crystallization peaks, in which the first is broader and the second is sharper, owing to the wide molecular weight distribution. With decreasing polylactone chain length in LAVE, the crystallization temperature (Tc) increases slightly from −5 to −1 °C. In addition to SEC and DSC, further characterization of EG-CV-X was conducted using Fourier transform infrared spectroscopy (FTIR) (Fig. 1B and Fig. S3†). The appearance of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1735 cm−1 alongside a decrease in the O–H stretch at 3300 cm−1 (from EG and terminal hydroxy) confirms the formation of the ester bond during the ROP of lactones with EG.
O stretch at 1735 cm−1 alongside a decrease in the O–H stretch at 3300 cm−1 (from EG and terminal hydroxy) confirms the formation of the ester bond during the ROP of lactones with EG.
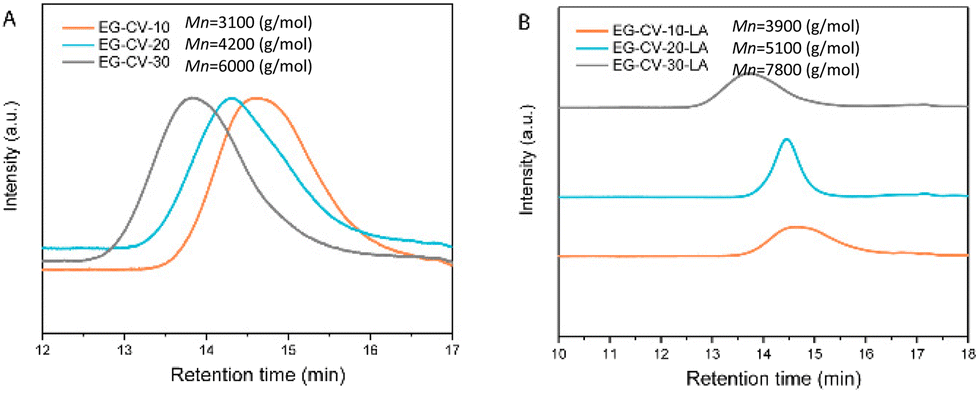 | ||
| Fig. 2 (A) SEC elugrams of EG-CV-X; (B) SEC elugrams of EG-CV-X-LA. | ||
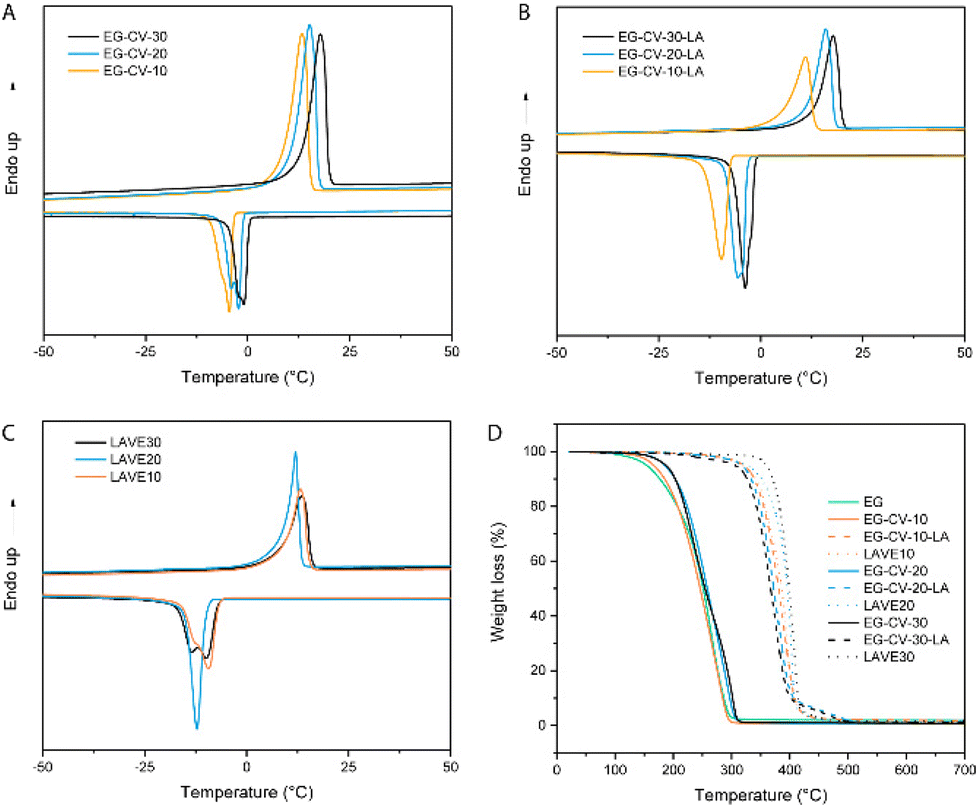 | ||
| Fig. 3 DSC analyses of the (A) EG-CV-X, (B) EG-CV-X-LA and (C) LAVE systems. (D) Comparative thermogravimetric analyses of EG-CV-X, EG-CV-X-LA, and LAVE. | ||
In the second step, the remaining hydroxy groups of EG-CV-X were used to react with the carboxylic group from LA. Since LA has a heat-sensitive five-membered cyclic disulfide ring, Steglich esterification was performed using dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) to avoid using a high reaction temperature. In this reaction, DCC acts as a dehydrating agent and hydrates to form dicyclohexylurea (DCU), a compound nearly insoluble in most organic solvents and water. It is worth noting that the hydroxy groups were both fully and partially substituted. Fig. 1A shows a comparison of the partial and full substitution of EG-CV-10 (EG-CV-10-LA-P and EG-CV-10-LA-F), with some hydroxy left in the partly substituted polymers (peaks at 3.60 ppm representing –CH2–OH). The signals at 3.55 and 3.15 ppm confirmed the successful esterification and enabled the determination of the degree of substitution (DS) of EG-CV-10-P (DS = 1.4). The gel fraction showed that partial substitution (70%) of the hydroxy group could result in a stable crosslinked network (Fig. S4†). Therefore, for all EG-CV-X-LA samples, we used 0.7 equiv. of the carboxylic group per hydroxy groups to ensure complete crosslinking, while preserving sufficient dangling polylactone chains.30 An appropriate proportion of dangling chains can significantly reduce the material's modulus by preventing the formation of entanglements,36 increasing the flowability of the network. The resulting EG-CV-X-LA are viscous yellowish liquids, and their structure is depicted in Scheme 1. Since LA only represents a small portion of EG-CV-X-LA, no visible changes were observed in FTIR (Fig. 1B and Fig. S3†). The post esterification marginally increases the molecular weight of the polymers, which is visible in SEC elugrams (Fig. 2B and Table S2†). The molecular weight from SEC was well correlated with that derived from the theoretical (Mw (theo)) and 1H-NMR calculation (Mw (NMR)). To our surprise, the polydispersity of EG-CV-X-LA was greatly improved compared to that of EG-CV-X, probably due to the washing steps after the Steglich esterification. A decreased Tc was observed after esterification, likely due to the introduction of LA, which inhibited PCL-co-PVL crystallization. With increased M/I ratios, a decreased Tc was also observed. However, the changes in Tm do not follow any visible rules.
In the third step, EG-CV-X-LA was crosslinked by a UV lamp at a wavelength of 365 nm, without an initiator and premixing step, resulting in the production of LAVEX (X = 10, 20 or 30, e.g., LAVE10 refers to the elastomer made of EG-CV-10-LA). After UV irradiation, the slightly yellow, oily mixture EG-CV-X-LA turned into a transparent film LAVEX displaying a rubbery behavior with no visual sign of flowing. Even though LA itself is able to self-polymerize above its Tm,37 EG-CV-X-LA is unable to crosslink when heated in our investigation. Gel fraction measurements (Fig. S4†) were performed to monitor the potential leaching of noncrosslinked material. The higher the gel fraction, the higher the crosslinking density, indicating a more stable polymer network. In this study, the uncured EG-CV-X-LA was fully soluble in acetone; therefore, the gel fraction was zero. However, the cured films of EG-CV-X-LA showed excellent crosslinking and stability, with gel fractions ranging from 85% to 95% depending on the M/I ratios. The marginal increase in gel fraction with increasing M/I ratios can be attributed to the increased flexibility of primers, which facilitates crosslinking. This is consistent with the curing efficiency of EG-CV-X-LA. In contrast to the system described by Choi et al.,28 in which the gel fraction shows a decreasing trend as the molecular weight of the primer increases. Our system, EG-CV-30-LA was fully cured after 20 min, while EG-CV-20-LA and EG-CV-10-LA needed 1 hour to be fully cured. This was probably due to the limited flexibility of EG-CV-10-LA and EG-CV-20-LA, which leads to slow polymerization and a lower gel fraction. Nonetheless, LAVE10 still possessed a high gel fraction of 85%, indicating that most EG-CV-X-LA was linked into the network. However, LAVE remains more stable in a solvent environment than commercially available EcoFlex (gel fraction of 60%).38
To further confirm proper network integrity, a frequency sweep in the linear viscoelastic regime (Fig. 4A and Fig. S5†) was performed to monitor the viscoelasticity of the materials before and after crosslinking. All LAVE samples showed a central plateau in G′ and a multidecade separation between G′ and G′′, convincingly resembling rubber. On the other hand, the modulus of uncured DE-CV-X-LA vastly increased with increasing frequency, further confirming successful curing. Next, we investigated the thermal properties of LAVE. Thermogravimetric analysis (TGA, Fig. 3D) showed a lower degradation profile for EG than for its derivatives. After ROP and esterification, the thermal stability of the primer significantly improves. Once crosslinked, the thermal stability reaches its maximum. LAVE starts to degrade at ∼300 °C with a maximal burn rate at ∼400 °C (Table S3†), which is beyond the requirements for most applications, such as wearables, grips, and biomedical devices.39 There is little difference between LAVE produced using different PCL-co-PVL chain lengths. DSC (Fig. 3C) analyses of the elastomers showed that all LAVEs possess a Tm below room temperature. However, no Tg was observed within the temperature range of −75 to 150 °C.
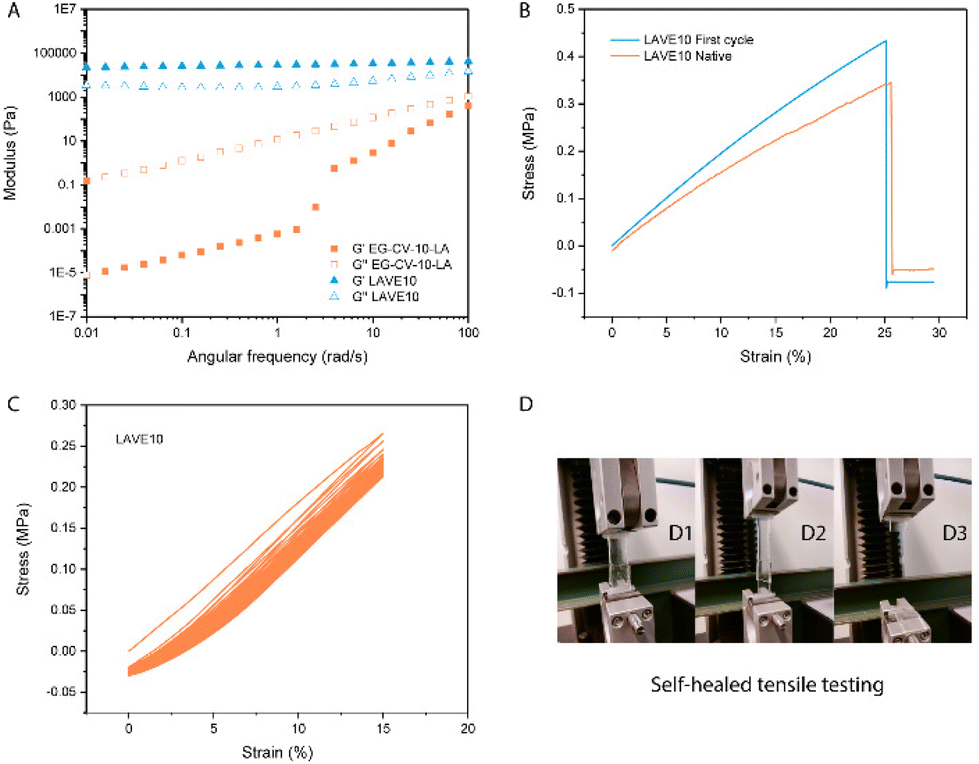 | ||
| Fig. 4 (A) Oscillatory rheology measurements on EG-CV-10-LA and LAVE10; (B) tensile stress–strain curves of native and first cycle healed LAVE10; (C) cycling test of LAVE10; (D) photograph of healed LAVE30 during tensile testing. | ||
The results of the tensile tests conducted on the various LAVEs revealed interesting insights into the mechanical properties of these elastomers. As shown in Fig. 4B and Fig. S6,† the LAVEs exhibited characteristic behavior of elastomeric materials, with a high degree of strain at break. With an increase in the number of polylactone chains in LAVE, the strain at break increased from 25% to 180%. At the same time, the stress at break increased from 0.34 to 1.41 MPa (Table S4†). It is worth noting that the stress–strain curve follows a strict linear increase in tensile stress with increasing strain, which is also a typical behavior of DCNs, without the involvement of physical bonding and entanglements.24 The exceptional stretchability and mechanical strength of LAVE can be attributed to two mechanisms: (1) dangling polylactone chains and (2) the two-phase network having disulfide as the crosslinks. To verify their reparability, the elastomers were cut into two pieces, joined back together and self-healed under UV irradiation. The ability of the LAVEs to self-heal under UV irradiation was further confirmed through force loading–unloading (Fig. 4C and Fig. S7†). The repaired samples showed no significant change in their mechanical properties, even after 50 cycles of repeated stretching and relaxation. This contrasts with thermoplastic elastomers, which exhibit severe hysteresis in stretching–relaxation cycles, resulting in mechanical loss under cyclic loading.40 The excellent mechanical stability of LAVEs after self-healing makes them ideal candidates for applications in which sustained mechanical performance is critical, such as in soft robotics or wearable electronics. Additionally, the adhesive properties of the primer used in our study contribute to the potential of the final products to function as an excellent bioglue.41 To conclude, LAVA demonstrated excellent stretchability and self-healing properties, making it an idea candidate for sustainable and durable materials. Our materials have advantages over 100% natural polymers such as gelatin and cellulose, due to their mechanical tunability. Meanwhile, the primer (EG-CV-X-LA) can be crosslinked and self-healed via disulfide exchange without the addition of initiator or catalyst.
Conclusions
This study showed the straightforward synthesis of a novel LA-based vitrimer-like elastomer with excellent stretchability and self-healing properties. The attributes of LAVE, such as molecular weight and mechanical and thermal properties, can be simply adjusted by varying the M/I ratios, allowing for material customization. Without premixing or the inclusion of any initiator or catalyst, LAVE films can be straightforwardly prepared through UV irradiation, making it an environmentally benign elastomer. As a result of its elastic, adjustable, and sustainable properties, LAVE has the potential to revolutionize the field of self-healing materials and has tremendous potential in a wide range of applications, such as sustainable wearables, human–machine interfaces, and food-contact materials. Further research in this field will focus on synthesizing a variety of LA-based primers with the possibility of application in 3D printing electronics. Hoping to address the challenges of sustainability in material science and pave the way for the development of a new generation of materials.Experimental
Materials
Ethylene glycol (EG, anhydrous, 99.8%), ε-caprolactone (ε-CL, 97%), δ-valerolactone (δ-VL, technical grade), stannous octoate (Sn(Oct)2, 92.5–100%), (±)-α-lipoic acid (LA, ≥98.0%), N,N′-dicyclohexylcarbodiimide (DCC, 99%), and 4-(dimethylamino)pyridine (DMAP, ≥99%) were purchased from Sigma-Aldrich. All reagents were used as received, except [ε-CL, δ-VL, and Sn(Oct)2], which were dried over MgSO4 for 30 min and filtered before use. Deuterated chloroform (CDCl3) was purchased from Sigma-Aldrich. All other solvents were from Avantor and were of analytical grade.Instrumentation
1H-NMR spectra were recorded at 25 °C on a Bruker Avance NMR spectrometer operating at 600 MHz. The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. Size exclusion chromatography (SEC) was performed on a GPCMax system from Viscotek equipped with a triple detector, consisting of a Malvern Dual detector and a Schambeck RI2012 refractive index detector, as well as two PLgel (both 5 μm 30 cm) from Agilent Technologies at 35 °C. The columns and detectors were maintained at a temperature of 35 °C. FTIR spectra were collected on a Bruker Vertex 70 infrared spectrometer using a diamond with an attenuated total reflectance probe. Differential scanning calorimetry (DSC) thermograms were recorded on a TA instruments Q1000 under nitrogen at heating and cooling rates of 10 °C min−1. A TA-instrumental 5500 under a nitrogen atmosphere was used for the thermogravimetric analyses (TGA) operating at 10 °C min−1. Uniaxial tensile and cyclic strain tests were performed on an Instron 5565 with a force of 100 N. The frequency sweep experiments were carried out on an Anton-Paar Physica MCR 302e rheometer with a plate–plate geometry of 10 mm using a strain of 1%.Synthesis of ethylene glycol-initiated polymers (EG-CV-X)
To a 20 ml vial equipped with a stir bar, EG (0.23 g, 7.5 mmol hydroxy group) as the initiator was introduced first and a mixture of ε-CL and δ-VL as monomers (with ε-CL: δ-VL = 1:1) as well as dried Sn(Oct)2 as the catalyst were added later. The mixture was stirred at room temperature for 10 min to achieve complete dissolution. The mixture was then immersed in a preheated oil bath at 110 °C. The conversion of monomer was evaluated by 1H-NMR by removing 0.1 ml aliquots from the system at appropriate time intervals. Once the desired conversion was achieved, the vessel was removed from the oil bath and cooled over an ice bath. The viscous diol-primer EG-CV-X were used afterward.
Yield: 99%. 1H-NMR (600 MHz, CDCL3, δ) 4.27 (t, 0.5n × 2H; –OC–CH2–CH2–CH2–CH2–O–), 4.27 (m, 2H; –O–CH2–CH2–O–), 4.16 (t, 0.5n × 2H; –OC–CH2–CH2–CH2–CH2–CH2–CH2–O–), 4.03 (m, n × 2H; nCH2), 3.60 (m, 2H; –CH2–OH), 2.57 (t, 0.5n × 2H; –O–CH2–CH2–CH2–CH2–CH2–CO–), 2.48 (t, 0.5n × 2H; –O–CH2–CH2–CH2–CH2–CO–), 2.28 (m, n × 2H; nCH2), 1.83–1.79 (m, 2n × 2H; –OC–CH2–CH2–CH2–CH2–CH2–O– or –OC–CH2–CH2–CH2–CH2–O–), 1.69 (m, 0.5n × 2H; –OC–CH2–CH2–CH2–CH2–CH2–O–), 1.61 (m, 2n × 2H; 2nCH2), 1.35 (m, 0.5n × 2H; 0.5nCH2). n denotes the molecular ratio of lactone monomers to hydroxy groups.
Synthesis of lipoic acid esterified EG-CV-X (EG-CV-X-LA)
The modification process was conducted using an esterification method according to a previously described protocol.41 EG-CV-X (1.0 eq.) and DMAP (1.4 eq.) were first dissolved in 20 ml of DCM. After complete dissolution, a mixture of α-LA (1.4 eq., 0.29 g) and DCC (1.4 eq., 0.29 g) in 10 ml of DCM was added followed by stirring for 24 h. The formed byproduct was filtered out, and the supernatant was precipitated in cold methanol. The primer was redissolved in DCM and precipitated in methanol three more times. Finally, the solution in DCM was precipitated in petroleum ether. After descanting and evaporating petroleum ether, a yellow oil-like material was obtained.Yield: 86%. 1H-NMR (600 MHz, CDCL3, δ) 4.27 (m, 2H; –O–CH2–CH2–O–), 4.03 (m, n × 2H; nCH2), 3.60 (m, 2H; –CH2–OH), 3.55 (m, H; –S–CH–CH2–CH2–S–), 3.18–3.07 (m, 2H; –S–CH–CH2–CH2–S–), 2.45 (m, 0.5 × 2H; –S–CH–CH2–CH2–S–), 2.28 (m, n × 2H; nCH2), 1.90 (m, 0.5 × 2H; –S–CH–CH2–CH2–S–), 1.61 (m, 2n × 2H; 2nCH2), 1.35 (m, 0.5n × 2H; 0.5nCH2). n denotes the molecular ratio of lactone monomers to hydroxy groups.
UV-mediated synthesis and self-healing of LAVE
LAVE were formed using solvent-casting and UV-curing techniques. For example, to produce LAVE30, EG-CV-30-LA (4.00 g) was dissolved in DCM and vigorously stirred for several minutes to create a homogeneous solution. The solution was then casted into a Teflon mold and exposed to UV light (365 nm, 8 Watt, UVP 3UV™ Lamp) for 20 min. During UV exposure, the yellow solution transformed into a transparent solid. To test the self-healing capabilities of the films, films were peeled off from the Teflon mode and cut into two pieces. Then, damaged samples were partly stacked on top of one another, sandwiched between two glass slides, and allowed to heal under UV irradiation for an hour.Gel fraction measurement
The gel fraction of LAVE was analyzed by swelling experiments in acetone for 24 h at ambient temperature, with another 24 h for solvent evaporation. This process was repeated twice to verify that all linear polymer was removed. The gel fraction was calculated by dividing the dry weight following extraction ma by that of the original sample mb, as shown in eqn (1):| G = ma/mb × 100 | (1) |
Author contributions
The manuscript was written through the contributions of all authors. All authors have given their approval to the final version of the manuscript. Conceptualization, X. L., and L. B.; methodology, X. L., L. B., and T. P.; investigation, X. L.; writing – original draft, X. L.; writing – review & editing, X. L., L. B., T. P., C. Y., F. S., and K. L.; funding acquisition, K. L.; resources, K. L.; supervision, K. L.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge the financial support from IVA 7/1d ZIM Postdoc (101617) and ZIM 201340 CICE (201340). This research received funding from the Dutch Research Council (NWO) in the framework of the ENW PPP Fund for the top sectors and from the Ministry of Economic Affairs in the framework of the ‘PPS-Toeslagregeling’. We are also deeply grateful to Jur van Dijken for his support on the thermal and mechanical analysis and Albert J. J. Woortman for his help with SEC analysis.References
- D. Jayabalakrishnan, D. N. Muruga, K. Bhaskar, P. Pavan, K. Balaji, P. Rajakumar, C. Priya, R. Deepa, S. Sendilvelan and M. Prabhahar, Mater. Today: Proc., 2021, 45, 7195–7199 CAS.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- M. Röttger, T. Domenech, R. van Der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS.
- X. Feng and G. Li, Chem. Eng. J., 2021, 417, 129132 CrossRef CAS.
- C. He, S. Shi, D. Wang, B. A. Helms and T. P. Russell, J. Am. Chem. Soc, 2019, 141, 13753–13757 Search PubMed.
- S. Engelen, A. A. Wróblewska, K. De Bruycker, R. Aksakal, V. Ladmiral, S. Caillol and F. E. Du Prez, Polym. Chem., 2022, 13, 2665–2673 RSC.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078–6083 CrossRef CAS PubMed.
- N. Boehnke, C. Cam, E. Bat, T. Segura and H. D. Maynard, Biomacromolecules, 2015, 16, 2101–2108 CrossRef CAS PubMed.
- P. Kovaricek and J.-M. Lehn, J. Am. Chem. Soc., 2012, 134, 9446–9455 CrossRef CAS PubMed.
- G. Zhang, W. Peng, J. Wu, Q. Zhao and T. Xie, Nat. Commun., 2018, 9, 4002 CrossRef PubMed.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- Y.-X. Lu and Z. Guan, J. Am. Chem. Soc., 2012, 134, 14226–14231 CrossRef CAS PubMed.
- C. Ye, V. S. Voet, R. Folkersma and K. Loos, Adv. Mater., 2021, 33, 2008460 CrossRef CAS PubMed.
- C. Ye, F. Yan, X. Lan, P. Rudolf, V. S. Voet, R. Folkersma and K. Loos, Appl. Mater. Today, 2022, 29, 101683 CrossRef.
- Q. Shi, K. Yu, X. Kuang, X. Mu, C. K. Dunn, M. L. Dunn, T. Wang and H. J. Qi, Mater. Horiz., 2017, 598–607 RSC.
- E. Rossegger, R. Höller, D. Reisinger, M. Fleisch, J. Strasser, V. Wieser, T. Griesser and S. Schlögl, Polymer, 2021, 221, 123631 CrossRef CAS.
- B. Zhang, K. Kowsari, A. Serjouei, M. L. Dunn and Q. Ge, Nat. Commun., 2018, 9, 1831 CrossRef PubMed.
- Y. Yang, Z. Pei, Z. Li, Y. Wei and Y. Ji, J. Am. Chem. Soc., 2016, 138, 2118–2121 CrossRef CAS PubMed.
- Z. Pei, Y. Yang, Q. Chen, E. M. Terentjev, Y. Wei and Y. Ji, Nat. Mater., 2014, 13, 36–41 CrossRef CAS.
- Y. Liu, Z. Tang, S. Wu and B. Guo, ACS Macro Lett., 2019, 8, 193–199 CrossRef CAS PubMed.
- T. Liu, S. Zhang, C. Hao, C. Verdi, W. Liu, H. Liu and J. Zhang, Macromol. Rapid Commun., 2019, 40, 1800889 CrossRef PubMed.
- Q. Zhang, C.-Y. Shi, D.-H. Qu, Y.-T. Long, B. L. Feringa and H. Tian, Sci. Adv., 2018, 4, eaat8192 CrossRef CAS PubMed.
- C. Dang, M. Wang, J. Yu, Y. Chen, S. Zhou, X. Feng, D. Liu and H. Qi, Adv. Funct. Mater., 2019, 29, 1902467 CrossRef.
- G. M. Scheutz, J. L. Rowell, S. T. Ellison, J. B. Garrison, T. E. Angelini and B. S. Sumerlin, Macromolecules, 2020, 53, 4038–4046 CrossRef CAS.
- L. Song, B. Zhang, G. Gao, C. Xiao and G. Li, Eur. Polym. J., 2019, 115, 346–355 CrossRef CAS.
- C. Choi, J. L. Self, Y. Okayama, A. E. Levi, M. Gerst, J. C. Speros, C. J. Hawker, J. Read de Alaniz and C. M. Bates, J. Am. Chem. Soc., 2021, 143, 9866–9871 CrossRef CAS PubMed.
- C. Choi, Y. Okayama, P. T. Morris, L. L. Robinson, M. Gerst, J. C. Speros, C. J. Hawker, J. Read de Alaniz and C. M. Bates, Adv. Funct. Mater., 2022, 32, 2200883 CrossRef CAS.
- X. Lan, W. Li, C. Ye, L. Boetje, T. Pelras, F. Silvianti, Q. Chen, Y. Pei and K. Loos, ACS Appl. Mater. Interfaces, 2022, 15, 4398–4407 CrossRef PubMed.
- T. Wu, Z. Wei, Y. Ren, Y. Yu, X. Leng and Y. Li, Polym. Degrad. Stab., 2018, 155, 173–182 CrossRef CAS.
- M. Müllner, T. Lunkenbein, M. Schieder, A. H. Gröschel, N. Miyajima, M. Förtsch, J. Breu, F. Caruso and A. H. E. Müller, Macromolecules, 2012, 45, 6981–6988 CrossRef.
- M. C. D'Alterio, I. D'Auria, L. Gaeta, C. Tedesco, S. Brenna and C. Pellecchia, Macromolecules, 2022, 55, 5115–5122 CrossRef.
- S. Kim, K. I. Wittek and Y. Lee, Chem. Sci., 2020, 11, 4882–4886 RSC.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed.
- L.-H. Cai, T. E. Kodger, R. E. Guerra, A. F. Pegoraro, M. Rubinstein and D. A. Weitz, Adv. Mater., 2015, 27, 5132–5140 CrossRef CAS PubMed.
- A. Kisanuki, Y. Kimpara, Y. Oikado, N. Kado, M. Matsumoto and K. Endo, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5247–5253 CrossRef CAS.
- J. Vaicekauskaite, P. Mazurek, S. Vudayagiri and A. L. Skov, J. Mater. Chem. C, 2020, 8, 1273–1279 RSC.
- J. Mofokeng and A. Luyt, Polym. Test., 2015, 45, 93–100 CrossRef CAS.
- D. Gu, S. Tan, C. Xu, A. J. O'Connor and G. G. Qiao, Chem. Commun., 2017, 53, 6756–6759 RSC.
- C. Liu, R. Lu, M. Jia, X. Xiao, Y. Chen, P. Li and S. Zhang, Chem. Matter., 2023, 35, 2588–2599 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00883e |
| This journal is © The Royal Society of Chemistry 2023 |